
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sterically demanding pyridine-quinoline anchoring ligands as building blocks for copper(I)-based dye-sensitized solar cell (DSSC) complexes†
Anastasios
Peppas
a,
Demetrios
Sokalis
a,
Dorothea
Perganti
b,
Gregor
Schnakenburg
c,
Polycarpos
Falaras
 *b and
Athanassios I.
Philippopoulos
*a
*b and
Athanassios I.
Philippopoulos
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis Zografou 15771, Athens, Greece. E-mail: p.falaras@inn.demokritos.gr; Tel: +302107274697
bInstitute of Nanoscience and Nanotechnology, National Center for Scientific Research “Demokritos”, 15341 Agia Paraskevi, Attica, Greece. E-mail: atphilip@chem.uoa.gr
cInstitut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Straße 1, D-53121 Bonn, Germany
First published on 29th August 2022
Abstract
The Pfitzinger condensation reaction was employed to synthesise N^N sterically demanding ligands bearing carboxylic acid anchoring groups, namely 2,2′-pyridyl-quinoline-4-carboxylic acid (pqca), 6′-methyl-2,2′-pyridyl-quinoline-4-carboxylic acid (6′-Mepqca), 8-methyl-2,2′-pyridyl-quinoline-4-carboxylic acid (8-Mepqca) and 8,6′-dimethyl-2,2′-pyridyl-quinoline-4-carboxylic acid (8,6′-Me2pqca). Preparation of the methyl ester analogues 6′-Mepqcame, 8-Mepqcame and 8,6′-Me2pqcame is also described. All ligands were fully characterised including the X-ray structures of pqca, 6′-Mepqca and 8-Mepqca. We also describe the synthesis and characterisation of seven homoleptic copper(I) complexes of the formula [Cu(N^N)2][PF6] (N^N = pqca (1), 6′-Mepqca (2), 8-Mepqca (3), 8,6′-Me2pqca (4), 6′-Mepqcame (6), 8-Mepqcame (7) and 8,6′-Me2pqcame (8)). Characterisation of the copper(I) complexes includes FT-IR, elemental analyses, multinuclear NMR spectroscopy, UV-vis spectroscopy, cyclic voltammetry, and a single-crystal X-ray diffraction study. The molecular structures of 1·DMSO, 2{2·Me2CO·0.5H2O}, 4, 6·CHCl3·0.13H2O, 2{7·C5H12}·CHCl3 and 8 have been determined, revealing that these complexes adopt a distorted tetrahedral geometry. These are the first crystallographically characterised examples of copper(I)-based coordination compounds incorporating the above mentioned N^N pyridyl-quinoline ligands. In solution, the new complexes are purple to red colored, while 2 displayed excellent stability in acetone at ambient temperature over a month. The absorption spectra of 1–8 display a main broad MLCT band with values of λmax at ∼530 nm and ε values ranging from 1800 to approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dm3 mol−1 cm−1. The photovoltaic performance of the prepared compounds was evaluated on mesoporous nanocrystalline TiO2 dye-sensitized solar cells (DSSCs), and compared with that of the [Cu(dmdcbpy)2][PF6] dye (dmdcbpy = 6,6′-dimethyl-2,2′-bipyridine-4,4′-dicarboxylic acid) (5), that has been used as standard, under the same experimental conditions. From a combination of electrochemical and absorption spectroscopy experiments, the MLCT energy levels of 2 are appropriate for electron injection onto the titania conduction band. Upon optimisation of the semiconductor's architecture, 2 proved to be the most efficient dye, reaching a conversion efficiency of η = 1.20%, which is slightly higher than that of 5 (η = 1.05%), mainly attributed to higher Voc values.
000 dm3 mol−1 cm−1. The photovoltaic performance of the prepared compounds was evaluated on mesoporous nanocrystalline TiO2 dye-sensitized solar cells (DSSCs), and compared with that of the [Cu(dmdcbpy)2][PF6] dye (dmdcbpy = 6,6′-dimethyl-2,2′-bipyridine-4,4′-dicarboxylic acid) (5), that has been used as standard, under the same experimental conditions. From a combination of electrochemical and absorption spectroscopy experiments, the MLCT energy levels of 2 are appropriate for electron injection onto the titania conduction band. Upon optimisation of the semiconductor's architecture, 2 proved to be the most efficient dye, reaching a conversion efficiency of η = 1.20%, which is slightly higher than that of 5 (η = 1.05%), mainly attributed to higher Voc values.
Introduction
In the field of solar cells, pioneering studies by Grätzel and co-workers trace back to the early 1990s, leading to a landmark discovery with a power conversion efficiency of solar energy to electricity of approximately 10%.1,2 This is a field of continuous interest, rendering dye sensitized solar cells (DSSCs) a realistic technology for harnessing the sun energy with power conversion efficiencies exceeding 12%.3,4 To this end, a 14.3% efficiency has been recorded at the lab-scale,5 denoting their potency as feasible alternatives to standard silicon-based solar cells.In short, DSSCs consist of an n-type semiconductor electrode such as TiO2 that is sensitized by dye molecules, with the classical I−/I3− redox mediator dissolved in a high boiling point nitrile or other solvent6 along with the counter electrode, consisting of Pt nanoparticles.7 The sensitization mechanism and the principles of cell operation have been well-established. At the heart of this device, an organic–inorganic dye, mainly a ruthenium(II) polypyridyl complex, is chemisorbed on the titania film.8 Among the plethora of ruthenium dyes,9,10 N719 (cis-[Ru(dcbpyH2)2(NCS)2][NBu4]2, dcbpyH2 = 2,2′-pyridine-4,4′-dicarboxylic acid) and Z907 constitute the state-of-the-art standard prototypes to comparatively evaluate the photoelectrochemical characteristics of a new molecular sensitizer.11
During the last decade, copper(I) complexes have emerged as alternatives to the standard ruthenium(II) dyes.12 The low cost and high natural abundance of copper as compared with expensive ruthenium13 constitute the basic reasons for this choice. In addition copper(I)-polypyridyl complexes display similar photochemical and photophysical properties14 in comparison with ruthenium(II) congeners. The potential use of copper(I) with a carboxylate substituted 1,10-phenanthroline ligand in DSSCs has previously been reported by Sauvage and co-workers.15 A substantial contribution to this topic has arisen from the seminal work of Housecroft, Constable and co-workers, with solar conversion efficiencies exceeding 2%.13,16 In particular, for the synthesis of heteroleptic bis(diimine) copper(I) dyes this group has developed the ‘surfaces-as-ligands, surfaces-as-complexes’ approach,17–19 while Odobel et al., following the HETPHEN approach, have isolated a number of [CuLL′]+ dyes (L = anchoring, L′ = ancillary ligand),20 with increased conversion efficiencies.21 The progress of copper(I) complexes in light harvesting/energy conversion applications is highlighted in a recent review article.22
Given the continuous interest in copper(I) polypyridyl complexes that could be potentially applied in the field of DSSCs, we set up to synthesize new homoleptic copper(I) complexes incorporating appropriate sterically demanding anchoring ligands, to prevent geometric changes upon the oxidation of copper(I) to copper(II), which take place during the redox process.12,23 The latter one is a prerequisite towards the design of copper(I)-based dyes. Following this strategy, we wanted to investigate the effect of varying the steric demands at the 6′ and 8-substituents within the class of 4-carboxy-2-(pyridin-2-yl)quinoline (pqca), on the performance of copper(I)-based DSSC devices. Well-described research on the ruthenium(II) coordination chemistry of pqca and its methyl ester analogue (pqcame) has provided us with the required knowledge to perform the current research.24–26 Methylation at the 6′ and 8 positions of the anchoring ligand may act as a protecting scaffold for the copper(I) metal center. In this respect, stereochemical protection can be potentially offered by the benzo ring of the quinoline moiety, as was reported for [Cu(RDABdipp)(LCOOMe)][PF6] (RDABdipp = 1,4-diaza-1,3-butadienes, R = H, Me; dipp = 2,6-diisopropylphenyl; LCOOMe = methyl-2-(pyridin-2-yl)-quinoline-4-carboxylate) complexes,27 and for the case of a number of ruthenium(II) complexes.15,28,29
Accordingly, simple organic diimine ligand precursors, analogues of pqca, substituted with methyl at the 6′ (6′-Mepqca), the 8 (8-Mepqca), and both the 8 and 6′ positions (8,6′-Me2pqca) of the pyridyl ring and the quinoline moiety, constitute the basis of this work. The synthesis of the methyl ester analogues 6′-Mepqcame, 8-Mepqcame and 8,6′-Me2pqcame, where the H atom of the COOH group was replaced by Me, is also reported. Herein, we focus on the preparation and characterisation of seven new homoleptic copper(I) dyes 1–4 and 6–8 of the type [Cu(N^N)2][PF6], where N^N is one of the diimines reported above (N^N = pqca (1), 6′-Mepqca (2), 8-Mepqca (3), 8,6′-Me2pqca (4), 6′-Mepqcame (6), 8-Mepqcame (7) and 8,6′-Me2pqcame (8)), aiming to test them as molecular sensitizers for dye-sensitized solar cells. The [Cu(dmdcbpy)2][PF6] dye (5), where dmdcbpy stands for 6,6′-dimethyl-2,2′-bipyridine-4,4′-dicarboxylic acid, was prepared independently in a one-pot reaction, serving as a copper(I) DSSC prototype for comparison of the performance of the device.12
Results and discussion
Synthesis and spectroscopic and structural characterisation of sterically demanding anchoring ligands
The 4-carboxy-2-(pyridin-2-yl)quinoline (pqca) precursor was prepared with slight modifications of the published procedure, following the Pfitzinger condensation reaction of isatin with 2-acetyl-pyridine, in a refluxing ethanol and aqueous KOH mixture.30 According to this strategy, we extend the series of pyridyl-quinoline derivatives to the sterically demanding compounds 6′-Mepqca, 8-Mepqca and 8,6′-Me2pqca bearing methyl groups at the pyridyl ring, the quinoline moiety or both rings (Scheme 1).31 Although the synthesis of the 8-Mepqca ligand has already been reported,32 in this study we present a complete spectroscopic characterisation, along with the crystal structure determination of this compound (vide infra).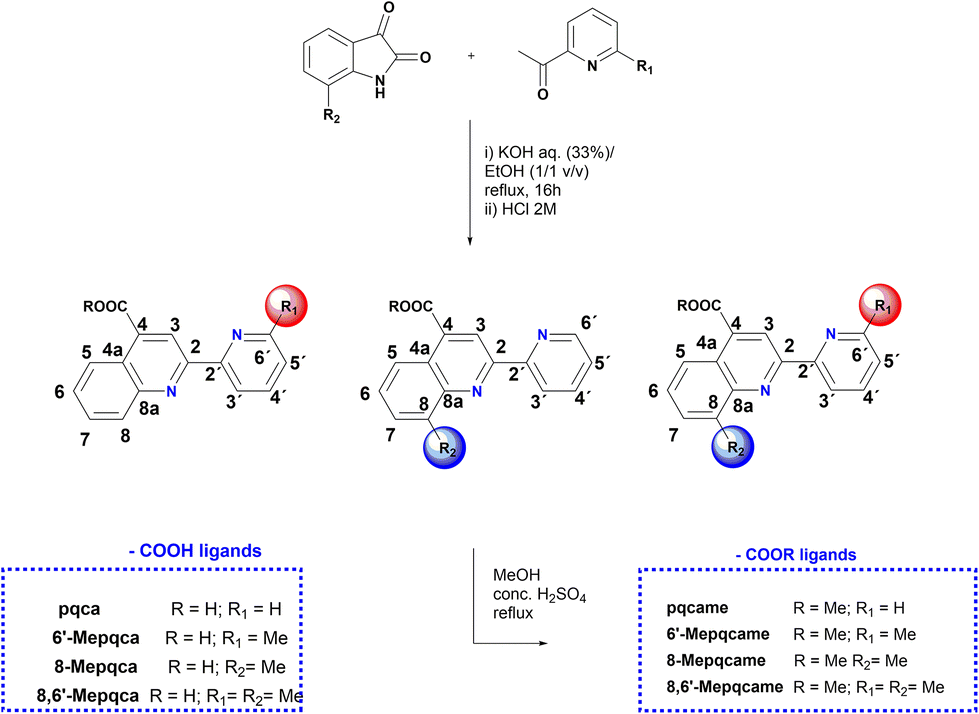 | ||
| Scheme 1 Synthesis of the N^N anchoring ligands containing COOH groups and their methyl ester analogues. | ||
The new organic precursors were obtained in high yields, as easily tractable materials that can be prepared on a gram scale in a one-pot procedure. They can be handled in air and stored for months without decomposition, showing high thermal stability, and decompose in temperatures ranging from 218 °C to 295 °C. Their solubility in most common organic solvents is poor; however, 8,6′-Me2pqca dissolves in Me2CO, methanol and DMSO. The FT-IR spectrum of the precursors is dominated by the asymmetric stretching vibration mode of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond at approximately 1690 cm−1,31,33 along with typical absorption bands for the CC and CN vibration modes, in the region of 1590–1450 cm−1.31 The 1H and 13C NMR spectra were in accord with the structures shown in Scheme 1; assignment was made with the help of 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC (ESI, Fig. S1–S9†) respectively. Within the series of ligand precursors, the singlet resonance signal of the methyl groups appear at δ 2.63 (6′-Mepqca), δ 2.8 (8-Mepqca) and δ 2.64, 2.87 ppm (8,6′-Me2pqca) respectively.
O bond at approximately 1690 cm−1,31,33 along with typical absorption bands for the CC and CN vibration modes, in the region of 1590–1450 cm−1.31 The 1H and 13C NMR spectra were in accord with the structures shown in Scheme 1; assignment was made with the help of 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC (ESI, Fig. S1–S9†) respectively. Within the series of ligand precursors, the singlet resonance signal of the methyl groups appear at δ 2.63 (6′-Mepqca), δ 2.8 (8-Mepqca) and δ 2.64, 2.87 ppm (8,6′-Me2pqca) respectively.
The synthesis of the methyl ester analogues 6′-Mepqcame, 8-Mepqcame and 8,6′-Me2pqcame is also reported; these generally display higher solubility in common organic solvents (Scheme 1). They were isolated as low melting solids (126 °C–154 °C), according to the improved synthesis reported for the pqcame ligand.27 The 1H NMR spectra of 6′-Mepqcame, 8-Mepqcame and 8,6′-Me2pqcame recorded in CDCl3 are included in the ESI (Fig. S10–S18†), displaying the characteristic singlet resonance for the methyl group protons at δ 2.71 (6′-Me), 2.92 (8-Me) and 2.92/2.69 ppm (8,6′-Me) respectively. Assignment was based on a combination of 1H–1H COSY, 1H–13C HSQC and 1H–13C, HMBC.
Colorless single crystals of pqca and 6′-Mepqca, suitable for X-ray diffraction, were grown upon slow evaporation of a DMSO solution of the compounds at ambient temperature. Low-quality yellowish needles, of 8-Mepqca, were obtained upon crystallisation of complex 3 in a mixture of Me2CO/Et2O. Confirmation of the gross structural features of 8-Mepqca was possible, despite the poor dataset refinement. As a result, we present only the molecular structure of this compound without describing its structural bonding parameters. Notably, a CSD search in the crystallographic data center revealed that the structures of these simple molecules had not previously been reported. Their molecular structures are depicted in Fig. 1–3.
