
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntioxidant activity of SSeCAHK in HepG2 cells: a selenopeptide identified from selenium-enriched soybean protein hydrolysates
Jian Zhang ,
Qiyue Zhang,
He Li*,
Xinwei Chen,
Wanlu Liu and
Xinqi Liu*
,
Qiyue Zhang,
He Li*,
Xinwei Chen,
Wanlu Liu and
Xinqi Liu*
National Soybean Processing Industry Technology Innovation Center, Beijing Advanced Innovation Center for Food Nutrition and Human Health, Beijing Technology and Business University, Beijing 100048, China. E-mail: lihe@btbu.edu.cn; liuxinqi@btbu.edu.cn
First published on 18th October 2021
Abstract
This paper is aimed at purifying and identifying selenium (Se)-containing antioxidative peptides from Se-enriched soybean peptides (SSP). In this work, the SSP was separated into five fractions (F1 to F5). Fraction F4, displaying the highest antioxidative activity, was further separated, and sub-fractions F4-1 to F4-5 were selected for antioxidative activity evaluation using 1,1-diphenyl-2-picrylhydrazyl (DPPH), 2,2-azino-bis-(3-ethylbenzo-thiazoline-6-sulphonic acid)diammonium salt (ABTS), and OH− radical scavenging assays. The Se-containing antioxidative peptides with sequence Ser–SeC–Ala–His–Lys (SSeCAHK) were identified in sub-fraction F4-1 and chemically synthesized. This Se-containing pentapeptide showed a preventive effect against hydrogen peroxide (H2O2)-induced oxidative stress in HepG2 cells. Pretreating the cells for 2 h with SSeCAHK (0.13–0.50 mg mL−1) induced strong intracellular, reactive oxygen species (ROS) scavenging activity while preventing a decrease in reduced glutathione (GSH) and an increase in malondialdehyde (MDA). Therefore, SSeCAHK treatment improved H2O2-induced oxidative stress in HepG2 cells, demonstrating the significant potential of SSeCAHK as a natural antioxidative functional material for dietary supplementation.
1. Introduction
Reactive oxygen species (ROS) are produced during cellular oxygen metabolism and include superoxide anions (O2−), hydrogen peroxide (H2O2), hydroxyl radicals (OH−), and singlet oxygen (1O2).1,2 ROS plays a vital role in the human physiological processes involved in cell differentiation, proliferation and migration, cell signaling, and homeostasis.3–5 ROS may be beneficial to human health at low concentrations by acting as a cellular signaling molecule to defend against infectious agents and mitogen response.6 In normal physiological conditions, ROS can be effectively scavenged by the antioxidant defense systems.7 However, when the body suffers from fatigue, illness, or harmful stimulation, the in vivo redox equilibrium can be disrupted.8,9 In this case, the excess production of ROS by the organism exceeds its oxidative scavenging capacity, leading to excessive ROS accumulation.10 Furthermore, oxidative stress, which is regarded as the primary cause of aging and chronic degenerative disorders, is caused by the excessive accumulation of ROS.11 Increasing research has confirmed that oxidative damage is correlated with the pathological development of cancer, atherosclerosis, diabetes, immune imbalance, endocrine dysfunction, and other diseases.12 Therefore, maintaining the redox balance of cells is critical for human health.Studies have shown that organisms have endogenous defense mechanisms, including enzymatic and non-enzymatic antioxidant defense systems, protecting cells from oxidative damage by eliminating ROS.13,14 The enzymatic antioxidant system consists of catalase (CAT), glutathione peroxidase (GSH-Px), and superoxide dismutase (SOD), while the non-enzymatic antioxidant system mainly includes glutathione (GSH) and ascorbic acid.15–18 In addition, many natural antioxidants may play a protective role in the pathogenesis of various diseases related to oxidative stress by removing excess ROS or inhibiting ROS generation.14,15,17,19
Selenium (Se), an essential trace element for human health, exhibits various benefits, such as antioxidant, immune-enhancing, and antitumor activity.20 In addition, Se is an essential component of many enzymes in the body, such as GSH-Px, thioredoxin reductase (TrxR), and the thyroid hormone deiodinase (DIO).21 Se exists in both inorganic and organic forms. The inorganic forms mainly include selenate and selenite, while the organic forms primarily consist of selenoamino acid, selenopeptide, and selenoprotein.22 Organic Se is more suitable for consumption as part of a balanced diet than inorganic Se due to its bioavailability and toxicity advantages.23 Due to the safety concerns regarding synthetic antioxidants and chemical drugs, natural antioxidants have attracted increasing attention.24 The natural antioxidant peptides from plants present benefits, such as abundant raw materials, high efficiency, and safety.25 Increasing evidence has suggested that the peptides found in plants inhibit oxidative stress by scavenging free radicals and increasing intracellular antioxidant enzyme activity and GSH levels.26–32 Moreover, Zhang et al. confirmed that Se and peptides have a synergistic effect, rendering Se-enriched soybean peptides (SSP) desirable antioxidants.33 Furthermore, it is suggested that SSP can be absorbed well and metabolized into selenoproteins, such as GSH-Px, to resist oxidative stress.34 Our previous studies also revealed that a faster absorption rate of SSP, which provided a scientific basis for the better functional activity of selenium in the body.35 Therefore, new strategies for exploring Se-containing peptides as antioxidant supplements to promote human health are emerging.
This study aimed to prepare Se-containing soybean antioxidant peptides and determine their roles in oxidative stress in vitro. A pentapeptide was isolated in the SSP, and the amino acid sequence was determined via nano LC-MS/MS. The synthetic peptide was then used to evaluate its preventive effect against H2O2-induced oxidative stress in HepG2 cells. The cell viability and several markers of oxidative damage, e.g., ROS generation, as well as the GSH, GSSG, and MDA concentrations, were evaluated. It is expected that this study will provide new insight into Se nutrient supplementation and oxidative damage prevention.
2. Materials and methods
2.1 Materials and chemicals
SSeCAHK peptides (purity > 98%) were synthesized by the Nanjing TGpeptide Biotechnology Co., Ltd (Shanghai, China). LCMS grade acetonitrile, formic acid, and methanol were purchased from Sigma (St. Louis, USA). Human hepatoma (HepG2) cells were obtained from Peking Union Medical College (Beijing, China). The H2O2 solution was purchased from the China National Pharmaceutical Group (Beijing, China). Dulbecco's Modified Eagle Medium (DMEM), penicillin, and streptomycin were purchased from Gibco (Grand Island, USA). Fetal bovine serum (FBS) was acquired from Excell Bio (Shanghai, China). The cell-counting kit-8 (CCK-8) assay was obtained from Dojindo (Kumamoto, Japan). The ROS, MDA, and GSH assay kits were obtained from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The bicinchoninic acid (BCA) protein assay kits were purchased from Thermo Fisher Scientific (Rockford, USA). All other chemicals and reagents were of analytical grade.2.2 Preparation of the SSP
The Se-enriched soybeans were supplied by Enshi Se-Run Health Tech Development Co., Ltd (Enshi, China). The SSP was prepared as described in a previous report.35 Briefly, the Se-enriched soybean protein product was obtained via alkali dissolution and acid precipitation from the Se-enriched soybeans. Then, the Se-enriched soybean protein was digested with alkaline protease, neutral protease, and papain at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. The degree of hydrolysis in optimal conditions was 68.53%. The hydrolysate was centrifuged at 4000 rpm for 15 min, and the supernatant was freeze-dried to obtain the SSP. The 85.10% protein content of the SSP was determined using the Kjeldahl method (Kjeltec 8000, FOSS Analytical A/S, Denmark). The ÄKTA pure system was operated via gel filtration chromatography to determine the molecular weight distribution of the SSP, while the molecular weight ratio <3000 Da was 88.1%. According to the GB 5009.93-2010 national standard, the total Se content in the SSP was 110.4 mg kg−1 and tested using Hydride Generation-Atomic Fluorescence Spectrometry (LC-AFS6500, Beijing Haiguang Instrument Co., Ltd, Beijing, China). The SSP amino acid composition was assayed using an amino acid analyzer (Biochrom 30+, BioChrom Ltd, England). The essential amino acids (Thr, Val, Met, Ile, Leu, Phe, His, and Lys) accounted for 37.92% of the total SSP amino acid content. The SSP methionine content was 189.32 μmol g−1, while the cystine content was 47.09 μmol g−1.
