
Synthesis of amorphous low Tg polyesters with multiple COOH side groups and their utilization for elastomeric vitrimers based on post-polymerization cross-linking†
Mikihiro
Hayashi
 *,
Ryoto
Yano
and
Akinori
Takasu
*,
Ryoto
Yano
and
Akinori
Takasu
Department of Life Science and Applied Chemistry, Graduate school of Engineering, Nagoya Institute of Technology, Gokiso-cho, Shouwa-ku, Nagoya 466-8555, Japan. E-mail: hayashi.mikihiro@nitech.ac.jp
First published on 18th March 2019
Abstract
In this research, we demonstrate the preparation and unique functions of elastomeric polyester vitrimer materials with different cross-link densities based on a post-polymerization cross-linking method. First, we synthesize polyesters with thiol (SH) groups at the side groups via melt polycondensation of pentanediol, adipic acid, and thiomalic acid. The SH-attached polyester is reacted with acrylic acid through the Michael addition reaction between the SH side groups of the polyester and the vinyl groups of acrylic acid, providing viscous amorphous polyesters with multiple COOH side groups (PE-COOH). PE-COOH polymers are cross-linked through the reaction with a diepoxy cross-linker (1,4-butanediol diglycidyl ether) in the presence of a trans-esterification catalyst Zn(OAc)2 at 120 °C. This cross-linking via the COOH–epoxy reaction generates free OH groups in the network, which act as the carriers for trans-esterification with ester bonds in the polyester strands. In this design, the COOH group equivalent weight (MCOOH) in PE-COOH is easily tuned by the initial polycondensation feed ratio between adipic acid and thiomalic acid. This also enables the tuning of the cross-link density and the fraction of free OH groups in the final vitrimer materials as reflected in the tensile and swelling properties. The representative vitrimeric properties, including the softening temperature (Tsoft) and stress-relaxation at high temperature, are all affected by the cross-link density (i.e., MCOOH in PE-COOH). Interestingly, a lower Tsoft and a shorter relaxation time were attained for the sample with a larger cross-link density. This is because the sample with the larger cross-link density possesses a larger fraction of the bond-exchange carriers, i.e., free OH groups, in the present system, which enables the activation of trans-esterification based bond-exchange with lower temperature and higher frequency.
Introduction
Cross-link formation is one of the proven methods to modify or enhance the physical properties of materials.1–3 The introduction of covalently bonded cross-links is essential to produce practical network materials, such as elastomers, gels, and thermosetting resin.4–7 However, the covalently bonded cross-links have an irreversible nature, and thus most of the materials cross-linked via covalent bonds suffer from the lack of recyclability and re-shapeability.Recently, there have been various methods to assess the above conventional problems in covalently cross-linked materials. A representative method to solve the problem is utilization of dynamic covalent chemistry.8–11 As an example, A. Takahara and H. Otsuka et al. have proposed cross-linked materials that possess the capability of bond reshuffling based on the chemical equilibrium between bond breaking and reforming.12–16 The external stimuli, such as heat and light, displace the equilibrium toward bond-breaking and bond-reshuffling, which thus adds unique functions to the materials, e.g., self-healing ability and re-processability.
Another close but different system with the essence of dynamic covalent chemistry has been reported by C. N. Bowman et al. in 2005,17 where they prepared a novel cross-linked material that could show multiple useful functions, such as plasticity and shape changes without residual stress. This was achieved by utilizing exchange reactions in the network based on radical addition–fragmentation upon exposure to light. Very recently, L. Leibler's group has also developed functional network materials, so-called vitrimers.18 In the first vitrimer report from Leibler's group, a diepoxy compound (diglycidyl ether of bisphenol A) and Pripol 1040 (a mixture of a tri-carboxylic acid compound and a di-carboxylic acid compound) were used as the components. By simply reacting these components in the presence of a catalyst zinc acetate Zn(OAc)2 at a high temperature, the cross-linked vitrimer materials were obtained. In this system, multiple free OH groups and esters were generated in the reaction between epoxy and carboxylic acid (COOH) groups, which worked as triggers to induce intermolecular trans-esterification in the presence of the trans-esterification catalyst, Zn(OAc)2. The obtained materials behaved as tough resins at room temperature, while the materials showed softening and re-processability at high temperatures due to the catalytically activated intermolecular trans-esterification. The similarity between the above two materials from Bowman's group and Leibler's group is that the key to achieve plasticity is the network topology alteration due to the bond-exchange processes, where it should be noted that the network connectivity remains unchanged. In this sense, the concept was very different from those of the conventional dynamic covalent chemistry systems that rely on the chemical equilibrium between bond breaking and reforming or supramolecular chemistry systems utilizing dissociation and re-association of weak cross-links.19–28
This pioneering work on vitrimers and their following reports have motivated many other research groups to create other types of vitrimer materials by utilizing not only trans-esterification,29–38 but also various bond-exchangeable functional groups. In particular, vitrimer materials that do not require any catalysts for bond-exchange have been developed by utilizing trans-amination,39–42 trans-alkylation,43–45 silyl ether exchange,46 metathesis reactions,47 conjugate addition–elimination reactions,48etc. These catalyst-free systems have a benefit of avoiding the issues derived from the deactivation of catalytic properties upon many cycles of usage. Recently, vitrimer materials with dual cross-links have also been reported to control both the mechanical properties at room temperature and the vitrimeric properties at high temperatures.49,50 As another trend, the vitrimer concept has been applied to composite systems with inorganic fillers,51–54 where the surface functionality of fillers affects the vitrimeric properties.