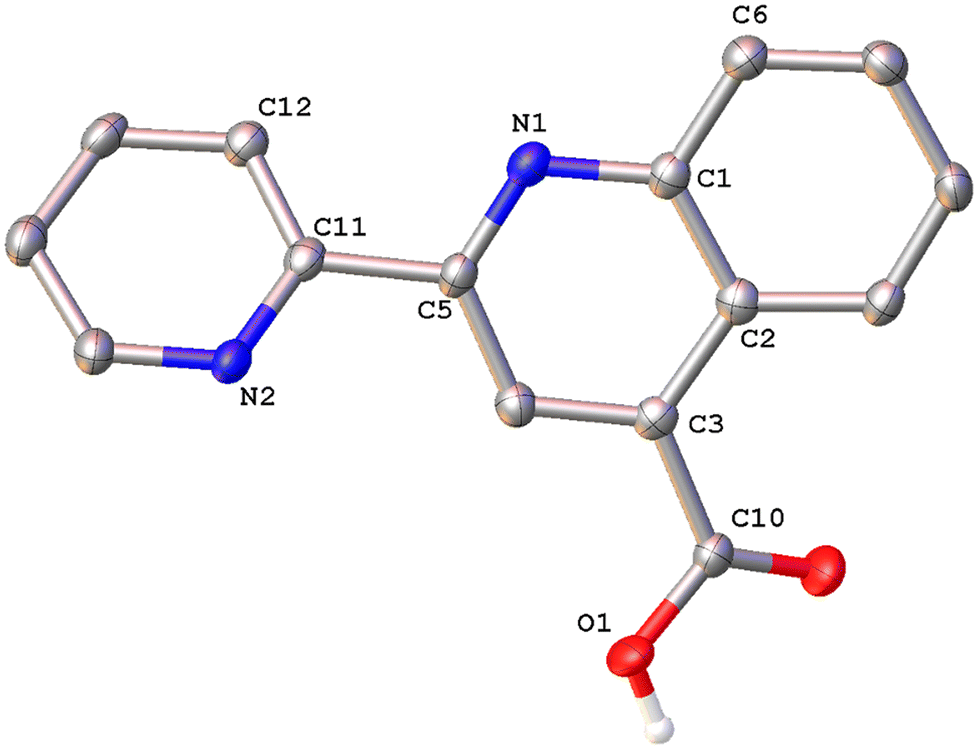 | ||
| Fig. 1 Molecular structure of pqca; ellipsoids are plotted at the 50% probability level. H atoms except those in the COOH groups are omitted. Selected bond lengths (Å) and angles (°): O(1)–C(10) 1.317(3); O(2)–C(10) 1.214(3); N(1)–C(5) 1.311(4); N(2)–C(11) 1.337(4); C(5)–C(11) 1.503(4); C(5)–N(1)–C(1) 117.9(2); C(11)–N(2)–C(15) 118.5(2); O(2)–C(10)–O(1) 124.1(3). | ||
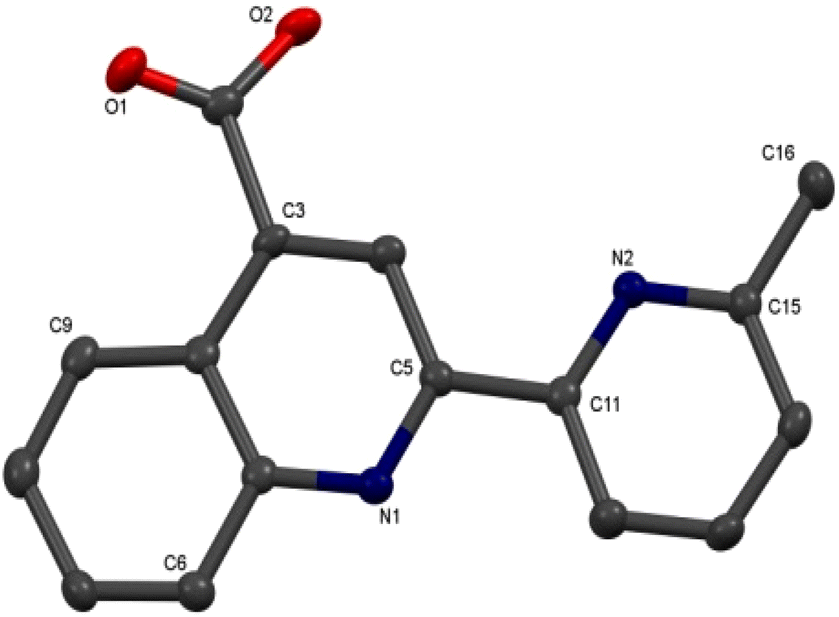 | ||
| Fig. 2 Molecular structure of 6′-Mepqca; ellipsoids are plotted at the 50% probability level. H atoms are omitted. Selected bond lengths (Å) and angles (°): O(1)–C(10) 1.216(4); O(2)–C(10) 1.322(4); N(1)–C(5) 1.329(4); N(2)–C(11) 1.353(4); C(5)–C(11) 1.492(4); C(15)–C(16) 1.496(4); C(5)–N(1)–C(1) 118.4(2); C(15)–N(2)–C(11) 118.7(2); O(1)–C(10)–O(2) 123.9(3). | ||
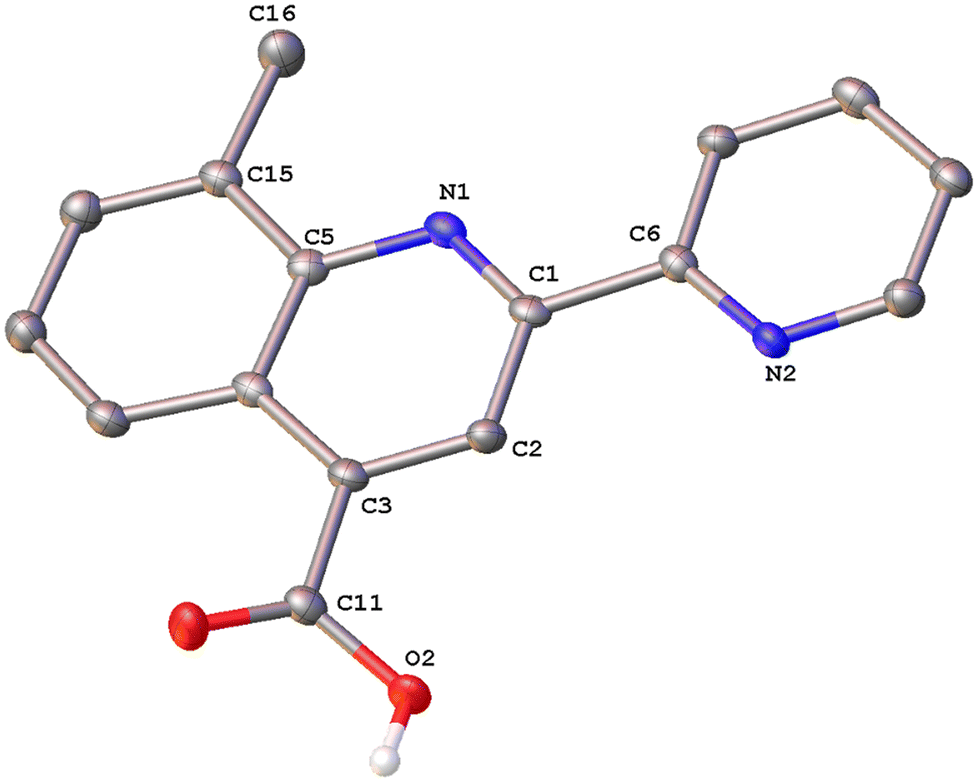 | ||
| Fig. 3 Molecular structure of 8-Mepqca; ellipsoids are plotted at the 50% probability level. H atoms, except those in the COOH groups are omitted. | ||
As expected, the two nitrogen atoms about the biaryl bond adopt a trans-configuration.13,16Pqca crystallises in the monoclinic crystallographic system, P21/n space group and 6′-Mepqca in the orthorhombic, P212121 space group. In pqca the pyridine ring close to the quinoline deviates from planarity, as the angle between the least-squares planes of the rings containing atoms N1 and N2 is 39.96°. For 6′-Mepqca, however, this angle drops to 0.76°, indicating that the molecule is planar. The asymmetric unit of pqca contains four molecules that are stabilised by non-classical intermolecular hydrogen bonding interactions (distance of O2⋯H14–C14 = 2.884 Å, bond angle = 124.73°). The bonding parameters are in accord with those reported for the methyl 2-(2-pyridyl)quinoline-4-carboxylate, pcame analogue.34
The packing in the crystal of 6′-Mepqca is such that a pair of molecules is stabilised by weak intermolecular π–π stacking interactions,35 between the pyridine/quinoline and quinoline/phenyl rings (distance between centroids (N2–C11–C15)/(N1–C1–C5) and (N1–C1–C5)/(C1–C7––C9) of 3.760 Å and 3.631 Å respectively) as shown in Fig. S19 of the ESI.† Lastly, a third molecule, almost orthogonal to this pair, displays typical intermolecular hydrogen bonding interactions via its N2 atom (distance of N2⋯H2–O2 = 1.911 Å, bond angle of 165.62°).36
Synthesis and characterisation of the homoleptic copper(I) complexes 1–4
The copper(I) complexes 1–4 were selectively prepared from the reaction of [Cu(MeCN)4][PF6] with two molar equivalents of the appropriate ligand (pqca, 6′-Mepqca, 8-Mepqca and 8,6′-Me2pqca) at ambient temperature. The synthetic route and reaction conditions are depicted in Scheme 2a.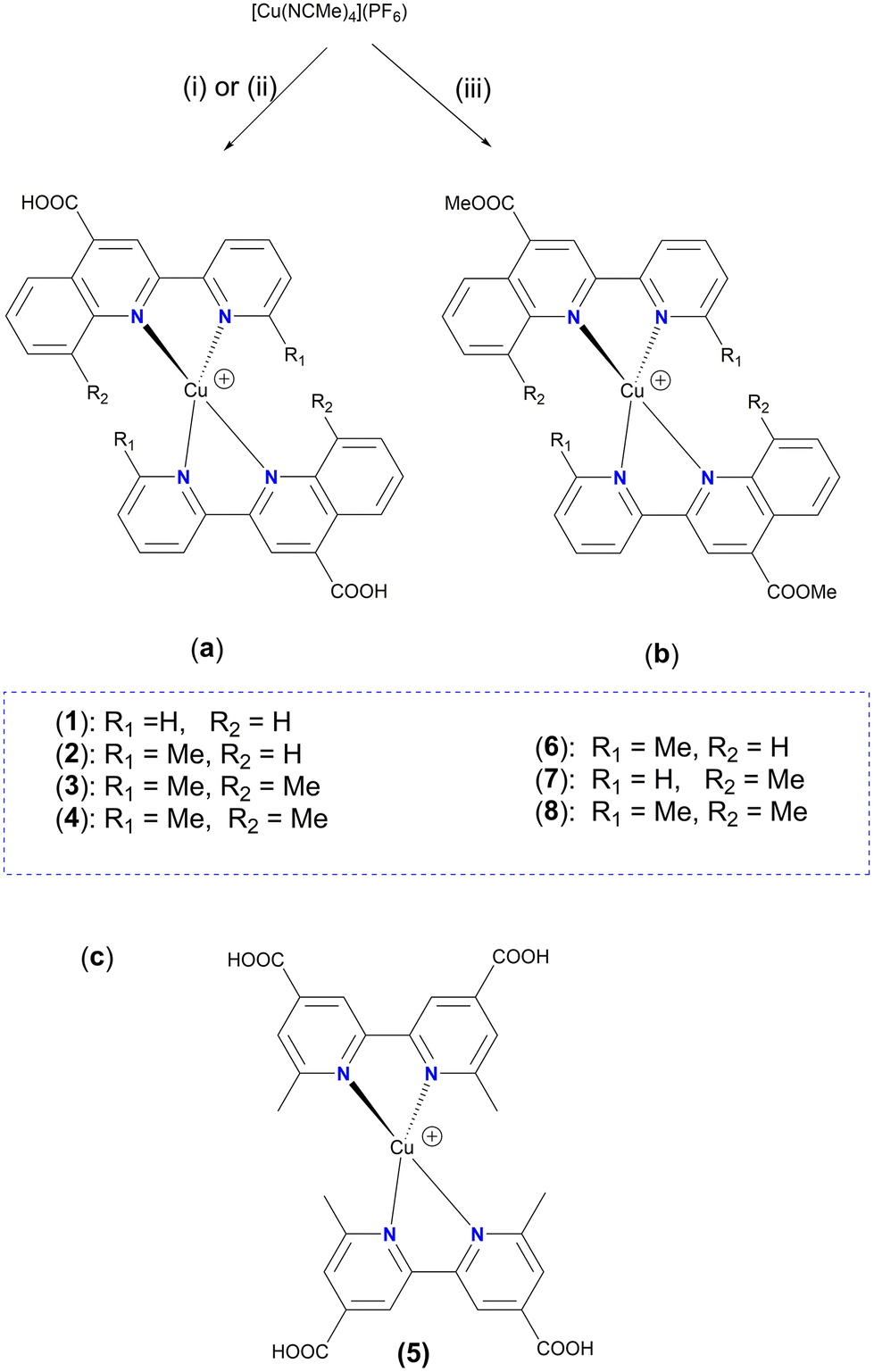 | ||
| Scheme 2 Reaction scheme of the copper(I) cations of complexes 1–4 (a) and 6–8 (b); (i) pqca or 6′-Mepqca, DMSO; (ii) 8-Mepqca or 8,6′-Me2pqca, Me2CO; (iii) 6′-Mepqcame, 8-Mepqcame, 8,6′-Me2pqcame, CH2Cl2. (c) Molecular structure of 5. | ||
The poor solubility of the pqca, 6′-Mepqca, 8-Mepqca and 8,6′-Me2pqca ligands, incorporating a carboxylic acid anchoring group, in common organic solvents is a drawback for the one-pot preparation of the relevant homoleptic copper(I) dyes. In our approach we wanted to avoid the classic two-step synthesis that includes the reduction of copper sulfate with ascorbic acid under basic conditions, and subsequent acidification, which generally suffers from low synthetic yields and purification problems of the final product.37
We realized that in DMSO, a smooth, one-pot reaction occurs immediately, affording 1·0.6DMSO and 2·0.9DMSO as air- and moisture-stable purple-black powders, in 72% and 80% yields. The presence of co-crystallized DMSO is confirmed by NMR (1H, 13C) spectroscopy and by elemental analysis. Both complexes are thermally stable and decompose upon heating at temperatures higher than 206 °C and 182 °C. Initial attempts to prepare 3 following the route reported above were unsuccessful since an unknown grayish solid was obtained, accompanied by ligand elimination (ligand formation was proved by 1H NMR spectroscopy, in solution). We then turned to acetone, where 3 was finally isolated as a black powder in 75% yield, which decomposes at 210 °C. Stability in this medium is rather limited and upon standing for approximately 20 min, precipitation of a black insoluble material was observed, and the color of the solution faded.
