
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical recycling of bromine-terminated polymers synthesized by ATRP†
Stella Afroditi
Mountaki
,
Richard
Whitfield
 ,
Kostas
Parkatzidis
,
Maria-Nefeli
Antonopoulou
,
Nghia P.
Truong
and
Athina
Anastasaki
*
,
Kostas
Parkatzidis
,
Maria-Nefeli
Antonopoulou
,
Nghia P.
Truong
and
Athina
Anastasaki
*
Laboratory of Polymeric Materials, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg-5, 8093, Zurich, Switzerland. E-mail: athina.anastasaki@mat.ethz.ch
First published on 26th January 2024
Abstract
Chemical recycling of polymers is one of the biggest challenges in materials science. Recently, remarkable achievements have been made by utilizing polymers prepared by controlled radical polymerization to trigger low-temperature depolymerization. However, in the case of atom transfer radical polymerization (ATRP), depolymerization has nearly exclusively focused on chlorine-terminated polymers, even though the overwhelming majority of polymeric materials synthesized with this method possess a bromine end-group. Herein, we report an efficient depolymerization strategy for bromine-terminated polymethacrylates which employs an inexpensive and environmentally friendly iron catalyst (FeBr2/L). The effect of various solvents and the concentration of metal salt and ligand on the depolymerization are judiciously explored and optimized, allowing for a depolymerization efficiency of up to 86% to be achieved in just 3 minutes. Notably, the versatility of this depolymerization is exemplified by its compatibility with chlorinated and non-chlorinated solvents, and both Fe(II) and Fe(III) salts. This work significantly expands the scope of ATRP materials compatible with depolymerization and creates many future opportunities in applications where the depolymerization of bromine-terminated polymers is desired.
Introduction
Over the last century, it is undeniable that polymers have been established as essential materials for a whole host of applications.1–5 In contrast, recycling efforts have lagged far behind, resulting in the accumulation of polymers within the environment and the necessity that new degradation and depolymerization methods are developed.6–13 An alternative sustainable life cycle for polymers is idealized, where starting monomers can be directly reobtained from polymers, and then used to synthesize a new batch of materials.14–17 However, developing these chemical recycling approaches is of considerable challenge, as the vast majority of polymers produced globally contain very strong all-carbon backbones and initial depolymerization strategies such as pyrolysis require very high temperatures (>400 °C) for monomer regeneration to be triggered.18,19 The essential step of these processes was a random repeat unit or backbone scission step within free radical or anionic polymers, which generated a backbone radical that was then capable of unzipping the polymer chain.20More recently, with the advent of controlled radical polymerization, and in particular reversible addition–fragmentation chain transfer (RAFT) polymerization21–24 and atom transfer radical polymerization (ATRP),25–27 it has become commonplace to prepare polymers containing labile end-groups,28–32 which can subsequently be activated at far lower temperatures to trigger an efficient depolymerization.33–35 In the area of RAFT, these end-group unzipping processes were first demonstrated by Gramlich and co-workers, who showed that bulky bottlebrush polymers could be deconstructed in solution to partially recover the monomer (∼20–30%) at 70 °C.36 This has inspired significant further research and several methods have been developed, enabling high methacrylate monomer regeneration at relatively low temperatures (<120 °C). Our group has showed that near-quantitative depolymerization of various linear, bulky and functional polymethacrylates could be achieved at 120 °C.37–39 In addition, both our laboratory and that of Sumerlin demonstrated that the combined stimuli of heat and light could accelerate the depolymerization, while successfully obtaining high depolymerization yields.40–42 The key to the success of these strategies is the low-temperature activation of the RAFT end-group (typically a dithiobenzoate or a trithiocarbonate) to enable radical generation combined with a high polymer dilution (5 mM repeat unit concentration) which creates favorable thermodynamic conditions for depolymerization to occur.39 Upon switching to slightly less diluted systems (i.e. 25 mM and 100 mM), a more controlled depolymerization was observed whereby a gradual decrease in molecular weight could be observed thus mirroring the reverse process of controlled polymerization.35,41,43 However, much lower conversions were reached on these occasions, thus compromising the overall depolymerization efficiency. It is highlighted that the vast majority of these works operate exclusively at high dilutions, although bulk depolymerization of RAFT-synthesized materials has also been realized albeit at significantly higher temperatures (i.e. 230 °C) by leveraging the in situ removal of the monomer during the depolymerization.44–46
Instead, the depolymerization of ATRP-synthesized materials can be realized at much higher concentrations and as such is currently attracting significant attention.47 Raus and co-workers initially showed that the depolymerization of ATRP bottlebrushes was possible at 90 °C,48 while Ouchi and coworkers first demonstrated that around 25% of the non-bulky methyl methacrylate (MMA) monomer could be recovered in the presence of a ruthenium catalyst.49 Impressively, the Matyjaszewski group was able to significantly boost the depolymerization conversions to 70–80%, by performing reactions at 170 °C and utilizing either a copper or iron-based catalyst.50–52 On the photo-thermal front, our group recently reported that photocatalytic ATRP depolymerization could also be successful under blue light irradiation and iron catalysis,53 also reaching high monomer recovery (up to 90%). It is noted that the depolymerization of the aforementioned ATRP-made materials can take place in up to 2 M concentration with the exception of a recent report that showcases a higher temperature bulk depolymerization.54 However, current literature reports, including both solution and bulk depolymerization strategies, have focused nearly exclusively on chlorine-terminated polymers,45,49–54 even though the vast majority of polymers prepared by ATRP have a bromine end-group. Therefore, the development of a single-step method to depolymerize bromine-terminated polymers yielding high conversions would significantly expand the scope of materials compatible with chemical recycling.
