
Bromine anion mediated epitaxial growth of core–shell Pd@Ag towards efficient electrochemical CO2 reduction†
Yinqiong
Wu‡
,
Jiachang
Zeng‡
,
Yang
Yang
,
Zhaojie
Li
,
Wenbiao
Zhang
,
Dan
Li
and
Qingsheng
Gao
 *
*
College of Chemistry and Materials Science, and Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou 510632, P. R. China. E-mail: tqsgao@jnu.edu.cn
First published on 8th April 2021
Abstract
Noble-metal core–shell nanostructures are promising electrocatalysts for CO2 reduction thanks to their interfacial configurations which are beneficial for electrocatalytic kinetics, but are severely limited to a few couples that require tiny lattice mismatches (<3%) between two metals for classical epitaxial growth. Herein, non-classical epitaxial growth mediated by bromine anions (Br−) is proposed to surmount the thermodynamic limitation of largely mismatched Ag and Pd lattices (5.012%), resulting in core–shell Pd@Ag cubes that are unavailable via conventional strategies. As evidenced, Br− strongly bonded on the surface of Pd seeds serves as a buffer layer (possibly AgBr) to relieve interfacial strains and enable the epitaxial growth of Ag lattices. Due to interfacial electron-transfer, the engineered Ag sites enhance efficient CO2 reduction to CO (faradaic efficiency ∼85% at −0.8 V vs. RHE in 0.5 M KHCO3), superior to the counterparts of Pd octahedrons, Ag nanoparticles and Pd–Ag nanoalloys. Providing new insights into the epitaxial growth of core–shell nanocrystals, this study is anticipated to inspire new design strategies for active and selective electrocatalysts.
Introduction
The electrochemical synthesis of value-added feedback via carbon dioxide (CO2) reduction is an appealing approach for the permanent storage of renewable electricity and closing the carbon neutral cycle.1–4 There is high requirement for electrocatalysts to address the huge kinetic barriers and rationalize the reaction pathways.5–7 For instance, noble-metal elements, e.g., Pd, Au, and Ag, can efficiently reduce CO2 to CO or formate, relying on surface atom arrangements and electronic configurations.8–11 Further recent progress has been made on their core–shell nanostructures (e.g., Pd@PdAu12 and Au@Pd13) that effectively ameliorate intermediate bindings via either the strain effect induced by the structure or the electron transfer between two components.14,15 Therefore, the rational construction of core–shell electrocatalysts with a particular emphasis on their interfacial interactions has attracted extensive attention.16–19 Although kinetic control over nucleation and growth is important to avoid the formation of two individual nanocrystals,20–22 the thermodynamics of epitaxial growth is a prerequisite for viable core–shell nanostructures. According to the layer-by-layer Frank–van der Merwe epitaxial growth model, a thermodynamic prerequisite is a small lattice mismatch (<3%) between core and shell elements,23–25 which unfortunately limits the variety of core–shell electrocatalysts.Thanks to the strong electronic interactions and the synergic enhancement between two metals, nanostructured Pd–Ag alloys have proved to have prominent electrocatalytic performances.26–32 However, core–shell nanostructures of Pd@Ag have rarely been reported, because the great lattice mismatch (5.012%) between Pd and Ag restricts epitaxial growth.20,33 Recently, binary PdAg alloys and Pd–H hydrides, instead of pure metal cores, were taken to synthesize PdAg@Ag octahedrons (Octs) and Pd@Ag nanosheets, respectively.34,35 The alloying of Pd with Ag or H can effectively expand the lattices on the surface to alleviate the mismatch with Ag shells and thus reduce the interfacial strain, highlighting the mediated epitaxial growth by the surface modification of seeds. Considering that most nanocrystal seeds are surface bonded with capping agents (e.g., oleylamine and halide anions) after wet-chemistry fabrication,36,37 the residual adsorbents that are difficult to remove would play the role of middlemen to buffer the lattice mismatch between core and shell metals. However, relevant reports are scarce. An unexpected growth of Ag shells on Pd cubes was always ascribed to the edge dislocation and stacking fault defects on Pd–Ag interfaces,38 which, however, have not been sufficiently understood in the context of ignoring mediation by surface capping agents.
Here, a bromine anion (Br−) mediated epitaxial growth of Ag shells on Pd Octs is proposed towards well-defined Pd@Ag core–shell cubes. Evidence is shown that the Br− strongly bonded on the surface of Pd Octs benefits the formation of a buffer layer (possibly AgBr) to relieve the strain and mediate the further epitaxial growth of Ag lattices, resulting in core–shell Pd@Ag cubes enclosed predominantly by Ag{100} facets. The electron transfer from Ag to Pd is detectable, which will upshift the d-band centre of the engineered Ag sites, and thereby enhance the binding with *COOH, a key intermediate in CO2 reduction.39 Pd@Ag cubes afford prominent electrocatalytic performance for CO2 reduction to CO with a high faradaic efficiency (FE) of ∼85% at −0.8 V vs. RHE in 0.5 M KHCO3, outperforming the counterparts of Pd Octs, Ag nanoparticles and Pd–Ag nanoalloys. Elucidating non-classical epitaxial growth of core–shell nanocrystals, this work will inspire the design of high-performance catalysts to be used in energy and environmental fields.
Experimental
Materials synthesis
Pd Octs as seeds for constructing Pd@Ag were synthesized according to previous reports.37 7 mL of a mixed solution containing 115 mmol L−1 cetyltrimethylammonium chloride (CTAC), 45 mmol L−1 citric acid and 50 mmol L−1 ascorbic acid (AA) was preheated to 100 °C in an oil bath under magnetic stirring (500 rpm). Quickly 4 mL of Na2PdCl4 (50 mmol L−1) was injected into the above solution, and the reaction was allowed to proceed for 3 h. After cooling to room temperature, the Pd Octs were collected by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min, and then washed with ultrapure water.
000 rpm for 10 min, and then washed with ultrapure water.
To fabricate core–shell Pd@Ag, 50 μL of Pd Octs solution (1 mg mL−1) and 250 μL of cetyltrimethylammonium (CTAB, 20 mmol L−1) were dispersed in 10 mL of ultrapure water and heated to 50 °C. The concentration of CTAB in solution is about 0.5 mmol L−1. And then, 150 μL of AgNO3 (10 mmol L−1) was added into the above solution that was stood still for more 30 minutes. After injecting AA (450 μL, 0.1 mol L−1), the solution was stirred at 50 °C for 6 h. Finally, Pd@Ag cubes were collected by centrifugation at 4500 rpm for 10 min.