Solubility of 8,6′-Me2pqca in acetone is satisfactory, permitting the formation of complex 4 in quite high yield (>92%) as a dark red solid, which decomposes at 195 °C. Homoleptic complex 5 (Scheme 2c), bearing the dmdcbpy ligand, has previously been prepared in 27% yield.12 Herein, we managed to isolate complex 5 in high yield (70%), according to the procedure reported for 1·0.6DMSO and 2·0.9DMSO. Our 1H NMR and UV-vis spectroscopic data agree with those reported in the literature.12
The composition of the copper(I) complexes 1–4 was confirmed by elemental analyses, and spectroscopic characterisation was examined by FT-IR, UV-vis, and NMR spectroscopy (1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC methods). The molecular composition of 1 and 2 was further confirmed by electro-spray mass spectral data, exhibiting peak envelopes corresponding to [M – PF6]+ and [M + H]+ respectively, with the correct isotope pattern. The FT-IR spectra of 1–4 are almost identical from 4000 to 400 cm−1, as expected for compounds that have similar molecular structures, indicating also successful coordination of the ligands.25,26 All complexes exhibit the typical strong absorption for the νas(CO) at ∼1718–1707 cm−1, indicating the presence of the carboxylic acid anchoring group in the corresponding ligands. This further suggests that the COOH group is not involved in co-ordination with the copper(I) centre. The very strong bands at approximately 842 cm−1 and 558 cm−1 can be readily assigned to the ν3(P–F) and ν4(P–F) vibration modes of the PF6− anion.38
The 1H and 13C NMR spectra of 1, 2, 3 and 4 in Me2CO-d6, were assigned by 2D routine techniques (Fig. S20–S30†). The 1H NMR spectra of 1 and 2 display sharp and well resolved peaks, attributed to the protons of pqca and 6′-Mepqca ligands. The intense singlet resonance at δ 9.20 (1) and 9.22 ppm (2) is typical of the H3 proton of the quinoline ligand, which is uncoupled. Co-crystallized DMSO is verified by the singlet resonance at δ 2.67 (1) and 2.62 (2) ppm (see Fig. S20†). The 13C NMR spectra of 1 and 2 are very informative, displaying typical resonance signals for carbon C6′, where the relevant hydrogen atom has been substituted by a methyl group. Ongoing from 1 (δ 150.3 ppm) to 2, this resonance is shifted toward lower fields (Δδ ∼ 9 ppm). Moreover, for 2, the singlet resonance at δ 41.1 ppm is attributed to the methyl carbon of co-crystallised DMSO. Notably, 2 proved to be very stable in Me2CO-d6 at ambient temperature, as its 1H NMR spectrum remained unaltered for 36 days (Fig. S24b†). In contrast, the less constrained compound 1 was stable for a period of 14 days (Fig. S21†). The 1H NMR spectrum of 3 (Fig. S27†) that was recorded at ambient temperature immediately after dissolution displays rather broad resonance signals, with an integration that is in accord with the proposed structure. The low stability of 3 in Me2CO-d6 solution did not permit us to record an appropriate 13C NMR spectrum of this complex. The 1H NMR spectrum of 4, in Me2CO-d6, displays the expected resonance signals that are slightly broadened (Fig. S28†), implying that steric hindrance at the C6′ and C8 positions of the pyridine/quinoline rings is more effective. This is in accord with the structural features obtained from single-crystal crystallography. Analyzing the 1H NMR spectrum of 4, the singlet resonance at δ 9.02 ppm is attributed to H3, and the pseudo-triplet resonance at δ 8.26 ppm to proton H4′. Characteristics are the two high field signals at δ 2.71 and δ 2.14 ppm, assigned to the methyl groups of the quinoline and pyridine moieties, respectively.
Synthesis and characterisation of the copper(I) complexes 6–8
To stabilize the oxidation state around the copper(I) centre, electron-withdrawing substituents, such as methyl ester functionalities, have been considered. The homoleptic copper(I) complexes 6–8 (Scheme 2b), were prepared from the reaction of [Cu(MeCN)4][PF6] with each of the methyl esters 6′-Mepqcame, 8-Mepqcame and 8,6′-Me2pqcame at ambient temperature in CH2Cl2. They are isolated as air-stable purple to red solids that dissolve better in common organic solvents than the acid congeners. The latter characteristic helped us to record well-resolved NMR spectra and characterise the new compounds appropriately. As a result, assignment of the NMR spectra of the less soluble complexes 2–4, with carboxylic acids in their periphery, became possible. Complexes 6–8 were prepared aiming to test their possible application in a DSSC. In general, the electron-withdrawing character of the ester functionality may shift the characteristic MLCT absorption band towards the lower energies,39 which may be beneficial for the performance of a DSSC.The FT-IR spectra of 6–8 are characterised by a strong asymmetric stretching vibration typical of the νas(CO) of the ester moiety at ∼1730 cm−1, shifted to higher wave numbers compared with 1–4, which is in good agreement with the vibration mode reported for the [Cu(pqcame)2][PF6] complex, at 1726 cm−1.27
The NMR spectra of 6–8 were recorded in CDCl3 and assignment was based on routine two-dimensional (2D) NMR techniques (ESI, Fig. S31–S39†).
X–ray
The solid-state structures of 1·DMSO, 2{2·Me2CO·0.5H2O}2, 4, 6·CHCl3·0.13H2O, {7·C5H12}2·CHCl3 and 8 were unambiguously determined by single-crystal X-ray diffraction. Suitable single crystals of 1·DMSO were obtained by slow diffusion of pentane into an acetone solution of the compound, while crystals of 2·Me2CO·0.5H2O were grown from an acetone solution of 2 layered with n-hexane. Very low-quality red plates of 4 were isolated when diethyl ether was diffused into an acetone solution of the complex. Crystals of 6·CHCl3·0.13H2O, 2{7·C5H12}·CHCl3 and 8 were obtained upon diffusion of pentane into a chloroform solution of the relevant complex (6, 7, 8), at ambient temperature.The molecular structures of all cations of complexes are depicted in Fig. 4–8 and the bond parameters are listed in Table S1.† Hydrogen atoms, the PF6− anion and the co-crystallized solvents are omitted for clarity.
 | ||
| Fig. 4 Molecular structure of the cation of complex ([Cu(pqca)2][PF6]·DMSO (1·DMSO)); ellipsoids are plotted at the 50% probability level. H atoms, except those in the COOH groups, and the co-crystallized solvents are omitted for clarity. | ||
 | ||
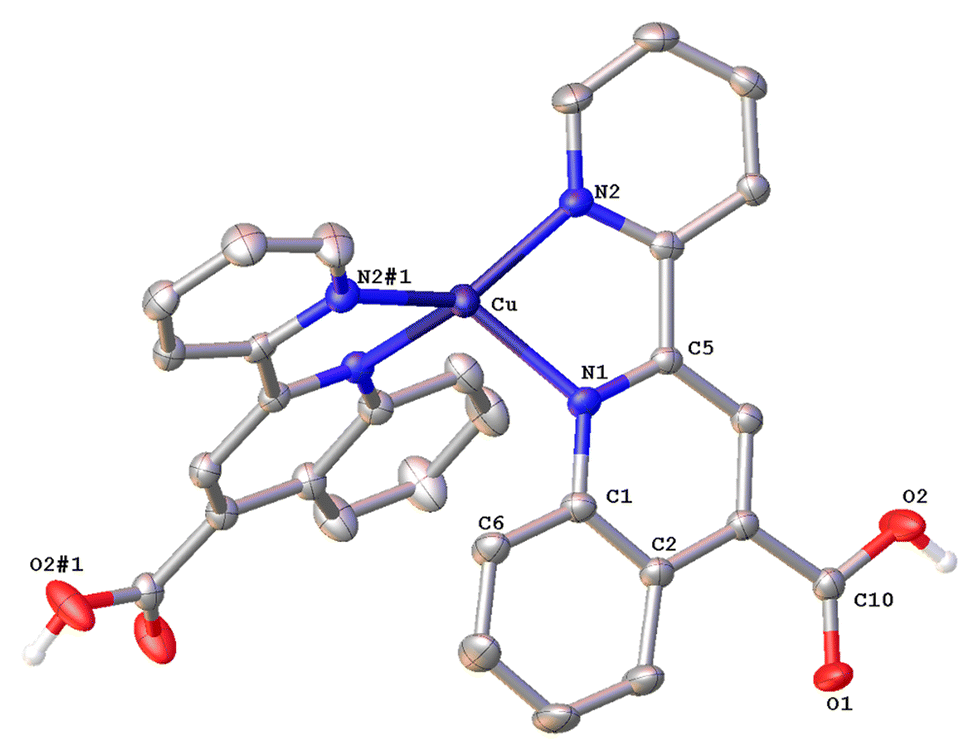
| Fig. 5 Molecular structure of the cation of complex 2{[Cu(6′-Mepqca)2][PF6]·Me2CO·0.5H2O} (2{2·Me2CO·0.5H2O}); ellipsoids are plotted at the 50% probability level. H atoms, except those in the COOH groups, and the co-crystallized solvents are omitted for clarity. | ||
 | ||
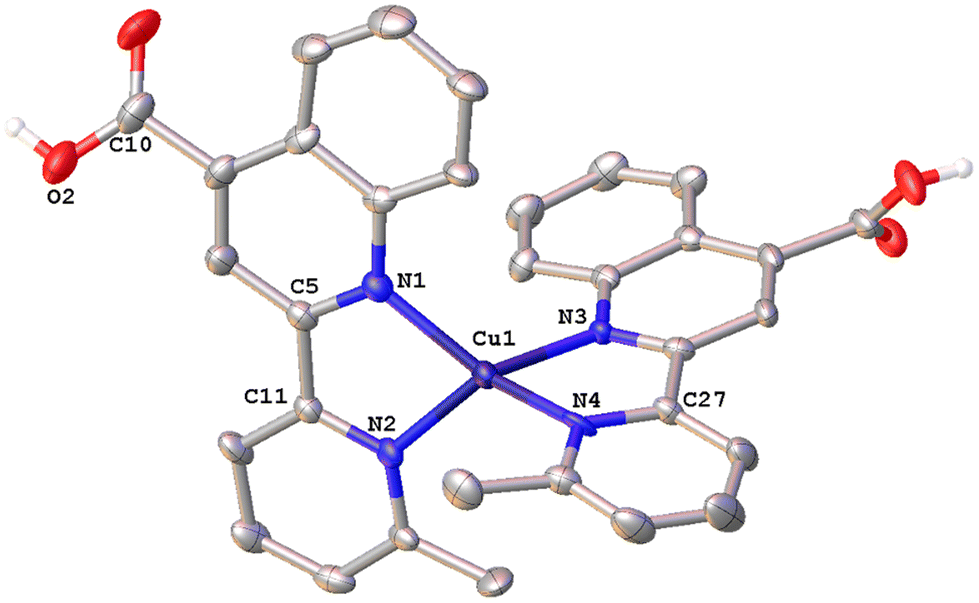
| Fig. 6 Molecular structure of the cation of complex [Cu(6′-Mepqcame)2][PF6]·CHCl3·0.13H2O (6·CHCl3·0.13H2O); ellipsoids are plotted at the 30% probability level. H atoms, except those in the COOH groups, and the co-crystallized solvents are omitted for clarity. | ||
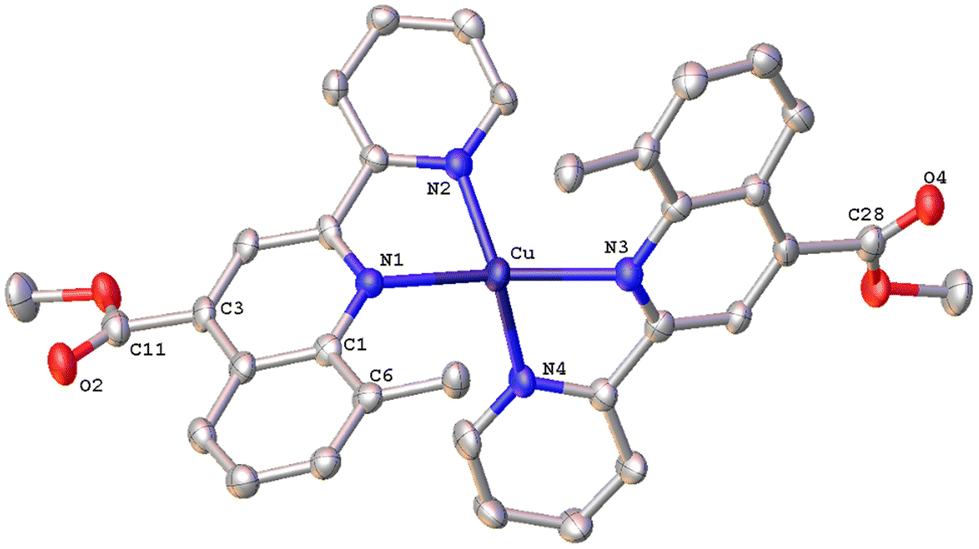 | ||
| Fig. 7 Molecular structure of the cation of complex [Cu(8-Mepqcame)2][PF6] (2{7·C5H12}·CHCl3); ellipsoids are plotted at the 50% probability level. H atoms, except those in the COOH groups, and the co-crystallized solvents are omitted for clarity. | ||
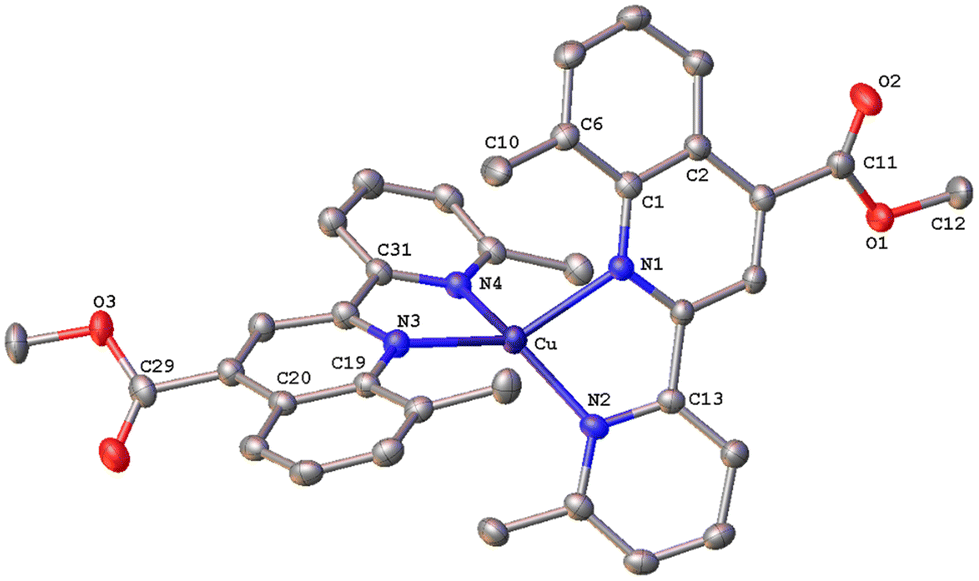 | ||
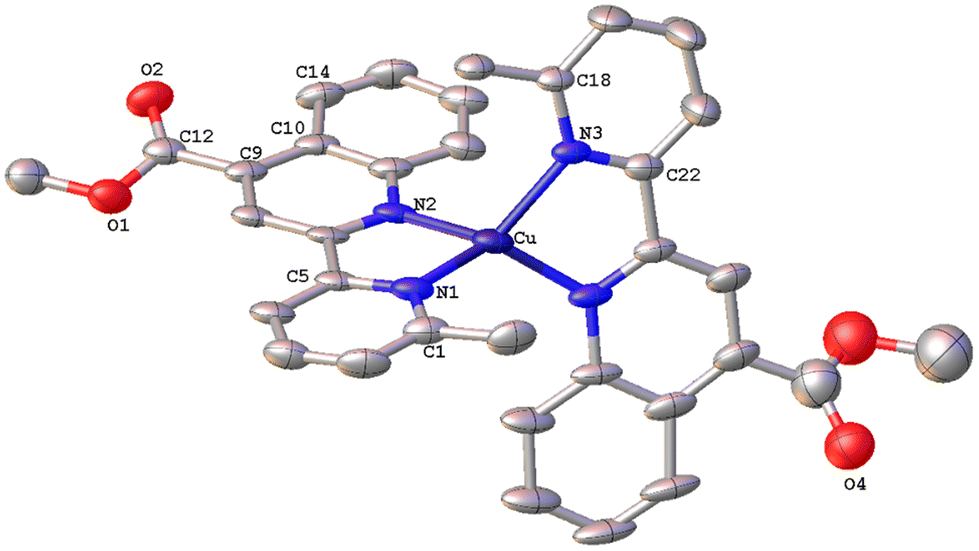
| Fig. 8 Molecular structure of the cation of complex [Cu(8,6′-Me2pqcame)2][PF6] (8); ellipsoids are plotted at the 50% probability level. H atoms, except those in the COOH groups, and the co-crystallized solvents are omitted for clarity. | ||
All complexes possess a four-coordinated copper(I) metal centre with coordination geometries varying from slightly distorted tetrahedral to that of a distorted tetrahedron. To quantify this distortion, the τ4 geometry index introduced by Houser was employed.40 In general, the values of τ4 range from 1.00 (ideal tetrahedral geometry) to 0.00 (ideal square-planar geometry) and can be calculated by the formula τ4 = [360 – (α + β)]/141, where α and β are the largest θ angles of a four-coordinated complex. Accordingly, the calculated τ4 values for complexes 1, 2, 6, 7 and 8 range between 0.49–0.73, which is indicative of significant distortion from the ideal tetrahedral geometry. Our results compare well with the τ4 values of 0.67 and 0.66 reported for [Cu(L)(phen)]+ and [Cu(L)(dmp)]+ (L = 6,6′-dimesityl-2,2′-bipyridine; dmp = 2,9-dimethyl-1,10-phenanthroline) respectively, where a distorted trigonal pyramidal geometry has been proposed.41 This is also in good agreement with the reported τ4 value of 0.59, for [Cu(pqcame)2][PF6],27 albeit the new compounds are slightly less distorted. Notably, complex 7, with a methyl group at the C8 of the quinoline ring, displays a geometry index τ4 = 0.49 implying a severe distortion from the ideal tetrahedral geometry.
