
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and magnetic properties of two bis(oxalato)cuprate(II) salts with pyridinium type counter ions†
Cyrielle L. F.
Dazem
a,
A. Ken
Inge
 b,
Dominique
Luneau
c,
Lars
Öhrström
*d and
Justin
Nenwa
*a
b,
Dominique
Luneau
c,
Lars
Öhrström
*d and
Justin
Nenwa
*a
aInorganic Chemistry Department, Faculty of Science, University of Yaoundé I, PO. Box 812, Yaoundé, Cameroon. E-mail: cyrielle_leyla@yahoo.fr; jnenwa@yahoo.fr
bDepartment of Materials and Environmental Chemistry, Stockholm University, Stockholm, SE-106 91 Sweden
cLaboratoire des Multimatériaux et Interfaces (UMR 5615), Université Claude Bernard de Lyon 1, Campus de la Doua, 69622 Villeurbanne, France. E-mail: dominique.luneau@univ-lyon1.fr
dChemistry and Biochemistry, Dept. of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Göteborg, Sweden. E-mail: ohrstrom@chalmers.se
First published on 16th December 2022
Abstract
Mono- and a di-protonated pyridine type cations have been used for the synthesis of two bis(oxalato)cuprate(II) salts, namely, (C5H7N2O)2[Cu(C2O4)2] (C5H7N2O = 2-amino-3-hydroxypyridinium) 1 and C13H16N2[Cu(C2O4)2] (C13H16N2 = 4,4′-trimethylenedipyridinium) 2. In the crystal structures, C2O42− adopts different coordination modes: the common bidentate chelating oxalate in 1 and the relatively scarce μ-oxalato-κ3O1,O2:O1′ in 2. X-ray diffraction also revealed that [Cu(C2O4)2]2− anions do not polymerize in 1 (a phenomenon hardly observed in such salts), while in 2, the polymerization of [Cu(C2O4)2]2− units occurs via the Cu–Oaxial contact forming a zigzag Cu(II) chain. In complex 1, the emerging building blocks are linked into 2D supramolecular layers via N–H⋯O and O–H⋯O hydrogen bonds and weaker C–H⋯O interaction to form a 3D net. In contrast to 2, the emerging building blocks are linked into 1D chains via N–H⋯O hydrogen bonds, which further extend to form a 3D supramolecular framework through Cu–Oaxial and other C–H⋯O interactions. The structural diversities show that iminium counterions play key roles in the construction of various architectures. Thermal analyses showed no weight loss for either system in the temperature range of 20–180 °C, which indicates that none of these complexes possess solvation water molecules. Magnetic studies indicate the presence of antiferromagnetic coupling between the spin centres in 1 and 2.
1. Introduction
Oxalates are ubiquitous. They are found in numerous plants but may also form less pleasant kidney and bladder stones in humans. The latter comprise calcium oxalates, and other oxalate complexes with metal ions are used, for example, in artistic chemical photography processes and the anticancer drug oxaliplatin (elotaxin).1However, oxalates are abundant also in today's research, and we have lately reported on oxalate networks and on naturally occurring metal–organic frameworks (MOFs).2 Recently, metal oxalates have also been featured in the production of the medical radioisotope 89Zr,3 and freestanding oxalate ions were reported as crystalline molecular rotors in salts with [M(en)3]2+ serving as a molecular stator.4 Perhaps the more obvious work is on oxalate MOFs5 and copper oxalates for magnetic devices for chemical sensing.6
Over the past years, great efforts have been devoted to the self-assembly of organic–inorganic molecules in the solid state for different reasons: i) it offers extraordinary structural versatility and allows the formation of fascinating structures7,8 – in fact the framework flexibility has been highlighted as an advantage for high-performance molecular recognition, separation and sensing application,9 ii) it extends the range of designing functional solid materials from neutral or ionic molecular-based building blocks properties,10,11 and iii) it allows the search of new structural topologies and connectivities in the solid state.2
Research activities mostly involve organic cations with metal complex anions as an inorganic entity, organic–inorganic salts. The versatile coordination modes and the ability to transmit the electronic effects between magnetic centres separated by more than 5 Å12,13 and to form interesting hydrogen-bonded solid state networks could justify the special attention paid towards the investigation of salts based on the oxalate(2-) ligand.14
Among the various anionic transition metal oxalates, the copper(II) ones have been considered attractive building blocks for the construction of organic–inorganic salts in recent years.15 One of the most interesting organic cation–copper oxalate combinations is that of pyridinium derivatives and anionic bis(oxalato)cuprate(II) complexes. Compared to many Cu(II) oxalate organic–inorganic salts based on ammonium-based counterions, pyridinium ones are still less explored. Moreover, with the structural variability observed in the reported pyridinium salts, the understanding of their basic properties is still ongoing. Therefore, more work is required to understand the factors that influence the resulting crystal structure. With regards to the wide range of pyridinium-based cations, there is still a broad chemical space for synthesizing new organic–inorganic compounds with unusual structures and unknown properties.