Although vitrimer materials have been developed as novel functional cross-linked materials as introduced above, there is further requirement of accumulating the knowledge on controlling the vitrimeric physical properties in order to practicalize them. For the vitrimer materials with a catalytically activated bond-exchange mechanism, it has been reported that control of the physical properties, such as the softening temperature and stress-relaxation dynamics, can be possible by considering the kind of catalyst and the catalyst amount.55–57 However, in most of the past reports, there has been little mention about the effects of the cross-link density on the vitrimeric physical properties, although the cross-link density is one of the dominant molecular parameters to control the macro-physical properties of network materials.
In this report, we thus demonstrate the preparation of elastomer-type vitrimer materials with tunable cross-link density for investigation of cross-link density effects on the elastomeric and vitrimeric physical properties (Fig. 1). So far, we have reported the synthesis of linear polyesters with multiple reactive side groups,28,58 which here leads us to synthesize model polyester precursors for vitrimer preparation. Concretely, we first synthesize low glass transition amorphous polyesters with multiple COOH side groups (PE-COOH) by melt polycondensation and the subsequent Michael addition reaction (Scheme 1). In the first melt polycondensation, the monomers, 1,6-pentanediol, adipic acid, and thiomalic acid were reacted in the presence of a scandium(III) trifluoromethanesulfonate (Sc(OTf)3) catalyst, which provides the polyester with multiple SH groups thanks to the usage of thiomalic acid.28,58 Since SH groups are known to show high reactivity with double bonds of various vinyl monomers,59,60 we reacted acrylic acid with the SH-attached polyester via the Michael addition reaction, providing PE-COOH. In this way, we prepare PE-COOH polymers with different fractions of COOH groups by varying the initial monomer feed ratios, where the fraction of COOH groups per chain is the primal key to control the cross-link density of the final cross-linked materials. PE-COOH chains are then thermally cross-linked in the presence of diepoxy cross-linkers (1,4-butanediol diglycidyl ether) and trans-esterification catalysts (Zn(OAc)2). During the cross-linking, free OH groups are generated by an epoxy-opening reaction between the diepoxy cross-linkers and the COOH groups in PE-COOH, and thus the obtained cross-linked material can possess bond-exchange ability via trans-esterification between the multiple OH groups and the ester bonds in the network (see Fig. 1). The cross-link density is investigated by tensile tests, and the network quality is further evaluated based on the modulus by the model analyses. We then systematically investigate the representative vitrimeric properties, including softening temperatures, stress-relaxation dynamics, and activation energy for the bond-exchange processes, for the model materials with two different cross-link densities to attain insights into the correlation between the cross-link density and the physical properties. The unique functions of the present vitrimer materials are also shown in the end of the paper.
 | ||
| Fig. 1 Schematic of the present molecular design. Free OH groups are generated by the epoxy-opening reaction with COOH groups in PE-COOH during the cross-linking reaction. The pictures of samples before and after cross-linking are also shown. | ||
 | ||
| Scheme 1 Synthesis of PE-COOH. | ||
Experimental section
Materials
Thiomalic acid (ThA), 1,5-pentanediol (PD), acrylic acid (AA), Sc(OTf)3, and other organic solvents were purchased from TCI. Adipic acid (AdA) and Zn(OAc)2 were purchased from Wako Pure Chemical Industries. The diepoxy cross-linker, 1,4-butanediol diglycidyl ether (BDE), was purchased from Aldrich. These chemicals were all used as received.Synthesis of PE-COOH
For preparation of the vitrimer materials, polyesters with multiple COOH groups at the side groups (PE-COOH) were firstly synthesized. We performed melt polycondensation of PD, AdA, and ThA with a small amount of the Sc(OTf)3 catalyst.58 In the polycondensation reaction, the stoichiometric ratio between COOH and OH was kept as unity while the molar ratio of ThA to AdA was varied to obtain the polyester with different fractions of the thiol (SH) group. Concretely, the feed molar ratios of PD/AdA/ThA were set as 1/0.7/0.3 and 1/0.3/0.7. The polycondensation was carried out at 80 °C for approximately 15 hours with vacuum stripping. Purification was conducted by precipitating its THF solution into methanol repeatedly. Since the SH group is known to exhibit high reactivity with the double bonds of various vinyl monomers,59,60 we reacted acrylic acid with the polyesters having multiple SH groups via the Michael addition reaction to obtain PE-COOH. The polyesters with SH groups were dissolved in DMF, and then acrylic acid (3 eq. to SH groups in the polyester) and triethylamine (0.33 eq. to SH groups in the polyester) were added. The reaction was conducted at room temperature overnight. Unreacted acrylic acid was removed by re-precipitation of the THF solution into methanol repeatedly. The characterization of various PE-COOH polymers, including the number average molecular weight (Mn), the dispersity index (PDI), the average number of COOH groups per chain (NCOOH), and the COOH group equivalent weight (MCOOH = Mn/NCOOH), is summarized in Table 1. The obtained PE-COOH polymers were coded as PE-X, where the X in the code represents the MCOOH for each sample.| Code | PD/AdA/ThA in feeda | M n | PDIb | N COOH | M COOH |