Complex 1·DMSO (Fig. 4) adopts a distorted tetrahedral geometry. Flattening is confirmed by the dihedral angle of 55.34° between the two five-membered chelate rings including the copper(I) centre (Cu–N(1)–C(5)–C(11)–N(2)) and (Cu–N(1)–C(5)–C(11)–N(2)) respectively. This structural feature is in accord to the relative τ4 index of 0.68, calculated above for this complex. In this conformation, the pyridine ring is twisted 13.35° out of the plane of the relative quinoline moiety that is bonded.
The angle between the least-squares planes of the pyridine and quinoline unit is 7.23°, as reported for other copper(I) complexes incorporating the 2-(pyridin-2-yl)quinoline moiety.42 Also, the bite angle of the bipyridine unit is 81.21°, while other N–Cu–N bond angles are within the range of 102.58(15) to 132.70° (Table S1†). Complex 1·DMSO exhibits characteristic C–H⋯π interactions (Fig. S40a†) between the H(15) hydrogen atom of the pyridine ring and the phenyl group centroid of quinoline, from an adjacent molecule (distance of (C15–H15)⋯centroid (ring (C6–C9)) = 3.549 Å.
In addition, the crystal packing in the structure of 1·DMSO is such that symmetry-related molecules are separated by typical π–π stacking interactions, including the phenyl rings of the quinoline moieties from adjacent molecules, at a distance of 3.899 Å (view along the a-axis). Additional stabilization offers the non-classical hydrogen bonding between the aromatic hydrogen atom H(8) and the O(1) atom of the adjacent carbonyl group (C(8)–H(8)⋯O(1) = 2.543 Å, bond angle = 171.52°). To this end, the co-crystallized DMSO solvent molecules and the PF6− counter anions (in all directions) participate in extensive classical and non-classical hydrogen bonding interactions (Fig. S40b†).36,43 Typical hydrogen bonds include the O(3) oxygen atom of DMSO and H(2) of the carboxylic acid group (O(2)–H(2)⋯O(3) = 1.813 Å, bond angle = 167.32°), while non-classical hydrogen bonding includes the F atoms of the PF6− counter anions and the hydrogen atoms of methyl groups from DMSO (C(16)–H(16A)⋯F(1)–P) = 2.829 (Å) as well as the aromatic hydrogen atoms of adjacent molecules. Bond lengths are in the range of 2.480–2.690 Å, as expected for other similar copper(I) polypyridine complexes.42
The asymmetric unit of 2{2·Me2CO·0.5H2O} (Fig. 5) consists of two crystallographically independent molecules (Cu1 and Cu1′) with marginally different bonding parameters. Both molecules reveal a distorted tetrahedral coordination geometry, as shown by the N(1)–Cu1–N(2), N(4)–Cu1–N(1) angles of 81.6(2)° and 135.2(2)° for the Cu1 atom, and N(3)′–Cu1′–N(2)′, N(4)′–Cu1′–N(2)′ angles of 81.2(2)° and 124.9(2)° for the Cu1′ atom, which differ from those of a perfect tetrahedron. In the crystal structure, it is clearly seen that the methyl group at the C6′ sufficiently protects the metal centre, leading to a less flattened structure (τ4 = 0.71). This becomes evident from the dihedral angle of 83.41° between the planes of the five-membered chelate rings Cu(1)–N(1)–C(5)–C(11)–N(2) and Cu(1)–N(3)–C(21)–C(27)–N(4) and that of 85.07° between the planes Cu(1′)–N(1′)–C(5′)–C(11′)–N(2′) and Cu(1′)–N(3′)–C(21′)–C(27′)–N(4′), respectively. These angles are closer to 90°, as compared with that of 1·DMSO (55.34°), and are in accordance with the literature reports.16 In the asymmetric unit, both molecules are stabilised by characteristic intermolecular C–H⋯π interactions between hydrogen atoms (H32D, H32E) of the pyridine ring from the Cu1′ complex molecule, and the ring centroids of the quinoline entity from the Cu1 centre (distance of C32D–H32D⋯centroid (C17–C23) = 2.859 Å, C32E–H32E⋯centroid (N3–C21) = 2.975 Å, and distance of C32E–H32E⋯centroid (C17–C23) = 3.467 Å).
The resulting network is reinforced by intermolecular hydrogen bonding interactions where water and acetone molecules are involved (O7–H7B⋯O5 = 2.020 Å, bond angle = 146.27°; O7–H7A⋯O36 = 2.236 Å, bond angle = 135.45°).
The solid-state structure of 4 is depicted in Fig. S41.† Due to the low quality of the structure, structural features are not described in detail. However, these data provide the required information about the nature of the molecule in the crystal.
The crystal structure of 6·CHCl3·0.13H2O (Fig. 6) is less distorted from the tetrahedral coordination geometry, when compared with [Cu(pqcame)2][PF6],27 due to the insertion of the methyl group (C6′). The dihedral angle of 80.82° between the least-squares planes of the two five-membered chelate rings is similar to that of complex 2 and compares well with those reported in the literature for other copper(I) complexes.16 Typical intermolecular π–π stacking, C–H⋯π and C–H⋯O contacts, in the crystal packing of 6·CHCl3·0.13H2O, are shown in Fig. S42.†
Complex 2{7·C5H12}·CHCl3 (Fig. 7) crystallizes in the monoclinic space group C2/c, with two independent molecules (Cu and Cu′) which display significant distortion from that of an ideal tetrahedron. In contrast to complex 6·CHCl3·0.13H2O with a methyl at the 6′, the copper(I) complex cation in 7 posses a more flattened structure, as the angles between the least-squares planes containing Cu or Cu′ and each of the chelated rings are 60.35° and 61.01° respectively. The two ligands around the copper centre are not planar and the angles between the planes defined by the pyridine and quinoline entities range from 25.27° to 32.45°. The crystal packing is such that a pair of molecules is stabilised by an intermolecular π–π stacking interaction between the centroids of the N3-containing rings of adjacent quinoline moieties, at a separation of 3.827 Å (Fig. S43†), and non-classical C–H⋯π contacts (distance of (C27–H27C)⋯centroid (C18′–23′) = 3.454 Å).35 Finally, the crystal network is reinforced by non-classical intermolecular hydrogen bonding interactions (C17′–H17′⋯O4′ = 2.535 Å).43
The solid-state structure of 8 (Fig. 8) reveals a distorted tetrahedral coordination geometry, as reported previously for the similar complexes 6 and 7. The angle between the least-squares planes including a copper(I) centre and each of the 8,6′-Me2pqcame bidentate ligands is 68.22°, indicative of a less flattened structure compared with that of 7. Due to the addition of methyl groups at the 8,6′-positions of the ligand periphery, the quinoline and pyridine moieties of each ligand are not coplanar. Deviation from planarity becomes evident from the angles of 20.95° (between the planes of the rings including N1/N2) and 23.43° (for N3/N4) respectively. The crystal packing of 8 involves typical C–H⋯π interactions. Thus, the hydrogen atom H30A of the methyl ester group displays a characteristic contact with the centroid of the adjacent benzo ring of quinoline (distance of (C30–H30A)⋯centroid(C6) = 2.891 Å). Also the distance of (C36–H36C)⋯centroid(C35–N4) = 3.326 Å, describing the interaction of H36C from a methyl group with the centroid of the pyridine moiety of an adjacent molecule. Notably, a Cambridge Structural Database search revealed that there are very few reports related to copper(I) complexes comprising these ligands.
Electronic spectra
The solution absorption spectra of the homoleptic copper(I) complexes 1–4 are shown in Fig. 9, while those of 5–8 are included in Fig. S44.† All compounds show a main and broad MLCT absorption band centered between 506 nm and 532 nm, while the bands in the region of 333–339 nm are typical of ligand-centered π–π* transitions.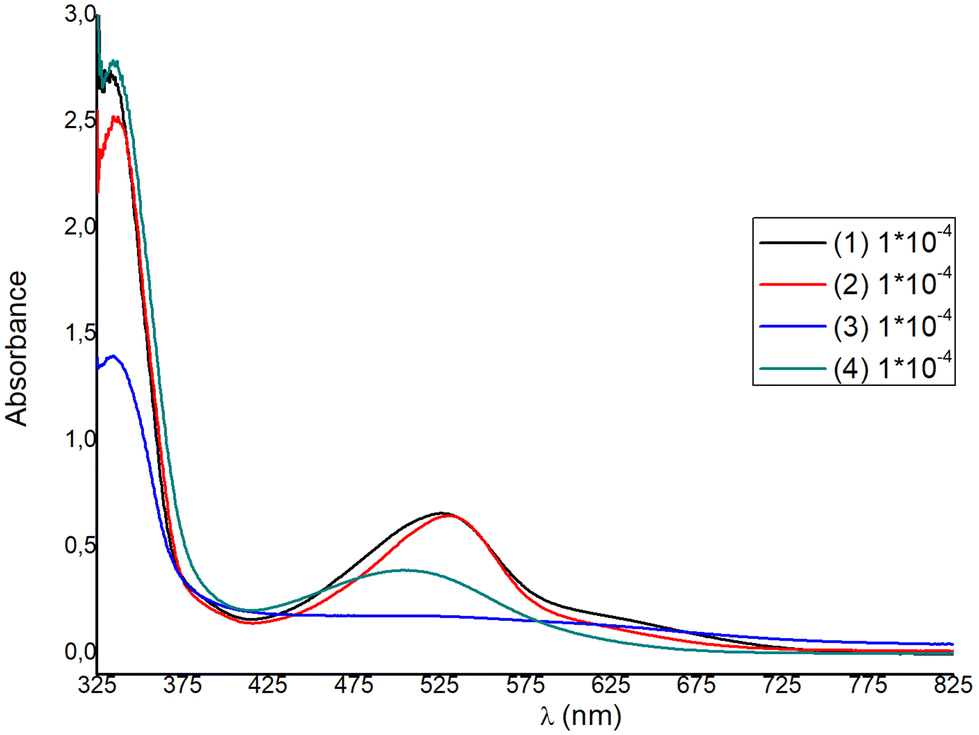 | ||
| Fig. 9 Absorption spectra of compounds 1–4 in Me2CO at 298 K. | ||
Ongoing from 1 to 2 a small red shift is observed for the main MLCT band from 527 nm (1) to 530 nm (2), with ε values at approximately 6600 dm3 mol−1 cm−1, significantly higher than those of other similar copper(I) diimine complexes.42 For complexes 3 and 4, the λmax of the MLCT band blue-shifts to 519 nm and 506 nm respectively, indicating an increase of the ΔE band gap. Blue-shifting is accompanied by an approximate four- and two-fold decrease of the extinction coefficient respectively. Within the homoleptic complexes 1–4, ε values range from 1800 to 6600 dm3 mol−1 cm−1 (ε3 < ε4 < ε1 ≈ ε2), while ongoing to the methyl ester analogues 6–8, the extinction coefficient increases significantly, reaching a value of ∼10000 dm3 mol−1 cm−1, for 6. This is in accord to the ε values reported for [Cu(pqcame)2][PF6]27 and other similar complexes in the literature.44,45
For DSSC application, dyes 1 and 2 are superior to 3 and 4, as the spectral window covered by the MLCT absorption band is broader and the extinction coefficients are much higher. For each complex a second, broad shoulder-like, MLCT band is observed around 630 nm, with very low ε values. This is a typical feature reported in the absorption spectra of copper(I) phenanthroline complexes, where generally three bands are observed, which are abbreviated as band I, band II, and band III.15,44,45 Taking this nomenclature into consideration, we may propose that the broad MLCT band can be attributed to band II, alongside band III, which is typically hidden within band II. The lowest energy MLCT broad band above 600 nm can be potentially attributed to band I, which is indicative of a distorted tetrahedral geometry. This further corroborates with the structural features obtained from the X-ray crystal structure analysis of the compounds studied, indicating a strong distortion from that of the ideal tetrahedral geometry (vide infra in Fig. 4–6). In addition, very recent reports further suggest that the highest energy MLCT band (band II) is attributed to an S0 → S2 transition (Franck–Condon level), while band I is attributed to an S0 → S1 charge transfer.45,46 In particular, the energy and the intensity of the S0 → S1 and S0 → S2 transitions can shift according to geometrical changes, owing to the steric hindrance of the ligands used. Accordingly, S0 → S2 energy shifting may be attributed to a change of the Cu–N bond lengths, while the S0 → S1 transition receives lower values for D2d symmetric compounds.45,46
Based on the poor optical characteristics derived from the solution absorption spectrum of 3, no further studies were undertaken with this complex. We emphasized dyes 1, 2 with carboxylic acid anchoring ligands and 6 and [Cu(pqcame2][PF6] (A) that bear a methyl ester moiety. Sample solutions and dyed films are displayed in Fig. S45.† From visual inspection it becomes evident that 1 and 2 adsorb strongly on the titania surface, but 6 and (A) rather slightly. Interestingly, complex 6 incorporating the ester 6′-Mepqcame gave better dye-modified surfaces compared with that of (A), which bears the less sterically demanding pqcame ligand.
From the solid-state absorption spectra presented in Fig. 10, it is easily seen that the absorption intensity of the electrode with dye 2 is higher than that of 1, indicating that 2 is more efficiently adsorbed on the TiO2 surface. Upon grafting of both dyes onto the titania surface the absorption maxima are blue shifted (22 nm for 1 and 16 nm for 2) compared with the data obtained in solution. This is an indirect proof of chemisorption of the dyes and may be attributed to deprotonation of the carboxylic acid functionalities, which shifts the orbitals of the anchoring ligand.26,47 Grafting of 6 onto TiO2 film is less efficient (Fig. S46†).