Physical measurements
Transition electronic microscopy (TEM), energy dispersive spectroscopy (EDS) and the corresponding elemental mapping were collected on a JEOL 2100F instrument. X-ray diffraction (XRD) analysis was performed on Bruker D8 diffractometer using Cu Kα radiation (λ = 1.54056 Å). X-ray photoelectron spectroscopy (XPS) was conducted on Thermo Scientific (Escalab 250Xi), using C 1s (284.8 eV) as a reference.Electrochemical measurements
All of the electrochemical measurements were conducted on an electrochemical workstation (CHI 760, Shanghai Chenhua), using a saturated calomel electrode (SCE) as the reference electrode, and a platinum (CV sweep) or a graphite electrode (CO2RR) as the counter electrode. All potentials were converted to the RHE using the equation: E (vs. RHE) = E (vs. SCE) + 0.241 V + 0.0591 × pH. For the CV sweep to clean the Pd surface, 20 μL of Pd Octs (1 mg L−1) solution were loaded onto a glassy carbon electrode (GCE) and tested in 0.5 M H2SO4. We also added 100 μL of CTAB (2 × 10−2 mol L−1) to a Pd Octs solution (2 mL, 1 mg L−1), 20 μL of which was then deposited onto a GCE for a control experiment. A CV scan between 0.1 and 1.2 V vs. RHE was taken to clean the surface of Pd, in which PdO formation/reduction and hydrogen/bisulfate adsorption/desorption could cause the removal of residual surfactants. For the CO2RR performance, catalysts were loaded onto GCEs and tested in CO2-saturated 0.5 M KHCO3 solution. Typically, 0.76 mg (Pd@Ag:Vulcan XC-72R carbon black = 1:4) of catalyst and 15.0 μL of Nafion solution were dispersed in 360 μL of water–ethanol (volume ratio = 4:1) by sonication for 30 min to form a homogeneous ink. Then 20 μL of ink was loaded onto a GCE of 5 mm in diameter (geometric area: 0.196 cm−2). The CO2RR test was conducted in a gas-tight two-compartment electrochemical cell equipped with a piece of cation exchange membrane (Nafion 117, Sigma-Aldrich) as the compartment separator. Each compartment contained 45 mL of electrolyte. Before electrolysis, the electrolyte in the cathodic compartment was purged by CO2 for at least 30 min. CO2 was delivered into the cathodic compartment at a rate of 36 mL min−1 and the quantification of gaseous products was conducted on a gas chromatograph (GC). Gas-phase products were sampled every 30 min using high-purity nitrogen (N2, 99.999%) as the carrier gas. The column effluent (separated gas mixtures) was first passed through a thermal conductivity detector (TCD) where hydrogen was quantified; then CO was converted to methane by passing through a methanizer and subsequently quantified by FID. According to the peak areas in GC, the partial current densities and FEs of CO and H2 were calculated using the following equations (eqn (1)–(3)):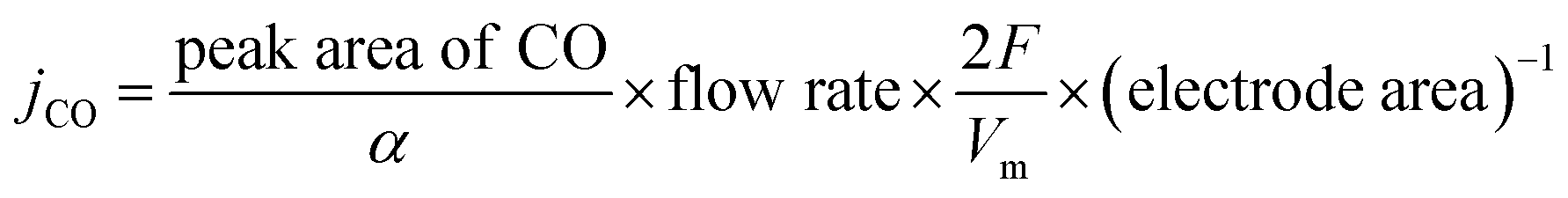 | (1) |
