
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid and efficient synthesis of formamidines in a catalyst-free and solvent-free system†
Zitong Zhoua,
Yu Zhaoa,
Donghua Zhoua,
Li Lia,
Hui Luo*ab,
Liao Cui*a and
Weiguang Yang *ac
*ac
aGuangdong Key Laboratory for Research and Development of Natural Drugs, The Marine Biomedical Research Institute, Guangdong Medical University, Zhanjiang, 524023, China. E-mail: luohui@gdmu.edu.cn; cuiliao@163.com; 09ywg@163.com
bSouthern Marine Science and Engineering Guangdong Laboratory (Zhanjiang), Zhanjiang, Guangdong 524023, China
cThe Marine Biomedical Research Institute of Guangdong Zhanjiang, Zhanjiang, Guangdong 524023, China
First published on 18th October 2021
Abstract
An operationally rapid and efficient synthesis of N-sulfonyl formamidines that proceeds under mild conditions was achieved by reaction of a mixture of an amine, a sulfonyl azide, and a terminal ynone under catalyst-free and solvent-free conditions. Terminal ynones provide the C source to formamidines via complete cleavage of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C.
C.
Amidines are ubiquitous in nature and have critical roles in pharmaceutical and agrochemical industries.1 In addition, amidine derivatives have been documented as key scaffolds in drug discovery2 and crucial fragments in numerous biologically relevant molecules.3 As a unique class of amidines, N-sulfonyl formamidines are widespread in insecticidal/acaricidal drugs4 or biologically active molecules.5 As shown in Fig. 1, acaricidal drugs include ebrotidine (a),6 inhibitors of steroid sulfatase (b),7 antagonists/inverse agonists (c)8 and antimicrobial agents (d).9 Therefore, development of novel, efficient and sustainable methods for the synthesis of N-sulfonyl formamidines has been a consistent focus of synthetic chemists.
 | ||
| Fig. 1 Some formamidines drugs or drug candidates. | ||
Four main synthetic methods for preparation of N-sulfonyl formamidines have been reported. The first is the condensation of sulfonamides with formamides using an oxidant to generate a reactive electrophilic intermediate (Scheme 1a).10 The second method is the reaction of sulfonamides with amines involving oxidation of the amine to an iminium cation generated electrochemically11,12 or by reaction of Cu(II) (generated in situ by aerial oxidation of CuBr at 100 °C) (Scheme 1b).13 The third method is cycloaddition of a sulfonyl azide to an enamine intermediate generated by oxidation of a trialkylamine followed by retrocycloaddition to generate a diazoalkane concomitantly (Scheme 1c).14–16 The fourth method is a three-component reaction, reported previously by our research team, between a sulfonyl azide, a terminal ynone (3-butyn-2-one) and an amide involving the formation of a highly reactive intermediate N-sulfonyl acetylketenimine reacting with the amide in a [2 + 2] cycloaddition followed by a [2 + 2] cycloreversion with the concomitant generation of 4-oxo-3-buren-2-one (Scheme 1d).17 Recently, Wusiman and colleagues proposed a straightforward one-pot multicomponent approach, but heating and solvent addition are required.18 Each of the methods described above has considerable advantages, including mild reaction conditions and high efficiency in the assembly of N-sulfonyl formamidines. However, their synthetic utility is impaired by the requirement of environmentally unfriendly solvents, metal catalysts and/or relatively long reaction times or high temperatures.
 | ||
| Scheme 1 Synthesis of N-sulfonylformamidines. | ||
Scheme 1 reveals an operationally simple protocol involving stirring a mixture of an amine, a sulfonyl azide, and a terminal ynone without a catalyst or solvent.