|---|---|---|---|---|---|
| a The feed ratio of 1,5-pentanediol (PD)/adipic acid (AdA)/thiomalic acid (ThA) in the initial polycondensation. b Number average molecular weights (Mn) and dispersity indices (PDI), which were determined by SEC in DMF with LiBr. PMMA standards were used for the estimation. c The average number of COOH groups per chain determined by SEC and 1H-NMR (see the calculation procedure in the ESI). d The COOH group equivalent weight calculated from Mn/NCOOH. | |||||
| PE-720 | 1/0.7/0.3 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
1.3 | 24 | 720 |
| PE-310 | 1/0.3/0.7 | 14300 |
1.5 | 46 | 310 |
Cross-linking reaction
To obtain a homogeneous mixture for the cross-linking reaction, we first prepared solutions of PE-COOH, the diepoxy cross-linker (BDE), and the trans-esterification catalyst (Zn(OAc)2) using CHCl3/MeOH (9/1 by vol.), separately. The solutions were then mixed in a Teflon-made mold (W × D × H = 6 cm × 4 cm × 2 cm), and the solvents were evaporated by room temperature casting and subsequently by drying in vacuo. In the blends, the functional group molar ratio between the COOH groups of PE-COOH and the epoxy groups of BDE was fixed to be unity while the molar ratio of Zn(OAc)2 to COOH groups was fixed to be 0.2 (20 mol%). The cross-linking was performed at 120 °C for 4 hours under vacuum. The samples after the cross-linking reaction were coded as CL-PE-X, where PE-X in the code represents PE-X used in the cross-linking. In a similar way, we also prepared a control sample by performing a cross-linking reaction with the diepoxy cross-linker but without adding Zn(OAc)2. We performed swelling tests, and the test protocol and results are provided in the ESI.†Polymer characterization
The dispersity indices, PDIs, of polymers were determined by size exclusion chromatography (SEC). The measurements were conducted by using Shodex KD803 + 804 columns, combined with a Tosoh DP8020 pump system and an RI (Tosoh RI-8020) detector. The eluent was dimethylformamide (DMF) with LiBr (0.05 wt%), and the flow rate was 0.5 ml min−1. The elution volume was calibrated with a standard series of poly(methyl methacrylate)s. The fraction of monomer units in a chain was determined by 1H-NMR in d-DMSO by using a Bruker Analytik DPX400 spectrometer (400 MHz). The progress of the Michael addition reaction was also checked by 1H-NMR in d-DMSO with a tiny amount of trifluoroacetic acid (TFA).Spectroscopy
Fourier transform infrared spectroscopy (FT-IR) measurements were conducted to monitor the progress of the cross-linking reaction by focusing on the signals from the COOH groups and epoxy groups. The measurements were performed at room temperature using a FT/IR 430 spectrometer combined with an ATR attachment (JASCO Co.).Thermal properties
The glass transition temperatures (Tgs) were evaluated by differential scanning calorimetry (DSC) using a DSC8000 (PerkinElmer) for cross-linked samples. The data were collected from the second heating thermogram from −80 to 200 °C at a temperature ramp rate of 10 °C min−1. Thermogravimetric analysis (TGA) was also carried out to investigate the thermal decomposition temperature using a TG/DTA7300 (HITACHI). The temperature ramp rate was 10 °C min−1. All the above thermal investigations were performed for approximately 10 mg samples under N2 gas flow (20 ml min−1).Scattering measurements
Small-angle X-ray scattering (SAXS) measurements were performed at BL40B2 in SPring-8, Hyogo, Japan, equipped with a Pilatus detector. The sample-to-detector distance was 2.2 m and the X-ray wavelength was 0.15 nm. The measurements were carried out for cross-linked samples at room temperature.Mechanical properties
Tensile tests for the cross-linked samples were performed with INSTRON5582 (INSTRON) to confirm the difference of cross-link densities. For the tests, dogbone-shaped samples were prepared with a cutting die, where the thickness, gauge length, and gauge width were 0.6 mm, 13 mm, and 4 mm, respectively. The tests were carried out at an elongation rate of 100 mm min−1 at room temperature. The stress–strain curves were presented with nominal stress and nominal strain.The softening temperature was measured by thermal mechanical analysis with TMA7100 (HITACHI). These measurements were performed for strip-shaped samples with a 10 mm length, 4 mm width, and 0.6 mm thickness. To estimate the softening temperature, the sample length was monitored with increasing temperatures at 3 °C min−1 under a very small constant tension (30 mN). The stress-relaxation was measured with an ARES-G2 shear rheometer (TA instruments) by using 8 mm parallel plates. The cross-linked samples were cut into disc-shaped films with 8 mm diameter and 0.6 mm thickness by using a cutting die. The tests were carried out by applying 3% strain and subsequently by monitoring the residual stress against time. The measurements were performed at various temperatures. The activation energy for the bond-exchange was estimated from Arrhenius plots between the relaxation time and the temperature, which will be explained in a later section. Uniaxial rheological measurements with dynamic temperature sweep modes were also performed with an RSA-G2 rheometer by using strip-shaped samples (please see the measurement conditions in the ESI†). All the above mechanical measurements were performed under a N2 gas atmosphere.