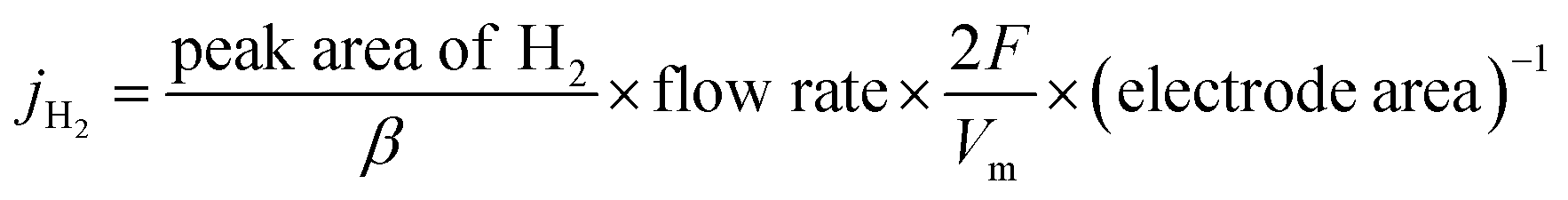 | (2) |
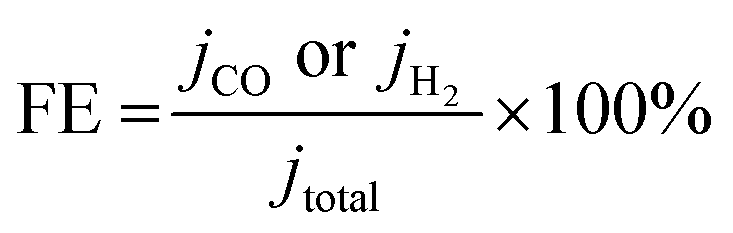 | (3) |
485 C mol−1).
Results and discussion
Pd Octs were used as the seeds for fabricating core–shell Pd@Ag. With an average edge length of 20 nm, Pd Octs specifically expose facets of {111} (Fig. 1a). The lattice fringe of 2.29 Å observed in high-resolution TEM (HR-TEM) corresponds to the (111) plane of an fcc Pd crystal. To further grow Ag on such Pd seeds, AgNO3 was reduced by AA in the presence of CTAB, producing cubic nanocrystals with a size of about 40 nm (Fig. 1b). A closer observation shows clear octahedral outlines inside and moiré fringes due to overlapping lattices (Fig. 1c), indicating the successful growth of Ag on Pd Octs. The clear grain boundary between a Pd core and an Ag shell (Fig. 1d) is believed to accommodate the large lattice mismatch between the two metals.38 Such Pd@Ag cubes mainly expose the Ag{100} facets on the shell, similar to previously reported Ag cubic nanocrystals.20 As a capping agent, CTAB reduces the growth rate of the Ag{100}, stabilizes these facets, and guides the morphology, leading to the final formation of cubic shells. In addition, the corresponding elemental mapping confirms that a Pd octahedron (red) is inside the cube, and Ag (green) is distributed outside (Fig. 1e and f). We collected a series of EDS profiles from a few Pd@Ag cubes, and gained an average Pd/Ag atomic ratio of 0.33.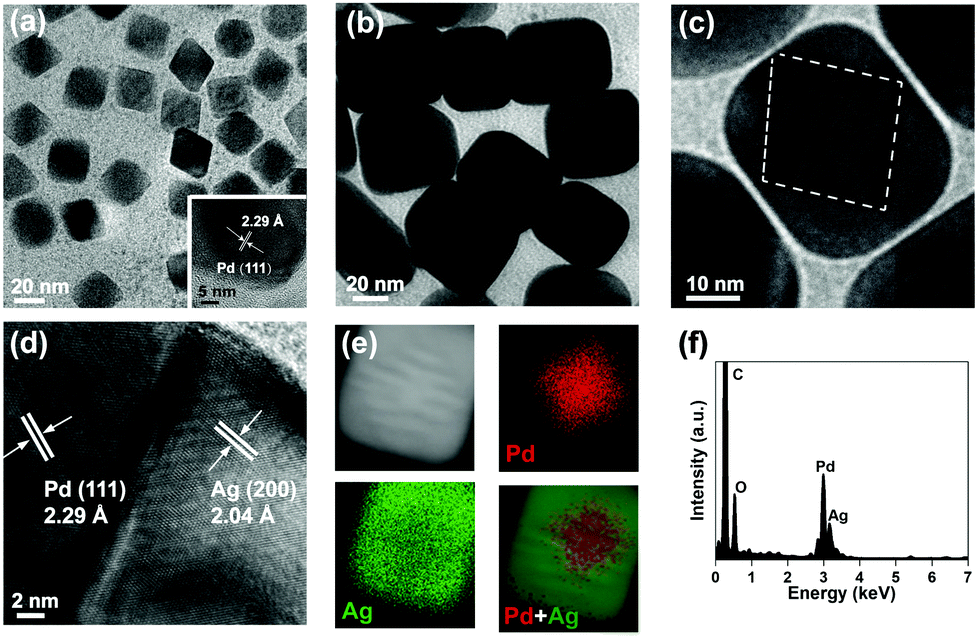 | ||
| Fig. 1 Structural characterization of Pd Octs and Pd@Ag cubes. (a) TEM image of Pd Octs. (b and c) TEM and (d) HR-TEM images, (e) elemental mapping (raw data are displayed as Fig. S1, ESI†) and (f) the corresponding EDS profile of a Pd@Ag cube. | ||
XRD and XPS investigations were further conducted to analyse Pd@Ag core–shell nanostructures. As depicted in the XRD patterns (Fig. 2a), Pd Octs exhibit the characteristic diffraction peaks indexed to the (111) and (200) planes of an fcc Pd crystal (JCPDS No. 46-1043). The main diffraction peaks of Pd@Ag cubes correspond to the (111) and (200) of Ag (JCPDS No. 04-0783), proving the effective reduction of Ag ions to crystallized Ag. In comparison with Pd Octs, the Pd(111) peak in Pd@Ag is obviously weakened, because the Pd core is wrapped in an Ag shell. Fig. 2b and c depict the XPS profiles of Pd and Ag, respectively, which clearly present varied chemical states owing to the core–shell structure. The Pd 3d profiles of Pd Octs can be deconvolved to three couples that are ascribed to metallic Pd0, Pd2+ and PdCl2 species, respectively. The presence of PdCl2 is due to the high concentration of Cl− required for fabrication. By contrast, the Pd 3d of Pd@Ag cubes can only be deconvolved to two couples associated with Pd0 and Pd2+. More importantly, the binding energies of Pd0 3d3/2 and 3d5/2 in Pd@Ag are 341.13 and 335.20 eV,40 obviously lower than those of Pd Octs, which indicates the electron transfer from Ag to Pd according to their different electronegativities (Pd: 2.20 > Ag: 1.93). Accordingly, the presence of Ag is confirmed by the Ag 3d profile of Pd@Ag, which shows visible signals for Ag+ species owing to the charge redistribution on Pd–Ag interfaces (Fig. 2c). Moreover, the atomic ratio of Pd to Ag on the surface of Pd@Ag is quantified as 1:18.4 by XPS survey spectra (Fig. S2, ESI†), agreeing with the core–shell structure.