:1:1. The degree of hydrolysis in optimal conditions was 68.53%. The hydrolysate was centrifuged at 4000 rpm for 15 min, and the supernatant was freeze-dried to obtain the SSP. The 85.10% protein content of the SSP was determined using the Kjeldahl method (Kjeltec 8000, FOSS Analytical A/S, Denmark). The ÄKTA pure system was operated via gel filtration chromatography to determine the molecular weight distribution of the SSP, while the molecular weight ratio <3000 Da was 88.1%. According to the GB 5009.93-2010 national standard, the total Se content in the SSP was 110.4 mg kg−1 and tested using Hydride Generation-Atomic Fluorescence Spectrometry (LC-AFS6500, Beijing Haiguang Instrument Co., Ltd, Beijing, China). The SSP amino acid composition was assayed using an amino acid analyzer (Biochrom 30+, BioChrom Ltd, England). The essential amino acids (Thr, Val, Met, Ile, Leu, Phe, His, and Lys) accounted for 37.92% of the total SSP amino acid content. The SSP methionine content was 189.32 μmol g−1, while the cystine content was 47.09 μmol g−1.
2.3 Purification of the SSP
The SSP was dissolved in deionized water and then loaded onto a Superdex 200 gel filtration column (10 × 300 mm), which was pre-equilibrated with deionized water. The column was eluted with deionized water at a flow rate of 0.5 mL min−1, and the eluted solution was collected every 1 mL. The absorbance was measured at 220 mm using an online spectrophotometer to determine the elution profile of the sample. Five major fractions (labeled F1, F2, F3, F4, and F5) were observed.The fraction displaying higher antioxidative activity than the others was further purified on a diethylaminoethyl cellulose (DEAE) Sepharose Fast Flow (FF) column (16 × 100 mm) equilibrated by 20 mmol L−1 Tris–HCl (pH 8.0) buffer. The column was eluted at a 1 mL min−1 flow rate, with a linear NaCl gradient (0–1.0 mol L−1) in a 20 mmol L−1 Tris–HCl buffer (pH 8.0). A 1 mL aliquot of the effluent was collected in each tube, and the absorbance at 220 nm was monitored as described above. The sub-fractions were concentrated and freeze-dried to determine the antioxidative activity.
2.4 Determination of the antioxidant activity in vitro
Here, 1,1-diphenyl-2-picrylhydrazyl (DPPH), 2,2-azino-bis-(3-ethylbenzo-thiazoline-6-sulphonic acid)diammonium salt (ABTS), and OH− free radical scavenging assays were applied to determine the antioxidative activity of the peptide fractions that were collected from the Superdex 200 gel filtration column and the DEAE Sepharose FF column elution.| DPPH radical scavenging activity (%) = [1 − (A1 − A2)/A0] × 100% |
| OH− scavenging activity (%) = (Acontrol − Asample)/Acontrol × 100% |
2.5 Nano LC-MS/MS analysis
Sub-fraction F4-1 from the DEAE Sepharose FF column displayed relatively high antioxidative activity and was analyzed using an Ultimate 3000 system coupled with a Q Exactive™ Hybrid Quadrupole-Orbitrap™ Mass Spectrometer (Thermo Fisher Scientific, USA). The sample was analyzed on a 150 μm × 15 cm in-house manufactured column packed with reversed-phase ReproSil-Pur C18-AQ resin (1.9 μm, 100 Å, Dr. Maisch GmbH, Germany) at a constant flow rate of 600 nL min−1. The binary gradient elution system consisted of ultrapure water (with 0.1% formic acid, A) and acetonitrile (with 0.1% formic acid, B). The gradient elution conditions were as follows: from 4% to 8% B for 2 min, from 8% to 28% B for 43 min, from 28% to 40% B for 10 min, from 40% to 95% B for 1 min, and from 95% to 95% B for 10 min. The injection volume was 5 μL. The MS parameters were as follows: resolution: 70000; AGC target: 3e6; maximum IT: 40 ms; and scan range: 100 m/z to 1500 m/z. MS/MS parameters were as follows: resolution: 17500; AGC target: 1e5; maximum IT: 60 ms; TopN: 20; and NCE/stepped NCE: 27. The raw data produced by mass spectrometry were used to identify the peptide sequence using the de novo method.
2.6 Synthesis of the Se-containing peptide, SSeCAHK
Se-containing peptides with high de novo scores were selected for the solid-phase reaction according to the nano LC-MS/MS results. The synthetic peptides were stored at −20 °C. The Nanjing TGpeptide Biotechnology Co., Ltd (Nanjing, China) synthesized the required peptide segments.2.7 Cellular antioxidant activity assays
2.8 Statistical analysis
Data were expressed as the mean ± standard deviation (SD). The results were analyzed using one-way analysis of variance (ANOVA) followed by Tukey's method using SPSS 23 software, while the graphs were created using the Graph Pad Prism 7 software.3. Results and discussion
3.1 Isolation and purification of the SSP
To purify Se-containing antioxidative soybean peptides, the SSP was initially separated into five fractions (F1 to F5, Fig. 1a) via size-exclusion chromatography on a Superdex 200 gel filtration column. Each fraction was collected, lyophilized, and then evaluated for antioxidant activity by comparing it with vitamin C (Vc). As shown in Fig. 1b, the DPPH radical scavenging activity of F1, F2, F3, F4, F5, and Vc at a concentration of 0.05 mg mL−1 was 11.8%, 14.3%, 19.7%, 24.9%, 3.0%, and 62.9%, respectively. The DPPH radical scavenging activity of fractions F1 to F4 increased as the molecular weights of the peptides decreased. However, fraction F5 exhibited lower DPPH radical scavenging activity than the other fractions. These results are consistent with a report by Liu et al.38 Fig. 1b shows that fractions F2 to F4 displayed higher radical scavenging activity than the other fractions. Therefore, the radical scavenging activity of ABTS and OH− (Fig. 1c and d) were used to further investigate the antioxidative ability of the three fractions (F2, F3, and F4). According to the results, F4 showed higher ABTS and OH− scavenging activity than the other two fractions (Fig. 1c and d), while the antioxidant ability of the three fractions was F4 > F2 > F3 in descending order. The antioxidative activity of peptides is reportedly related to their amino acid composition and molecular weights. In general, peptides with low molecular weights demonstrate high antioxidative activity.41,42 Gel filtration involves the separation of molecules in accordance with their size. It is conjectured that fraction F4 with a lower molecular weight can more easily react with free radicals, converting them to more stable products and terminating the radical chain reactions.25 | ||
| Fig. 1 The purification profiles of the Se-containing antioxidant peptides and their radical scavenging activity. (a) Superdex 200 gel filtration chromatography of the enzymatic hydrolysates. (b) DPPH radical scavenging activity of the fractions. (c) ABTS radical scavenging activity of the fractions. (d) OH− radical scavenging activity of the fractions. | ||
Fig. 1b–d shows that fraction F4 displayed the best radical scavenging activity of all the fractions. Therefore, fraction F4 was further separated on a DEAE Sepharose FF ion-exchange column (Fig. 2a). Five sub-fractions (F4-1 to F4-5) were separated according to differences in the charge of the peptides. Each fraction was pooled, lyophilized, and evaluated for antioxidative activity, including their DPPH, ABTS, and OH− radical scavenging ability at 0.05 mg mL−1. As demonstrated in Fig. 2b–d, a clear difference was evident in the DPPH, ABTS, and OH− radical scavenging activity of the five sub-fractions. All five fractions were able to scavenge all three free radicals. The highest DPPH (74.6%) and ABTS (189.3 μmol TEAC per mg peptide) radical scavenging activity was exhibited by sub-fraction F4-1. However, F4-4 was best at scavenging OH− free radicals, with a clearance rate of 14.5%. The DPPH and ABTS assays were based on electron transfer, measuring the antioxidant capacity in a colored oxidant reduction.43 Many studies have indicated that both the peptide and organic Se content influence antioxidant capacity.44–46 The DPPH and ABTS scavenging properties of sub-fraction F4-1 can mainly be ascribed to the presence of CH2–SeH cysteinyl residue, providing electrons and hydrogen to directly or indirectly react with free radicals to form stable substances.47 Schöneich et al. indicated that the OH− reaction with the peptides mainly involved the methionine moiety.48 Therefore, it is speculated that sub-fraction F4-5 contains SeMet and can eliminate OH−. Although DPPH and ABTS have been questioned due to their lack of biological relevance, these assays indicate that sub-fraction F4-1 exhibited excellent free radical scavenging ability. Considering all the above, sub-fraction F4-1 was selected for further sequence analysis.