Result
Synthesis and characterization of PE-COOH
We firstly synthesized the network precursor polymer, PE-COOH, by melt polycondensation and the subsequent Michael addition reaction as shown in Scheme 1. In the polycondensation, the monomers, PD, AdA, and ThA, were reacted at 80 °C in the presence of a small amount of the Sc(OTf)3 catalyst. Since the polycondensation was carried out at a relatively low temperature with vacuum stripping, the cross-linking via sulfide-coupling did not occur.28,58 The monomer feed ratio, PD/AdA/ThA, was varied to obtain polyesters with different fractions of SH groups. 1H-NMR measurements for the polymers after the purification revealed that the fraction of PD/AdA/ThA units nicely followed the feed molar ratio (see Fig. S2 and Table S1 in the ESI†). We then introduced COOH groups into the side groups of the chains by reacting the SH-attached polyesters with acrylic acid via the Michael addition reaction. The perfect conversion of the SH groups into COOH groups was confirmed by 1H-NMR as shown in Fig. S4.† Since the COOH groups were not clear from 1H-NMR with d-DMSO due to the use of TFA, we confirmed the existence of COOH groups by performing FT-IR (Fig. S5†). The characterization results of various PE-COOH polymers are summarized in Table 1. Since X (310 or 720) in the code PE-X represents the MCOOH for each sample, the sample with a larger X has a larger average distance between neighboring COOH groups. The synthesized PE-COOH polymers are all viscous at room temperature as shown in Fig. 1, regardless of the MCOOH values.Cross-linking reaction
We first prepared homogeneous mixtures of PE-COOH, the diepoxy cross-linker (BDE), and Zn(OAc)2 by solvent casting methods. After drying, the mixtures were heated at 120 °C under vacuum for 4 hours. Note that the decomposition temperatures for both PE-COOH polymers are much higher than 120 °C as revealed by TGA measurements (Fig. S6a†). The progress of cross-linking was monitored by FT-IR spectroscopy. Fig. S7† shows changes of the characteristic peaks for the sample before and after cross-linking, where the peak from epoxy groups at a region from 840 to 950 cm−1 and the peak from COOH or OH groups at a region from 2800 to 4000 cm−1 are focused on. The sample before cross-linking possesses a distinct epoxy peak at 915 cm−1 from the epoxy group while it was not recognized after cross-linking.61 In addition, the sample before cross-linking showed a peak from COOH groups at 3250 cm−1, which disappeared and a new peak from OH groups appeared at 3500 cm−1 in CL-PE-X.61 PE-310 and PE-720 showed the same changes of FT-IR spectra after the cross-linking reaction. These results indicate that the cross-linking sufficiently progressed under the present conditions, and the cross-linked material contained multiple OH groups in the network as we designed. In fact, the FT-IR spectra did not change significantly with a longer cross-linking reaction time than 2 hours as shown in Fig. S8.† The swelling tests in CHCl3 also revealed that the present cross-linking reaction time was sufficient, and the gel fraction reached nearly 100% after 2 h cross-linking (see Fig. S9†). The average swelling ratio in three time tests, which was determined simply from the ratio of the swollen mass to initial mass, was 420% for CL-PE-310. This value was similar to the values from room temperature swelling tests for other well cross-linked vitrimer systems.37,41 The swelling ratio was 710% for CL-PE-720, indicating the lower cross-link density than that of CL-PE-310. Since typical vitrimer preparation involves the step growth polymerization of low molar mass monomers and cross-linking formation simultaneously, the characterization of the network component polymer, such as the molecular weight and number of functional groups, is difficult. On the other hand, the present post-polymerization cross-linking method has a benefit of correlating the vitrimer physical properties and molecular characteristics easily.Macroscopically, the sample after the cross-linking reaction was elastomeric and highly transparent as shown in Fig. 1. DSC thermograms in Fig. S10† reveal that the cross-linked materials were all amorphous. In both cross-linked samples, the Tg was much lower than room temperature. Concretely, the Tg value is −39 °C and −18 °C for CL-PE-720 and CL-PE-310, respectively, showing that the cross-linked samples with larger fractions of cross-linkable functional groups (i.e., COOH groups) attained a higher Tg. The cross-linked sample showed a stable rubbery plateau at temperatures above the Tg in the rheological measurement with a temperature sweep mode as shown in Fig. S11.† The plateau values were within the same range as the rubbery plateau values (1–10 MPa) for other various-type vitrimers.30,37,41,43,46 We also confirmed by SAXS measurements that there were no nm-scale or larger assemblies in the network (see Fig. S12†).