Herein, we report the rapid depolymerization of bromine-terminated polymethacrylates triggered by an iron catalyst (Scheme 1). A comprehensive study is performed, evaluating a range of different depolymerization conditions with the aim to achieve high monomer regeneration. The effect of the solvent and both metal salt and ligand concentrations are critically evaluated before kinetic evaluation of the optimized conditions is performed. Finally, the versatility of the method is demonstrated by achieving high depolymerization conversions under a wide-range of compatible reaction conditions.
 | ||
| Scheme 1 A comparison between previous approaches and the current method for the depolymerization of polymers obtained by ATRP. | ||
Experimental
Materials
Benzyl methacrylate (>98.0%) and tetraethylene glycol dimethyl ether were obtained from Tokyo Chemical Industry. Ethyl α-bromophenyl acetate (97%) and 1,2,4-trichlorobenzene were obtained from Acros Organics. Anhydrous iron(II) bromide (98%) and iron(III) bromide (98%) were obtained from Alfa Aesar. N,N-Dimethylformamide (>99%) and 1,3-dichlorobenzene (98%) were obtained from Fluka. Tetrabutylammonium bromide (99%) and anisole (99%) were obtained from Sigma Aldrich and the polystyrene standard (average Mn 180 kDa) was purchased from Supelco. All chemicals were used as received, except the monomer that was passed through a column of basic alumina prior to usage.Instrumentation
1H NMR spectra were measured on a Bruker DPX – 300 spectrometer. The depolymerization conversions were assessed by comparing the integrals of the monomeric vinyl protons (5.49–5.97 ppm) to the integrals of both the monomer and polymer peaks (4.81–5.06 ppm). Size Exclusion Chromatography (SEC) was performed using a Shimadzu modular system consisting of a CBM-20A system controller, an SIL-20A automatic injector, a 10.0 μm bead size guard column (50 × 7.5 mm), followed by three KF-805L columns (300 × 8 mm, bead size: 10 μm, pore size maximum: 5000 Å) and an RID-20A differential refractive-index detector. N,N-Dimethylacetamide (HPLC grade, with 0.03% w/v LiBr) served as the eluent and the flow rate was maintained at 1 mL min−1 using an LC-20AD pump. A calibration curve for molecular weight was generated using commercially available narrow molar mass distribution poly(methyl methacrylate) standards with molecular weights spanning from 5000 to 1.5 × 106 Da.Depolymerization of PBzMA-Br with FeBr2/TBABr (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1:1)
:1:1)
In a 15 mL glass vial, 3 mg (1.39 × 10−5 mol, 1 eq.) of FeBr2 were placed. The vial was promptly sealed with a rubber septum and then purged with nitrogen for 30 minutes. In the same vial and under nitrogen, 440 μL of deoxygenated TEGDME was added and incubated at 170 °C for 2 minutes until all FeBr2 was dissolved. Following this, 8.8 mL of deoxygenated TCB was introduced (95% v/v), resulting in a slightly orange solution. In a separate 15 mL glass tube equipped with a stirring bar, 9.2 mg (1.56 × 10−6 mol, 1 eq.) of PBzMA-Br, 4.6 mg (0.5 eq.) of PS-H internal standard and 0.50 mg (1.56 × 10−6 mol, 1 eq.) of TBABr were added. The tube was sealed with a rubber septum and degassed with nitrogen for five minutes. Under nitrogen, 1 mL (0.34 mg FeBr2, 1.56 × 10−6, 1 eq.) of the catalyst stock solution was introduced to the reaction tube and it was stirred vigorously to dissolve all solids. A small aliquot of the reaction was collected, under a nitrogen blanket with nitrogen-purged needles, for 1H NMR and SEC analysis, before the reaction tube was placed into the oil bath at 170 °C. Additional samples were collected after 5, 15, and 60 minutes of reaction time. This procedure remained consistent for all the various conditions employed. 1H NMR analysis was performed in deuterated acetone and SEC analysis was carried out in dimethylacetamide, after samples had been passed through a column of basic alumina and a 0.45 μm PTFE filter.
Results and discussion
A bromine-terminated polymethacrylate was initially synthesized by photo-ATRP. Benzyl methacrylate was selected as the model monomer, ethyl α-bromophenyl acetate (EBPA) as the initiator and iron(III) bromide/tetrabutylammonium bromide (FeBr3/TBABr) as the catalyst (Scheme S1†).55–57 Polymerization was conducted under blue light irradiation and after a rigorous purification, a well-defined polymer was successfully obtained (Fig. S1 and S2†). Inspired by previous work from our group in collaboration with the Matyjaszewski group, where the photo-thermal depolymerization of chlorine-terminated polymers had been successfully achieved using an iron-based catalytic system, we also started our investigation here using iron as a catalyst.50,53 In particular, iron has the benefits of being lower in cost and toxicity, and greater in abundance in comparison to more widely used copper catalysts. Rather than selecting iron(II) chloride as the metal salt, instead iron(II) bromide (4 equiv.) was chosen while keeping triethylene glycol methyl diether (TEGDME) as both the solvent and ligand.50,58 Depolymerization reactions were performed at 170 °C, utilizing a 50 mM repeat unit concentration of polymer and a ratio of polymer to FeBr2 of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. Under these conditions, approximately 20% of depolymerization was achieved in just 5 minutes, as evidenced by the appearance of vinyl peaks in the 1H Nuclear Magnetic Resonance (NMR) spectrum (Fig. 1, pink & S3, S4 and Table S1,† entries 1–4). Prior to proceeding with further experiments, it was important to understand what the maximum threshold of depolymerization achievable could be. To investigate this, it was necessary to quantify the end-group fidelity of the starting polymer. Clearly, it would only be possible to depolymerize those chains that contained the bromine end-group, so a chain extension experiment was conducted and detailed calculations were performed to assess the livingness.50,59 Pleasingly, a clear shift in the molar mass distribution to higher molecular weights was observed, suggesting that the polymer had high end-group fidelity, but there was also a slight increase in it's dispersity value (Đ = 1.31 and Mn = 24100, Fig. S5†) indicating partial loss of the end-group. The livingness was calculated based on the deconvolution of the SEC traces and the chain-end functionality was determined to be 85% (Fig. S6†). As such, 20% of depolymerization suggests an overall efficiency of 23% which although still relatively low, demonstrates that it was possible for this catalytic system to successfully activate the carbon–bromine chain-end and trigger a low-temperature depolymerization, but this very limited conversion could not be exceeded even after a longer reaction time (e.g. 1 hour). The low conversion was attributed to the relatively inactive catalyst employed resulting in insufficient chain-end activation. It was thus hypothesized that the employment of a higher-activity catalyst would improve our depolymerization results, as faster activation of the C–Br would potentially minimize any effect from the instability of the C–Br bond towards termination events like lactonization.60 TBABr was subsequently selected as an alternative ligand source (i.e. to supply additional coordinating bromide), as this has previously been shown to be very effective for controlling polymerization.61 Depolymerization was performed with 4 equivalents of TBABr with respect to polymer and all other reaction conditions were kept consistent with the previous experiment ([P-Br]:[FeBr2]:[TBABr] = 1:4:4). Pleasingly, after 5 minutes of depolymerization, as much as 49% monomer regeneration was achieved (or 57% of depolymerization efficiency) as evidenced by 1H NMR (Fig. 1a, blue & S7 and Table S1,† entries 5–8). This conclusion was validated by size exclusion chromatography (SEC), as a significant reduction in the intensity of the polymer peak (50% by area) was observed in comparison to a PS-H internal standard (Fig. 1b, blue & S8†). This reduction was uniform and no low molecular weight polymer formation occurred, suggesting that after the C–Br bond was activated forming the chain-end radical, the polymer chains fully unzipped to monomer. On closer analysis of the SEC traces, the formation of high molecular weight polymer chains could also be observed, indicating that significant termination via combination had occurred. This suggested that too high a concentration of radicals had formed during the reaction and termination events (both combination and disproportionation) were competing with the depolymerization, potentially limiting the extent of monomer generation (Fig. 1b & S8†). Encouraged by this data, we sought to further optimize the reaction conditions, so the amount of monomer regeneration could be further increased.