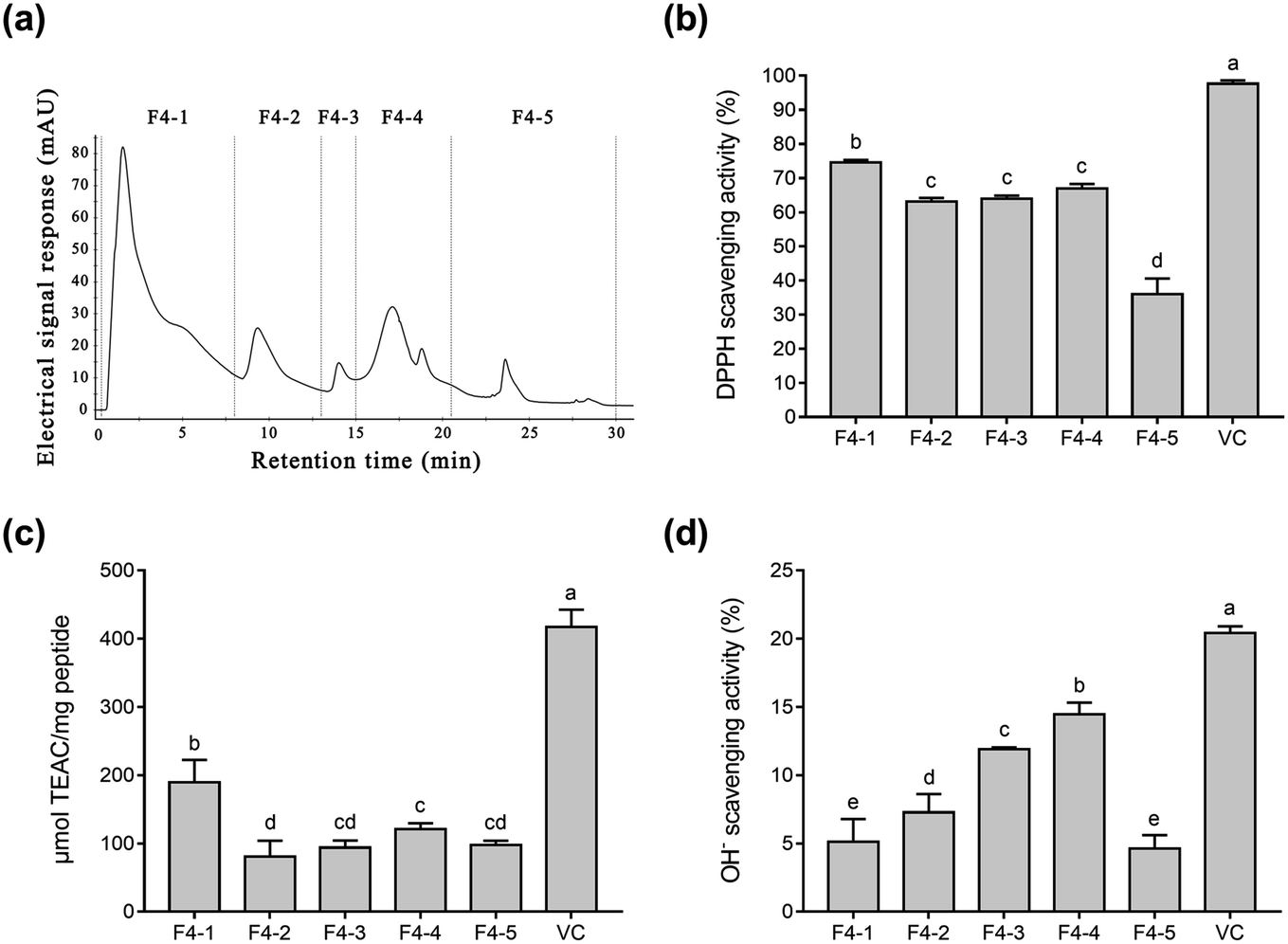 | ||
| Fig. 2 The purification profiles of the Se-containing antioxidant peptides and their radical scavenging activity. (a) DEAE-Sepharose-FF chromatography of fraction F4, eluted from the Superdex 200 column. (b) DPPH radical scavenging activity of the sub-fractions. (c) ABTS radical scavenging activity of the sub-fractions. (d) OH− radical scavenging activity of the sub-fractions. | ||
3.2 Identification of the peptides of SSP
Sub-fraction F4-1 obtained from the DEAE Sepharose FF column displayed relatively high antioxidative activity and was further analyzed via nano LC-MS/MS for molecular mass determination and peptide characterization. The b and y ions represent the primary fragment ions at a collision energy of <200 eV. The MS/MS data of ions b and y were used to identify the peptides, which were processed by de novo sequencing. The MS and MS/MS spectra of a single charged ion at m/z 593.156 are shown in Fig. 3 (only y and b ions are shown). As shown in Fig. 3a, the full-scan spectrum of sub-fraction F4-1 revealed the presence of monoselenized peptides at m/z 593.156. The molecular mass of the peptide was determined to be 592.187 Da with a retention time of 3.98 min. This Se compound was selected for CID and further MS analysis to obtain its structural information. The product ion spectra of the precursor ion, m/z 593.156, are shown in Fig. 3b. The product ions were differentiated into b-ions and y-ions via MS/MS. The subsequent peptide sequence is shown as an inset in Fig. 3b, and it is speculated that SSeCAHK represents a novel Se-containing peptide.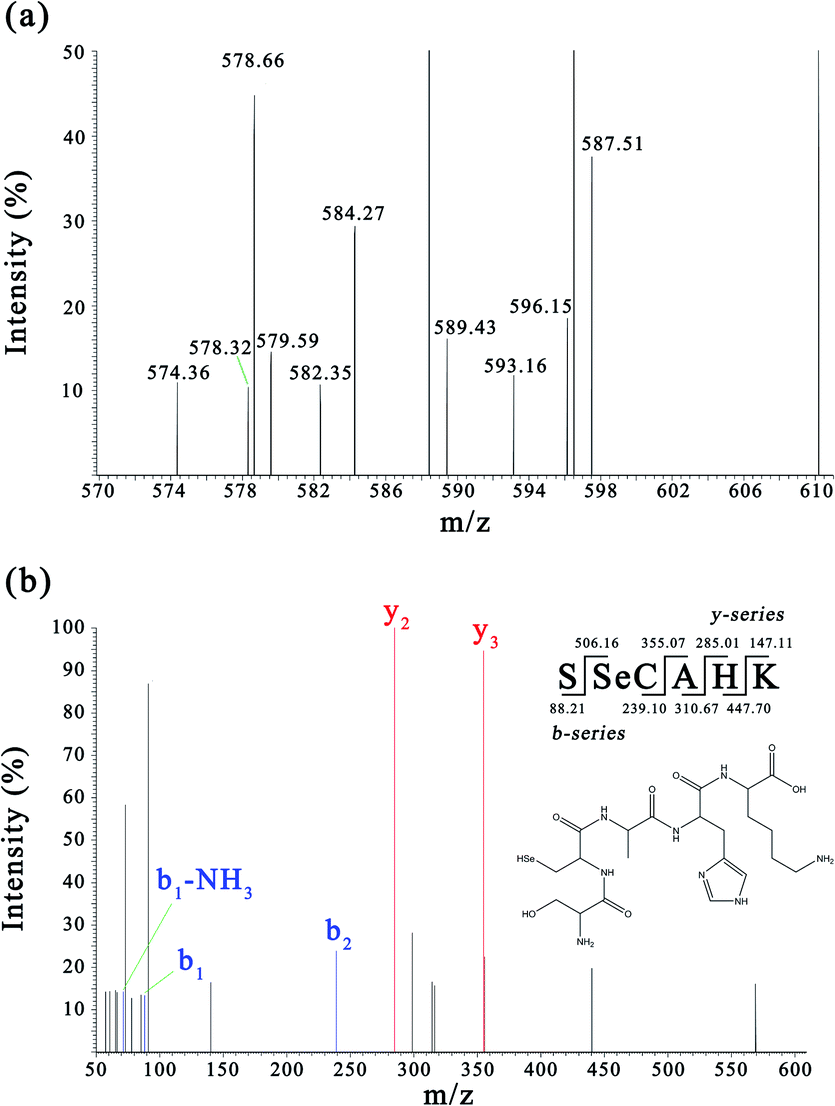 | ||
| Fig. 3 MS and MS/MS spectra of the peptides (m/z = 593.156). (a) Full scan MS spectrum of fraction F4-1. (b) CID spectrum of the analyte at m/z 593.156. | ||
Zhou et al. demonstrated that Ser regulated the epigenetic expression of the GSH synthesis-related genes essential in the defense against oxidative damage.49 Ser was found at the N-terminal of SSeCAHK, possibly contributing significantly to the antioxidation activity. SeC was also found in the SSeCAHK, whereas SeC is typically found as the main form of Se in mammalian proteins in foods of animal origin which is believed to be significant in radical scavenging activity.44,50 The radical scavenging activity of peptides is related to the hydrophobic amino acid content.51 The identified Se-containing antioxidative peptide, SSeCAHK, contains Ala