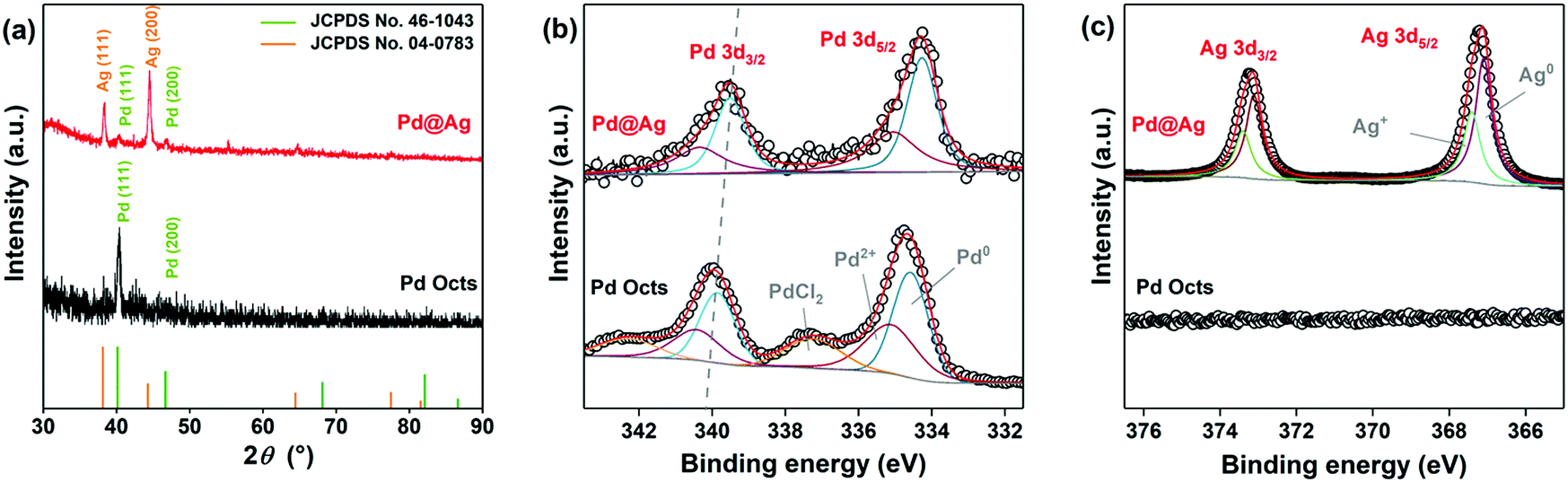 | ||
| Fig. 2 (a) XRD patterns and XPS profiles of (b) Pd 3d and (c) Ag 3d of Pd@Ag cubes and Pd Octs. | ||
The above characterizations have identified the successful formation of core–shell Pd@Ag that is, however, unavailable according to the Frank–van der Merwe epitaxial growth model. The lattice mismatch of Pd and Ag (5.012%) is larger than the threshold value of 3%, which makes it difficult for Ag to grow directly into a uniform single crystal shell on Pd. We found that adding CTAB is indispensable for epitaxial growth. As indicated by the TEM image (Fig. 3a), Pd Octs are well dispersed with clear octahedral outlines. When only AgNO3 and AA are added into a clean solution of Pd Octs with heating, the Octs are severely agglomerated and no core–shell structure can be obtained (Fig. 3b). The residual Cl− on the Pd surface, which was previously used as the capping agent to direct the formation of octahedral Pd seeds, will react with Ag+ to produce AgCl colloids. In the presence of a suitable amount of CTAB, well-defined Pd@Ag cubes can be successfully achieved and the reaction solution turns yellow-brown owing to the surface plasmon resonance of nanostructured Ag (Fig. 3c).41 This indicates the key role of Br− for the non-classical epitaxial growth of Ag on Pd.
 | ||
| Fig. 3 TEM images of (a) Pd Octs, and samples obtained (b) without and (c) with CTAB. Insets correspond to the digital photos of the final solutions after preparation. | ||
The functionality of Br− was always implied by previously reported Pd@Ag cubes that were prepared with CTAB stabilized Pd seeds.38 Although the formation of an Ag shell was ascribed to the edge dislocation and stacking fault defects on Pd–Ag interfaces, the mediation by the residual Br− on the Pd surface deserved more concern. By contrast, the seeds used to fabricate Pd@Ag in this work are the Pd Octs with surface Cl− adsorbates that are possibly exchanged with the Br− derived from CTAB owing to its strong binding on Pd(111). In this regard, we employed a CV sweep in 0.5 M H2SO4 to remove the residual surfactants on the Pd surface, and CV was also used as a probe to examine the cleaning effect, inquiring into the difference between Cl− and Br− bindings on Pd Octs. At the beginning (Fig. 4a), Pd Octs show severely suppressed current peaks related to surface PdO formation/reduction and hydrogen/bisulfate adsorption/desorption, indicating Cl− adsorbate remaining on Pd surface.37 After 100 scans, the peak currents increase significantly, confirming the successful removal of adsorbents from Pd{111}. The unbroken crystal facets are also proved by the characteristic peaks of CVs which are consistent with the voltammetric features of single-crystal Pd electrodes.42 In a control experiment, we added CTAB to Pd Octs, and the same CV sweep was performed (Fig. 4b). Even after 100 cycles, the current densities in CVs are still suppressed, without visible characteristic peaks associated with Pd(111). As indicated, the binding of Br− is stronger than that of Cl−, which drives the exchange of Cl− by Br− on the Pd Octs surface. The weak binding of Cl− on the Pd surface can be further used to interpret the easy formation of AgCl precipitate when Cl−-capped Pd Octs are exposed to AgNO3. The easily-exchangable Cl− can desorb from the Pd surface to react with Ag+. By contrast, Br− strongly adsorbed on Pd Octs would direct the growth of the Ag shell. Further evidence is acquired from XPS analysis. Before CVs, there are the peaks for both Cl 2p and PdCl2 in the XPS profile (Fig. 4c), but they both disappear after CVs, which proves once again that Cl− has been removed from the Pd Octs surface. In comparison, after introducing CTAB to Pd Octs, the satellite peaks of PdCl2 are significantly weakened. The signals for both Cl and PdCl2 disappear after the sweep, while the peak of Br 3d still remains. It is confirmed that Br− has replaced Cl− and was strongly bonded on the surface of Pd seeds, which will benefit the formation of a buffer layer (possibly AgBr) between a Pd core and an Ag shell. The buffer layer relieves the strain and mediates the further epitaxial growth of Ag lattices, resulting in a non-classical manner of epitaxial growth for constructing core–shell Pd@Ag.