We then performed tensile tests for the cross-linked samples at room temperature (Fig. 2). The Young's modulus (EY) determined from the initial slope within 10% strain, the elongation at break (εb), and the maximum stress during elongation (σmax) are summarized in Table 2. The sample with a smaller MCOOH showed the larger EY, indicating that the cross-link density is larger for the sample with the smaller MCOOH. This is because the network precursor PE-X with a smaller X has a smaller distance between neighboring COOH groups. There is another tendency that εb is smaller for the sample with a smaller MCOOH, which is also an indicator of tighter network formation. We also evaluated the quality of the cross-linked network based on the model analyses62 for the experimental modulus (please see Fig. S13 in the ESI†). The model analyses revealed that the observed modulus was somewhat lower than the value expected if all the COOH groups were used for forming intermolecular cross-links, although the present design was suitable to control the cross-link density with the fraction of COOH groups in PE-COOH. Such imperfection should be primarily due to the defects of the network, such as dangling chains and loop formation, and partially due to the formation of microgels during solvent casting. Other possible causes may be the unexpected intramolecular epoxy–COOH reaction63 or the sub-reaction of epoxy groups with water or an alcohol during the casting process before the cross-linking reaction.64
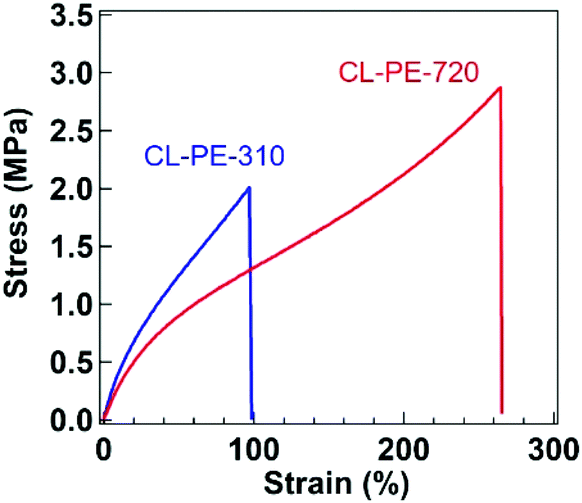 | ||
| Fig. 2 Stress–strain curves for CL-PE-720 (red) and CL-PE-310 (blue). | ||
Investigation of softening temperatures
According to the past reports,18 vitrimer materials show an inflection of the linear expansion coefficient not only at the Tg point but also at a high temperature during the temperature increase. Fig. 3a presents the changing behavior of the sample length during the temperature increase, where the y-axis indicates the sample length from measurements normalized by the length at 100 °C. Although both samples were amorphous and had a Tg lower than room temperature, a deviation from the constant length change was observed in each sample. It should also be noted that the decomposition temperature was much higher than 200 °C for both samples (see Fig. S6b†). Moreover, in the control experiment for cross-linked samples without any trans-esterification catalysts, such a deviation was not observed (see Fig. S14†). Therefore, this deviation can be attributed to the activation of the bond-exchange processes. Fig. 3b presents the difference (ΔLexp–const) between the experimental normalized sample length change and the possible constant sample length change that was indicated by the dotted line in Fig. 3a. Here we tentatively define the softening temperature (Tsoft) as the temperature at which ΔLexp–const reaches 0.005. The values of Tsoft are 138 °C for CL-PE-310 and 144 °C for CL-PE-720, respectively, showing that Tsoft was lower for the sample with a higher cross-link density (i.e., smaller MCOOH). Interestingly, this result is opposite to the general Tg change for cross-linked materials, where Tg is known to be higher for the sample with a higher cross-link density due to the larger degree of chain mobility restriction.64 In the case of the Tsoft in the present design, the sample with a smaller MCOOH possesses a larger fraction of the bond-exchange carriers, i.e., free OH groups. Therefore, the bond-exchange is activated more easily, which is the reason for the observed Tsoft difference between CL-PE-310 and CL-PE-720. | ||
