
In situ transmission electron microscopy study of the electrochemical sodiation process for a single CuO nanowire electrode†
Abstract
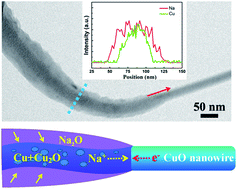
In this work, an in situ transmission electron microscopy (TEM) study of the electrochemically driven sodiation and desodiation of a CuO nanowire (NW) was performed. Upon sodiation, Na ions first reacted with CuO, yielding a mixture of Cu, Cu2O, and Na2O, and then some Cu2O was subsequently reduced into nanocrystal Cu. The final sodiation product was nanocrystalline Cu mixed with Na2O and Cu2O. Upon extraction of Na+, the nanocrystalline Cu first oxidized into Cu2O and finally transformed back to nanocrystalline CuO. The volume of the CuO NW was found to expand markedly during the first sodiation, but the morphology of the NW remained unchanged during the following cycles. The mechanism by which high-performance CuO NW anodes are used for rechargeable Na ion batteries was analyzed by carrying out an in situ TEM technique based on our results, and this study may be beneficial for designing the optimal structure of the CuO anode, improving its cycle performance, and making CuO more feasible for Na ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