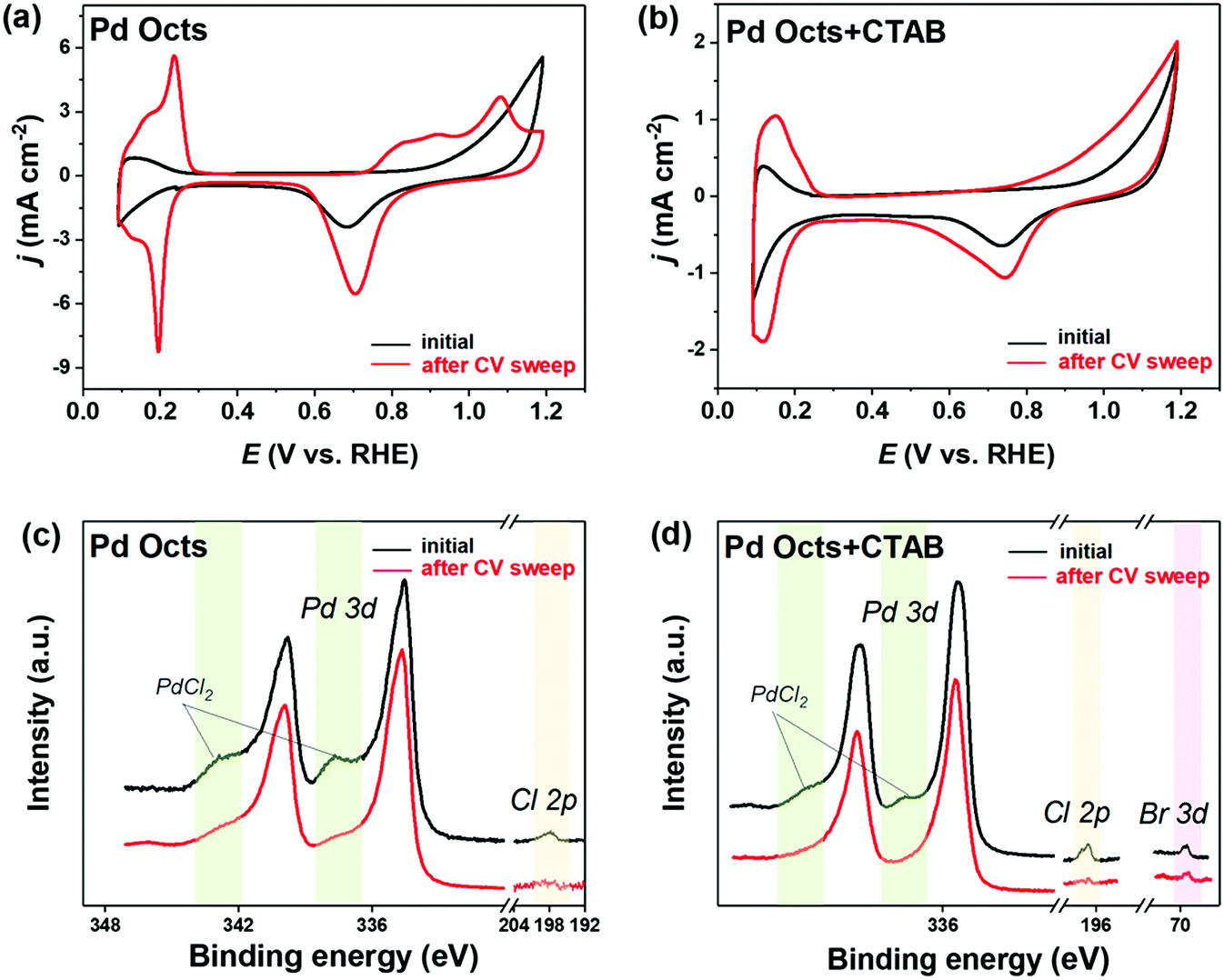 | ||
| Fig. 4 Comparison of electrochemical surface cleaning for Pd Octs and Pd Octs with CTAB in 0.5 M H2SO4. (a and b) CVs and (c and d) XPS profiles of (a and c) Pd Octs and (b and d) Pd Octs with CTAB, before and after surface cleaning by CV sweeping. | ||
Such epitaxial growth depends highly on the surface Br− adsorbent. When excessive CTAB (1.0 mmol L−1) is introduced, there is a considerable amount of Br− and CTA+ in solution that unfortunately favours the formation of individual Ag nanoparticles, rather than epitaxial growth on Pd seeds (Fig. S3, ESI†). In the meantime, the CTA+ long-chain is also important to produce well-dispersed core–shell nanocrystals, because it can prevent the agglomeration of nanocrystals.43 When KBr is used to prepare Pd@Ag, only white colloid AgBr precipitates are obtained, which would disturb the growth of Pd@Ag (Fig. S4, ESI†). In addition, the surfactant of CTAB will direct the growth of Ag crystals towards cubic nanostructures mainly exposing {100} facets.
On the basis of the above analysis, we propose a mechanism for the unexpected manner of epitaxial growth towards core–shell Pd@Ag (Fig. 5). When CTAB is introduced into the reaction, the exchange of Cl− adsorbents by Br− on the surface of Pd seeds is enabled owing to the stronger binding of the latter. To keep the surface charge balance, CTA+ long chains will wrap up the seeds and stabilize them for further reactions. Owing to the low solubility product constant of AgBr (5.0 × 10−13), Ag+ will be deposited on the Pd surface, possibly in the form of AgBr, resulting in a buffer layer to alleviate the mismatch and thus reduce the interfacial strains between a Pd core and an Ag shell. Thereby, this non-classical epitaxial growth of Ag on Pd is achieved via key mediation by Br−, which is only available on a surface that can effectively adsorb and stabilize Br−.