| Fig. 3 (a) Changing behavior of the sample length during the temperature increase under a constant tension (30 mN). The y-axis indicates the sample length from measurements normalized by the length at 100 °C. The curve of CL-PE-310 was shifted vertically for a clearer comparison. (b) Temperature-dependent changes of the difference (ΔLexp–const) between the experimental normalized sample length and the possible constant sample length which is indicated by the dotted lines in (a). The dotted line in (b) represents the point at which ΔLexp–const = 0.005. | ||
Stress-relaxation properties
The stress-relaxation capability at high temperatures is one of the most unique properties for vitrimer materials.18 Such stress-relaxation is known to be induced by the bond-exchange processes in the network.18Fig. 4a and b present the stress-relaxation curves for CL-PE-310 and CL-PE-720 at various temperatures, where the stress (σ) normalized by the initial stress (σ0) was plotted as a function of time. CL-PE-310 and CL-PE-720 samples show great stress-relaxation at these temperatures, while they showed negligible stress-relaxation at room temperature. From the above results, the great stress-relaxation observed for CL-PE-X was found to be due to the bond-exchange via trans-esterification in the network. The relaxation became faster at higher temperature, meaning that the trans-esterification was more activated. We also revealed that the experimental stress-relaxation spectra were well fitted with the Kohlrausch–Williams–Watts (KWW) function65 with a certain distribution of the relaxation time as summarized in Fig. S15.† | ||
| Fig. 4 Stress-relaxation spectra for (a) CL-PE-720 and (b) CL-PE-310, where the stress (σ) is normalized by the initial stress (σ0). The dotted lines in (a) and (b) indicate the point at σ/σ0 = 1/e (=0.3679). (c) Arrhenius plots of τ as a function of inverse temperatures. | ||
Fig. 4c summarizes the relaxation time (τ) which is defined as the time at σ/σ0 = 1/e (=0.368) as a function of temperature. For example, the values of τ at 160 °C and 180 °C for CL-PE-310 were 179 s and 66 s while those for CL-PE-720 were 331 s and 120 s, respectively. These values are relatively higher than those of other non-polyester-based vitrimers with a trans-esterification mechanism,34,37 which may be due to the large fraction of ester bonds in the polyester main-chain. Interestingly, the one with a larger cross-link density (i.e., CL-PE-310) showed a shorter τ at any temperatures in the measured temperature range, which can be again explained as follows: in the present system, the samples with larger cross-link densities (i.e., smaller MCOOH) possess larger fractions of free OH groups in the network. The OH groups work as bond-exchange carriers, and thus the more rapid and efficient bond-exchange can be realized.
We then estimated the activation energy (Ea) for the bond-exchange from the slope of the Arrhenius type relationship, eqn (1), between τ and 1/T.18
 | (1) |
In eqn (1), τ0 is the pre-exponential factor, R is the gas constant, and T is the measurement temperature. The values of Ea are determined from the slope of the dotted lines in Fig. 4c. The Ea for CL-PE-310 was 81 kJ mol−1 while that for CL-PE-720 was 83 kJ mol−1. The Ea values were found to be unaffected by the cross-link density in the present system, but the values are lower than those of other polyester-based vitrimers with the trans-esterification mechanism and similar catalyst conditions (∼130 kJ mol−1).33 Although the reason for the relatively low Ea in the present system is not clarified yet, we will carry out more systematic studies to clarify the reason in the future by using the present model system.