:4. Under these conditions, approximately 20% of depolymerization was achieved in just 5 minutes, as evidenced by the appearance of vinyl peaks in the 1H Nuclear Magnetic Resonance (NMR) spectrum (Fig. 1, pink & S3, S4 and Table S1,† entries 1–4). Prior to proceeding with further experiments, it was important to understand what the maximum threshold of depolymerization achievable could be. To investigate this, it was necessary to quantify the end-group fidelity of the starting polymer. Clearly, it would only be possible to depolymerize those chains that contained the bromine end-group, so a chain extension experiment was conducted and detailed calculations were performed to assess the livingness.50,59 Pleasingly, a clear shift in the molar mass distribution to higher molecular weights was observed, suggesting that the polymer had high end-group fidelity, but there was also a slight increase in it's dispersity value (Đ = 1.31 and Mn = 24100, Fig. S5†) indicating partial loss of the end-group. The livingness was calculated based on the deconvolution of the SEC traces and the chain-end functionality was determined to be 85% (Fig. S6†). As such, 20% of depolymerization suggests an overall efficiency of 23% which although still relatively low, demonstrates that it was possible for this catalytic system to successfully activate the carbon–bromine chain-end and trigger a low-temperature depolymerization, but this very limited conversion could not be exceeded even after a longer reaction time (e.g. 1 hour). The low conversion was attributed to the relatively inactive catalyst employed resulting in insufficient chain-end activation. It was thus hypothesized that the employment of a higher-activity catalyst would improve our depolymerization results, as faster activation of the C–Br would potentially minimize any effect from the instability of the C–Br bond towards termination events like lactonization.60 TBABr was subsequently selected as an alternative ligand source (i.e. to supply additional coordinating bromide), as this has previously been shown to be very effective for controlling polymerization.61 Depolymerization was performed with 4 equivalents of TBABr with respect to polymer and all other reaction conditions were kept consistent with the previous experiment ([P-Br]:[FeBr2]:[TBABr] = 1:4:4). Pleasingly, after 5 minutes of depolymerization, as much as 49% monomer regeneration was achieved (or 57% of depolymerization efficiency) as evidenced by 1H NMR (Fig. 1a, blue & S7 and Table S1,† entries 5–8). This conclusion was validated by size exclusion chromatography (SEC), as a significant reduction in the intensity of the polymer peak (50% by area) was observed in comparison to a PS-H internal standard (Fig. 1b, blue & S8†). This reduction was uniform and no low molecular weight polymer formation occurred, suggesting that after the C–Br bond was activated forming the chain-end radical, the polymer chains fully unzipped to monomer. On closer analysis of the SEC traces, the formation of high molecular weight polymer chains could also be observed, indicating that significant termination via combination had occurred. This suggested that too high a concentration of radicals had formed during the reaction and termination events (both combination and disproportionation) were competing with the depolymerization, potentially limiting the extent of monomer generation (Fig. 1b & S8†). Encouraged by this data, we sought to further optimize the reaction conditions, so the amount of monomer regeneration could be further increased.
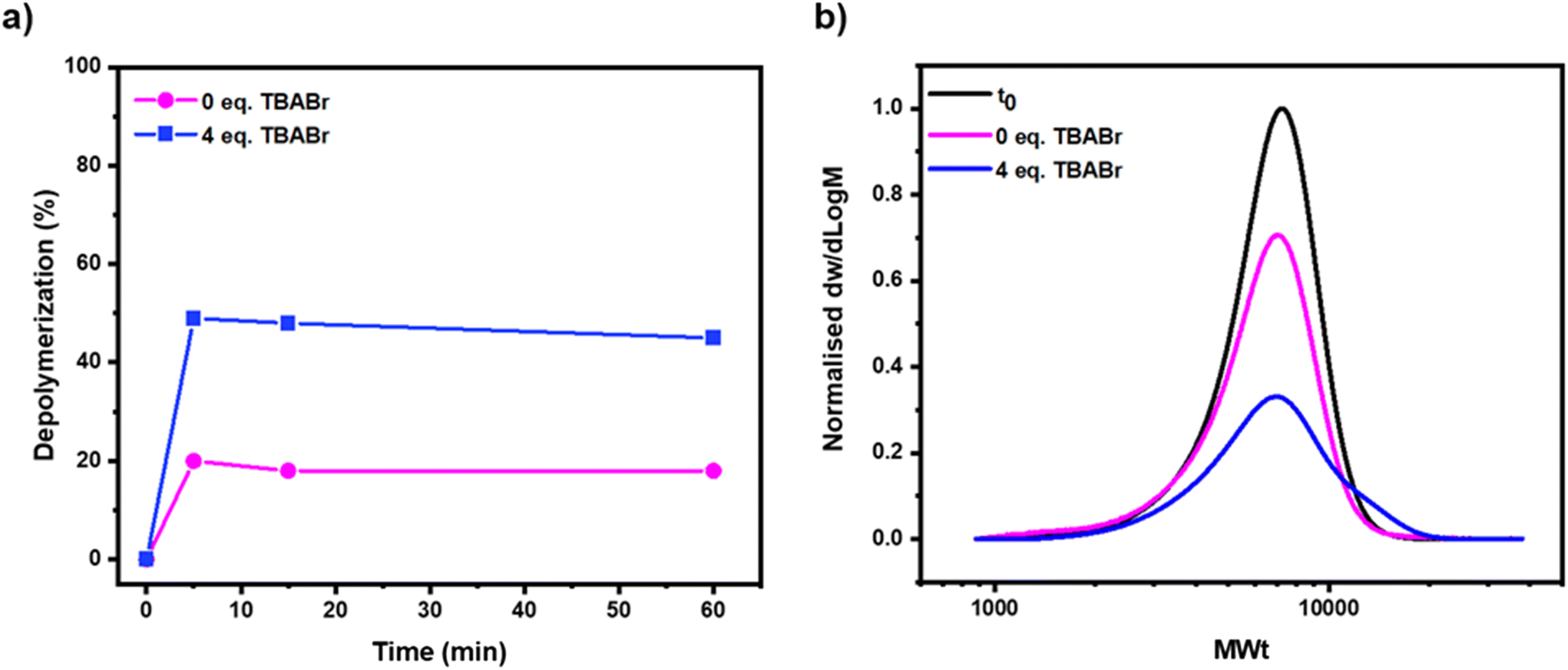 | ||