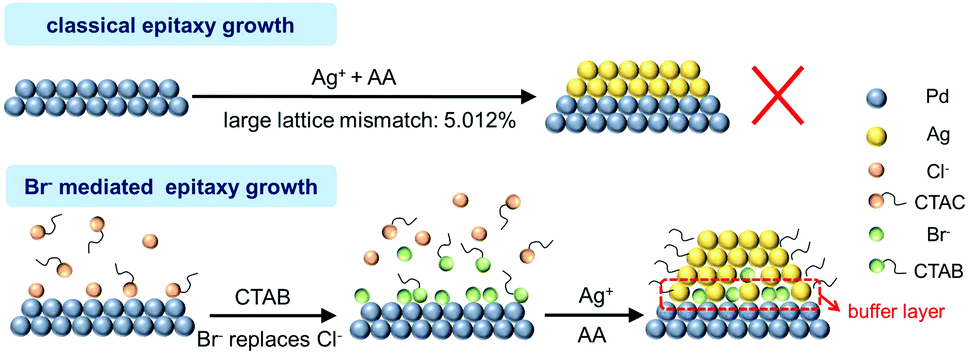 | ||
| Fig. 5 Schematic illustration for the Br− mediated epitaxial growth towards Pd@Ag cubes. | ||
Noble-metal core–shell nanostructures can effectively ameliorate intermediate bindings and then cut down reaction energy barriers for CO2RR.44,45 Before CO2RR tests, Pd Octs and Pd@Ag cubes were examined by CVs (Fig. 6a). Pd Octs show obvious characteristic peaks that disappear when Pd is coated with Ag. This proves the formation of a core–shell structure that exposes an Ag shell outside Pd@Ag. The CO2RR activity of carbon (Vulcan XC-72R) supported Pd@Ag cubes, Ag nanoparticles, Pd–Ag nanoalloys (Pd/Ag = 0.33), and Pd Octs was then investigated in an H-cell with CO2-saturated 0.5 M KHCO3 aqueous solution. All the samples afford a higher current density with CO2 saturation than the cases saturated with N2, indicating the reduction of CO2 (Fig. S5, ESI†). Fig. 6b displays the linear sweeping voltammetry (LSV) curves, in which Pd@Ag shows the highest current density and the lowest overpotential. Over a wide potential range from −0.9 V to −0.6 V (vs. RHE), Pd@Ag cubes achieve a high CO FE of 85%, which is better than the counterparts (Fig. 6c). Furthermore, a durability experiment over Pd@Ag was investigated at −0.8 V for 24 h. It affords stable current output and CO FE (Fig. 6d), confirming the satisfactory long-term durability of this core–shell electrocatalyst. Accordingly, TEM observation confirms the well-maintained core–shell nanostructures of Pd@Ag after a long-term test (inset of Fig. 6d). Regarding the CO2 reduction occurring on the catalyst surface, we can deduce that the Ag shell as the electrocatalytically active sites is effectively engineered by the Pd core via strong electronic interactions. The proven electron transfer from Ag to Pd by XPS (Fig. 2) leads to the upshifted d-band centre of Ag, and consequently enhances the binding with *COOH intermediates. Such enhancement can overcome the high energy barrier of generating *COOH that is typically considered as the potential determining step for CO2RR over Ag catalysts.31,46 It is noteworthy that compressive strain of shell Ag lattices is possible,18 but this effect would be negligible due to the thick Ag shell. Therefore, the interfacial interactions in Pd@Ag cubes, mainly electronic effects, account for the obviously boosted CO2RR performance.
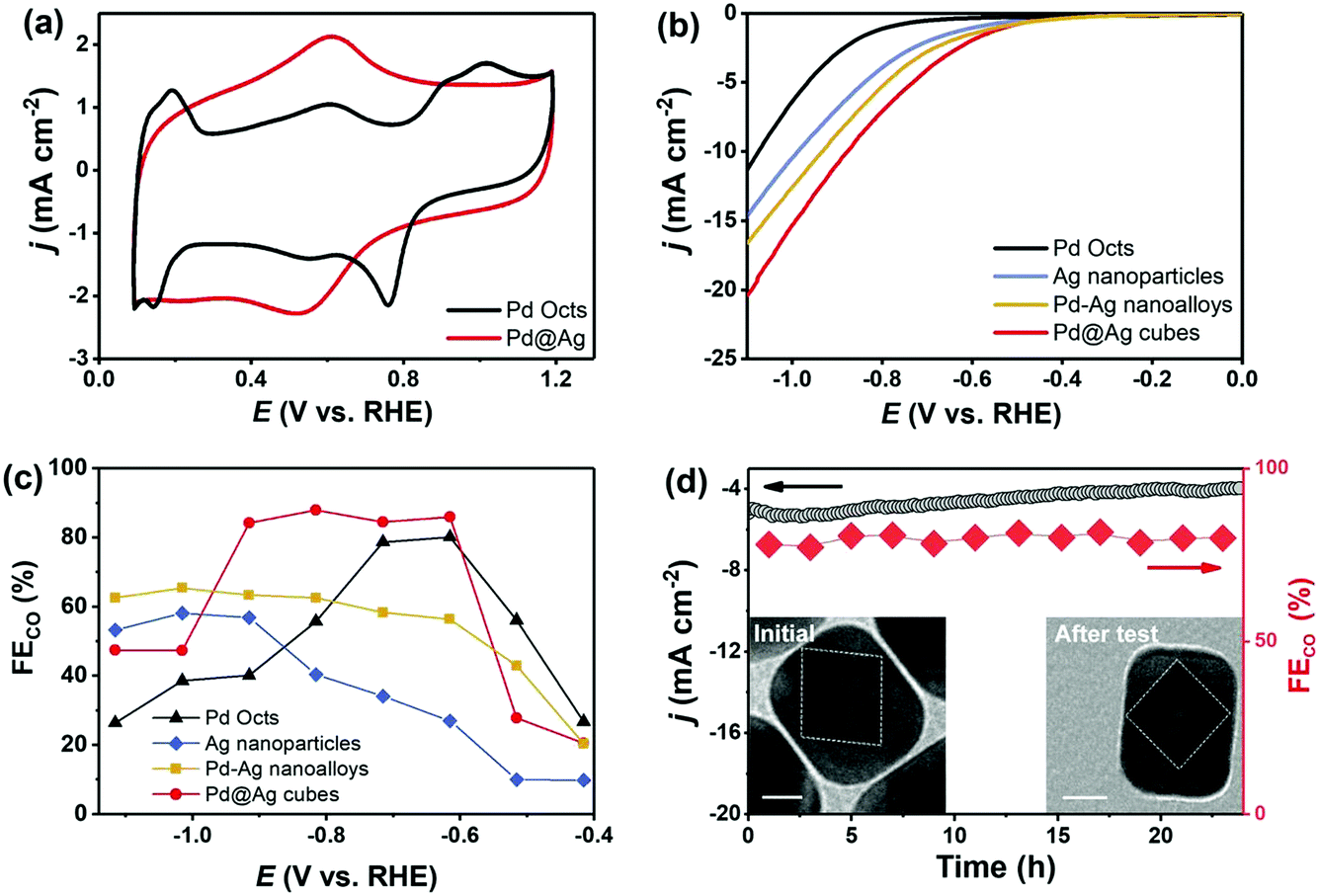 | ||
| Fig. 6 CO2RR performance in CO2-saturated 0.5 M KHCO3. (a) CV curves without bubbling CO2; (b) polarization curves; (c) FE of CO at various applied potentials; (d) long-term i–t curve and CO FE, along with TEM images before and after electrolysis (scale bar: 20 nm). | ||
Conclusions
In summary, a Br− mediated epitaxial growth is successfully introduced to synthesize Pd@Ag core–shell cubes with Pd Octs as the seeds. Control experiments and characterizations show that Br− can be adsorbed on the Pd surface and form an AgBr buffer layer to effectively overcome the large lattice mismatch between Pd and Ag. As a result, non-classical epitaxial growth is enabled towards core–shell nanostructures that are unavailable via conventional strategies. Such unique Pd@Ag core–shell nanostructures prove to have obviously improved CO2RR performance owing to the engineered Ag sites via interfacial electron-transfer. In CO2-saturated 0.5 M KHCO3, they afford better activity and higher efficiency (CO FE of 85% at −0.8 V vs. RHE) than Pd Octs, Ag nanoparticles or even Pd–Ag nanoalloys. Providing a new understanding of the epitaxial growth of core–shell nanocrystals, this work will inspire the exploitation of high-performance electrocatalysts to be used in energy and environmental fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (grant no.: 21773093), Guangdong Natural Science Foundation (2021A1515012351) and China Postdoctoral Science Foundation funded project (grant no.: 2020M673056).References