as the hydrophobic amino acid residue. Protein hydrolysates exhibit strong antioxidant activity and often contain basic amino acids in their molecular composition. His–Lys is found at the C-terminal of SSeCAHK and can possibly be used as a proton donor to improve the antioxidant capacity of the peptide. Therefore, SSeCAHK may exhibit significant antioxidant activity. SSeCAHK was synthesized using the solid-phase synthesis method.
3.3 The effect of H2O2 on cell viability and ROS generation
The protective effect of SSeCAHK against H2O2-induced oxidative stress in HepG2 cells was investigated. H2O2, a considerably active oxygen molecule with relatively stable properties, is widely used with in vitro models to investigate oxidative stress.15,52 Cell viability is often used as an indicator of cytotoxicity.53 A decline in cell viability represents an indicator of the successful establishment of an oxidative stress model. As shown in Fig. 4a, the HepG2 cell viability decreased in a concentration-dependent manner in conjunction with an increase in the H2O2 concentration. The cell viability reached 72.63% after 600 μM H2O2 treatment for 2 h, decreasing significantly (P < 0.05) when the H2O2 concentration exceeded 600 μM. Moreover, the cell viability of the H2O2-treated group exhibited a substantially more significant decline than the control group (P < 0.05), indicating the successful establishment of the oxidative stress model. | ||
| Fig. 4 (a) The effect of H2O2 on the viability of the HepG2 cells. (b) The effect of SSeCAHK on the viability of the HepG2 cells. (c) The effect of H2O2 on ROS generation. | ||
The production and clearance of ROS in normal cells are in a dynamic equilibrium. The oxidative injury environment can lead to an overexpression of intracellular ROS,54 followed by excessive ROS accumulation, inducing severe cell injuries, such as apoptosis, necrosis, and DNA damage.55,56 The intracellular ROS production was measured to evaluate the cellular oxidative stress generated by H2O2. As shown in Fig. 4c, the intracellular ROS generation in the HepG2 cells was positively correlated with the increased H2O2 concentration (P < 0.05). ROS generation was highest when the H2O2 concentration exceeded 600 μM. Treatment with 600 μM H2O2 resulted in significantly decreased cell viability and higher accumulated ROS in the HepG2 cells. Therefore, 600 μM was selected as the final H2O2 concentration to induce oxidative stress for further assays.