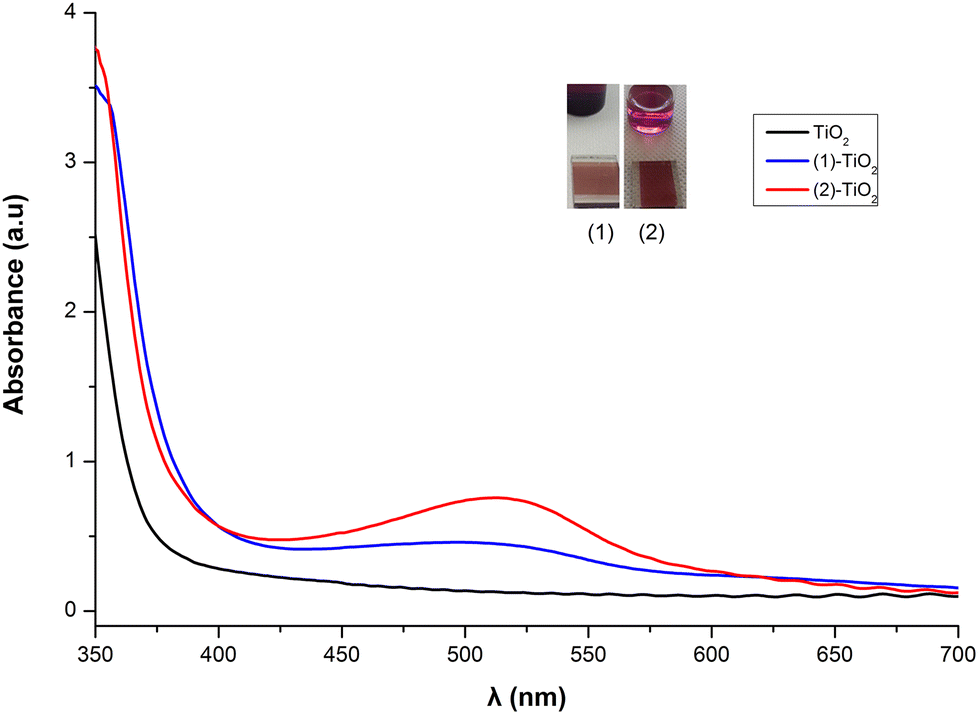 | ||
| Fig. 10 Dyes 1 and 2 adsorbed on TiO2. | ||
Electrochemical studies
The electrochemical characteristics of dyes 1, 2 and 4 were measured by means of cyclic voltammetry under argon (Fig. S47†), and the relative data are summarized in Table 1.| Complex | E pa (mV) | E pc (mV) | E 1/2/V (ΔE = Epc − Epa/mV) |
|---|---|---|---|
| a No reduction process observed within the solvent accessible window. | |||
| (1) | — | — | — |
| (2) | +155 | –69 | +0.43 (224) |
| (4) | +209 | — | |
Cyclic voltammograms were recorded in Me2CO due to the high solubility and chemical stability of the complexes in this solvent. It was not possible to measure complex 3 in this solvent within the time scale of the measurement, due to partial decomposition. The relevant methyl ester analogues 6–8 were not measured since they chemisorb slightly on the titania surface, while 2 and 4 exhibit a copper-centered oxidation. The oxidation potential for 2 (+0.43 V) is within the range reported for copper(I)-bipyridyl complexes,18 while the peak separation of 224 mV implies slow electron transfer kinetics. In accord with the literature reports, chemical irreversibility of the redox process is also observed for both complexes (2 and 4).48 In short, the electrochemical behavior of complexes 1, 2 and 4 follows the expected trend, with complex 2, containing a methyl substituent at the 6′ position of quinoline, being electrochemically the most stable.16,49
The electrochemical characteristics of dye 2 absorbed onto TiO2 were also measured by means of cyclic voltammetry. A three-electrode, one-compartment electrochemical cell was used for the cyclic voltammetry (CV) measurements with a supporting electrolyte of 0.1 M [nBu4N][PF6] in acetonitrile (MeCN). Metrohm platinum wire was used as the counterelectrode, Ag/AgCl (3 M KCl) as the reference electrode and sensitized TiO2 films as the working electrodes.
Potentials referred are quoted versus Ag/AgCl or +0.77 V vs. the standard hydrogen electrode (SHE), where the potential Ag/AgCl (3 M KCl) is +0.21 V. The applied scan rate was 20 mV s−1.
The cyclic voltammogram presented in Fig. 11 displays a characteristic oxidation and reduction wave (Epa = +0.64 V, Epc = +0.47 V) which is attributed to the Cu(I)/Cu(II) system. The peak separation of ∼175 mV showed quite slow electron transfer kinetics, so the electrochemical behavior is described as quasi-reversible.18,50 The half wave potential, E1/2, deduced from the cyclic voltammogram curve, determines the highest occupied molecular orbital (HOMO) level of the dye. Thus, the E1/2 for the 2 complex is +0.56 V vs. Ag/AgCl or +0.77 V vs. a standard hydrogen electrode (SHE). This value is sufficiently positive compared with the I–/I3− redox potential (+0.33 V vs. SHE), suggesting efficient dye regeneration51 (vide infra in the DSSC evaluation).
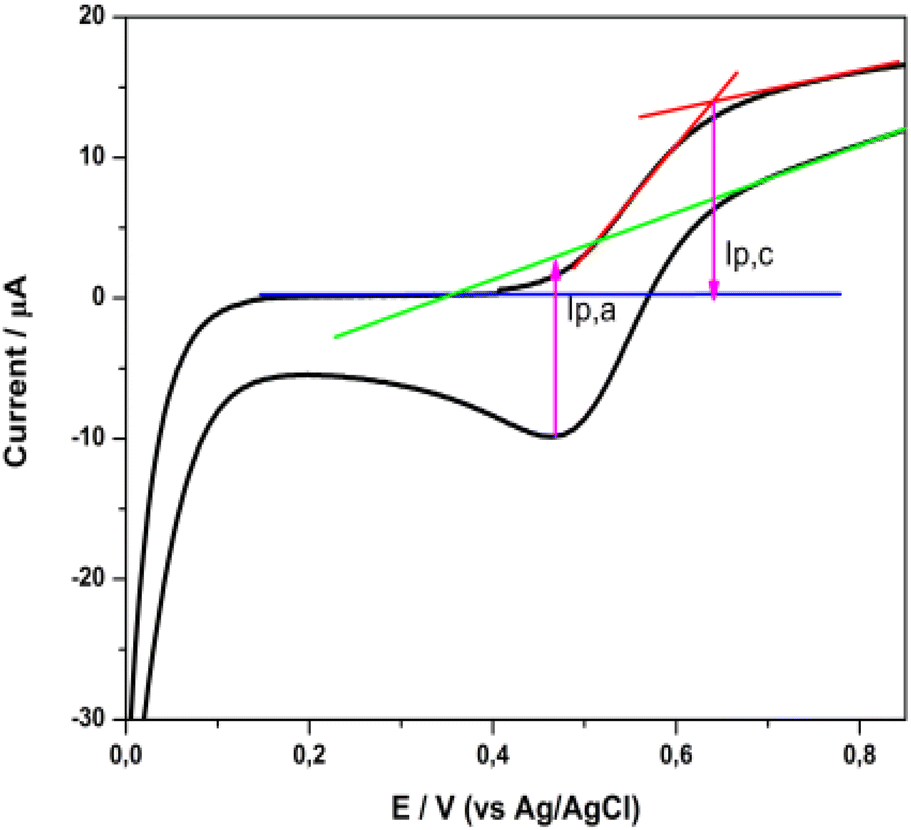 | ||
| Fig. 11 Cyclic voltammogram of a 2 sensitized titania film, in a three-electrode electrochemical cell. | ||
The lowest unoccupied molecular orbital (LUMO) level of the dye was estimated from the HOMO level and the excitation energy of the dye adsorbed on TiO2. The latter was determined from the difference Eox – Eg, where Eox stands for the redox potential of the Cu(I)/Cu(II) redox couple and Eg is the absorption threshold of the low-energy tail of the TiO2/dye absorption spectrum (Fig. S48†), which in the case of 2 is at 2.03 eV (612 nm).52,53 The LUMO level of 2, calculated at −1.26 V vs. NHE, is sufficiently negative compared with the conduction band (CB) edge of TiO2 (−0.5 V vs. SHE),54 implying efficient electron injection into the CB of titania.
Based on cyclic voltammetry and UV-vis data, the energy diagram of the interface including the HOMO and LUMO energy levels of dye 2 has been estimated and is depicted in Fig. 12. This would enable us to justify the photovoltaic performance of this complex.
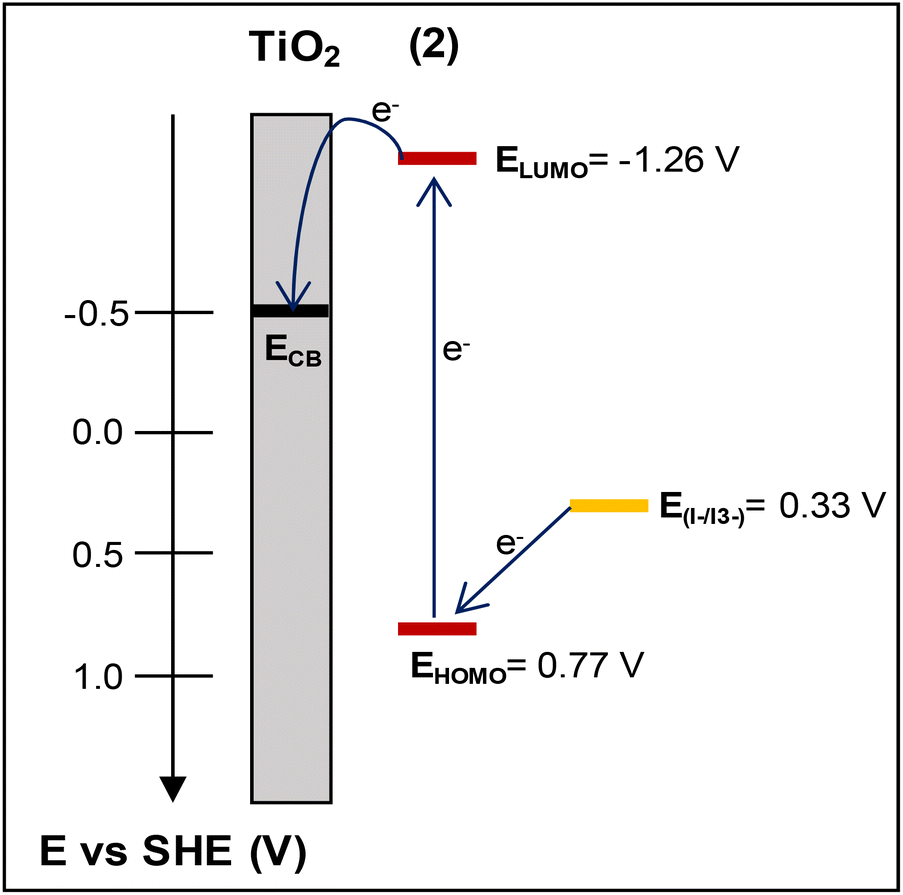 | ||
| Fig. 12 The energy diagram of the photoelectrode electrolyte interface depicting the HOMO–LUMO energy levels of dye (2) vs. the conduction band (CB) level of TiO2 and the redox potential of I–/I3−. | ||
FT-IR and Raman spectroscopic data of the sensitized films
The corresponding FT-IR spectra of the dye-sensitized titania nano-powdered films are dominated by the anatase TiO2 bands, while the dye contribution is relatively poor (weak intensity bands). The broad band centered at ∼3500 cm−1 corresponds to the characteristic O–H stretching vibration produced by the hydroxyl groups and chemisorbed water on the titania film.56 The rather sharp band located in the region of 1635–1620 cm−1 is due to the bending modes of water (H–O–H) that is overlapped with the Ti–O stretching vibration mode.52,53 The CO shift (∼1725 cm−1 (1), 1725 cm−1 (2), 1727 cm−1 (4)) was attributed to chemisorption of the dyes onto the titania surface, via ester-type bonding.55 The TiO2 nanoparticles treated with the new dyes display a number of weak but characteristic IR bands in the typical region of 1565–1300 cm−1 that correspond to the pyridine ring modes. This further suggests efficient dye grafting on the titania surface. The absorption band at the lowest part of the spectrum ∼700–450 cm−1 is related to the Ti–O vibration.55
In this respect adsorption of dye 2, the most prominent for DSSCs, onto the titania film becomes apparent by comparing the micro-Raman spectra of 2 in a powder form and that chemisorbed onto the titania film (Fig. S50†).
The two bands at the lower frequency region (638 cm−1 and 143 cm−1) of the spectrum are related to stretching vibrations of the semiconductor. Among them, the most intense band (143 cm−1) is attributed to O–Ti–O vibrations (E1g).57 Representative fingerprint bands of dye 2 are shown in the 1600–1300 cm−1 range: a strong band at 1443 cm−1 for C–H bending and the v(CN) and v(CC) pyridine modes at 1473, 1512, 1550 and 1551 cm−1. The strong band located at 1365 cm−1 is attributed to a combination of the in-plane C–H stretching vibrations and the CC stretching vibrations of the aromatic carbons.25,57 These bands are also observed in the spectrum of 2 in the powder form.
In comparison with the bare complex (powder form), slight differences either in the peak position or width were observed, implying weak electronic coupling of complex 2 onto the semiconductor surface. This could be attributed to the single carboxylic acid group present in this molecular dye.26,47
Photovoltaic performance of the dyes in DSSCs
The photovoltaic performance of nanocrystalline TiO2 solar cells sensitized by dyes 1 and 2 containing carboxylic acid functionalities, and that of the methyl ester analogues 6 and (A), has been evaluated under the standard AM 1.5 irradiation conditions.26,48 The performance of the homoleptic copper(I) dye 5 that served as a reference herein12 has been also reported by other research groups.37,39 For sensitization, the working electrodes were immersed in a solution of the dye and left undisturbed overnight (see the experimental part).The influence of the architecture of the modified electrodes (photoanodes) on the resulting performance of the photovoltaic device, assembled with the most promising dyes, was investigated. Typical photocurrent density–photovoltage J–V curves of 1, 2, A and 6-sensitized solar cells are depicted in Fig. 13. The lower case letters (a–d) highlight the different architectures of the semiconductor material of the anode, dyed with 1 and 2, as reported in Table 2 (vide infra). A comparison of the best performing film (2d in Table 2) with the 5 cell reference is shown in Fig. S51.†
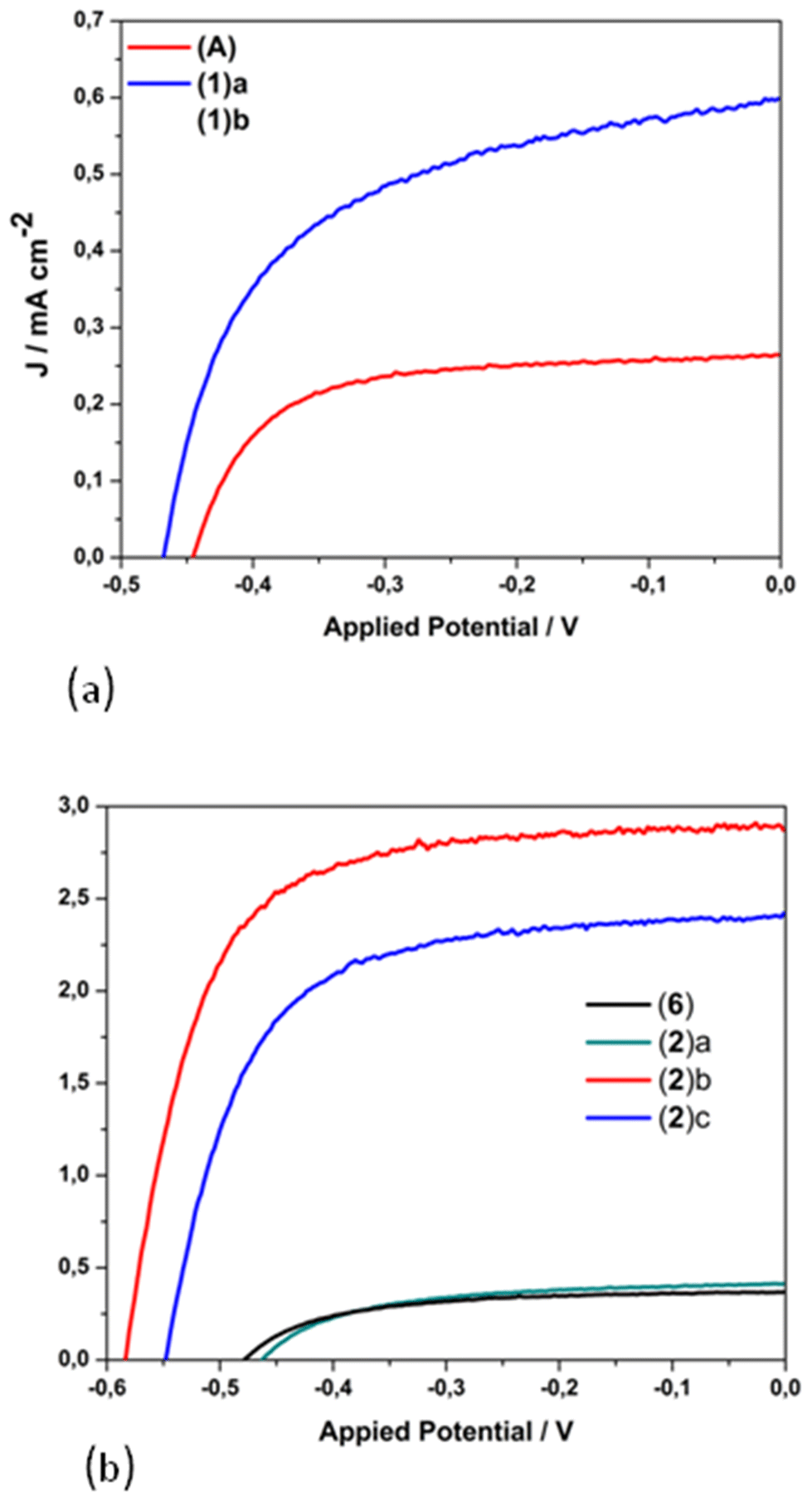 | ||
| Fig. 13 J–V curves for the copper(I)-based dyes 1, [Cu(pqcame2][PF6] (A) (a); 2 and 6 (b). Lower case letters highlight the different architecture of the semiconductor material of the anode, dyed with 1 and 2, as reported in Table 2. | ||
| Anode-dye (entry) | Film (+post-treatment) | J sc/mA cm−2 | V oc/mV | FF | η/% |
|---|---|---|---|---|---|
| NRT = transparent titania paste, NRAO = active opaque titania paste, WER4 = scattering layer, CL = compact layer, DSL = transparent titania paste (3 μm layer). | |||||
| 1(a) | NRT | 0.26 | 446 | 0.66 | 0.08 |
| 1(b) | NRAO–WER4 | 0.60 | 470 | 0.55 | 0.15 |
| 2(a) | NRT | 0.41 | 465 | 0.56 | 0.11 |
| 2(b) | NRAO–WER4 | 2.87 | 585 | 0.68 | 1.15 |
| 2(c) | DSL–NRAO–WER4 | 2.42 | 549 | 0.64 | 0.85 |
| 2(d) | CL–NRAO–WER4 | 2.94 | 591 | 0.69 | 1.20 |
| A | NRAO–WER4 | 0.28 | 443 | 0.43 | 0.05 |
| 5 | CL–NRAO–WER4 | 2.90 | 576 | 0.63 | 1.05 |
| 6 | NRAO–WER4 | 0.37 | 480 | 0.57 | 0.10 |
The corresponding electrical parameters or cell characteristics (short-circuit current density (Jsc), open-circuit voltage (Voc); fill factor (FF) and power conversion efficiency (η)) of all the fabricated DSSCs are reported in Table 2 (the mean value of three essays). From this Table it is clearly seen that photoanodes consisting of only one transparent titania layer (NRT, entry a), dyed with 1 and 2, display a very poor performance of approximately 0.1% (1(a), η = 0.08% and 2(a), η = 0.11%). On the other hand, the combination of an active layer (NRAO) and a scattering layer (WER4) leads to a substantial tenfold increase in the power conversion efficiency of dye 2 (η = 1.15%, entry 2(b)), while that of 1 is doubled (η = 0.15%, entry 1(b)).