| Fig. 1 The effect of TBABr on the depolymerization of bromine-terminated polymers synthesized by ATRP. In (a) kinetics illustrate the depolymerization conversion obtained from 1H NMR and in (b) SEC traces under the various conditions are illustrated after 60 minutes of depolymerization. Both reactions were performed with a 50 mM repeat unit concentration of polymer at 170 °C in TEGDME. | ||
To start, the importance of the choice of solvent for the depolymerization was investigated (Fig. 2a). Our group previously demonstrated that the solvent greatly impacted the extent of depolymerization for RAFT polymers, and the effect on ATRP depolymerization could feasibly be more significant, given the variance of catalyst activity and solubility in different solvents.39,62,63 The two main criteria that it was necessary to consider were the boiling point of the solvent and its propensity to dissolve all the reaction components. Dimethylformamide (DMF), anisole, 1,2-dichlorobenzene (DCB) and 1,2,4-trichlorobenzene (TCB) were selected as these all had boiling points higher than 150 °C and with 5% v/v TEGDME they gave high solubility of both polymer and catalyst. When depolymerization was performed in either DCB, DMF or anisole, around 40% of the monomer was regenerated, a similar percentage to the results obtained in TEGDME (Fig. 2b & S9–S11 and Table S2,† entries 1–12). In all cases, the depolymerization was complete after just 5 minutes, but similarly to when TEGDME had been used, in both DCB and anisole, alongside the monomer production, high molecular weight polymer chain formation was also evidenced by SEC, suggesting that termination was again competing with the depolymerization. When TCB was selected as the solvent, it was possible to significantly improve the depolymerization, achieving a conversion as high as 61% in 5 minutes and a depolymerization efficiency of 71% (Fig. 2b & S9–S11 and Table S2,† entries 13–16). In a similar vein to when the other solvents were utilized, no further depolymerization was observed after either 15 or 60 minutes and all the SEC traces overlapped, illustrating that the reaction had already reached its maximum possible value. The high molecular weight polymer formation was much less pronounced in TCB, suggesting that less termination had occurred during the depolymerization, most likely due to a slower rate of radical activation in this solvent (Fig. 2c). This suppressed termination and allowed a larger number of the chain-end radicals to gradually form and unzip, yielding a greater final amount of monomer. TCB was therefore selected for all subsequent experiments, alongside a 5-minute reaction time.
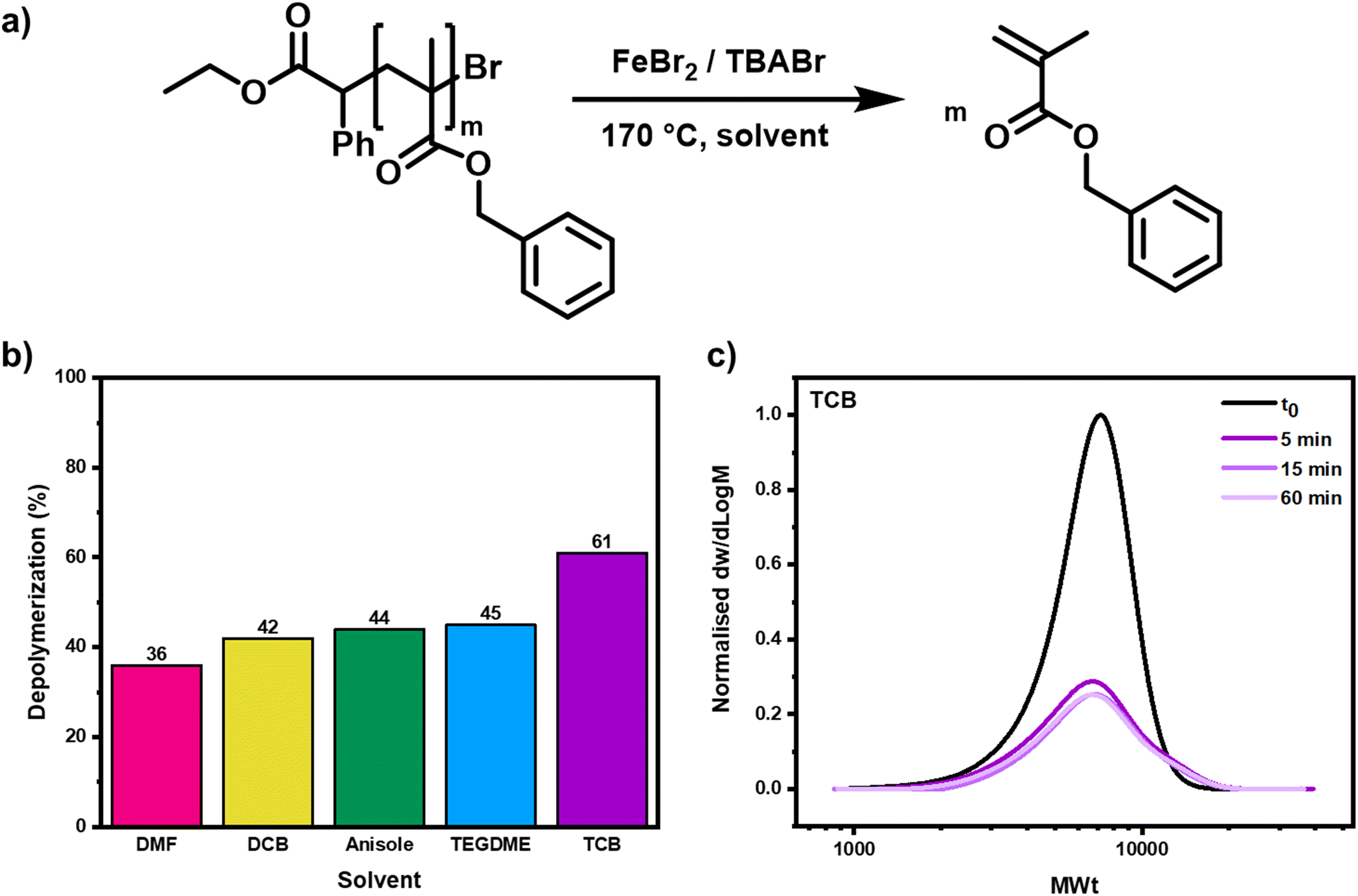 | ||
| Fig. 2 The effect of solvent on the depolymerization. In (a) a scheme of the depolymerization is shown, in (b) a bar graph shows the relative depolymerization conversions in the various solvents after 60 minutes and in (c) the SEC traces for the depolymerization in the optimal solvent, TCB, are shown at various timepoints. | ||
Next, we wanted to systematically investigate the effect of both the concentration of the metal salt and TBABr, with the aim of promoting further depolymerization. First, the effect of the metal salt concentration was investigated, while keeping the amount of polymer and TBABr constant ([P-Br]:[FeBr2]:[TBABr] = 1:X:4, Fig. 3a). We hypothesized that by lowering the metal salt concentration, the rate at which chain-end radical generation was occurring would be lowered, resulting in a consistently lower radical concentration throughout the depolymerization and as a result an even lower extent of termination. On decreasing the FeBr2 concentration from 4 equivalents to 1 equivalent, the depolymerization reached 72%, the highest thus far and no high molecular weight chains could be evidenced in the SEC, implying that termination events had been significantly