3.4 The effect of SSeCAHK on cell viability
Before determining the cytoprotective effect of SSeCAHK, it was necessary to identify the optimal SSeCAHK concentration. HepG2 cells were treated with different SSeCAHK concentrations (0.13 mg mL−1, 0.25 mg mL−1, 0.50 mg mL−1, 1.00 mg mL−1, 2.00 mg mL−1, and 4.00 mg mL−1) for 24 h, and the cell viability was measured using the CCK-8 method. As shown in Fig. 4b, exposure to SSeCAHK at a concentration below 0.50 mg mL−1 over a 24 h incubation period did not alter cell viability, compared to the control. The cell viability significantly decreased at SSeCAHK concentrations of 1.00 mg mL−1, 2.00 mg mL−1, and 4.00 mg mL−1, indicating severe HepG2 cell damage. Therefore, the appropriate SSeCAHK concentration was 0.50 mg mL−1. Moreover, two additional experimental groups were established using concentrations of 0.25 mg mL−1 and 0.13 mg mL−1 to further investigate the protective effect of different SSeCAHK concentrations.3.5 The preventive effect of SSeCAHK on ROS generation
The ROS levels were determined to analyze the cytoprotective effect of SSeCAHK against H2O2-induced oxidative stress in HepG2 cells. As shown in Fig. 5a, the ROS level significantly increased in the H2O2-treated group compared to the control group. The intracellular ROS levels increased dramatically in the HepG2 cells after incubation with 600 μM H2O2 for 2 h (fluorescence intensity increased from 5.32 ± 0.27 to 16.8 ± 0.84), indicating enhanced oxidative stress. However, SSeCAHK treatment significantly reduced the ROS level. Compared with the H2O2-treated group, the level of ROS in the SSeCAHK group (0.13 mg mL−1, 0.25 mg mL−1, and 0.50 mg mL−1) decreased by 87.5%, 34.5%, and 33.3%, respectively (p < 0.05). Remarkably, 0.13 mg mL−1 SSeCAHK displayed more significant protective effects than the others treatments. The results of the ROS assay indicated that SSeCAHK effectively protected against oxidative damage by decreasing the ROS production in H2O2-induced HepG2 cells. Several studies have demonstrated that antioxidant peptides derived from plants can protect HepG2 cells from oxidative stress damage by scavenging ROS.57–59 Wang et al. found that the corn gluten peptide fractions exhibited excellent intracellular ROS scavenging capacities in oxidized HepG2 cells induced by H2O2.26 Yarnpakdee et al. also found that the increase of ROS due to H2O2 was attenuated after treatment with peptides from tilapia hydrolysate in HepG2 cells.7 The ROS values of the SSeCAHK in our study are comparable to those that have been reported. Furthermore, peptides with higher ratios of hydrophobic amino acids (His, Pro, Met, Cys, Tyr, Trp, Phe or Met) are considered more effective in eliminating peroxyl radicals.60 SSeCAHK may exert ROS scavenging ability based on the hydrophobic amino acid composition. On the other hand, ROS are effectively eliminated by the non-enzymatic factors, such as GSH. Se is an active center of GSH-Px, Se supplementation could increase the activities of GSH-Px.61 SSeCAHK could act as a Se donor for Se enzymes such as GSH-Px, which can catalyze GSH to scavenging ROS.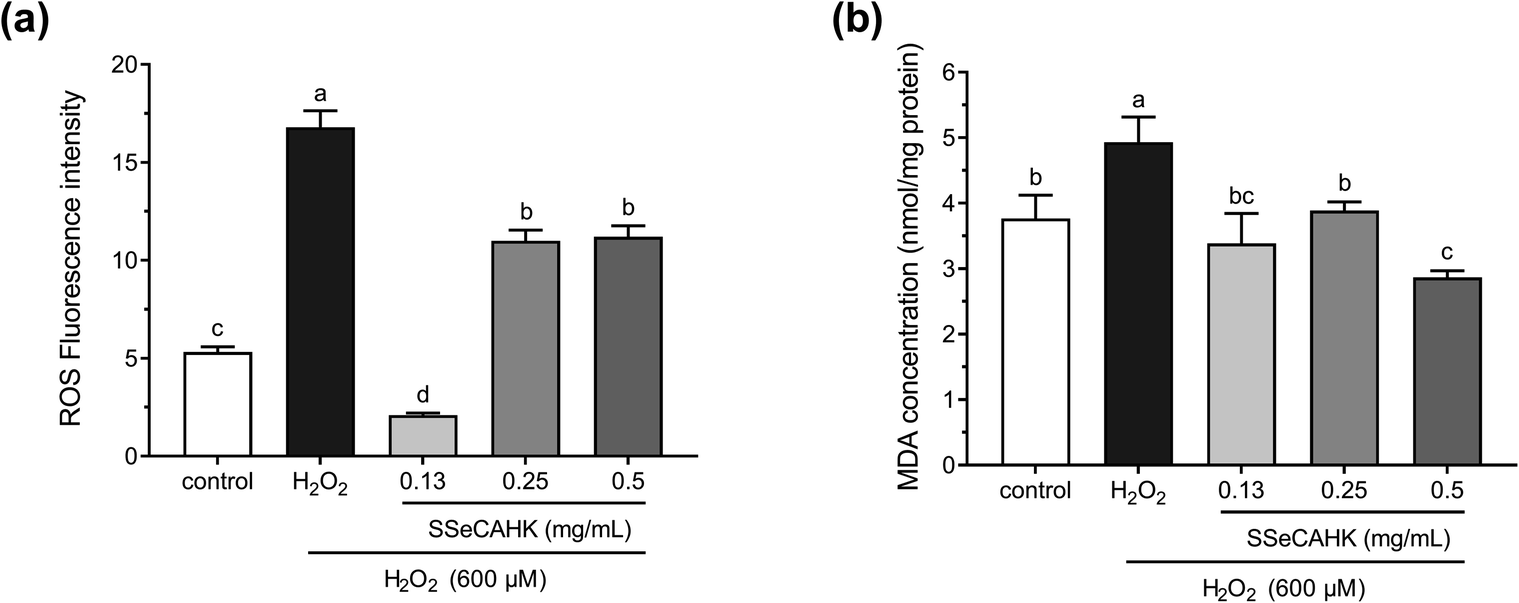 | ||
| Fig. 5 (a) Intracellular ROS scavenging capacity of different SSeCAHK concentrations in H2O2-induced oxidative stress conditions in the HepG2 cells. (b) The effect of different SSeCAHK concentrations on intracellular MDA. | ||
3.6 MDA levels
H2O2 is the primary precursor of the highly reactive free radicals, O2− and OH−, and can cause lipid peroxidation.62 As a primary lipid oxidation product, MDA can aggravate membrane damage in senescence physiology and physiological resistance.2 Intracellular MDA is a direct result of unsaturated fatty acid peroxidation in the membrane and is often regarded as a biomarker of oxidative stress.24 Therefore, the MDA concentrations in the HepG2 cells were investigated further. As shown in Fig. 5b, treating HepG2 cells with 600 μM H2O2 for 2 h significantly increased the MDA concentration, compared with the control group. However, a 24 h pretreatment with SSeCAHK substantially inhibited the increase in MDA levels induced by H2O2. Additionally, no significant differences were observed among the low- and medium-level treatment groups, compared with the control group, with the 0.50 mg mL−1 group displaying the most noticeable difference. The results showed that SSeCAHK pretreatment could restore the MDA concentration to normal level in oxidized HepG2 cells, while the concentration of MDA was not dose-dependent of SSeCAHK in the range of 0.13 mg mL−1 to 0.25 mg mL−1. These results indicated that SSeCAHK treatment significantly inhibited MDA release in the HepG2 cells induced by H2O2. Similar findings were obtained for the soybean peptides, Se-containing rice peptides, and corn peptides, which significantly reduced the MDA content.58,63,64 Furthermore, ROS and lipid peroxidation are the most typical indicators of oxidative damage.54 An essential step in cell membrane degradation is the ROS reaction with the double bonds of polyunsaturated fatty acids to yield lipid hydroperoxides.53 SSeCAHK may inhibit lipid peroxidation by reducing ROS production, decreasing the MDA concentration.3.7 Reduced GSH, GSSG, and total GSH (T-GSH) concentrations