For five years now, our research group has been investigating pyridinium-type counterion salts with bis(oxalato)cuprate(II) anions, with a view not only to producing new functional materials but also to unraveling the subtle structural features that characterize these solids. Accordingly, we previously successfully contributed to these endeavours with three novel pyridinium derivatives of Cu(II) oxalate salts, namely, bis(3-aminopyridinium) bis(oxalato)cuprate(II) dihydrate, bis(2-amino-4,6-dimethylpyridinium) bis(oxalato)cuprate(II)pentahydrate,16 and bis(2-amino-pyridinium) bis(oxalato)cuprate(II) trihydrate.17 Recently ternary co-crystal salts, namely, bis(3-aminopyridinium) bis(oxalato)cuprate(II) dioxalic acid and bis(4-dimethylaminopyridinium) bis(oxalato)diaquacuprate(II) oxalic acid, were also prepared and structurally characterised.18
Encouraged by these results we continued with this type of salt and investigated the self-assembly of bis(oxalato)cuprate(II) anionic units with pyridinium derivatives containing amino(–NH2) and hydroxyl (–OH) functional groups and an alkyl (–CH2–)n group between two pyridinium ions (Fig. 1). We report here the synthetic and structural characterization of (C5H7N2O)2[Cu(C2O4)2] 1 and C13H16N2[Cu(C2O4)2] 2, as well as their spectroscopic analysis and magnetic properties (C5H7N2O = 2-amino-3-hydroxypyridinium and C13H16N2 = 4,4′-trimethylenedipyridyl).
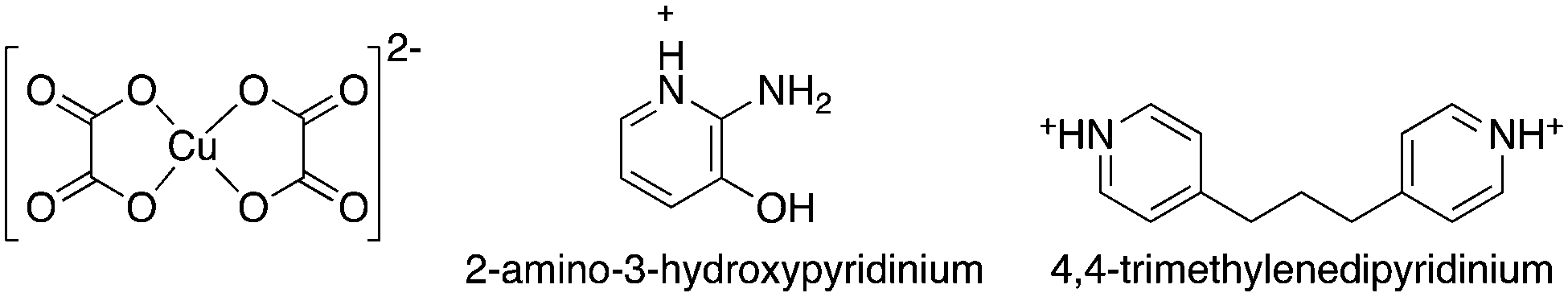 | ||
| Fig. 1 Building blocks of (C5H7N2O)2[Cu(C2O4)2] (1) and C13H16N2[Cu(C2O4)2] (2). | ||
2. Experimental
2.1 Materials and methods