suppressed (Fig. 3a, d & S12 and Table S3,† entries 2 and 3). This gave a remarkable depolymerization efficiency of 84%, demonstrating that it was possible to depolymerize almost every chain that had originally contained a bromine end-group. By further lowering the concentration of FeBr2 concentration to 0.25, 0.10 and 0.05 equivalents, a gradual decrease in the amount of regenerated monomer was observed with 72%, 53% and 41% of depolymerization obtained respectively (Fig. 3a, d & S12 and Table S3,† entries 4–7). This suggests that at lower catalyst concentrations there was either not sufficient catalyst available to activate all of the polymer chains or that the rate of activation was so low that other side reactions, for example lactonization, were more pronounced potentially eliminating the C–Br end-group.50,52,64 Altogether, our data highlight that a fine balance is required to minimize the various termination and side reactions so as to maximize the depolymerization, and to achieve this, a 1:1:4 ratio between polymer, FeBr2 and TBABr was found to be the optimal.
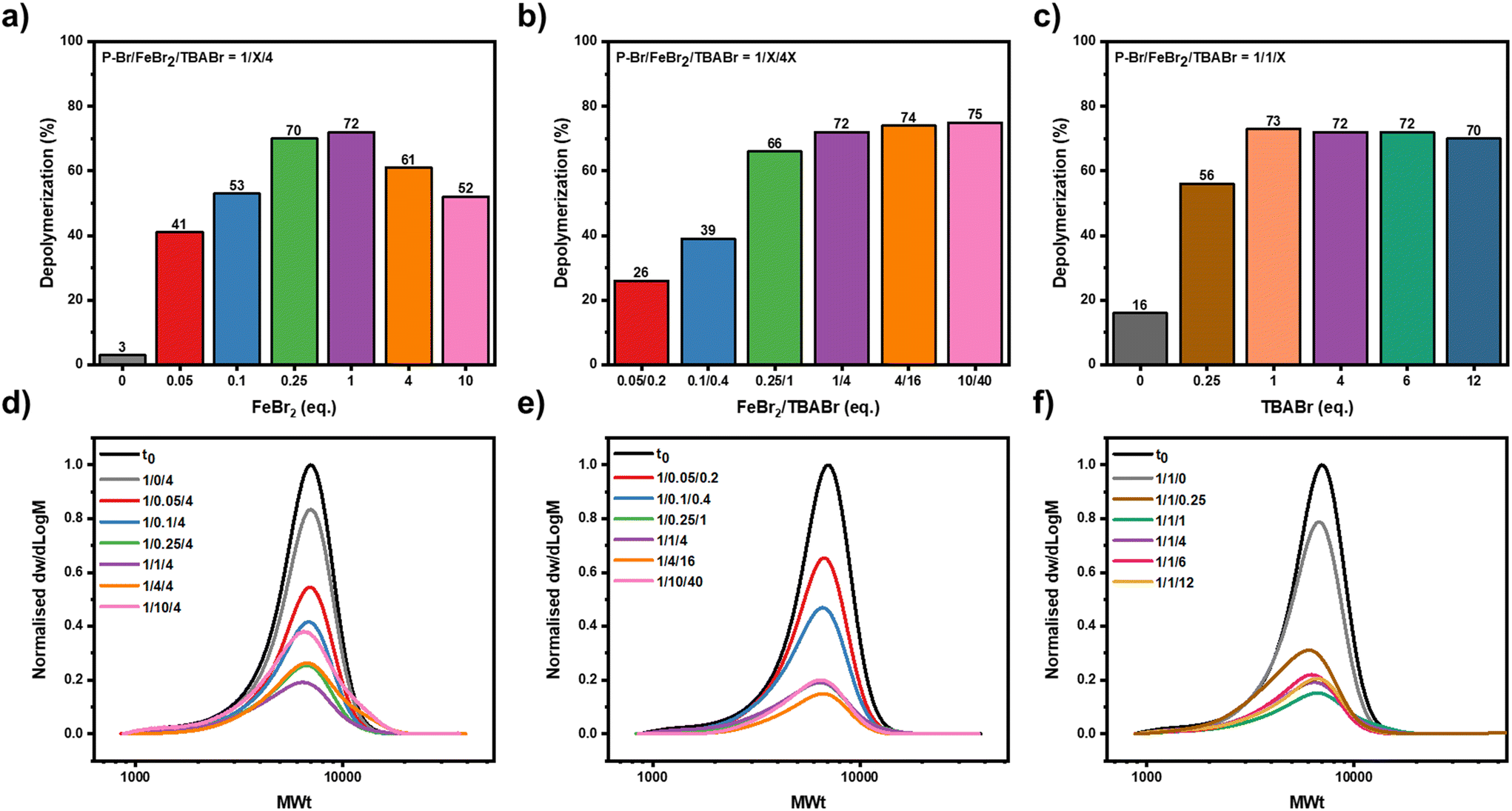 | ||
| Fig. 3 The effect of the metal salt and TBABr concentration on the depolymerization. In (a) a bar graph shows the relative depolymerization conversions with various metal salt concentrations. In (b) a bar graph shows the relative depolymerization conversions with various FeBr2/TBABr concentrations (X:4X). In (c) a bar graph shows the relative depolymerization conversions with various TBABr concentrations. (d–f) Show the SEC traces for the corresponding reactions for (a–c). All reactions were performed with a 50 mM repeat unit concentration of polymer, FeBr2/TBABr as the catalyst in TCB at 170 °C. Samples were taken after 5 minutes. | ||
Our next aim was to understand the importance of this 1:4 FeBr2 to TBABr ratio (Fig. 3b). Several depolymerization experiments were performed where we scaled up and down the concentration of the FeBr2 and the TBABr, while maintaining this 1:4 ratio. Similarly, to the previous results, when the concentration of catalyst was decreased the amount of depolymerization also decreased (Fig. 3b, e & S13 and Table S4,† entries 3–6). For example, using a ratio of [P-Br]:[FeBr2]:[TBABr] of 1:0.25:1 resulted in 66% of depolymerization compared to the 72% with a ratio of 1:1:4 (Table S4,† entries 3 and 4). This trend was further demonstrated with 39% and 26% depolymerization obtained when ratios of [P-Br]:[FeBr2]:[TBABr] equal to 1:0.10:0.40 and 1:0.05:0.20 were used, respectively (Table S4,† entries 5 and 6). As such, it was concluded that at least 1 equivalent of FeBr2/TBABr was necessary to maximize the depolymerization. Instead, at higher concentrations of metal salt and TBABr ([P-Br]:[FeBr2]:[TBABr] = 1:4:16 and 1:10:40), the depolymerization yield was maintained constant, with 74 and 75% achieved, respectively (Fig. 3b, e & S13 and Table S4,† entries 1 and 2). This contrasted the results when a [P-Br]:[FeBr2]:[TBABr] of 1:10:4 was utilized, where much lower depolymerization occurred and just 52% of the starting monomer was obtained (Fig. 3a and Table S3,† entry 1). Together this suggests that so long as the TBABr is in excess to the metal salt, radical termination reactions can be suppressed, even at high catalyst loadings. A polymer-to-metal salt ratio ([P-Br]:[FeBr2] of 1:1) is though preferred to minimize the amount of catalyst required.