Studies have shown that cells are well-equipped with defense mechanisms against oxidative stress-induced cell damage.65 GSH is widely used as an index for intracellular non-enzymatic antioxidant defense.16 GSH depletion and GSSG production reflect the degree of intracellular oxidation.66 Therefore, the effect of SSeCAHK on the GSH levels in the HepG2 cells was evaluated. As shown in Fig. 6a and b, 600 μM H2O2 induced a dramatic decrease in the T-GSH and GSH levels, compared to the control, indicating HepG2 cell oxidation. However, 0.25 mg mL−1 SSeCAHK pretreatment for 24 h altered the GSH levels, preventing T-GSH and GSH depletion to near control levels. Pretreatment with 0.13 mg mL−1 SSeCAHK increased the T-GSH and GSH levels. Compared with the 0.50 mg mL−1 SSeCAHK group, the SSeCAHK concentrations of 0.13 mg mL−1 and 0.25 mg mL−1 displayed stronger antioxidant capacity. It is suggested that the non-enzymatic antioxidant defense regulation of SSeCAHK in HepG2 cells may be dose-dependent. In addition, as shown in Fig. 6c, H2O2 significantly reduced the GSSH level. The GSSG concentrations were not significantly different from those of the control group compared to the SSeCAHK (0.13 mg mL−1 and 0.25 mg mL−1) group. The T-GSH, GSH, and GSSH levels of the HepG2 cells in the H2O2 group decreased significantly due to oxidative stress injury.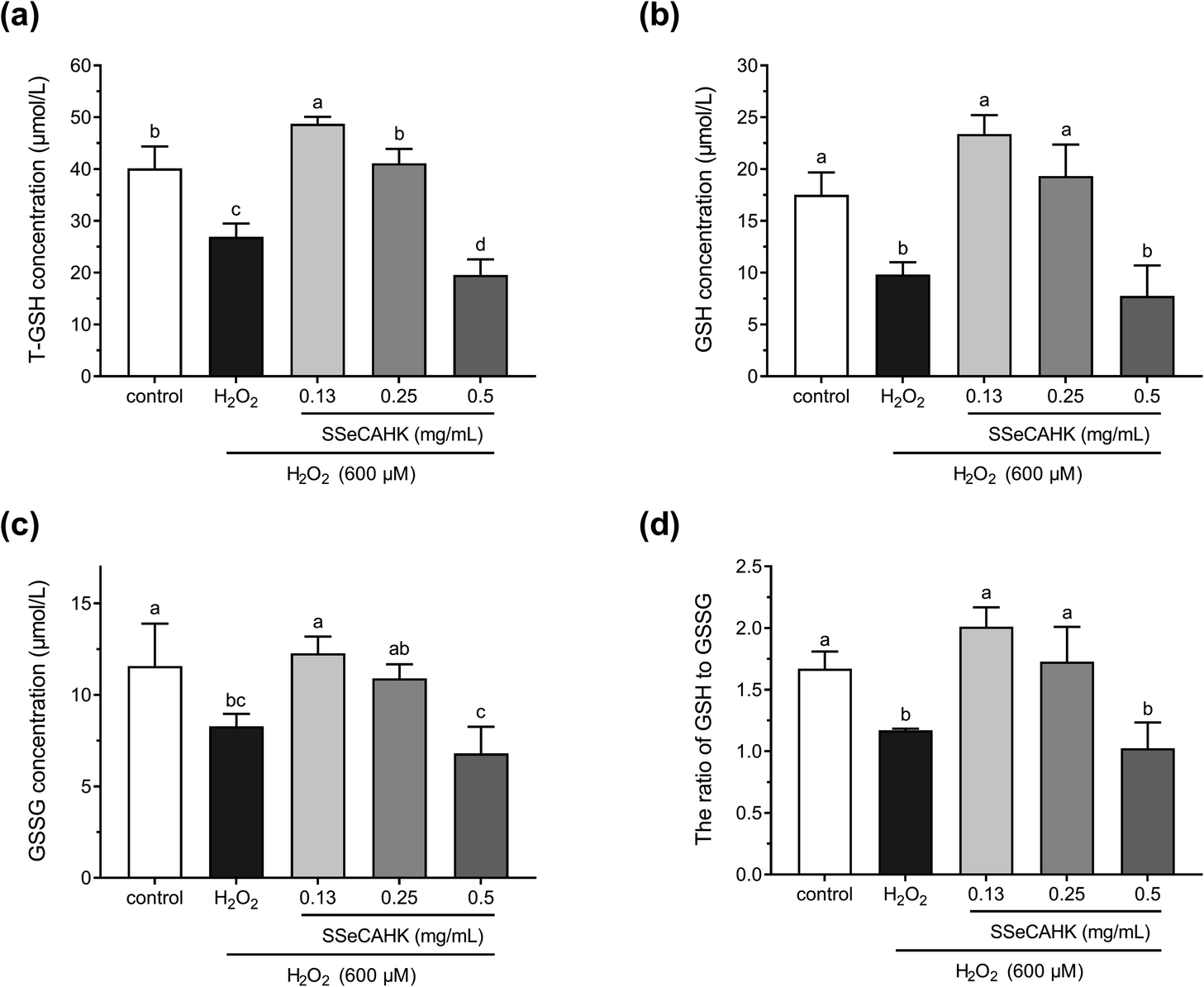 | ||
| Fig. 6 The effect of different SSeCAHK concentrations on the intracellular T-GSH (a), GSH (b), GSSG (c), and GSH to GSSG ratio (d). | ||
GSH/GSSG denotes the primary cellular redox buffer and is a representative indicator of the redox cell environment.67,68 The GSH to GSSG ratio was also assessed to further investigate the effect of SSeCAHK on HepG2 cells. As shown in Fig. 6d, the GSH/GSSG ratio was suppressed following H2O2 exposure. However, a significant difference (P < 0.05) was found between the low- and middle-level treatment groups of SSeCAHK and H2O2. SSeCAHK treatment significantly enhanced the GSH/GSSG ratio, with the 0.13 mg mL−1 SSeCAHK group being the highest. These results suggest that 0.13 mg mL−1 SSeCAHK pretreatment is optimal in protecting HepG2 cells from oxidative stress injury induced by H2O2. While the GSH/GSSG ratio in 0.5 mg mL−1 SSeCAHK group was significantly lower than 0.13 mg mL−1 SSeCAHK group. This condition may be caused by the negative effects of high Se concentrations in 0.5 mg mL−1 SSeCAHK. Se is one of the essential mineral elements able to preserve from health damage. The organic Se has been found to have greater potency in terms of bioavailability and effects on health than inorganic Se.69 Nevertheless, Se can become toxic when it is elevated above a threshold concentration.70 High concentration of Se may produces free radicals, which will have toxic effects on the body. Meanwhile, Zhao et al. found that Se affects the synthesis of protein by a biphasic mechanism. The high concentrations of Se could inhibited the synthesis of protein and total amino acids.71 In this paper, adverse result caused by 0.5 mg mL−1 SSeCAHK is not only decrease in GSH/GSSG ratio, but also reduction of concentrations of T-GSH, GSH and GSSG. The synthesis of GSH in the high-level treatment group of SSeCAHK may be blocked due to the high Se concentration. Meanwhile, GSH was also oxidized to GSSG as a substrate to resist oxidative stress. Therefore, the GSH/GSSG ratio in the high-level treatment group of SSeCAHK was lower than that in the low-level treatment group. The results suggest that although SSeCAHK can resist oxidative stress, it need to be ingested in appropriate concentrations to avoid the negative effects of high Se concentrations. Although SSeCAHK displays strong antioxidant capacity, the molecular mechanism by which it exerts its antioxidant activity remains elusive. However, the results of previous animal studies have shown that Se-enriched protein from Ganoderma lucidum can increase the GSH level in mice, playing a vital role in the antioxidant defense system.72 Liu et al. found that the GSH level could be improved by Se-biofortified soybean peptide treatment, decreasing the MDA content in rats.73 These results provided some basis for the antioxidant effect SSeCAHK and have important significance for further understanding the biological activity of SSP.