(C5H7N2O)2[Cu(C2O4)2] 1. A light brown solution containing oxalic acid dihydrate (126 mg, 1 mmol) and 2-amino-3-hydroxypyridine (220 mg, 2 mmol) in 20 mL of distilled water was stirred at 50 °C for 1 h. Subsequently, a blue aqueous solution of (NH4)2[Cu(C2O4)2]·2H2O (311 mg, 1 mmol) was added into the aforementioned solution and stirred for an additional 30 min at a constant temperature of 50 °C. After the reaction was stopped and cooled, the resulting solution was filtered and stored at room temperature to produce dark green crystals of 1 after 14 days: yield ca. 96% [based on (NH4)2[Cu(C2O4)2]·2H2O]. X-ray powder diffraction was performed to confirm the purity of the sample. Anal. calcd (Found%) for C14H14CuN4O10 (Mr = 461.84): C, 36.38 (35.10); H, 3.03 (2.90); N, 12.13 (11.50). IR (KBr pellet, cm−1): 3468(w), 3423(w), 3345(w), 3101(w), 1708(s), 1629(s), 1571(s), 1288(s), 1244(s), 795(s), 895(w), 586(w). UV-vis (H2O solution, nm) 440; 713.
C13H16N2[Cu(C2O4)2] 2. A colourless solution containing oxalic acid dihydrate (126 mg, 1 mmol) and 4,4′-trimethylenedipyridine (200 mg, 1 mmol) in 20 mL of distilled water was stirred at 50 °C for 1 h. Subsequently, a blue aqueous solution of (NH4)2[Cu(C2O4)2]·2H2O (311 mg, 1 mmol) was added into the aforementioned solution and stirred for an additional 30 min at a constant temperature of 50 °C. After the reaction was stopped and cooled, the resulting solution was filtered and stored at room temperature to produce dark blue crystals of 2 after 16 days: yield ca. 93% [based on (NH4)2[Cu(C2O4)2]·2H2O]. X-ray powder diffraction was performed to confirm the purity of the sample. Anal. calcd (found%) for C17H16CuN2O8 (Mr = 439.87): C, 46.37 (45.90); H, 3.64 (3.50); N, 6.37 (6.20). IR (KBr pellet, cm−1): 3436(m), 1666(m), 1671(s), 1641(s), 1498(m), 1413(s), 1286(s), 1203(m), 824(m), 542(w), 488(m). UV-vis (H2O solution, nm): 721.