Our investigations began with examination of the synthesis of the parent and previously unreported system 4-methyl-N-(morpholinomethylene)benzenesulfonamide 4aa via morpholine 1a, p-tosyl azide 2a and but-3-yn-2-one 3a. We first detected this unconventional product 4aa in a CuAAC/ring-opening reaction system with a low yield (8%, Table 1, entry 1). After screening using AgTFA/MeCN as a catalyst condition, product 4aa was obtained in high yield of 90% (Table 1, entry 2). If AgTFA or MeCN were not added, the yield of the product decreased (Table 1, entries 3 and 4). To our delight, the reaction delivering product 4aa in highest yield was catalyst-free under a neat condition in 2 min (95%, Table 1, entry 5). Curiously, compound 4aa did not contain all the fragments of but-3-yn-2-one 3a. Therefore, different terminal alkynes were detected. Additional screening revealed that the terminal ynones 3b–3d gave comparable yields (Table 1, entries 6–8). For example, ethyl propiolate 3b gave a yield of 90% in 3 min and tert-butyl propiolate 3c gave a yield of 92% in 5 min. Phenylacetylene 3d was used, but it failed to produce the desired product (Table 1, entry 9). Considering atomic economy, speed and efficiency, the “parent reaction” was defined to the condition of Table 1, entry 5.
|
||||
|---|---|---|---|---|
| Entry | Alkyne 3 | Cat./solvent | Time | Yieldb (%) 4aa |
| a Reaction conditions: 1a (0.1 mmol), Cat. (2.0 mol%) in the solvent (3 mL) was added slowly to 2a (1.8 equiv.) and 3 (1.8 equiv.) in order, then stirred at room temperature for the corresponding time (detected by TLC or solidification).b Isolated yields. | ||||
| 1 | 3a | CuI/MeCN | 4 h | 8 |
| 2 | 3a | AgTFA/MeCN | 4 h | 90 |
| 3 | 3a | —/MeCN | 12 h | 79 |
| 4 | 3a | AgTFA/— | 1 min | 56 |
| 5 | 3a | —/— | 2 min | 95 |
| 6 | 3b | —/— | 3 min | 90 |
| 7 | 3c | —/— | 5 min | 92 |
| 8 | 3d | —/— | 6 min | 70 |
| 9 | 3e | —/— | 12 h | 0 |

Under the optimized conditions, the capacity of this reaction to affect the coupling of a range of different substrates was investigated. As revealed in Table 2, various structurally and electronically distinct amines engaged readily in the desired reaction to give the anticipated products (4aa–4ax) in modest-to-good yields. Secondary amines of alkyl groups proved more effective substrates than their aromatic counterparts (4aa–4ae, 75–98% yield vs. 4af–4am, 26–98% yield) with the best “performing” member of the latter group being 1,2,3,4-tetrahydroquinoline and affording the anticipated product 4ah. Likewise, the structure of primary amines could be varied to a reasonable degree to elicit products (4aj–4ax), but showed lower efficiency than that of secondary amines. Most aliphatic primary amines elicited modest yields whereas the substrate benzylamine gave the products 4ak in high yield of 94%. Electron-withdrawing and electron-donating groups attached to the aromatic ring of aromatic primary amines of the general form 4 (e.g., methyl, halogen, ester, 2-naphthyl and nitro) were tolerated, as evidenced by formation of the anticipated products, 4ao–4ax, in serviceable yield. However, amines derived from tertiary amines, heterocyclic and inorganic ammonium salts failed to transfer into the corresponding products under the optimized conditions.