4. Conclusions
In conclusion, a Se-containing pentapeptide, SSeCAHK, is successfully identified for the first time via LC-MS/MS in SSP. The findings of this study indicate that SSeCAHK protects HepG2 cells against H2O2-induced oxidative damage by scavenging intracellular ROS and enhancing the GSH system. In addition, SSeCAHK prevents an increase in MDA levels, reducing the level of H2O2-induced lipid peroxidation in HepG2 cells. SSeCAHK may be used as an antioxidative bioactive ingredient during the production and development of functional foods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2021YFC2101400).References
- J. N. Moloney and T. G. Cotter, Semin. Cell Dev. Biol., 2018, 80, 50–64, DOI:10.1016/j.semcdb.2017.05.023.
- H. Wang, J. Liu, X. Liu and Z. Liu, Br. Food J., 2019, 121, 2809–2820, DOI:10.1108/bfj-07-2018-0434.
- I. Mironczuk-Chodakowska, A. M. Witkowska and M. E. Zujko, Adv. Med. Sci., 2018, 63, 68–78, DOI:10.1016/j.advms.2017.05.005.
- L. Zhao, J. Su, L. Li, J. Chen, S. Hu, X. Zhang and T. Chen, Food Res. Int., 2014, 66, 186–196, DOI:10.1016/j.foodres.2014.08.051.
- J. T. Hancock, R. Desikan and S. J. Neill, Biochem. Soc. Trans., 2001, 29, 345–350, DOI:10.1042/bst0290345.
- R. Del Pino-Garcia, M. D. Rivero-Perez, M. L. Gonzalez-SanJose, K. D. Croft and P. Muniz, Food Funct., 2017, 8, 2444–2454, 10.1039/c7fo00390k.
- S. Yarnpakdee, S. Benjakul, H. G. Kristinsson and H. E. Bakken, J. Food Sci. Technol., 2015, 52, 6194–6205, DOI:10.1007/s13197-014-1672-4.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84, DOI:10.1016/j.biocel.2006.07.001.
- W. Droge, Physiol. Rev., 2002, 82, 47–95, DOI:10.1152/physrev.00018.2001.
- I. C. Jeung, D. Jee, C. R. Rho and S. Kang, Int. J. Med. Sci., 2016, 13, 139–146, DOI:10.7150/ijms.13861.
- C. Simioni, G. Zauli, A. M. Martelli, M. Vitale, G. Sacchetti, A. Gonelli and L. M. Neri, Oncotarget, 2018, 9, 17181–17198, DOI:10.18632/oncotarget.24729.
- L. Signorini, S. Granata, A. Lupo and G. Zaza, Int. J. Mol. Sci., 2017, 18, 1481, DOI:10.3390/ijms18071481.
- H. H. Kwok, W. Y. Ng, M. S. M. Yang, N. K. Mak, R. N. S. Wong and P. Y. K. Yue, Free Radical Biol. Med., 2010, 48, 437–445, DOI:10.1016/j.freeradbiomed.2009.11.013.
- G. Shui, Y. M. Bao, J. Bo and L. J. An, Eur. J. Pharmacol., 2006, 538, 73–79, DOI:10.1016/j.ejphar.2006.03.065.
- M. Zhao, Q. Yang, L. Lin, B. Sun and Y. Wang, J. Funct. Foods, 2017, 31, 160–171, DOI:10.1016/j.jff.2017.01.046.
- M. Alia, S. Ramos, R. Mateos, A. B. Granado-Serrano, L. Bravo and L. Goya, Toxicol. Appl. Pharmacol., 2006, 212, 110–118, DOI:10.1016/j.taap.2005.07.014.
- G. Qi, Y. Mi, R. Fan, R. Li, Y. Wang, X. Li, S. Huang and X. Liu, RSC Adv., 2017, 7, 32198–32208, 10.1039/c7ra05000c.
- H. Zhang, S. Zhang, J. Wang and B. Sun, J. Funct. Foods, 2017, 29, 53–59, DOI:10.1016/j.jff.2016.12.009.
- G. Kim, Y. Lee, J. Song and H. Jang, Food Sci. Biotechnol., 2013, 22, 241–247, DOI:10.1007/s10068-013-0033-9.
- F. Liu, Z. Y. Zhu, X. Sun, H. Gao and Y. M. Zhang, Int. J. Biol. Macromol., 2017, 99, 196–204, DOI:10.1016/j.ijbiomac.2017.02.064.
- L. Vanda Papp, J. Lu, A. Holmgren and K. Kum Khanna, Antioxid. Redox Signaling, 2007, 9, 775–806, DOI:10.1089/ars.2007.1528.
- J. Zhang, H. Zhou, H. Li, Z. Ying and X. Liu, Food Funct., 2021, 12, 1390–1401, 10.1039/d0fo02236e.
- U. Tinggi, Toxicol. Lett., 2003, 137, 103–110, DOI:10.1016/s0378-4274(02)00384-3.
- S. M. Lee, Y. Choi, J. Sung, Y. Kim, H. S. Jeong and J. Lee, Prev. Nutr. Food Sci., 2014, 19, 348–352, DOI:10.3746/pnf.2014.19.4.348.
- T. Sun, S. Zhang, W. Yang, Z. Zhao and D. Yang, Molecules, 2019, 24, 4486, DOI:10.3390/molecules24244486.
- L. Wang, L. Ding, Z. Yu, T. Zhang, S. Ma and J. Liu, Food Res. Int., 2016, 90, 33–41, DOI:10.1016/j.foodres.2016.10.023.
- K. Suetsuna and J. R. Chen, Food Sci. Technol. Res., 2002, 8, 227–230, DOI:10.3136/fstr.8.227.
- S. Y. Kim, J. Y. Je and S. K. Kim, J. Nutr. Biochem., 2007, 18, 31–38, DOI:10.1016/j.jnutbio.2006.02.006.
- H. Zhuang, N. Tang and Y. Yuan, J. Funct. Foods, 2013, 5, 1810–1821, DOI:10.1016/j.jff.2013.08.013.
- L. J. Xing, Y. Y. Hu, H. Y. Hu, Q. F. Ge, G. H. Zhou and W. G. Zhang, Food Chem., 2016, 194, 951–958, DOI:10.1016/j.foodchem.2015.08.101.
- S. Sudhakar and R. A. Nazeer, J. Funct. Foods, 2015, 14, 502–512, DOI:10.1016/j.jff.2015.02.028.
- M. Abdel-Hamid, J. Otte, C. De Gobba, A. Osman and E. Hamad, Int. Dairy J., 2017, 66, 91–98, DOI:10.1016/j.idairyj.2016.11.006.
- X. Zhang, H. He, J. Xiang, B. Li, M. Zhao and T. Hou, Food Chem., 2021, 358, 129888, DOI:10.1016/j.foodchem.2021.129888.
- S. Saffari, S. Keyvanshokooh, M. Zakeri, S. A. Johari and H. Pasha-Zanoosi, Aquacult. Nutr., 2017, 23, 611–617, DOI:10.1111/anu.12428.
- S. Gao, J. Zhang, Q. Zhang, W. Li, H. Li, T. Yu and Q. Liu, Food Sci., 2021, 42, 165–172, DOI:10.7506/spkx1002-6630-20200102-014.
- H. M. Kang and M. E. Saltveit, J. Agric. Food Chem., 2002, 50, 513–518, DOI:10.1021/jf011124d.