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Recent advances in innovative strategies for the CO2 electroreduction reaction, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- D. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldan Cuenya, Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Designing materials for electrochemical carbon dioxide recycling, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, Understanding the roadmap for electrochemical reduction of CO2 to multi-carbon oxygenates and hydrocarbons on copper-based catalysts, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- J. Wu, Y. Huang, W. Ye and Y. Li, CO2 reduction: from the electrochemical to photochemical approach, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- A. D. Handoko, F. Wei, B. S. Yeo and Z. W. Seh, Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- D. Gao, H. Zhou, F. Cai, J. Wang, G. Wang and X. Bao, Pd-containing nanostructures for electrochemical CO2 reduction reaction, ACS Catal., 2018, 8, 1510–1519 CrossRef CAS.
- S. Thota, Y. Wang and J. Zhao, Colloidal Au-Cu alloy nanoparticles: synthesis, optical properties and applications, Mater. Chem. Front., 2018, 2, 1074–1089 RSC.
- X. Yuan, L. Zhang, L. Li, H. Dong, S. Chen, W. Zhu, C. Hu, W. Deng, Z.-J. Zhao and J. Gong, Ultrathin Pd–Au shells with controllable alloying degree on Pd nanocubes toward carbon dioxide reduction, J. Am. Chem. Soc., 2019, 141, 4791–4794 CrossRef CAS PubMed.
- S. Zhu, X. Qin, Q. Wang, T. Li, R. Tao, M. Gu and M. Shao, Composition-dependent CO2 electrochemical reduction activity and selectivity on Au–Pd core–shell nanoparticles, J. Mater. Chem. A, 2019, 7, 16954–16961 RSC.
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Surface and interface control in nanoparticle catalysis, Chem. Rev., 2019, 120, 1184–1249 CrossRef PubMed.
- X. Yin, L. Yang and Q. Gao, Core–shell nanostructured electrocatalysts for water splitting, Nanoscale, 2020, 12, 15944–15969 RSC.
- Y. W. Lee, M. Kim, Z. H. Kim and S. W. Han, One-step synthesis of Au@Pd core−shell nanooctahedron, J. Am. Chem. Soc., 2009, 131, 17036–17037 CrossRef CAS PubMed.
- J. Gu, Y.-W. Zhang and F. Tao, Shape control of bimetallic nanocatalysts through well-designed colloidal chemistry approaches, Chem. Soc. Rev., 2012, 41, 8050–8065 RSC.
- M. Luo and S. Guo, Strain-controlled electrocatalysis on multimetallic nanomaterials, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- Y. Ge, Z. Huang, C. Ling, B. Chen, G. Liu, M. Zhou, J. Liu, X. Zhang, H. Cheng, G. Liu, Y. Du, C.-J. Sun, C. Tan, J. Huang, P. Yin, Z. Fan, Y. Chen, N. Yang and H. Zhang, Phase-selective epitaxial growth of heterophase nanostructures on unconventional 2H-Pd nanoparticles, J. Am. Chem. Soc., 2020, 142, 18971–18980 CrossRef CAS PubMed.
- J. Zeng, C. Zhu, J. Tao, M. Jin, H. Zhang, Z.-Y. Li, Y. Zhu and Y. Xia, Controlling the nucleation and growth of silver on palladium nanocubes by manipulating the reaction kinetics, Angew. Chem., Int. Ed., 2012, 51, 2354–2358 CrossRef CAS PubMed.
- C. Fang, T. Bi, Q. Ding, Z. Cui, N. Yu, X. Xu and B. Geng, High-density Pd nanorod arrays on Au nanocrystals for high-performance ethanol electrooxidation, ACS Appl. Mater. Interfaces, 2019, 11, 20117–20124 CrossRef CAS PubMed.
- C. Fang, J. Zhao, R. Jiang, J. Wang, G. Zhao and B. Geng, Engineering of hollow PdPt nanocrystals via reduction kinetic control for their superior electrocatalytic performances, ACS Appl. Mater. Interfaces, 2018, 10, 29543–29551 CrossRef CAS PubMed.
- C. Zhu, J. Zeng, J. Tao, M. C. Johnson, I. Schmidt-Krey, L. Blubaugh, Y. Zhu, Z. Gu and Y. Xia, Kinetically controlled overgrowth of Ag or Au on Pd nanocrystal seeds: from hybrid dimers to nonconcentric and concentric bimetallic nanocrystals, J. Am. Chem. Soc., 2012, 134, 15822–15831 CrossRef CAS PubMed.
- E. Bauer and J. H. van der Merwe, Structure and growth of crystalline superlattices: From monolayer to superlattice, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 3657–3671 CrossRef CAS PubMed.