| a Reaction conditions: 1 (0.1 mmol) and 2a (1.8 equiv.) were added, then 3a (1.8 equiv.) was added slowly, and stirred at room temperature for the corresponding time (detected by TLC or solidification).b Reaction conditions: 1 (0.1 mmol) and AgTFA (2 mol%) in MeCN (3 mL) was added to 2a (1.8 equiv.) and 3a (1.8 equiv.), then stirred at 60 °C for the corresponding time (detected by TLC). |
|---|
 |
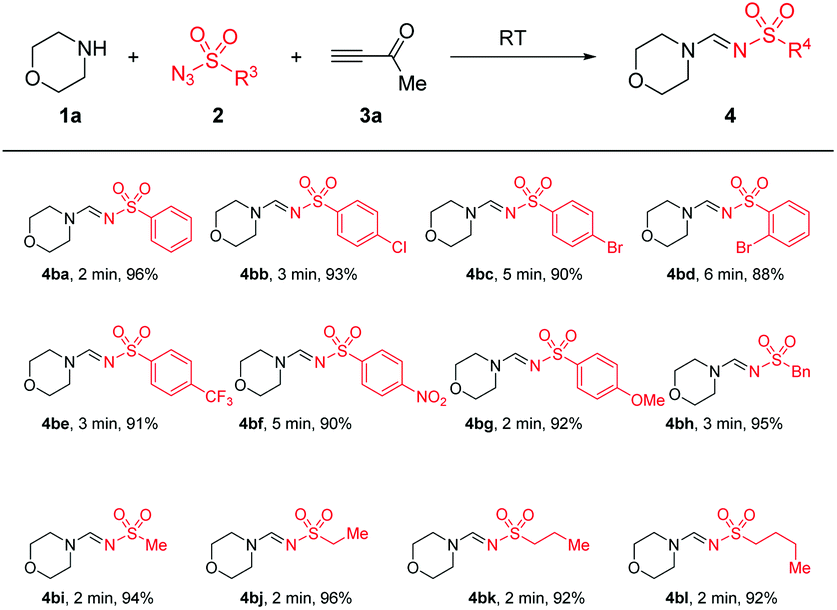
Next, the scope and limitation of substrates sulfonyl azides 2 were tested. Importantly, the substrates sulfonyl azides showed rapidity and efficiency in this reaction. Change of R3 by aromatic or aliphatic substituents, such as phenyl, -(4-CF3C6H4), -(4-NO2C6H4), -(4-OMeC6H4), -Me, -n−Bu or benzyl, enabled the reaction to run smoothly to give the anticipated products (4ba–4bl) in good yields of 88–96%. All products were produced mainly in the E-syn conguration (for details please see the spectra in the ESI†) (Table 3).
| a Reaction conditions: 1a (0.1 mmol) and 2 (1.8 equiv.) were added, then 3a (1.8 equiv.) was added slowly, and stirred at room temperature for the corresponding time (detected by TLC or solidification). |
|---|
 |
According to recent reports of complete cleavage of CC19 and the reaction characteristics of ynones,20 further experiments were carried out to gain deeper understanding of the mechanistic pathway (Scheme 2). We undertook the reaction of 2a with 4-morpholinobut-3-en-2-one 5, which was synthesized by 1a and 3a, under the standard conditions. The corresponding products 4aa were generated with a yield of 75%. The desired product could not be obtained without terminal ynones. Taken together, these results implied that the terminal ynones provided the C source to formamidines via complete cleavage of CC, and that compound 5 might be an intermediate.