| Compound | 1 | 2 |
|---|---|---|
| Empirical formula | C7H7Cu0.5N2O5 | C17H16CuN2O8 |
| Formula weight | 230.92 | 439.86 |
| Temperature/K | 99.9(4) | 100.0(2) |
| Crystal system | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 5.0894(3) | 10.5484(2) |
| b/Å | 7.1344(3) | 22.2344(6) |
| c/Å | 11.4050(4) | 7.22640(18) |
| α/° | 86.127(3) | 90 |
| β/° | 77.266(4) | 102.125(2) |
| γ/° | 81.424(4) | 90 |
| Volume/Å3 | 399.15(3) | 1657.05(7) |
| Z | 2 | 4 |
| ρ calcg/cm3 | 1.921 | 1.763 |
| μ/mm−1 | 2.623 | 2.362 |
| F (000) | 235.0 | 900.0 |
| Crystal size/mm3 | 0.1 × 0.023 × 0.015 | 0.107 × 0.056 × 0.042 |
| Radiation/Å | CuKα (λ = 1.54184) | CuKα (λ = 1.54184) |
| 2Θ range for data collection/° | 7.952 to 150.808 | 7.952 to 151.102 |
| Index ranges | −6 ≤ h ≤ 6, −8 ≤ k ≤ 8, −12 ≤ l ≤ 14 | −9 ≤ h ≤ 12, −27 ≤ k ≤ 26, −9 ≤ l ≤ 9 |
| Reflections collected | 6951 | 15792 |
| Independent reflections | 1571 [Rint = 0.0465, Rsigma = 0.0350] | 3303 [Rint = 0.0340, Rsigma = 0.0233] |
| Data/restraints/parameters | 1571/0/149 | 3303/0/261 |
| Goodness-of-fit on F2 | 1.092 | 1.069 |
| Final R indexes [I > = 2σ (I)] | R 1 = 0.0359, wR2 = 0.0975 | R 1 = 0.0263, wR2 = 0.0735 |
| Final R indexes [all data] | R 1 = 0.0393, wR2 = 0.0990 | R 1 = 0.0290, wR2 = 0.0747 |
| CCDC deposition numbers | CCDC 2217520 | CCDC 2217521 |
3. Results and discussion
3.1 Synthesis
Salts, namely, (C5H7N2O)2[Cu(C2O4)2] (1) and C13H16N2[Cu(C2O4)2] (2) (C5H7N2O = 2-amino-3-hydroxypyridinium; C13H16N2 = 4,4′-trimethylenedipyridinium) were obtained following two chemical processes similar to the one described by us for related compounds.16 First the imine groups of the pyridine derivatives (DerPy) are protonated by oxalic acid, producing iminium cations (DerPy-H)+. Second, an aqueous solution of (NH4)2[Cu(C2O4)2]·2H2O was added, and the target compounds crystallised and were collected after around 2 weeks.3.2 Single crystal X-ray structures
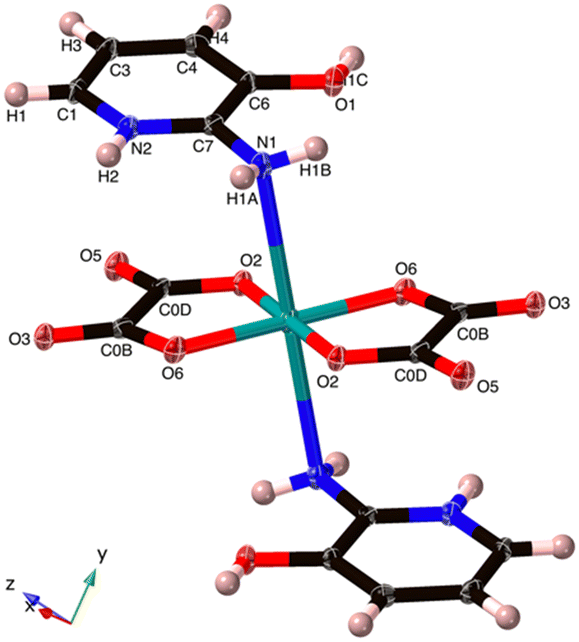 | ||
| Fig. 2 The Cu coordination in (C5H7N2O)2[Cu(C2O4)2] 1. The O5 hydroxyl group hydrogen bonds to O3 and the N1 amine. The axial Cu–N bond is 2.711(5) Å. | ||
The Cu2+ ion exhibits a square-planar geometry with four O atoms from two chelating oxalato2− ligands. However, there are also Cu–N interactions in the axial positions at 2.711(5) Å, and as the N1 geometry is distinctly non-planar with the N1 distance from the C7–H1A–H1B plane being 0.212 Å, we interpret this as a bonding interaction giving an overall octahedral configuration. We can thus also describe 1 as a neutral coordination compound. However, it needs to be noted that this Cu–N distance is a fair bit longer than the Cu–N peak of Jahn–Teller distorted compounds found in the CSD at 2.35–2.48 Å (Fig. S1†).