Functions as vitrimers
In the following, we demonstrate the unique functions of the present materials as vitrimers. First, Fig. 5a presents the re-shapeability; a film of CL-PE-310 was rolled in a metal thin rod and fixed with temperature-resistant tapes. The fixed sample was kept in an oven at 160 °C for 2 hours. After cooling to room temperature, a stable spiral-shaped sample was obtained. This indicates that the bond-exchange progressed for the rolled sample at high temperatures, and the sample was transformed to a new fixed equilibrated shape. Next, Fig. 5b shows the surface healing property. We made a scar of approximately 0.1 mm depth with a razor on the surface of CL-PE-310 and monitored the change of the scar at high temperature by using microscopy. By keeping the sample at 180 °C, the scar disappeared after 5 min. This suggests that the activation of bond-exchange at a high temperature enabled the re-ordering of chains near the surface. Fig. 5c also shows a unique property related to the surface, that is, self-adhesion. Two film samples of CL-PE-310 were overlapped in the area of 4 mm × 4 mm, and we placed a 50 g weight on the overlapped part. The sample with the weight on top was kept in an oven at 160 °C for 2 h. Strong adhesion was observed after the sample was removed from the oven. The adhered sample was stable against a 200 g weight, and a bulk rupture occurred with a 250 g weight. Note that the control sample without a trans-esterification catalyst showed no adhesion with the same test procedure. The result indicates that the bond-exchange progressed between the different surfaces. Finally, we tested the recyclability of the broken samples (Fig. 5d). CL-PE-310 was cut into small pieces, and these were pressed at 160 °C with a Teflon-made mold having a hole (8 mm diameter and 0.5 mm depth). The cut pieces were nicely merged, producing disc-shaped samples. By immersing the sample in CHCl3 (good solvent to the component polyester), the sample was swollen with keeping the disc-shape. We confirmed that the recycling could be repeated at least two times. The sol fraction was negligible, and FT-IR revealed that there were no changes in the chemical structures (see Fig. S16†). | ||
| Fig. 5 Functions of the present elastomeric vitrimer; (a) re-shapeability, (b) healing of the surface, (c) self-adhesion, and (d) recyclability. Please see each test protocol in the text. | ||
Conclusions
In this paper, we described the preparation and unique functions of elastomeric vitrimer materials with different cross-link densities based on post-polymerization cross-linking of polyesters with multiple COOH groups. We first prepared polyesters with COOH side groups, PE-COOH, via melt polycondensation and the subsequent Michael addition reaction. The fraction of COOH groups was varied well by changing the monomer feed ratio at the initial melt polycondensation. After characterization, diepoxy cross-linkers (1,4-butanediol diglycidyl ether) and the trans-esterification catalyst (Zn(OAc)2) were mixed with PE-COOH via solvent casting. FT-IR confirmed that the cross-linking efficiently progressed by 4 hour heating at 120 °C. In this molecular design, the free OH groups, which worked as the trigger of trans-esterification, were generated through the epoxy–COOH cross-linking reaction. The tensile test revealed that the cross-link density in the final vitrimer materials was tuned by the COOH group equivalent weight (MCOOH) of PE-COOH. We then investigated the correlation between the cross-link density and representative vitrimeric properties in terms of the softening temperature, stress-relaxation ability, and the activation energy for bond-exchange. A series of the data demonstrated that the lower Tsoft and shorter τ were observed for the sample with a larger cross-link density. This is because, in the present design, the samples with larger cross-link densities (i.e., smaller MCOOH) possess larger fractions of free OH groups in the network, which enables more efficient and rapid trans-esterification.So far, it has been reported that the macroscopic properties of vitrimer materials can be controlled by the kind of catalyst for bond-exchange and the amount of catalysts. The present knowledge could be thus important to control the vitrimeric properties, which can bring further development of vitrimer materials. Moreover, post-polymerization cross-linking methods as described in this work can be a versatile strategy to control vitrimer properties from the molecular level consideration. We are currently investigating the correlation between vitrimer properties and the molecular weights of component polymers, where the intrinsic dynamics of component polymers might affect the properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. K. Nagata from the Nagoya Institute of Technology and Prof. M. Tokita from the Tokyo Institute of Technology for their assistance in performing thermal mechanical analysis and TGA. The authors also thank Dr Y. Doi for his assistance in the measurements of the polymer density and SAXS. The authors also thank Mr M. Futamura from Nagoya Municipal Industrial Research Institute for his kind help in performing tensile tests. The authors also thank Mr T. Aikawa from TA Instruments for his kind help in performing stress-relaxation and uniaxial rheology. This work was supported by the JSPS KAKENHI Grant-in-Aid for Young Scientists (B) (No. 17k17708 for M. H.).References
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- D. L. Thomsen, P. Keller, J. Naciri, R. Pink, H. Jeon, D. Shenoy and B. R. Ratna, Macromolecules, 2001, 34, 5868–5875 CrossRef CAS.
- T. Chung, A. Rorno-Uribe and P. T. Mather, Macromolecules, 2008, 41, 184–192 CrossRef CAS.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673–1676 CrossRef PubMed.
- R. Gheneim, C. Perez-Berumen and A. Gandini, Macromolecules, 2002, 35, 7246–7253 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. I. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- N. Roy, B. Bruchmann and J. M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS PubMed.
- K. Imato, T. Kanehara, S. Nojima, T. Ohishi, Y. Higaki, A. Takahara and H. Otsuka, Chem. Commun., 2016, 52, 10482–10485 RSC.
- K. Imato, A. Takahara and H. Otsuka, Macromolecules, 2015, 48, 5632–5639 CrossRef CAS.
- K. Imato, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, J. Am. Chem. Soc., 2014, 136, 11839–11845 CrossRef CAS PubMed.
- K. Imato, J. C. Natterodt, J. Sapkota, R. Goseki, C. Weder, A. Takahara and H. Otsuka, Polym. Chem., 2017, 8, 2115–2122 RSC.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- M. Hayashi and F. Tournilhac, Polym. Chem., 2017, 8, 461–471 RSC.
- M. Hayashi, S. Matsushima, A. Noro and Y. Matsushita, Macromolecules, 2015, 48, 421–431 CrossRef CAS.