 | ||
| Scheme 2 Investigation of the reaction mechanism. | ||
Based on the results stated above and related precedent,19,21 a plausible mechanism for the formation of N-sulfonyl formamidines (4aa) from precursors 1a, 2a and 3a is shown in Scheme 3. First, the substrates 1a and 3a are expected to take Michael addition to form the intermediate 4-morpholinobut-3-en-2-one 5. This species is captured by tosyl azide 2a by participating in the [3 + 2] cycloadditions to generate the 1,2,3-triazoline intermediate 6, which itself undergoes retro-[3 + 2]-cycloaddition to yield the product 4aa, with elimination of the side-product 1-diazopropan-2-one 7.21
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
We developed an operationally rapid and efficient reaction for preparing N-sulfonyl formamidines from a mixture of mines, sulfonyl azides and terminal ynones with catalyst-free, solvent-free, mild conditions and a wide scope of substrates. From a mechanistic perspective, the terminal ynones have a critical role in cleavage, and provide the C source to formamidines via complete cleavage of CC. This methodology appears quite flexible and can generate forms of the title products that will be particularly useful in, for example, drug-development studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the following for support: Applied Basic Research Fund project of Guangdong Province of China (2019A1515110918); Medical Scientific Research Foundation of Guangdong Province (A2021037 and A2020202); Key Discipline Construction Project of Guangdong Medical University (4SG21004G); Innovation and Entrepreneurship Team Leads the Pilot Program of Zhanjiang (2020LHJH005); Science and Technology Program of Guangdong Province (2019B090905011).References
- (a) V. P. Charpe, A. Ragupathi, A. Sagadevan and K. C. Hwang, Green Chem., 2021, 23, 5024 RSC; (b) A. A. Aly, S. Brase and M. A.-M. Gomaa, ARKIVOC, 2018, 6, 85 Search PubMed; (c) R. Nazzareno, F. Marialuigia, M. Cristina, P. Roberto and A. Rosa, Curr. Enzyme Inhib., 2016, 12, 30 Search PubMed; (d) D. Oehlich, H. Prokopcova and H. J. M. Gijsen, Bioorg. Med. Chem. Lett., 2014, 24, 2033 CrossRef PubMed; (e) M. Cristina, F. Marialuigia and A. Rosa, Mini-Rev. Med. Chem., 2013, 13, 1305 CrossRef PubMed; (f) J. Y. Quek, T. P. Davis and A. B. Lowe, Chem. Soc. Rev., 2013, 42, 7326 RSC; (g) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109 RSC; (h) E. D. Anderson and D. L. Boger, J. Am. Chem. Soc., 2011, 133, 12285 CrossRef CAS PubMed; (i) G. M. Castanedo, P. S. Seng, N. Blaquiere, S. Trapp and S. T. Staben, J. Org. Chem., 2011, 76, 1177 CrossRef CAS PubMed; (j) Y. Ohta, Y. Tokimizu, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2010, 12, 3963 CrossRef CAS PubMed; (k) B. K. Singh, Int. J. ChemTech Res., 2009, 1, 250 CAS; (l) J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219 CrossRef CAS.
- (a) L. Pticek, L. Hok, P. Grbcic, F. Topic, M. Cetina, K. Rissanen, S. K. Pavelic, R. Vianello and L. Racane, Org. Biomol. Chem., 2021, 19, 2784 RSC; (b) L. Pticek, L. Hok, P. Grbcic, F. Topic, M. Cetina, K. Rissanen, S. K. Pavelic, R. Vianello and L. Racane, J. Am. Chem. Soc., 2019, 141, 15240 CrossRef PubMed; (c) J. H. K. Rozenfeld, E. L. Duarte, T. R. Oliveira and M. T. Lamy, Biophys. Rev., 2017, 9, 633 CrossRef CAS PubMed; (d) D. Schade, J. Kotthaus, L. Riebling, J. Kotthaus, H. Müller-Fielitz, W. Raasch, O. Koch, N. Seidel, M. Schmidtke and B. Clement, J. Med. Chem., 2014, 57, 759 CrossRef CAS PubMed; (e) X. Chen, B. A. Orser and J. F. MacDonald, Eur. J. Pharmacol., 2010, 648, 15 CrossRef CAS PubMed.