Cu1–O6 and Cu1–O2 are at two independent distances, namely, 1.936 (5) and 1.938 (5) Å, respectively. There are also two unique O6–Cu1–O2 bond angles of 84.80 (8) and 95.20 (8)°. These distances and angles are akin to those found in some reported Cu(II) oxalate salts featuring similar coordination motifs.23,24
The packing of 1 shows strong interlocking between complex anion and cationic sub-lattices through a multidirectional hydrogen bonding network. The organic cations, (C5H7N2O)+, link adjacent copper oxalate anions via hydrogen bonding interactions N2–H2⋯O2, N1–H1A⋯O6 and O1–H1C⋯O3 to form a 1D chain in the x-direction (Table 2).
| D | H | A | d(D–H)/Å | d(H–A)/Å | d(D–A)/Å | D–H–A/° |
|---|---|---|---|---|---|---|
| 13 − x, −y, 1 − z2; 1 − x, 1 − y, 1 − z3; 1 + x, +y, +z. | ||||||
| N2 | H2 | O21 | 0.96(3) | 1.81(3) | 2.764(2) | 177(3) |
| O1 | H1C | O32 | 0.85(4) | 1.75(4) | 2.602(2) | 178(4) |
| N1 | H1A | O63 | 0.91(3) | 1.98(3) | 2.862(3) | 162(3) |
These chains are further linked by the axial Cu–N bonds in the z-direction, and a pair of organic cations, (C5H7N2O)+, bridges two neighbouring [Cu(C2O4)2]2− units via weaker C–H⋯O hydrogen bonding interactions between C atoms from (C5H7N2O)+ and O atoms from [Cu(C2O4)2]2− (O3⋯H3 at 2.49 Å and O5⋯H1 at 2.43 Å; see Fig. 3), to generate a 3D supramolecular network.
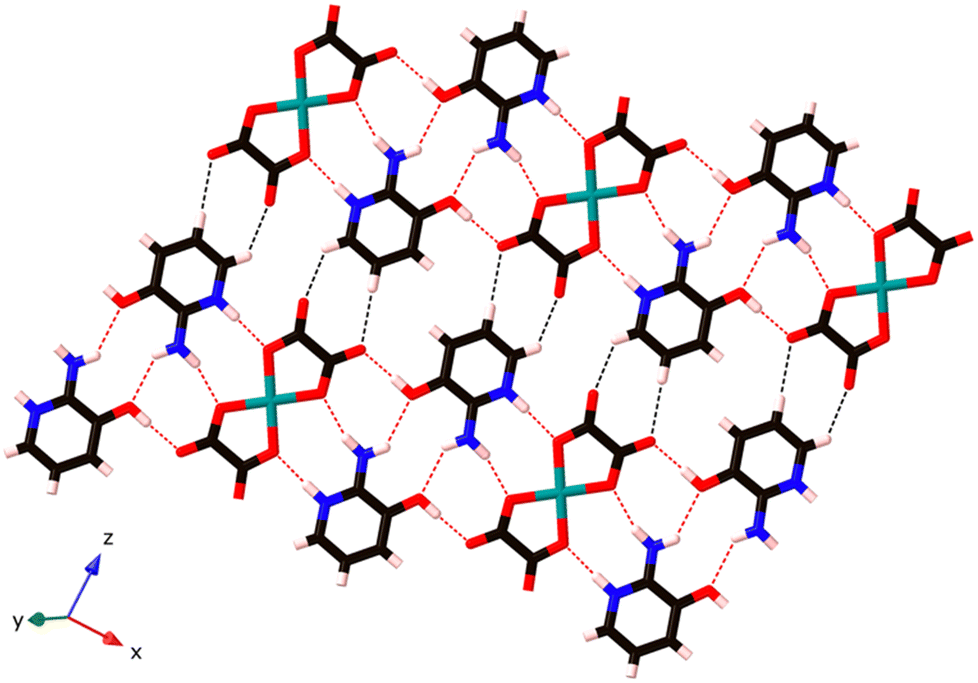 | ||
| Fig. 3 The hydrogen bonded sheets in (C5H7N2O)2[Cu(C2O4)2] 1. These are then connected to a 3D network by the weak axial Cu–N bonds; see Fig. 2. Strong hydrogen bonds in red and weak C–H hydrogen bonds in black. | ||
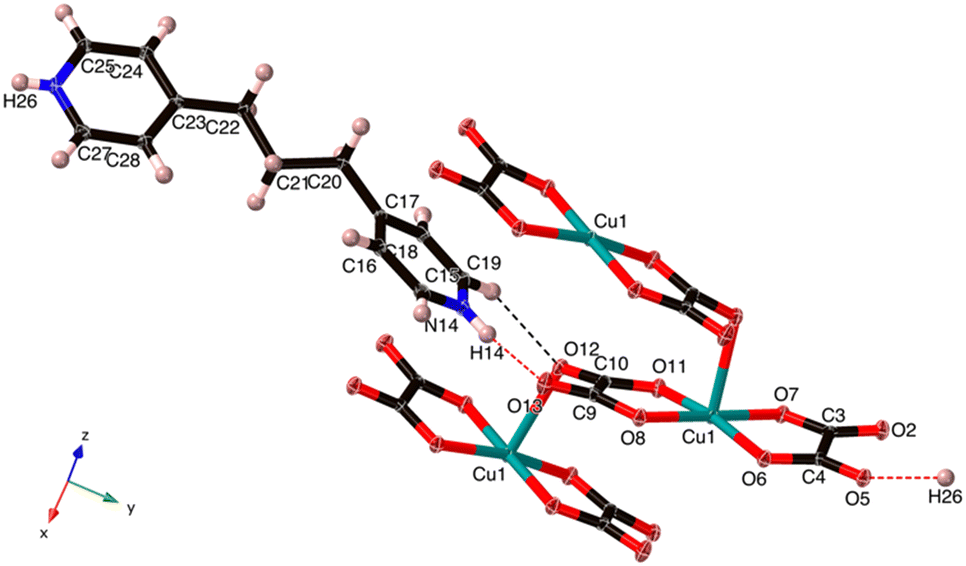 | ||
| Fig. 4 The asymmetric unit of C13H16N2[Cu(C2O4)2] (2) with one extra oxalate, one extra Cu and one additional N–H. The Cu–O7 is 2.538 Å, which is normal for an axial Jahn–Teller bond. | ||