- M. Gallego, L. Mora, M. Reig and F. Toldra, Food Res. Int., 2018, 105, 873–879, DOI:10.1016/j.foodres.2017.12.006.
- K. L. Liu, Y. Zhao, F. S. Chen and Y. Fang, Food Chem., 2015, 187, 424–430, DOI:10.1016/j.foodchem.2015.04.086.
- T. Fukuda, K. Yamagata, S. Fujiyama, T. Matsumoto, I. Koshida, K. Yoshimura, M. Mihara, M. Naitou, H. Endoh, T. Nakamura, C. Akimoto, Y. Yamamoto, T. Katagiri, C. Foulds, S. Takezawa, H. Kitagawa, K. I. Takeyama, B. W. O'Malley and S. Kato, Nat. Cell Biol., 2007, 9, 604–611, DOI:10.1038/ncb1577.
- H. Zhang, J. Wang, Y. Liu, L. Gong and B. Sun, J. Funct. Foods, 2016, 25, 333–340, DOI:10.1016/j.jff.2016.06.012.
- Y. Xia, F. Bamdad, M. Gaenzle and L. Chen, Food Chem., 2012, 134, 1509–1518, DOI:10.1016/j.foodchem.2012.03.063.
- S. Ranathunga, N. Rajapakse and S. K. Kim, Eur. Food Res. Technol., 2006, 222, 310–315, DOI:10.1007/s00217-005-0079-x.
- K. Liu, R. Du and F. Chen, LWT--Food Sci. Technol., 2019, 111, 555–560, DOI:10.1016/j.lwt.2019.05.076.
- X. Zhao, Q. Zhao, H. Chen and H. Xiong, Food Chem., 2019, 272, 201–209, DOI:10.1016/j.foodchem.2018.08.039.
- Y. Fang, X. Pan, E. Zhao, Y. Shi, X. Shen, J. Wu, F. Pei, Q. Hu and W. Qiu, Food Chem., 2019, 275, 696–702, DOI:10.1016/j.foodchem.2018.09.115.
- D. Guo, Y. Zhang, J. Zhao, H. He and T. Hou, J. Trace Elem. Med. Biol., 2019, 51, 57–64, DOI:10.1016/j.jtemb.2018.09.010.
- S. Zhu, C. Du, T. Yu, X. Cong, Y. Liu, S. Chen and Y. Li, J. Food Sci., 2019, 84, 3504–3511, DOI:10.1111/1750-3841.14843.
- C. Schöneich, F. Zhao, J. Yang and B. L. Miller, ACS Symp. Ser., 1997, 675, 79–89, DOI:10.1021/bk-1997-0675.ch004.
- X. Zhou, L. He, S. Zuo, Y. Zhang, D. Wan, C. Long, P. Huang, X. Wu, C. Wu, G. Liu and Y. Yin, Biochim. Biophys. Acta, Mol. Basis Dis., 2018, 1864, 488–498, DOI:10.1016/j.bbadis.2017.11.009.
- B. Hernandez-Ledesma, A. Davalos, B. Bartolome and L. Amigo, J. Agric. Food Chem., 2005, 53, 588–593, DOI:10.1021/jf048626m.
- N. Rajapakse, E. Mendis, W. K. Jung, J. Y. Je and S. K. Kim, Food Res. Int., 2005, 38, 175–182, DOI:10.1016/j.foodres.2004.10.002.
- B. O. Cho, H. W. Ryu, C. W. Lee, C. H. Jin, W. D. Seo, J. Ryu, D. S. Kim, S. Y. Kang, H. S. Yook and I. Y. Jeong, Food Sci. Biotechnol., 2015, 24, 643–650, DOI:10.1007/s10068-015-0084-1.
- J. Wu, J. Huo, M. Huang, M. Zhao, X. Luo and B. Sun, J. Agric. Food Chem., 2017, 65, 10495–10504, DOI:10.1021/acs.jafc.7b04815.
- J. Tan, P. Li, H. Xue and Q. Li, Biotechnol. Lett., 2020, 42, 2453–2466, DOI:10.1007/s10529-020-02982-2.
- T. Moriwaki, A. Yamasaki and Q. M. Zhang-Akiyama, Oxid. Med. Cell. Longevity, 2018, 2018, 3862070, DOI:10.1155/2018/3862070.
- L. Zhang and Y. Wang, Dose-Response, 2018, 16, 155932581878263, DOI:10.1177/1559325818782631.
- Y. Du, R. Esfandi, W. G. Willmore and A. Tsopmo, Antioxidants, 2016, 5, 39, DOI:10.3390/antiox5040039.
- G. Yi, J. U. Din, F. Zhao and X. Liu, Food Funct., 2020, 11, 2725–2737, 10.1039/c9fo01466g.
- H. Agrawal, R. Joshi and M. Gupta, Food Chem., 2016, 204, 365–372, DOI:10.1016/j.foodchem.2016.02.127.
- J. Ren, M. Zhao, J. Shi, J. Wang, Y. Jiang, C. Cui, Y. Kakuda and S. J. Xue, Food Chem., 2008, 108, 727–736, DOI:10.1016/j.foodchem.2007.11.010.
- X. Zhang, H. He, J. Xiang, H. Yin and T. Hou, J. Agric. Food Chem., 2020, 68, 15061–15073, DOI:10.1021/acs.jafc.0c05594.
- G. Li, J. Kang, X. Yao, Y. Xin, Q. Wang, Y. Ye, L. Luo and Z. Yin, Eur. Food Res. Technol., 2011, 233, 427–435, DOI:10.1007/s00217-011-1534-5.
- Y. Qin, P. Shi, W. Liu, D. Guo and H. He, Sci. Technol. Food Ind., 2017, 17, 305–309, DOI:10.13386/j.issn1002-0306.2017.17.060.
- X. She, F. Wang, J. Ma, X. Chen, D. Ren and J. Lu, Food Agric. Immunol., 2016, 27, 99–110, DOI:10.1080/09540105.2015.1079597.
- B. Halliwell and J. M. C. Gutteridge, Methods Enzymol., 1990, 186, 1–85, DOI:10.1016/0076-6879(90)86093-b.
- R. Liang, Z. Zhang and S. Lin, Food Chem., 2017, 237, 793–802, DOI:10.1016/j.foodchem.2017.05.144.
- H. Sies, Free Radical Biol. Med., 1999, 27, 916–921, DOI:10.1016/s0891-5849(99)00177-x.
- C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255–6285, DOI:10.1021/cr0406559.
- S. A. Rider, S. J. Davies, A. N. Jha, A. A. Fisher, J. Knight and J. W. Sweetman, Aquaculture, 2009, 295, 282–291, DOI:10.1016/j.aquaculture.2009.07.003.
- S. J. Hamilton, Sci. Total Environ., 2004, 326, 1–31, DOI:10.1016/j.scitotenv.2004.01.019.
- L. Zhao, G. H. Zhao, Z. D. Zhao, P. Chen, J. Y. Tong and X. S. Hu, J. Agric. Food Chem., 2004, 52, 3954–3959, DOI:10.1021/jf049965i.
- M. Guan, W. Tang, Z. Xu and J. Sun, Evidence-Based Complementary Altern. Med., 2014, 2014, 182817, DOI:10.1155/2014/182817.
- W. Liu, T. Hou, W. Shi, D. Guo and H. He, J. Funct. Foods, 2018, 50, 183–191, DOI:10.1016/j.jff.2018.09.034.
| This journal is © The Royal Society of Chemistry 2021 |