Given the importance of the TBABr concentration in controlling the extent of termination, it's optimal concentration for depolymerization was also explored (Fig. 3c).65 We therefore performed depolymerization reactions with various amounts of TBABr while keeping the amount of metal salt constant ([P-Br]:[FeBr2]:[TBABr] 1:1:X). These experimental results clearly fell into 2 categories; (i) when the amount of TBABr was equal or greater to the amount of metal salt (X = 1, 4, 6, 12), there was a reproducibly high depolymerization conversion (69–75%, Fig. 3c, f & S14 and Table S5,† entries 1–4), whereas (ii) when TBABr was not in excess (i.e. X = 0.25) a much lower extent of depolymerization was observed (52%, Fig. 3c, f & S14 and Table S5,† entries 5 and 6). A [P-Br]:[FeBr2]:[TBABr] ratio of 1:1:1 was therefore selected as the optimal condition as this maximized the extent of depolymerization, while minimized the amount of TBABr required.
Considering that the depolymerization conditions had been judiciously optimized, kinetic analysis was performed to further understand the rate of reaction (Fig. 4). It was quite remarkable that in all the previous experiments the maximum depolymerization conversion had been obtained within just 5 minutes, so a depolymerization was performed with the optimized conditions ([P-Br]:[FeBr2]:[TBABr] of 1:1:1) and the reaction was frequently sampled (Fig. 4 & S15 and Table S6†). In the early stages, only a small amount of depolymerization was observed, with 6% of monomer obtained in the first 40 seconds (Table S6,† entries 1–3). This was attributed to the time required for the reaction solution to reach 170 °C and for the catalyst to trigger radical activation of the bromine end-groups. There was subsequently, a rapid and linear increase in depolymerization, reaching 62% in just 90 seconds (Table S6,† entries 4–8). After this, the rate of depolymerization slowed down and the kinetic profile plateaued to a maximum depolymerization conversion of 73% (Table S6,† entry 9). In terms of molecular weight and dispersity evolution, there was a sharp drop in the polymer signal of the SEC profiles as the depolymerization proceeded, demonstrating that most of the polymer chains fully unzipped on activation (Fig. 4b & S15†). A slight decrease in the Mn from ∼6000 to ∼5000 and a small amount of low molecular weight tailing could be evidenced in SEC alongside a small increase in dispersity (Đ = 1.14–1.20). This suggests there was a small amount of termination occurring during the depolymerization and this is likely also demonstrated through the depolymerization efficiency of 85%. In addition, it was also feasible that a small amount of lactonization might be contributing to this loss of depolymerization efficiency over the timeframe of our experiments (Scheme S2†). To investigate this, we performed a depolymerization without any catalyst and after 5 minutes, the formation of benzyl bromide could clearly be observed in the 1H NMR (Fig. S16, S17 and Table S7,† entries 1 and 2). To confirm the loss of bromine chain-ends, we subsequently added the optimal amount of FeBr2/TBABr catalyst to the reaction and after a further 10 minutes only 25% of monomer regeneration had been achieved (Fig. S18, S19 and Table S7,† entries 3 and 4). Together this demonstrates that lactonization is a key competitor to depolymerization and the key to successful depolymerization is to use conditions that provide fast enough depolymerization to out-compete the lactonization, while not being so fast that the too high radical concentration triggers significant termination events. With the optimized depolymerization conditions these criteria are fulfilled, which allows the vast majority of the bromine-terminated polymers to be fully unzipped. This work significantly expands the scope of depolymerizable materials from ATRP, and also has the significant advantage of being much faster than many of the previously reported depolymerization methods.33,39,45,50,54
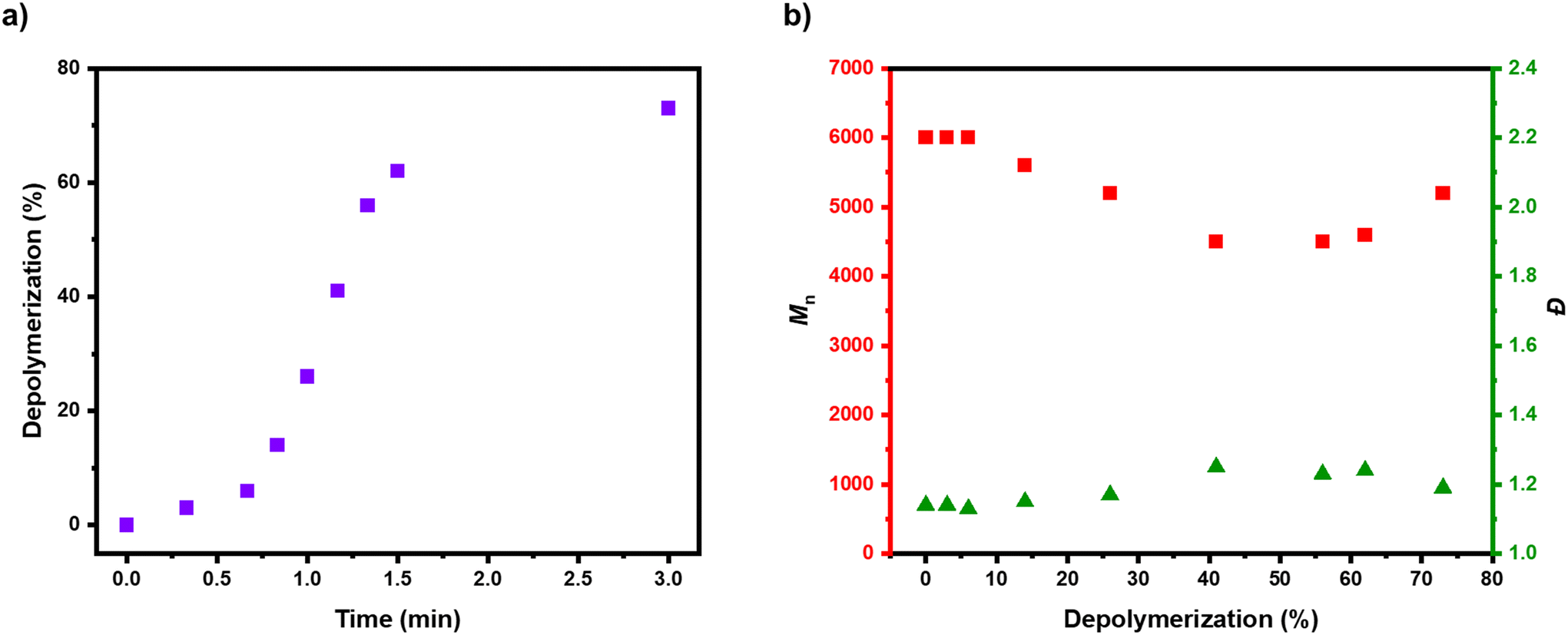 | ||