The Cu(II) centre in 2 shows a square pyramidal geometry with four oxygen atoms (O1, O4, O5, and O8) from two oxalate(2-) ligands in the equatorial plane and one terminal oxygen atom (O7) from the neighbouring [Cu(C2O4)2]2− unit in the apical position. The length of Cu–Oeq bonds (Cu1–O6 = 1.957(1), Cu1–O7 = 1.934(1), Cu1–O11 = 1.935(2) and Cu1–O8 = 1.943(1) Å) are in good agreement to those in related Cu(II) complexes of 2,6-bis(4′-pyridyl-1′-pyridiniumyl)pyrazine25 and aminoguanidinium;26 the length of the Cu–Oaxial bond (Cu–O7 = 2.454(2) Å) is consistent with the ones in known aminoguanidinium26 and potassium27 salts and normal for a Jahn–Teller Cu–O bond. The bite angles of the bidentate oxalate were within the expected bond angles.25
An extended analysis of this compound permitted us to notice that it is a single O-bridged copper coordination polymer constructed from oxalate ligands adopting a rare μ-oxalato-κ3O, 1O2:O1′ coordination mode.26,28 We also note, in contrast to 1, that the stacking of the anionic [Cu(C2O4)2]2− entities occurs through the weak interaction between the Cu(II) ion and terminal O12 atom, leading to a uniform zigzag Cu(II) chain that runs along the crystallographic z-axis, Cu⋯Cu = 5.348 Å (Fig. 4). Interestingly, in the 4-(pyridin-1-ium-4-yl)piperazin-1-ium compound the μ-oxalato-κ3O1,O2:O1′ coordination mode results in a different polymer chain, and the one –CH2– unit shorter 4,4′-dimethylenedipyridinium salt contains discrete [Cu(C2O4)2]2− units.28 Moreover, the aminoguanidinum compound, the second example of the μ-oxalato-κ3O1,O2:O1′ mode, adopts a staircase-type polymer chain.26
We also note that (H2CBpy)[Cu(C2O4)2]·2H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 has been found as a single O-bridged copper coordination polymer constructed from oxalate ligands. However, here we have a μ-oxalato-κ3O1,O2:O1 polymer, which is more commonly observed in these systems.