This is mainly attributed to the marginal increase of the Jsc (2.87 mA cm−2) and a rise of the open circuit potential by 115 mV. Apparently, the existence of the scattering layer plays a crucial role (increase of the optical path length) for the increased performance of photovoltaic devices sensitized with dye 2.58 Interestingly, a decrease in the performance of a device assembled with three layers of titania (entry 2c) was observed (absence of a compact layer), as the Jsc and Voc values drop to 2.42 mA cm−2 and 549 mV respectively. However, a further improvement in the performance of the cell device was obtained upon TiCl4 pre-treatment of the anode.59 This procedure proved beneficial for the optimization of the device, as indicated by a slight increase of the short circuit current and a slight enhancement of the Voc. As a result, the most efficient cell displayed Jsc, Voc and power energy-conversion efficiency (PCE-η) values of 2.94 mA cm−2, 591 mV and 1.20% respectively (entry 2(d)). To this end, the 2-based cell device is the most efficient, with a relative power conversion efficiency exceeding by 13% that of the reference 5, measured under the same experimental conditions (entry 5). Overall, the efficiency of 2 shows a gain in terms of the Voc value.
To test the viability of copper(I) complexes A and 6 with methyl ester functionalities in DSSCs and improve the initially poor photovoltaic characteristics reported above, we decided to use the protocol of He et al. for a Zn(II) sensitizer60 and that of Kee and coworkers for homoleptic copper(I) dyes.49 Accordingly, and prior to dye dipping, the photoanode was pre-treated with tBuOK/THF as described in eqn (1).
| –Ti-OH + tBuOK→ –Ti-OK + tBuOH | (1a) |
| –Ti-OK + Dye-COOR→ –Ti-OOC-Dye + ROK (R = Me) | (1b) |
However, the application of the protocol led to a degradation of the surface characteristics (Fig. S52†) with a concomitant decrease of the performances (<0.01%), to significantly lower than the performances reported for non-pre-treated photoelectrodes (Table 2). In any case, the photovoltaic performances of A (0.05%) and 6 (0.10%) were significantly higher than those reported for other copper(I) complexes incorporating methyl ester functionalities.49
To this end, these results are in favor of a great potency of the new copper(I) molecular compounds, as compared with ruthenium(II) congeners that display similar photovoltaic data, in terms of the significantly low cost and higher availability of the Earth-abundant copper-based precursors. Besides their potency for applications in window-glass building facades, investigation of the photosensitization process may be facilitated by their usage as copper redox mediators.61,62
Experimental
Materials and methods
All manipulations concerning the synthesis of the copper(I) complexes were carried out under argon using standard Schlenk techniques, while the synthesis of the ligands was performed under aerobic conditions. The solvents were dried by standard methods (MeCN and CH2Cl2 over P2O5, diethyl ether (Et2O) over Na wire, Me2CO over K2CO3) and distilled under argon. DMSO (Me2SO) was dried over freshly activated molecular sieves of 4 Å, and methanol over molecular sieves of 3 Å. The distilled solvents were stored over molecular sieves under an argon atmosphere. All solvents were thoroughly degassed by three freeze–pump–thaw cycles prior to use.The copper(I) precursor [Cu(MeCN)4][PF6] was synthesised according to the literature reports.63 The pqca ligand was prepared by a slight modification of the reported procedure64 while its methyl ester analogue (pqcame) was prepared according to the method reported in the literature.27 7-Methyl-isatin,65,66 6-methylpicolinic acid,67 ethyl-6-methylpicolinate,68 2-acetyl-6-methyl-pyridine,69 and 6,6′-dimethyl-2,2′-bipyridine-4,4′-dicarboxylic acid (dmdcbpy),12,70 used for the synthesis of the organic ligands, were prepared according to the literature methods and their purity was tested by FT-IR, 1H NMR and melting point measurements.
Elemental analyses were obtained from the Microanalysis Center of the Institut für Anorganische Chemie Universität Bonn. Infrared spectra were measured on a Shimadzu IRAffinity-1 spectrometer, as potassium bromide pellets in the spectral range of 4000–400 cm−1. 1H and 13C{1H } NMR spectra were recorded at 298 K on a Bruker Avance DRX 500 MHz, a Bruker Prodigy 500 MHz and a Bruker Avance Neo 400 MHz spectrometer in CDCl3, Me2CO-d6 and DMSO-d6. The 1H and 13C{1H} NMR spectra were calibrated against the internal residual proton and natural abundance 13C resonances of the deuterated solvent. J values are given in Hz. Spectra were assigned using the 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC methods. Melting or decomposition points were determined using a Sanyo Gallenkamp variable heater apparatus and are uncorrected. The samples were heated slowly until the compounds melted or decomposed. Micro-Raman spectra were measured in the backscattering configuration using a Renishaw inVia Reflex system equipped with an Ar laser emitting at 514.4 nm. The scattered light is filtered by a dielectric edge Rayleigh rejection filter with cutoff at 100 cm−1 and analyzed with an 1800 lines per mm diffraction grating. Laser power was set to 0.1% to avoid dye degradation. Absorption spectra were recorded with a Cary 3E UV-vis spectrometer in acetone or chloroform solution. A three-electrode, one-compartment electrochemical cell was used for the cyclic voltammetry (CV) measurements with a supporting electrolyte of 0.1 M tetrabutylammonium hexafluoridophosphate [TBA][PF6] in acetonitrile. Metrohm platinum wire was used as a counterelectrode, Ag/AgCl (3 M KCl) as the reference electrode and sensitised TiO2 films as the working electrodes. Potentials referred to are quoted versus the standard hydrogen electrode (SHE), where Ag/AgCl (3 M KCl) is +0.21 V.
Synthesis of organic precursors with COOH groups
O)), 1588 (s, ν(CC)), 1554 (m, ν(CC)), 1453 (s, ν(CN)), 1286 (s), 1261 (s), 901 (m, br, δ(O–H)), 795 (s, γ(C–H)out-of-plane), 773 (s, γ(C–H)out-of-plane), 732 (s, γ(C–H)out-of-plane), 654 (s, γ(C–H)out-of-plane). 1H NMR (500 MHz, DMSO-d6) δH/ppm 14.05 (1H, br s, COOH), 8.99 (1H, s, H3), 8.76 (1H, d, J = 10, H9), 8.43 (1H, d, J = 10 Hz, H3′), 8.19 (1H, d, J = 10, H6), 7.91 (1H, t, J = 10, H4′), 7.87 (1H, d, J = 10, H7), 7.74 (1H, t, J = 10, H8), 7.41 (1H, d, J = 10, H5′), 2.63 (3H, s, Me of pyridine ring). 13C{1H} NMR δC/ppm (126 MHz, DMSO-d6) 167.57 (COOH), 157.80 (C6′), 155.27 (C2′), 153.77 (C2), 148.16 (C10), 137.65 (C4′), 137.03 (C4), 130.11 (C7), 129.88 (C6), 128.22 (C8), 125.55 (C9), 124.44 (C5), 124.33 (C5′), 119.22, 118.17 (C3′), 24.19 (Me of pyridine ring); ESI-MS (MeOH, positive mode): m/z 279.1 [M + H]+ (calc. 279.3).
O)), 1599 (s, ν(CC)), 1556 (m, ν(CC)), 1480 (s, ν(CN)), 1269 (s, νs(C–O), 1244 (s, ν(C–O)), 916 (m, br, ν(O–H)), 794 (s, γ(C–H)out-of-plane), 772 (s, γ(C–H)out-of-plane), 740 (s, γ(C–H)out-of-plane), 716 (s, γ(C–H)out-of-plane), 648 (s, γ(C–H)out-of-plane). 1H NMR (400 MHz, DMSO-d6) δH/ppm 13.85 (1H, br s, COOH), 8.98 (1H, s, H3), 8.79 (1H, br s, H6′), 8.69 (1H, br s, H3′), 8.58 (1H, br s, H6), 8.07 (1H, br s, H4′), 7.73 (1H, br s, H8), 7.62 (1H, br s, H7), 7.55 (1H, br s, H5′), 2.87 (3H, s, Me of quinoline ring). 13C{1H} NMR(100 MHz, DMSO-d6) δC/ppm 167.80 (COOH), 154.89 (C2), 153.76 (C2′), 149.40 (C6′), 147.02 (C10), 137.90 (C4), 137.55 (C4′), 137.27 (C9), 130.15 (C8), 127.99 (C7), 124.88 (C5′), 124.49 (C5), 123.43 (C6), 121.08 (C3′), 118.61 (C3), 17.98 (Me of quinoline ring).
O)), 1591 (s, ν(CC)), 1561 (m, ν(CC)), 1457 (s, ν(CN)), 1284 (s), 1262 (s), 918 (m, br, δ(O–H)), 802 (s, γ(C–H)out-of-plane), 773 (s, γ(C–H)out-of-plane), 763 (s, γ(C–H)out-of-plane), 736 (s, γ(C–H)out-of-plane). 1H NMR (400 MHz, DMSO-d6) δH/ppm 8.94 (1H, s, H3), 8.54 (1H, d, J = 8, H6), 8.46 (1H, d, J = 8, H3′), 7.91 (1H, t, J = 8, H4′), 7.71 (1H, d, J = 8, H8), 7.60 (1H, t, J = 8, H7), 7.39 (1H, d, J = 8, H5′), 2.85 (3H, s, Me of quinoline ring), 2.62 (3H, s, Me of pyridine ring). 13C{1H} NMR (100 MHz, DMSO-d6) δC/ppm 167.80 (COOH), 157.72 (C6′), 154.11 (C2), 153.86 (C2′), 147.00 (C10), 137.65 (C4′), 137.40 (C4), 137.23 (C9), 130.08 (C8), 127.91 (C7), 124.38 (C5), 124.21 (C5′), 123.34 (C6), 118.64 (C3), 118.19 (C3′), 24.20 (Me of pyridine ring), 17.97 (Me of quinoline ring).
General synthetic procedure of the methyl ester analogues
In a round bottom flask, 1 mmol of the 8-Mepqca (6′-Mepqca or 8,6′-Me2pqca) precursor was dispersed in MeOH (25 mL). To this solution, 1.2 mL of conc. H2SO4 was added dropwise, and the slightly yellow solution was refluxed overnight. The organic solvent was evaporated, and the residue was diluted with water (15 mL). The pH of the mixture was adjusted to ∼8–9 (30% w/w NaOH(aq)), leading to the precipitation of a white solid, which was filtered, washed with distilled H2O (2 × 5 mL), and dried in vacuo for 24 h. Finally, the solid was dried in a vacuum desiccator under P2O5.O)), 1595 (s, ν(CC)), 1587 (s, ν(CC)), 1455 (s, ν(CN)), 1238 (s), 1220 (s), 1140 (s, νas(O–CH3)), 797 (vs, γ(C–H)out-of-plane), 773 (vs, γ(C–H)out-of-plane), 733 (s, γ(C–H)out-of-plane). 1H NMR (500 MHz, CDCl3) δH/ppm 9.08 (1H, s, H3), 8.76 (1H, d, J = 10, H9), 8.46 (1H, d, J = 10, H3′), 8.23 (1H, d, J = 10, H6), 7.77 (2H, t, J = 10, H7/H4′), 7.65 (1H, t, J = 10, H8), 7.24 (1H, d, J = 10, H5′), 4.09 (3H, s, COOMe), 2.71 (3H, s, Me of pyridine ring). 13C{1H} NMR (126 MHz, CDCl3) δC/ppm 167.16 (COOMe), 158.25 (C6′), 156.17 (C2′), 155.03 (C2), 148.99 (C10), 137.35 (C4′), 135.82 (C4), 130.51 (C6), 129.87 (C7), 128.29 (C8), 125.67 (C9), 125.12 (C5), 124.10 (C5′), 120.60 (C3), 118.88 (C3′), 52.78 (COOMe), 24.75 (Me of pyridine ring).
O)), 1588 (s, ν(CC)), 1558 (s, ν(CC)), 1477 (s, ν(CN)), 1266 (s), 1246 (s), 1105 (s, νas(O–CH3)), 797 (vs, γ(C–H)out-of-plane), 770 (vs, γ(C–H)out-of-plane), 740 (s, γ(C–H)out-of-plane). 1H NMR (400 MHz, CDCl3) δH/ppm 9.06 (1H, s, H3), 8.75–8.71 (2H, m, H3′/H6′), 8.59 (1H, d, J = 8, H6), 7.87 (1H, br s, H4′), 7.61 (1H, d, J = 8, H8), 7.52 (1H, br s, H7), 7.36 (1H, br s, H5′), 4.05 (3H, s, COOMe), 2.92 (3H, s, Me of quinoline ring). 13C{1H} NMR (100 MHz, CDCl3) δC/ppm 167.24 (COOMe), 156.07 (C2′), 154.17 (C2), 149.26 (C6′), 147.86 (C10), 138.20 (C9), 137.06 (C4′), 136.14 (C4), 130.09 (C8), 128.17 (C7), 125.20 (C5), 124.34 (C5′), 123.48 (C6), 121.78 (C3′), 119.73 (C3), 52.69 (COOMe), 18.53 (Me of quinoline ring).