- (a) K. D. Veeranna, K. K. Das and S. Baskaran, Org. Biomol. Chem., 2021, 19, 4054 RSC; (b) F. Orofino, G. I. Truglio, D. Fiorucci, I. D'Agostino, M. Borgini, F. Poggialini, C. Zamperini, E. Dreassi, L. Maccari, R. Torelli, C. Martini, M. Bernabei, J. F. Meis, N. K. Khandelwal, R. Prasad, M. Sanguinetti, F. Bugli and M. Botta, Int. J. Antimicrob. Agents, 2020, 55, 105865 CrossRef CAS PubMed; (c) W. Zhang, J. Chu, A. M. Cyr, H. Yueh, L. E. Brown, T. T. L. Wang, J. Pelletier and J. A. Porco, J. Am. Chem. Soc., 2019, 141, 12891 CrossRef CAS PubMed; (d) Z. Zhang, B. Huang, G. Qiao, L. Zhu, F. Xiao, F. Chen, B. Fu and Z. Zhang, Angew. Chem., Int. Ed., 2017, 56, 4320 CrossRef CAS PubMed; (e) S. Arya, N. Kumar, P. Roy and S. M. Sondhi, Eur. J. Med. Chem., 2013, 59, 7 CrossRef CAS PubMed.
- (a) M. Takata, S. Misato, F. Ozoe and Y. Ozoe, Pest Manage. Sci., 2020, 76, 3720 CrossRef CAS PubMed; (b) M. A. I. Ahmed and F. Matsumura, J. Med. Entomol., 2012, 49, 1405 CrossRef CAS PubMed; (c) M.-Y. Liu and J. F. W. Plapp Jr, J. Econ. Entomol., 1990, 83, 2181 CrossRef CAS PubMed; (d) R. M. Hollingworth, Environ. Health Perspect., 1976, 14, 57 CrossRef CAS PubMed.
- (a) J. D. Pino, P. V. Moyano-Cires, M. J. Anadon, M. J. Diaz, M. Lobo, M. A. Capo and M. T. Frejo, Chem. Res. Toxicol., 2015, 28, 1073 Search PubMed; (b) M. A. I. Ahmed, C. F. A. Vogel and F. Matsumura, Pestic. Biochem. Physiol., 2015, 120, 57 CrossRef CAS PubMed.
- (a) R. J. Andrade, M. I. Lucena, R. Martin-Vivaldi, M. C. Fernandez, F. Nogueras, G. Pelaez, A. Gomez-Outes, M. D. Garcia-Escano, V. Bellot, A. Hervás, F. Cardenas, F. Bermudez, M. Romero and J. Salmerón, J. Hepatol., 1999, 31, 641 CrossRef CAS PubMed; (b) B. L. Slomiany, J. Piotrowski and A. Slomiany, Arzneim.-Forsch., 1997, 47, 475 CAS; (c) P. C. Konturek, T. Brzozowski, S. I. Konturek, M. Marquez, J. Torres and J. A. Ortiz, Arzneim.-Forsch., 1997, 47, 578 CAS.
- H. A. M. Hejaz, L. W. L. Woo, A. Purohit, M. J. Reed and B. V. L. Potter, Bioorg. Med. Chem., 2004, 12, 2759 CrossRef CAS PubMed.
- H. Heitsch, R. H. Becker and H. W. Kleemann, Bioorg. Med. Chem., 1997, 5, 673 CrossRef CAS PubMed.
- M. S. El-Gaby, G. A. E.-H. Ali, A. A. El-Maghraby, M. T. A. El-Rahman and M. H. Helal, Eur. J. Med. Chem., 2009, 44, 4148 CrossRef CAS PubMed.
- (a) M. Gazvoda, M. Kočevar and S. Polanc, Eur. J. Org. Chem., 2013, 2013, 5381 CrossRef CAS; (b) S. Chen, Y. Xu and X. Wan, Org. Lett., 2011, 13, 6152 CrossRef CAS PubMed; (c) G. R. Pettit and R. E. Kadunce, J. Org. Chem., 1962, 27, 4566 CrossRef CAS.
- J. Chen, W. Long, S. Fan, Y. Yang and X. Wan, Chem. Commun., 2017, 53, 13256 RSC.
- B. Huang, C. Yang, J. Zhou and W. Xia, Chem. Commun., 2020, 56, 5010 RSC.