| Fig. 4 Depolymerization kinetics with the optimized conditions ([P-Br]:[FeBr2]:[TBABr] of 1:1:1). In (a) the evolution of depolymerization conversion with time is shown and in (b) the evolution of molecular weight and dispersity with depolymerization is presented. | ||
Our final aim was to further expand the scope of this depolymerization by investigating its compatibility with (i) a non-chlorinated solvent, and (ii) FeBr3 as an alternative metal salt rather than FeBr2. One potential disadvantage of the optimized conditions demonstrated thus far is that high depolymerization conversions were only obtainable when chlorinated solvents were used (Table S8,† entries 1–4). Anisole was therefore selected and pleasingly, under our optimized ratio of polymer to catalyst ([P-Br]:[FeBr2]:[TBABr] of 1:1:1), as much as 64% of the starting monomer could be regenerated (Fig. S20, S21 and Table S8,† entries 5–8). This amounted to a depolymerization efficiency of 80%, which was comparable to the extent of depolymerization obtained with TCB (86%). Next, we performed depolymerization with FeBr3/TBABr instead of FeBr2/TBABr as the catalyst. This had no noticeable negative impact on the depolymerization, with a similarly high depolymerization achieved in both cases (73% vs. 70%, Fig. S22, S23 and Table S9†). Interestingly, the depolymerization with FeBr3/TBABr could be performed in the dark (aluminum foil-wrapped) and an identical depolymerization conversion was achieved as when the reaction had been performed in fume hood light (Fig. S22, S24 and Table S10†). This suggests that light was not playing a role in reducing the Fe(III) to active Fe(II) and instead, most likely, a thermally induced reduction was occurring. This mechanism likely has significant complexity and will be the subject of a future publication. Altogether, these results demonstrate that excellent depolymerization of bromine-terminated polymers can be achieved under a versatile range of reaction conditions.
Conclusions
To summarize, we have demonstrated that bromine-terminated polymers obtained from ATRP can be successfully depolymerized with very high efficiencies (∼86%). This was achieved through careful optimization of the solvent and both the metal salt and TBABr concentrations. Key to the success was minimizing competing termination and lactonization, allowing efficient bromine chain-end activation and depolymerization. The versatility of the reaction conditions was expanded to include a range of catalyst concentrations, both chlorinated and non-chlorinated solvents, FeBr2 and FeBr3 salts, and both light and dark conditions. This significantly widens the scope of materials compatible with depolymerization and will create many future opportunities as chemical recycling methods develop.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
A. A. gratefully acknowledges ETH Zurich (Switzerland) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (DEPO: Grant No. 949219) for financial support. N. P. T. acknowledges the award of a DECRA Fellowship from the ARC (DE180100076). K. P. kindly thanks the Onassis Foundation for financial support: Scholarship ID: FZQ051-1/2020-2021.References
- H. Staudinger, Ber. Dtsch. Chem. Ges. B, 1920, 53, 1073–1085 CrossRef.
- D. Feldman, Des. Monomers Polym., 2008, 11, 1–15 CrossRef CAS.
- S. J. Rowan, ACS Macro Lett., 2020, 9, 122 CrossRef CAS PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- D. E. Fagnani, J. L. Tami, G. Copley, M. N. Clemons, Y. D. Y. L. Getzler and A. J. McNeil, ACS Macro Lett., 2021, 10, 41–53 CrossRef CAS PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- A. Adili, A. B. Korpusik, D. Seidel and B. S. Sumerlin, Angew. Chem., Int. Ed., 2022, 61, e202209085 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 12268–12277 CrossRef CAS PubMed.