O)), 1592 (s, ν(CC)), 1561 (s, ν(CC)), 1456 (s, ν(CN)), 1241 (s), 1222 (s), 1114 (s, νas(O–CH3)), 806 (s, γ(C–Hout-of-plane)), 772 (vs, γ(C–Hout--of-plane)), 736 (s, γ(C–Hout-of-plane)). 1H NMR (400 MHz, CDCl3) δH/ppm 9.07 (1H, s, H3), 8.56 (1H, d, J = 8, H6), 8.52 (1H, d, J = 8 Hz, H3′), 7.75 (1H, t, J = 8, H4′), 7.61 (1H, d, J = 8, H8), 7.52 (1H, t, J = 8, H7), 7.22 (1H, d, J = 8, H5′), 4.07 (3H, s, COOMe), 2.92 (3H, s, Me of quinoline), 2.69 (s, 3H, Me of pyridine ring). 13C{1H} NMR (100 MHz, CDCl3) δC/ppm 167.46 (COOMe), 158.08 (C6′), 155.45 (C2), 154.58 (C2′), 147.88 (C10), 138.21 (C9), 137.18 (C4′), 136.17 (C4), 129.99 (C8), 127.99 (C7), 125.08 (C5), 123.88 (C5′), 123.46 (C6), 119.83 (C3), 118.77 (C3′), 52.71 (COOCH3), 24.76 (Me of pyridine ring), 18.54 (Me of quinoline ring).
Synthesis of copper(I) complexes 1–4
O)), 1600 (s, ν(CC)), 1575 (m, ν(CC)), 1473 (s, ν(CN)), 1249 (s, νs(C–O)), 844 (vs, ν3(P–F)), 799 (s, γ(C–H)out-of-plane), 777 (s, γ(C–H)out-of-plane), 557 (s, ν4(P–F)). UV-vis (Me2CO, 1.0 × 10−4 mol dm−3): λmax/nm 334 (ε/dm3 mol−1 cm−1 27200), 527 (6600). 1H NMR (500 MHz, Me2CO-d6) δH/ppm 9.20 (1H, br s, H3), 9.05 (1H, br s, H3′), 8.94 (1H, br s, H9), 8.77 (1 H, br s, H6′), 8.38 (1H, br s, H4′), 8.09 (1H, br s, H6), 7.79 (1H, br s, H5′), 7.71 (1H, br s, H8), 7.54 (1H, br s, H7), 2.67 (3.6H, s, Me of DMSO). 13C{1H} NMR (126 MHz, Me2CO-d6) δC/ppm 154.33 (COOH), 153.33 (C2), 152.78 (C2′), 150.33 (C6′), 147.33 (C10), 140.44 (C4), 139.72 (C4′), 132.18 (C7), 130.62 (C8), 129.93 (C6), 128.56 (C5′), 126.96 (C9), 124.63 (C3′), 124.39 (C5), 121.53 (C3), 40.66 (Me of DMSO). ESI-MS (MeOH, positive mode): m/z 563.08 [M–PF6]+ (calc. 563.08, base peak).
O)), 1600 (s, ν(CC)), 1575 (m, ν(CC)), 1473 (s, ν(CN)), 1249 (s, νs(C–O)), 844 (vs, ν3(P–F)), 799 (s, γ(C–H)out-of-plane), 777 (s, γ(C–H)out-of-plane), 557 (s, ν4(P–F)). UV-vis (Me2CO, 1.0 × 10−4 mol dm−3): λmax/nm 334 (ε/dm3 mol−1 cm−1 25300), 530 (6500). 1H NMR (500 MHz, Me2CO-d6) δH/ppm 9.22 (1H, s, H3), 8.95 (1H, d, J = 10, H3′), 8.90 (1H, d, J = 10, H9), 8.32 (1H, t, J = 10, H4′), 8.06 (1H, d, J = 10, H6), 7.77 (1H, d, J = 10, H5′), 7.73 (1H, t, J = 10, H8), 7.55 (2H, t, J = 10, H7), 2.62 (5.4H s, Me of DMSO), 2.26 (3H, s, Me of pyridine ring). 13C{1H} NMR (126 MHz, Me2CO-d6) δC/ppm 167.20 (COOH), 159.16 (C6′), 153.56 (C2), 152.12 (C2′), 147.14 (C10), 140.06 (C4′), 139.10 (C4), 132.43 (C7), 130.72 (C8), 129.54 (C6), 128.27 (C5′), 127.54 (C5), 127.08 (C9), 122.26 (C3′), 121.71 (C3), 41.14 (Me of DMSO), 25.18 (CH3). ESI-MS (MeOH, positive mode): m/z 592.10 [M + H–PF6]+ (calc. 591.10, [M–PF6]+, base peak).
O)), 1599 (s, ν(CC)), 1550 (m, ν(CC)), 1479 (s, ν(CN)), 1260 (s, νs(C–O)), 842 (vs, ν3(P–F)), 771 (s, γ(C–H)out-of-plane), 742 (m, γ(C–H)out-of-plane), 558 (s, ν4(P–F). UV-vis (Me2CO,1.0 × 10−4 mol dm−3): λmax/nm 335 (ε/dm3 mol−1 cm−1 14000), 519 (1800). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 8.94–8.74 (3H, m, H-pqca), 8.41 (2H, m, H-pqca), 7.68 (3H, m, H-pqca), 2.83 (3H, s, Me of quinoline).
O)), 1602 (s, ν(CC)), 1572 (m, ν(CC)), 1473 (s, ν(CN)), 1262 (s, νs(C–O)), 843 (vs, ν3(P–F)), 773 (s, γ(C–H)out-of-plane), 740 (m, γ(C–H)out-of-plane), 557 (s, ν4(P–F)). UV-vis (Me2CO, 1.0 × 10−4 mol dm−3): λmax/nm 334 (ε/dm3 mol−1 cm−1 27900), 506 (4000). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 9.81 (1H, s, H3), 8.80 (1H, d, J = 8, H3′), 8.71 (1H, br d, H6), 8.31 (1H, br t, H4′), 7.74–7.66 (3H, m, H5′/H8/H7), 2.70 (3H, s, Me of quinoline ring), 2.12 (3H, s, Me of pyridine ring overlaped with the residual protons of Me2CO-d6). 13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 166.52 (COOH), 158.76 (C6′), 154.34 (C2), 153.13 (C2′), 147.46 (C10), 140.56 (C4′), 139.12 (C4), 137.04 (C9), 133.91 (C8), 129.90 (C7), 127.82 (C5′), 125.41(C5), 124.93 (C6), 122.84 (C3′), 122.20 (C3), 24.36 (Me of pyridine ring), 19.46 (Me of quinoline ring).
Synthesis of [Cu(dmdcbpy)2][PF6]·3DMSO (5·3DMSO)
In a Schlenk tube, 18 mg (0.066 mmol) of dmdcbpy was dissolved in warm DMSO (5 mL) and then [Cu(MeCN)4][PF6] (12 mg, 0.033 mmol) was added. The color of the solution turned to red, and the mixture was stirred at ambient temperature for about 1 h. The solvent was pumped down under reduced pressure and a mixture of acetone/diethyl ether was added (1/20, v/v) to afford a dark red solid. The solid was filtered, washed once with a mixture of acetone/diethyl ether (1/30, v/v) and dried under vacuum at ambient temperature for 24 h. Yield: (0.024 g, 75%). The spectroscopic data of 5 are in accordance with those reported in the literature.13General synthesis of copper(I) complexes 6–8
A Schlenk tube was charged with two molar equivalents of the methyl ester analogue 6′-Mepqca (8-Mepqca or 8,6′-Me2pqca) and one equivalent of [Cu(MeCN)4][PF6], and the solids were degassed under vacuum for 10 minutes. Dry CH2Cl2 (10 mL) was then added, and the reaction mixture was stirred at ambient temperature for 1.5 h. Subsequently, the solvent was evaporated to dryness; the residue washed with diethyl ether (4 × 7 mL) and pentane (2 × 7 mL) and was dried under vacuum for 24 h, affording compounds 6–8 as analytically pure solids.O)), 1600 (s, ν(CC)), 1571 (m, ν(CC)), 1473 (s, ν(CN)), 1252 (s, νs(C–O)), 1147 (m, νas(O–CH3)), 840 (vs, ν3(P–F)), 794 (s, γ(C–H)out-of-plane), 775 (s, γ(C–H)out-of-plane), 558 (s, ν4(P–F)). UV-vis (Me2CO, 1.0 × 10−4 mol dm−3): λmax/nm 333 (ε/dm3 mol−1 cm−1 40200), 532 (9900). 1H NMR (500 MHz, CDCl3) δH/ppm 9.05 (1H, s, H3), 8.88 (1H, br s, H9), 8.62 (1H, br s, H3′), 8.19 (1H, br s, H4′), 7.77 (1H, br s, H6), 7.65 (1H, br s, H8), 7.55 (1H, br s, H5′), 7.49 (2H, t, J = 10, H7), 4.21 (3H, s, COOMe), 2.15 (3H, s, Me). 13C{1H} NMR (126 MHz, CDCl3) δC/ppm 165.86 (COOMe), 158.11 (C6′), 152.15 (C2), 150.88 (C2′), 146.35 (C10), 139.45 (C4′), 137.27 (C4), 131.85 (C7), 130.24 (C8), 128.40 (C6), 127.59 (C5′), 126.81 (C5), 126.36 (C9), 121.37 (C3′), 120.97 (C3), 53.58 (COOMe), 25.21 (Me of pyridine ring). ESI-MS (MeOH, positive mode): m/z 619.14 [M–PF6]+ (calc. 619.14, base peak).
O)), 1595 (s, ν(CC)), 1548 (m, ν(CC)), 1476 (s, ν(CN)), 1263 (s, νs(C–O)), 1105 (m, νas(O–CH3)), 841 (vs, ν3(P–F)), 771 (s, γ(C–H)out-of-plane), 764 (s, γ(C–H)out-of-plane), 557 (s, ν4(P–F)). UV-vis (CHCl3, 1.0 × 10−4 mol dm−3): λmax/nm 338 (ε/dm3 mol−1 cm−1 22700), 513 (2800). 1H NMR (400 MHz, CDCl3) δH/ppm 8.77 (1H, s, H3), 8.67 (1H, d, J = 8, H6), 8.55 (1H, d, J = 8, H3′), 8.17 (1H, t, J = 8, H4′), 7.96 (1H, br s, H6′), 7.57 (1H, t, J = 8, H7), 7.51–7.48 (2H, m, H8/H5′), 4.15 (3H, s, COOMe), 2.68 (3H, s, Me of pyridine ring). 13C{1H} NMR (100 MHz, CDCl3) δC/ppm 166.07 (COOMe), 152.85 (C2′), 152.50 (C2), 148.32 (C6′), 146.24 (C10), 139.15 (C4′), 137.83 (C4), 135.63 (C9), 133.13 (C8), 129.29 (C7), 126.87 (C5′), 126.68 (C5), 124.40 (C6), 124.11 (C3′), 120.58 (C3), 53.36 (COOMe), 19.19 (Me of quinoline ring).
O)), 1601 (s, ν(CC)), 1573 (m, ν(CC)), 1472 (s, ν(CN)), 1266 (s, νs(C–O)), 1113 (m, νas(O–CH3)), 841 (vs, ν3(P–F)), 773 (s, γ(C–H)out-of-plane), 749 (s, γ(C–H)out-of-plane), 557 (s, ν4(P–F)). UV-vis (CHCl3, 1.0 × 10−4 mol dm−3): λmax/nm 339 (ε/dm3 mol−1 cm−1 25100), 513 (3950). 1H NMR (400 MHz, CDCl3) δH/ppm 8.81 (1H s, H3), 8.69 (1H, d, J = 8, H6), 8.46 (1H, d, J = 8, H3′), 8.14 (1H, t, J = 8, H4′), 7.58 (1H, t, J = 8, H7), 7.52–7.47 (2H, m, H8/H5′), 4.16 (3H, s, COOMe), 2.58 (3H, s, Me of quinoline ring), 1.94 (3H, s, Me of pyridine ring). 13C{1H} NMR (CDCl3, 100 MHz, 298 K) δC/ppm 166.07 (COOMe), 157.78 (C6′), 153.13 (C2), 151.97 (C2′), 146.42 (C10), 139.58 (C4′), 138.54 (C4), 135.59 (C9), 133.90 (C8), 129.38 (C7), 127.32 (C5′), 126.91 (C5), 124.76 (C6), 122.24 (C3′), 121.31 (C3), 53.58 (COOMe), 24.59 (Me of pyridine ring), 19.56 (Me of quinoline ring).
Crystallography
The data collection on pqca was performed on a Nonius KappaCCD diffractometer by using graphite monochromated Mo-Kα radiation (λ = 0.7103 Å), generated by a sealed tube. The data collection for 6′-Mepqca, 1·DMSO, 2{2·Me2CO·0.5H2O}, 6·CHCl3·0.13H2O was done using a Bruker D8 Venture diffractometer with Cu-Kα radiation (λ = 1.5418 Å) generated from an Ils Helios Optics source. For 8-Mepqca, 4, 2{7·C5H12}·CHCl3 and 8, a Bruker X8 Kappa ApexII diffractometer was used. The diffractometers were equipped with a low-temperature device (Bruker Kryoflex I, and Oxford Cryostream 800er series, both at 100(2) K). Intensities were measured by fine-slicing x and u-scans and corrected for background, polarization, and Lorentz effects.71,72The structures were solved by intrinsic phasing methods implemented in Sheldrick's XT program and refined anisotropically by the least-squares procedure implemented in the SHELX program system.72,73 Hydrogen atoms were included using the riding model on the bound carbon atoms. ORTEP-type diagrams and structure analysis used Mercury v. 3.0.
Crystallographic data and refinement conditions for compounds 1–4 are summarized below
821 reflections, 2652 unique, Rint = 0.1397, Parameters = 173, Final R indexes [I > 2σ(I)] R1 = 0.0765, Final R indexes [all data] wR2 = 0.2094. CCDC 2184654.†
142 reflections, 2230 unique, Rint = 0.0989, Parameters = 184, Final R indexes [I > 2σ(I)] R1 = 0.0405, Final R indexes [all data] wR2 = 0.0988. CCDC 2184655.†
400 reflections, 2944 unique, Rint = 0.1041, Parameters = 234, Final R indexes [I > σ(I)] R1 = 0.0538, Final R indexes [all data] wR2 = 0.1432. CCDC 2184656.†
596 reflections, 12559 unique, Rint = 0.0738, Parameters = 1074, Final R indexes [I > 2σ(I)] R1 = 0.0623, Final R indexes [all data] wR2 = 0.1718. CCDC 2184657.†
447 reflections, 6873 unique, Rint = 0.1200, Parameters = 532, Final R indexes [I > 2σ(I)] R1 = 0.0964, Final R indexes [all data] wR2 = 0.2710. CCDC 2184658.†
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 14.8494(11), b = 16.0090(13), c = 18.9006(13) Å, U = 3934.8(5) Å3, Z = 2, Dc = 1.514 g cm−3, μ = 0.773 mm−1, T = 100 K. Total 89442 reflections, 18851 unique, Rint = 0.2108, Parameters = 1122, Final R indexes [I > 2σ(I)] R1 = 0.0906, Final R indexes [all data] wR2 = 0.2781. CCDC 2184659.†
827 reflections, 8115 unique, Rint = 0.0880, Parameters = 475, Final R indexes [I > 2σ(I)] R1 = 0.0436, Final R indexes [all data] wR2 = 0.1497. CCDC 2184660.†
:15) was cast upon the sensitized film (photoanode). Sputtered platinum on FTO glass was used as a counterelectrode. The active area of the DSSCs was set to 0.25 cm2.
, a = 14.8494(11), b = 16.0090(13), c = 18.9006(13) Å, U = 3934.8(5) Å3, Z = 2, Dc = 1.514 g cm−3, μ = 0.773 mm−1, T = 100 K. Total 89442 reflections, 18851 unique, Rint = 0.2108, Parameters = 1122, Final R indexes [I > 2σ(I)] R1 = 0.0906, Final R indexes [all data] wR2 = 0.2781. CCDC 2184659.†
827 reflections, 8115 unique, Rint = 0.0880, Parameters = 475, Final R indexes [I > 2σ(I)] R1 = 0.0436, Final R indexes [all data] wR2 = 0.1497. CCDC 2184660.†
:15) was cast upon the sensitized film (photoanode). Sputtered platinum on FTO glass was used as a counterelectrode. The active area of the DSSCs was set to 0.25 cm2.