- K. A. Lozovoy, A. G. Korotaev, A. P. Kokhanenko, V. V. Dirko and A. V. Voitsekhovskii, Kinetics of epitaxial formation of nanostructures by Frank–van der Merwe, Volmer–Weber and Stranski–Krastanow growth modes, Surf. Coat. Technol., 2020, 384, 125289 CrossRef CAS.
- Y. Zhou, N. Han and Y. Li, Recent progress on Pd-based nanomaterials for electrochemical CO2 reduction, Acta Phys.-Chim. Sin., 2020, 36, 2001041 Search PubMed.
- M. Cui, G. Johnson, Z. Zhang, S. Li, S. Hwang, X. Zhang and S. Zhang, AgPd nanoparticles for electrocatalytic CO2 reduction: bimetallic composition-dependent ligand and ensemble effects, Nanoscale, 2020, 12, 14068–14075 RSC.
- J. H. Lee, S. Kattel, Z. Jiang, Z. Xie, S. Yao, B. M. Tackett, W. Xu, N. S. Marinkovic and J. G. Chen, Tuning the activity and selectivity of electroreduction of CO2 to synthesis gas using bimetallic catalysts, Nat. Commun., 2019, 10, 3724 CrossRef PubMed.
- M. Nazemi, P. Ou, A. Alabbady, L. Soule, A. Liu, J. Song, T. A. Sulchek, M. Liu and M. A. El-Sayed, Electrosynthesis of ammonia using porous bimetallic Pd-Ag nanocatalysts in liquid- and gas-phase systems, ACS Catal., 2020, 10, 10197–10206 CrossRef CAS.
- M. Yang, X. Lao, J. Sun, N. Ma, S. Wang, W. Ye and P. Guo, Assembly of bimetallic PdAg nanosheets and their enhanced electrocatalytic activity toward ethanol oxidation, Langmuir, 2020, 36, 11094–11101 CrossRef CAS PubMed.
- J. Zeng, W. Zhang, Y. Yang, D. Li, X. Yu and Q. Gao, Pd-Ag alloy electrocatalysts for CO2 reduction: Composition tuning to break the scaling relationship, ACS Appl. Mater. Interfaces, 2019, 11, 33074–33081 CrossRef CAS PubMed.
- C. Fang, J. Zhao, G. Zhao, L. Kuai and B. Geng, Simultaneous tunable structure and composition of PtAg alloyed nanocrystals as superior catalysts, Nanoscale, 2016, 8, 14971–14978 RSC.
- C. Zhu, J. Zeng, J. Tao, M. C. Johnson, I. Schmidt-Krey, L. Blubaugh, Y. Zhu, Z. Gu and Y. Xia, Kinetically controlled overgrowth of Ag or Au on Pd nanocrystal seeds: from hybrid dimers to nonconcentric and concentric bimetallic nanocrystals, J. Am. Chem. Soc., 2012, 134, 15822–15831 CrossRef CAS PubMed.
- B.-S. Choi, J. Song, M. Song, B. S. Goo, Y. W. Lee, Y. Kim, H. Yang and S. W. Han, Core–shell engineering of Pd–Ag bimetallic catalysts for efficient hydrogen production from formic acid decomposition, ACS Catal., 2018, 9, 819–826 CrossRef.
- X. Wang, B. Wu, G. Chen, Y. Zhao, P. Liu, Y. Dai and N. Zheng, A hydride-induced-reduction strategy for fabricating palladium-based core–shell bimetallic nanocrystals, Nanoscale, 2014, 6, 6798–6804 RSC.
- X.-Y. Ma, Y. Chen, H. Wang, Q.-X. Li, W.-F. Lin and W.-B. Cai, Electrocatalytic oxidation of ethanol and ethylene glycol on cubic, octahedral and rhombic dodecahedral palladium nanocrystals, Chem. Commun., 2018, 54, 2562–2565 RSC.
- Y. Yang, J. Zeng, Y. Shu and Q. Gao, Revealing facet effects of palladium nanocrystals on electrochemical biosensing, ACS Appl. Mater. Interfaces, 2020, 12, 15622–15630 CrossRef CAS PubMed.
- J. Gong, F. Zhou, Z. Li and Z. Tang, Controlled synthesis of non-epitaxially grown Pd@Ag core–shell nanocrystals of interesting optical performance, Chem. Commun., 2013, 49, 4379–4381 RSC.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction, Chem, 2018, 4, 1809–1831 CAS.
- W. Huang, X. Kang, C. Xu, J. Zhou, J. Deng, Y. Li and S. Cheng, 2D PdAg alloy nanodendrites for enhanced ethanol electroxidation, Adv. Mater., 2018, 30, 1706962 CrossRef PubMed.
- J. He, S. Li, D. Lyu, D. Zhang, X. Wu and Q.-H. Xu, Aggregation induced emission enhancement by plasmon coupling of noble metal nanoparticles, Mater. Chem. Front., 2019, 3, 2421–2427 RSC.
- M. Hara, U. Linke and T. Wandlowski, Preparation and electrochemical characterization of palladium single crystal electrodes in 0.1 M H2SO4 and HClO4: Part I. Low-index phases, Electrochim. Acta, 2007, 52, 5733–5748 CrossRef CAS.
- F. Wu, W. Wang, Z. Xu and F. Li, Bromide (Br)-based synthesis of Ag nanocubes with high-yield, Sci. Rep., 2015, 5, 10772 CrossRef PubMed.
- J. Fan, H. Du, Y. Zhao, Q. Wang, Y. Liu, D. Li and J. Feng, Recent progress on rational design of bimetallic Pd based catalysts and their advanced catalysis, ACS Catal., 2020, 10, 13560–13583 CrossRef CAS.
- S. Das, J. Perez-Ramirez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Core-shell structured catalysts for thermocatalytic, photocatalytic, and electrocatalytic conversion of CO2, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, Shape-dependent electrocatalytic reduction of CO2 to CO on triangular silver nanoplates, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Raw data of Pd@Ag cubes elemental mapping, XPS survey spectra of Pd@Ag cubes, TEM images of samples in control experiments, and LSV of Pd Octs, Ag nanoparticles, Pd–Ag nanoalloys and Pd@Ag in 0.5 M KHCO3 saturated by N2 and CO2. See DOI: 10.1039/d1qm00169h |
| ‡ These authors contribute equally to this work. |
| This journal is © the Partner Organisations 2021 |