- M. Hayashi, A. Noro and Y. Matsushita, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 755–764 CrossRef CAS.
- X. Z. Yan, T. R. Cook, J. B. Pollock, P. F. Wei, Y. Y. Zhang, Y. H. Yu, F. H. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 4460–4463 CrossRef CAS PubMed.
- S. C. Grindy, M. Lenz and N. Holten-Andersen, Macromolecules, 2016, 49, 8306–8312 CrossRef CAS.
- M. Burnworth, L. M. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- D. Mozhdehi, J. A. Neal, S. C. Grindy, Y. Cordeau, S. Ayala, N. Holten-Andersen and Z. B. Guan, Macromolecules, 2016, 49, 6310–6321 CrossRef CAS.
- M. Hayashi, K. Shibata, I. Kawarazaki and A. Takasu, Macromol. Chem. Phys., 2018, 219, 1800127 CrossRef.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Polymers, 2018, 10, 43 CrossRef.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, RSC Adv., 2016, 6, 88647–88655 RSC.
- Y. W. Zhou, J. G. P. Goossens, S. van den Bergen, R. P. Sijbesma and J. P. A. Heuts, Macromol. Rapid Commun., 2018, 39, 1800356 CrossRef PubMed.
- Y. W. Zhou, R. Groote, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Polym. Chem., 2019, 10, 136–144 RSC.
- Y. W. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2017, 50, 6742–6751 CrossRef CAS.
- G. B. Lyon, L. M. Cox, J. T. Goodrich, A. D. Baranek, Y. F. Ding and C. N. Bowman, Macromolecules, 2016, 49, 8905–8913 CrossRef CAS.
- Z. Q. Pei, Y. Yang, Q. M. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS PubMed.
- D. W. Hanzon, N. A. Traugutt, M. K. McBride, C. N. Bowman, C. M. Yakacki and K. Yu, Soft Matter, 2018, 14, 951–960 RSC.
- H. Zhang, C. Cai, W. X. Liu, D. D. Li, J. W. Zhang, N. Zhao and J. Xu, Sci. Rep., 2017, 7, 11833 CrossRef PubMed.
- A. Demongeot, R. Groote, H. Goossens, T. Hoeks, F. Tournilhac and L. Leibler, Macromolecules, 2017, 50, 6117–6127 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8., 14857 CrossRef PubMed.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- T. Stukenbroeker, W. D. Wang, J. M. Winne, F. E. Du Prez, R. Nicolaÿ and L. Leibler, Polym. Chem., 2017, 8, 6590–6593 RSC.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- Y. Nishimura, J. Chung, H. Muradyan and Z. B. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- M. Roöttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- J. S. A. Ishibashi and J. A. Kalow, ACS Macro Lett., 2018, 7, 482–486 CrossRef CAS.
- L. Q. Li, X. Chen, K. L. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- Y. Liu, Z. Tang, S. Wu and B. Guo, ACS Macro Lett., 2019, 8, 193–199 CrossRef CAS.
- X. Chen, L. Q. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- Z. H. Tang, Y. J. Liu, B. C. Guo and L. Q. Zhang, Macromolecules, 2017, 50, 7584–7592 CrossRef CAS.
- A. Legrand and C. Soulie-Ziakovic, Macromolecules, 2016, 49, 5893–5902 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS.
- K. Yamamoto and A. Takasu, Macromolecules, 2010, 43, 8519–8523 CrossRef CAS.
- G. Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- M. Le Neindre and R. Nicolaÿ, Polym. Chem., 2014, 5, 4601–4611 RSC.
- L. H. Sinh, N. N. Trung, B. T. Son, S. Shin, D. T. Thanh and J. Y. Bae, Polym. Eng. Sci., 2014, 54, 695–703 CrossRef CAS.
- M. Rubinstein and R. Colby, Polymer Physics, Oxford University Press, New York, 2003 Search PubMed.
- Y. Akagi, J. P. Gong, U. Chung and T. Sakai, Macromolecules, 2013, 46, 1035–1040 CrossRef CAS.
- J. R. Han, T. Liu, C. Hao, S. Zhang, B. H. Guo and J. W. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
- G. Williams, D. C. Watts, S. B. Dev and A. M. North, Trans. Faraday Soc., 1971, 67, 1323–1335 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR and FT-IR spectra for PE-COOH, TGA thermograms, FT-IR spectra for samples after a cross-linking reaction, changes of the FT-IR spectra after a long time cross-linking reaction, swelling tests, DSC curves, dynamic temperature ramp rheology, SAXS profiles, model analysis of network quality based on the Young's modulus, dilatometry test for the control sample with no catalysts, model analysis of stress-relaxation curves based on the KWW equation, and FT-IR spectra for recycled samples. See DOI: 10.1039/c9py00293f |
| This journal is © The Royal Society of Chemistry 2019 |