The DSSCs were illuminated under simulated solar light (1 sun, 1000 W m−2) from a 300 W, Xe source in combination with AM 1.5G optical filters (Oriel). The active area was set at 0.152 cm2, using a large black mask in front of the cells. Current density–voltage (J–V) measurements (under dark and light conditions) were recorded using linear sweep voltammetry on an Autolab PGSTAT30 potentiostat, working in a two-electrode mode, at a scan rate of 20 mV s−1.
Conclusions
In this paper, a series of new sterically demanding pyridine-quinoline ligands has been reported. These compounds were specially tailored to sterically protect the copper(I) metal centre of the homoleptic complexes 1–4 and 6–8, with carboxylic acid and methyl ester groups respectively in their periphery. Methyl substitution at the 6′ position efficiently protects the copper metal centre as indicated by the increased stability of 2 (36 days in acetone solution), checked by 1H NMR spectroscopy, compared with the non-substituted analogue 1 (14 days). The new copper(I) complexes have been applied as photosensitizers in DSSCs.Chemisorption of complexes 1, 2 onto the TiO2 surface became apparent by UV-vis and FT-IR spectroscopy, while the surface characteristics of dye 2 sensitized films were studied by Raman spectroscopy, implying weak electronic coupling of 2 onto the semiconductor surface. The photovoltaic performances of solar cell devices gave efficiencies ranging from 0.1% to 1.20%, while complexes with methyl esters did not perform efficiently as photosensitizers in DSSCs. After optimization of the photoanodes, a combination of dye 2 and a three-component anode (compact layer/opaque layer/scattering layer) afforded an overall conversion of η = 1.20%, higher than that of the 5-cell reference, which in our hands showed η = 1.05%. As a result, the 2-based device may be considered as a promising candidate, appropriate for cost-efficient building-integrated outdoor applications in DSSCs. Based on these results, the judicious design and development of some new copper(I) based molecular antennas is underway, rendering them candidates for the fabrication of highly performing photovoltaic devices based on DSSC technology.
Author contributions
Anastasios Peppas: investigation; formal analysis; writing – original draft. Demetrios Sokalis: investigation; formal analysis; Dorothea Perganti: investigation; formal analysis; Gregor Schnakenburg: resources; Polycarpos Falaras: supervision; resources; Athanassios I. Philippopoulos: conceptualization; funding acquisition; writing – review & editing; supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. I. Philippopoulos would like to thank Prof. Dr A. C. Filippou of the Chemistry Department of the University of Bonn for the elemental analyses and Mrs Charlotte Rödde from the X-ray Department of the same Institute. Dr A. Peppas would like to thank the University of Bonn for financial support within the programme entitled: “Forschungs stipendium aus Mitteln des Sonder forschungs bereichs (SFB) 813 “Chemie an Spinzentren”. This research work was supported by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), under the HFRI PhD Fellowship grant (GA. no. 2055/14475 for the Ph.D. candidate A. Peppas). This research has also been co-financed in part by the Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)-Research Funding Program: THALES, investing in knowledge society through the European Social Fund. Project title: “Innovative materials for nanocrystalline solar cells” (MIS: 377756). P. Falaras acknowledges co-financing of this research by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code:T1EDK-03547, MIS 5033808).References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 73 CrossRef.
- M. Grätzel, J. Photochem. Photobiol., A, 2004, 164, 3 CrossRef.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, Md. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, Md. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. Fujisawab and M. Hanaya, Chem. Commun., 2015, 51, 15894 RSC.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 664 CrossRef PubMed.
- S. Gauthier, B. Caro, F. Robin-Le Guen, N. Bhuvanesh, J. A. Gladysz, L. Wojcik, N. Le Poul, A. Planchat, Y. Pellegrin, E. Blart, D. Jacquemin and F. Odobel, Dalton Trans., 2014, 43, 11233 RSC.
- G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos and P. Falaras, Coord. Chem. Rev., 2011, 255, 2602 CrossRef CAS.
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386 RSC.
- N. Robertson, ChemSusChem, 2008, 1, 977 CrossRef CAS PubMed.
- N. Alonso-Vante, J.-F. Nierengarten and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1994, 1649 RSC.
- T. Bessho, E. C. Constable, M. Graetzel, A. Hernandez Redondo, C. E. Housecroft, W. Kylberg, M. K. Nazeeruddin, M. Neuburger and S. Schaffner, Chem. Commun., 2008, 3717 RSC.
- (a) E. C. Constable, A. H. Redondo, C. E. Housecroft, M. Neuburger and S. Schaffner, Dalton Trans., 2009, 6634 RSC; (b) B. Bozic-Weber, E. C. Constable and C. E. Housecroft, Coord. Chem. Rev., 2013, 257, 3089 CrossRef CAS.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69 CrossRef CAS.
- A. K. Ichinaga, J. R. Kirchhoff, D. R. McMillin, C. O. Dietrich-Buchecker, P. A. Marnot and J. P. Sauvage, Inorg. Chem., 1987, 26, 4290 CrossRef CAS.
- B. Bozic-Weber, E. C. Constable, C. E. Housecroft, P. Kopecky, M. Neuburger and J. A. Zampese, Dalton Trans., 2011, 40, 12584 RSC.
- S. Y. Brauchli, F. J. Malzner, E. C. Constable and C. E. Housecroft, RSC Adv., 2015, 5, 48516 RSC.
- F. J. Malzner, S. Y. Brauchli, E. C. Constable, C. E. Housecroft and M. Neuburger, RSC Adv., 2014, 4, 48712 RSC.
- (a) E. Schönhofer, B. Bozic-Weber, C. J. Martin, E. C. Constable, C. E. Housecroft and J. A. Zampese, Dyes Pigm., 2015, 115, 154 CrossRef; (b) G. Risi, M. Becker, C. E. Housecroft and E. C. Constable, Molecules, 2020, 25, 1528 CrossRef CAS.
- M. Sandroni, M. Kayanuma, A. Planchat, N. Szuwarski, E. Blart, Y. Pellegrin, C. Daniel, M. Boujtita and F. Odobel, Dalton Trans., 2013, 42, 10818 RSC.
- (a) M. Sandroni, L. Favereau, A. Planchat, H. Akdas-Kilig, N. Szuwarski, Y. Pellegrin, E. Blart, H. Le Bozec, M. Boujtita and F. Odobel, J. Mater. Chem. A, 2014, 2, 994 RSC; (b) M. S. Yann Pellegrin, E. Blart, A. Planchat, M. Evain, N. C. Bera, M. Kayanuma, M. Sliwa, M. Rebarz, O. Poizat, C. Daniel and F. Odobel, Inorg. Chem., 2011, 50, 11273 CrossRef.
- Y. Liu, S.-C. Yiu, C.-L. Ho and W.-Y. Wong, Coord Chem Rev, 2018, 375, 514 CrossRef CAS.
- (a) B. N. Mongal, A. Pal, T. K. Mandal, J. Datta and S. Naskar, Polyhedron, 2015, 102, 615 CrossRef CAS; (b) K. A. Wills, H. J. Mandujano-Ramirez, G. Merino, D. Mattia, T. Hewat, N. Robertson, G. Oskam, M. D. Jones, S. E. Lewis and P. J. Cameron, RSC Adv., 2013, 3, 23361 RSC; (c) R. Ilmi, I. Juma Al-busaidi, A. Haque and M. S. Khan, J. Coord. Chem., 2018, 71, 3045 CrossRef CAS.
- E. Kolovou, A. Peppas, N. Zacharopoulos, K. Koukoulakis, E. Bakeas, G. Schnakenburg and A. I. Philippopoulos, Inorg. Chem. Commun., 2018, 92, 64 CrossRef CAS.
- A. Philippopoulos, E. Chatzivasiloglou, O. Igglessi-Markopoulou, V. Likodimos, G.-C. Konti and P. Falaras, J. Coord. Chem., 2012, 65, 2535 CrossRef CAS.
- G. Konti, E. Chatzivasiloglou, V. Likodimos, G. Kantonis, A. G. Kontos, A. I. Philippopoulos and P. Falaras, Photochem. Photobiol. Sci., 2009, 8, 726 CrossRef CAS PubMed.
- A. Peppas, E. Papadaki, G. Schnakenburg, V. Magrioti and A. I. Philippopoulos, Polyhedron, 2019, 171, 412 CrossRef CAS.
- N. Zacharopoulos, E. Kolovou, A. Peppas, K. Koukoulakis, E. Bakeas, G. Schnakenburg and A. I. Philippopoulos, Polyhedron, 2018, 154, 27 CrossRef CAS.
- D. M. Classen, Inorg. Chem., 1976, 15, 3166 CrossRef.
- (a) A. Peppas Synthesis and characterization of homoleptic Copper(I) complexes. Application in third generation solar cells (Gratzel type). Master thesis, National and Kapodistrian University of Athens, Greece, 2015. Persistent URL: https://pergamos.lib.uoa.gr/uoa/dl/object/1320032 Search PubMed; (b) A. A. Farah, J. G. C. Veinot, M. Najman and W. J. Pietro, Macromol. Sci., Pure Appl. Chem., 2000, 37, 1507 CrossRef.
- A. Peppas “Synthesis and characterization of complexes of Cu(I) compounds for use in nanocrystalline solar cells”. Doctoral Dissertation, National and Kapodistrian University of Athens, Athens, Greece, 2019. Persistent URL: https://pergamos.lib.uoa.gr/uoa/dl/object/2884748 Search PubMed.
- G. J. Atwell, B. C. Baguley and W. A. Denny, J. Med. Chem., 1989, 32, 396 CrossRef CAS PubMed.
- R. F. Fernandes, P. H. F. Stroppa, G. R. Ferreira, A. D. da Silva, H. G. M. Edwards and L. F. C. de Oliveira, Vib. Spectrosc., 2016, 86, 128 CrossRef CAS.
- A. A. Farah and W. J. Pietro, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, E57, o677–o678 CrossRef.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- S. J. Grabowski, Crystals, 2016, 6, 59 CrossRef.
- (a) S. Sakaki, T. Kuroki and T. Hamada, J. Chem. Soc., Dalton Trans., 2002, 1649 Search PubMed; (b) A. Colombo, C. Dragonetti, D. Roberto, A. Valore, P. Biagini and F. Melchiorre, Inorg. Chim. Acta, 2013, 407, 204 CrossRef CAS.
- A. Milton, F. Benial, V. Ramakrishnan and R. Murugesan, Spectrochim. Acta, Part A, 2002, 58, 1703 CrossRef.
- T. E. Hewat, J. Yellowlees and N. Robertson, Dalton Trans., 2014, 43, 4127 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- M. G. Fraser, H. van der Salm, S. A. Cameron, A. G. Blackman and K. C. Gordon, Inorg. Chem., 2013, 52, 2980 CrossRef CAS PubMed.
- S. Keller, M. Alkan-Zambada, A. Prescimone, E. C. Constable and C. E. Housecroft, Crystals, 2020, 10, 255 CrossRef CAS.
- T. Steiner, Crystallogr. Rev., 2003, 9, 177 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113 RSC.
- Y. Liu, S.-C. Yiu, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2018, 375, 514 CrossRef CAS.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2 CrossRef CAS.
- A. I. Philippopoulos, A. Terzis, C. P. Raptopoulou, V. J. Catalano and P. Falaras, Eur. J. Inorg. Chem., 2007, 5633 CrossRef CAS.
- B. Bozic-Weber, S. Y. Brauchli, E. C. Constable, S. O. Furer, C. E. Housecroft, F. J. Malzner, I. A. Wright and J. A. Zampese, Dalton Trans., 2013, 42, 12293 RSC.
- J. W. Kee, Y. Y. Ng, S. A. Kulkarni, S. K. Muduli, K. Xu, R. Ganguly, Y. Lu, H. Hirao and H. S. Soo, Inorg. Chem. Front., 2016, 3, 651 RSC.
- K. A. Wills, H. J. Mandujano-Ramirez, G. Merino, D. Mattia, T. Hewat, N. Robertson, G. Oskam, M. D. Jones, S. E. Lewis and P. J. Cameron, RSC Adv., 2013, 3, 23361 RSC.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819 CrossRef CAS PubMed.
- Y. Zhou, Q. He, Y. Yang, H. Zhong, C. He, G. Sang, W. Liu, C. Yang, F. Bai and Y. Li, Adv. Funct. Mater., 2008, 18, 3299 CrossRef CAS.
- J. Jiang, J. A. Spies, J. R. Swierk, A. J. Matula, K. P. Regan, N. Romano, B. J. Brennan, R. H. Crabtree, V. S. Batista, C. A. Schmuttenmaer and G. W. Brudvig, J. Phys. Chem. C, 2018, 122, 13529 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- P. Falaras, Sol. Energy Mater. Sol. Cells, 1998, 53, 163 CrossRef CAS.
- C. Deng, P. F. James and P. V. Wright, J. Mater. Chem., 1998, 8, 153 RSC.
- G. C. Vougioukalakis, T. Stergiopoulos, A. G. Kontos, E. K. Pefkianakis, K. Papadopoulos and P. Falaras, Dalton Trans., 2013, 42, 6582 RSC.
- L. Zhu, Z. Yulong, X. P. Lin, X. Q. Gu and Y. Qiang, Superlattices Microstruct., 2014, 65, 152 CrossRef CAS.
- A. Burke, S. Ito, H. Snaith, U. Bach, J. Kwiatkowski and M. Grätzel, Nano Lett., 2008, 8, 977 CrossRef CAS.
- J. He, A. Hagfeldt, S.-E. Lindquist, H. Grennberg, F. Korodi, L. Sun and B. Åkermark, Langmuir, 2001, 17, 2743 CrossRef CAS.
- S. C. Pradhan, A. Hagfeldt and S. Soman, J. Mater. Chem. A, 2018, 6, 22204 RSC.
- A. Colombo, C. Dragonetti, D. Roberto and F. Fagnani, Molecules, 2021, 26, 194 CrossRef CAS.
- G. J. Kubas, B. Monzyk and A. L. Crumblis, Inorg. Synth., 2007, 28, 68 CrossRef.
- M. Y. Goikhman, I. V. Podeshvo, N. L. Loretsyan, T. D. Anan'eva, R. Y. Smyslov, T. N. Nekrasova, M. A. Smirnov, E. N. Popova and A. V. Yakimanskii, Polym. Sci., Ser. B, 2011, 53, 89 CrossRef CAS.
- T. Sandmeyer, Helv. Chim. Acta, 1919, 2, 234 CrossRef CAS.
- P. J. Montoya-Pelaez, Y.-S. Uh, C. Lata, M. P. Thompson, R. P. Lemieux and C. M. Crudden, J. Org. Chem., 2006, 71, 5921 CrossRef CAS PubMed.
- G. Black, E. Depp and B. B. Corson, J. Org. Chem., 1949, 14, 14 CrossRef CAS PubMed.
- F. J. McCarty, C. H. Tilford and M. G. Van Campen, J. Am. Chem. Soc., 1957, 79, 472 CrossRef CAS.
- H. B. Amin and R. Taylor, J. Chem. Soc., Perkin Trans. 1, 1979, 2, 624 RSC.
- V.-M. Mukkala and J. J. Kankare, Helv. Chim. Acta, 1992, 75, 1578 CrossRef CAS.
- Bruker SADABS-2014/5 (Bruker AXS Karlsruhe; 2014).
- G. M. Sheldrick, Acta Crystallogr., 2008, 3, A71 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, 3, C71 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and UV-vis spectra, cyclic voltammograms, crystal data, micro-Raman and IR spectra of the sensitized photoelectrodes. CCDC 2184654–2184660. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02382b |
| This journal is © The Royal Society of Chemistry 2022 |