- N. Zaquen, B. Wenn, K. Ranieri, J. Vandenbergh and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 178–187 CrossRef CAS.
- T. Shimizu, R. Whitfield, G. R. Jones, I. O. Raji, D. Konkolewicz, N. P. Truong and A. Anastasaki, Chem. Sci., 2023, 14, 13419–13428 RSC.
- E. W. Montroll and R. Simha, J. Chem. Phys., 2004, 8, 721–726 CrossRef.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- Z. Deng and E. R. Gillies, JACS Au, 2023, 3, 2436–2450 CrossRef CAS PubMed.
- W. Kaminsky and J. Franck, J. Anal. Appl. Pyrolysis, 1991, 19, 311–318 CrossRef CAS.
- C. B. Godiya, S. Gabrielli, S. Materazzi, M. S. Pianesi, N. Stefanini and E. Marcantoni, J. Environ. Manage., 2019, 231, 1012–1020 CrossRef CAS PubMed.
- L. E. Manring, Macromolecules, 1991, 24, 3304–3309 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT polymerization, John Wiley & Sons, 2008 Search PubMed.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- D. A. Shipp, J.-L. Wang and K. Matyjaszewski, Macromolecules, 1998, 31, 8005–8008 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS PubMed.
- M.-N. Antonopoulou, R. Whitfield, N. P. Truong, D. Wyers, S. Harrisson, T. Junkers and A. Anastasaki, Nat. Chem., 2022, 14, 304–312 CrossRef CAS PubMed.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 CAS.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- N. P. Truong, G. R. Jones, K. G. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed.
- T. Kimura, K. Kuroda, H. Kubota and M. Ouchi, ACS Macro Lett., 2021, 10, 1535–1539 CrossRef CAS PubMed.
- G. R. Jones, H. S. Wang, K. Parkatzidis, R. Whitfield, N. P. Truong and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 9898–9915 CrossRef CAS PubMed.
- T. Chen, H. Wang, Y. Chu, C. Boyer, J. Liu and J. Xu, ChemPhotoChem, 2019, 3, 1059–1076 CrossRef CAS.
- V. Lohmann, G. R. Jones, N. P. Truong and A. Anastasaki, Chem. Sci., 2024, 15, 832–853 RSC.
- M. J. Flanders and W. M. Gramlich, Polym. Chem., 2018, 9, 2328–2335 RSC.
- H. S. Wang, N. P. Truong, G. R. Jones and A. Anastasaki, ACS Macro Lett., 2022, 11, 1212–1216 CrossRef CAS PubMed.
- F. Häfliger, N. P. Truong, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2023, 12, 1207–1212 CrossRef PubMed.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS PubMed.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. De Alwis Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253–258 RSC.
- V. Bellotti, H. S. Wang, N. P. Truong, R. Simonutti and A. Anastasaki, Angew. Chem., Int. Ed., 2023, 62, e202313232 CrossRef CAS PubMed.
- J. B. Young, J. I. Bowman, C. B. Eades, A. J. Wong and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 1390–1395 CrossRef CAS PubMed.
- H. S. Wang, K. Parkatzidis, T. Junkers, N. P. Truong and A. Anastasaki, Chem, 2024, 10, 388–401 CAS.
- J. B. Young, R. W. Hughes, A. M. Tamura, L. S. Bailey, K. A. Stewart and B. S. Sumerlin, Chem, 2023, 9, 2669–2682 CAS.
- R. Whitfield, G. R. Jones, N. P. Truong, L. E. Manring and A. Anastasaki, Angew. Chem., Int. Ed., 2023, 62, e202309116 CrossRef CAS PubMed.
- B. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Aust. J. Chem., 2006, 59, 755–762 CrossRef CAS.
- M. R. Martinez and K. Matyjaszewski, CCS Chem., 2022, 4, 2176–2211 CrossRef CAS.
- V. Raus, E. Čadová, L. Starovoytova and M. Janata, Macromolecules, 2014, 47, 7311–7320 CrossRef CAS.
- Y. Sano, T. Konishi, M. Sawamoto and M. Ouchi, Eur. Polym. J., 2019, 120, 109181 CrossRef CAS.
- M. R. Martinez, D. Schild, F. De Luca Bossa and K. Matyjaszewski, Macromolecules, 2022, 55, 10590–10599 CrossRef CAS.
- M. R. Martinez, S. Dadashi-Silab, F. Lorandi, Y. Zhao and K. Matyjaszewski, Macromolecules, 2021, 54, 5526–5538 CrossRef CAS.
- M. R. Martinez, F. De Luca Bossa, M. Olszewski and K. Matyjaszewski, Macromolecules, 2021, 55, 78–87 CrossRef.
- K. Parkatzidis, N. P. Truong, K. Matyjaszewski and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 21146–21151 CrossRef CAS PubMed.
- F. De Luca Bossa, G. Yilmaz and K. Matyjaszewski, ACS Macro Lett., 2023, 12, 1173–1178 CrossRef CAS PubMed.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Macromolecules, 2017, 50, 7967–7977 CrossRef CAS.
- M. Rolland, N. P. Truong, R. Whitfield and A. Anastasaki, ACS Macro Lett., 2020, 9, 459–463 CrossRef CAS PubMed.
- K. Parkatzidis, S. Boner, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2022, 11, 841–846 CrossRef CAS PubMed.
- S. Dadashi-Silab and K. Matyjaszewski, Molecules, 2020, 25, 1648 CrossRef CAS PubMed.
- J. W. Bartels, S. I. Cauët, P. L. Billings, L. Y. Lin, J. Zhu, C. Fidge, D. J. Pochan and K. L. Wooley, Macromolecules, 2010, 43, 7128–7138 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, B. Parker and K. Matyjaszewski, Macromolecules, 2011, 44, 4022–4025 CrossRef CAS.
- M. Teodorescu, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 2000, 33, 2335–2339 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- J. L. de la Fuente, M. Fernández-Sanz, M. Fernández-García and E. L. Madruga, Macromol. Chem. Phys., 2001, 202, 2565–2571 CrossRef CAS.
- A. T. Jackson, A. Bunn, I. M. Priestnall, C. D. Borman and D. J. Irvine, Polymer, 2006, 47, 1044–1054 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00279a |
| This journal is © The Royal Society of Chemistry 2024 |