- Q. Gou, Z. Liu, T. Cao, X. Tan, W. Shi, M. Ran, F. Cheng and J. Qin, J. Org. Chem., 2020, 85, 2092 CrossRef CAS PubMed.
- (a) B. Kaboudin, S. Torabi, F. Kazemi and H. Aoyamab, RSC Adv., 2020, 10, 26701 RSC; (b) A. Rouzi, R. Hudabaierdi and A. Wusiman, Tetrahedron, 2018, 74, 2475 CrossRef CAS; (c) S. Shojaei, Z. Ghasemi and A. Shahrisa, Tetrahedron Lett., 2017, 58, 3957 CrossRef CAS; (d) X. Xu, Z. Ge, D. Cheng, L. Ma, C. Lu, Q. Zhang, N. Yao and X. Li, Org. Lett., 2010, 12, 897 CrossRef CAS PubMed; (e) X. Xu, X. Li, L. Ma, N. Ye and B. Weng, J. Am. Chem. Soc., 2008, 130, 14048 CrossRef CAS PubMed.
- (a) J. Gui, H. Xie, H. Jiang and W. Zeng, Org. Lett., 2019, 21, 2804 CrossRef CAS PubMed; (b) L. Zhang, J.-H. Su, S. Wang, C. Wan, Z. Zha, J. Du and Z. Wang, Chem. Commun., 2011, 47, 5488 RSC; (c) S. Wang, Z. Wang and X. Zheng, Chem. Commun., 2009, 47, 7372 RSC.
- (a) X. Zheng and J.-P. Wan, Adv. Synth. Catal., 2019, 361, 5690 CrossRef CAS; (b) A. Contini, E. Erba and P. Trimarco, ARKIVOC, 2008, 2008, 136 Search PubMed.
- W. Yang, D. Huang, X. Zeng, D. Luo, X. Wang and Y. Hu, Chem. Commun., 2018, 54, 8222 RSC.
- A.-R. Liu, L. Zhang, J. Lia and A. Wusiman, RSC Adv., 2021, 11, 15161 RSC.
- B. Liu, Y. Ning, M. Virelli, G. Zanoni, E. A. Anderson and X. Bi, J. Am. Chem. Soc., 2019, 141, 1593 CrossRef CAS PubMed.
- (a) L. Wang, H. Zhu, T. Peng and D. Yang, Org. Biomol. Chem., 2021, 19, 2110 RSC; (b) Z.-Y. Wang, K.-K. Wang, R. Chen, H. Liu and K. Chen, Eur. J. Org. Chem., 2020, 2020, 2456 CrossRef CAS; (c) C. Najera, L. K. Sydnes and M. Yus, Chem. Rev., 2019, 119, 11110 CrossRef CAS PubMed; (d) Y. Li, J. Yu, Y. Bi, G. Yan and D. Huang, Adv. Synth. Catal., 2019, 361, 4839 CrossRef CAS; (e) D. Tejedor, S. Lpez-Tosco, F. Cruz-Acosta, G. Mndez-Abt and F. Garcia-Tellado, Angew. Chem., Int. Ed., 2009, 48, 2090 CrossRef CAS PubMed.
- (a) Y. K. Kumar, G. R. Kumar, T. J. Reddy, B. Sridhar and M. S. Reddy, Org. Lett., 2015, 17, 2226 CrossRef CAS PubMed; (b) A. Contini, E. Erba and S. Pellegrino, Synlett, 2012, 23, 1523 CrossRef CAS; (c) X. Xu, Z. Ge, D. Cheng, L. Ma, C. Lu, Q. Zhang, N. Yao and X. Li, Org. Lett., 2010, 12, 897 CrossRef CAS PubMed; (d) X. Xu, X. Li, L. Ma, N. Ye and B. Weng, J. Am. Chem. Soc., 2008, 130, 14048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06809a |
| This journal is © The Royal Society of Chemistry 2021 |