29 has been found as a single O-bridged copper coordination polymer constructed from oxalate ligands. However, here we have a μ-oxalato-κ3O1,O2:O1 polymer, which is more commonly observed in these systems.
The organic and inorganic parts, arranged in an alternating manner, whichever x or y direction is chosen, are linked via N26–H26⋯O5, and N14–H14⋯O13 hydrogen bonds (Table 3 and Fig. 4) to form 1D cation-anion ribbons along the y-axis.
| D | H | A | d(D–H)/Å | d(H–A)/Å | d(D–A)/Å | D–H–A/° |
|---|---|---|---|---|---|---|
| 1 +x, −1 + y, +z. | ||||||
| N26 | H26 | O51 | 0.77(2) | 1.95(2) | 2.7066(18) | 165(2) |
| N14 | H14 | O13 | 0.82(2) | 1.91(2) | 2.6989(19) | 161(2) |
In addition, the 1D chains are extended through Jahn–Teller Cu–Oaxial bonds, generating sheets parallel to the yz plane. Finally, two pairs of weak hydrogen bonds C15–H15⋯O5 at 2.22(3) Å and C18–H18⋯O2 at 2.26(3) Å connect the aforementioned sheets in the xy-plane to form a 3D supramolecular network (Fig. S7†).
Another way to look at the packing in the xy-plane is to consider the charges, and as shown in Fig. S8,† opposite charges form a kind of a chess board pattern.
3.3 IR spectra
The IR spectra of 1 and 2 are presented in Fig. S2.† They exhibit sharp absorption bands at around 1708–1670 cm−1 and 1288–1244 cm−1, which can be attributed to the stretching modes of carboxyl COO− groups.30 Moreover, 1 has a weak absorption band at 3345 cm−1, which can be ascribed to the stretching vibrations of the phenol hydroxyl group.313.4 UV-vis spectra in solution
The UV spectrum of 1 in water (Fig. 5 left) presents two broad and weak absorption bands at 440 and 713 nm (ε = 25.438 mol−1 L cm−1), and that of 2 in water (Fig. 5 right) presents a broad and strong absorption band at 719 nm (ε = 48.245 mol−1 L cm−1). In the case of 1, the obtained bands are characteristic of Cu(II) metal centres with a square-planar geometry,32,33 while those for 2 could suggest a square pyramidal geometry.34,35 All these bands are due to the spin allowed intrametal (d–d) transition of the copper(II) center and are assigned to the envelope of the transitions from 2B1g (ground state to the excited 2A1g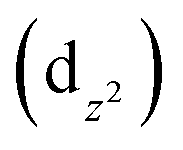 ), 2B2g (dxy) and 2Eg (dxz, dyz) states.
), 2B2g (dxy) and 2Eg (dxz, dyz) states.
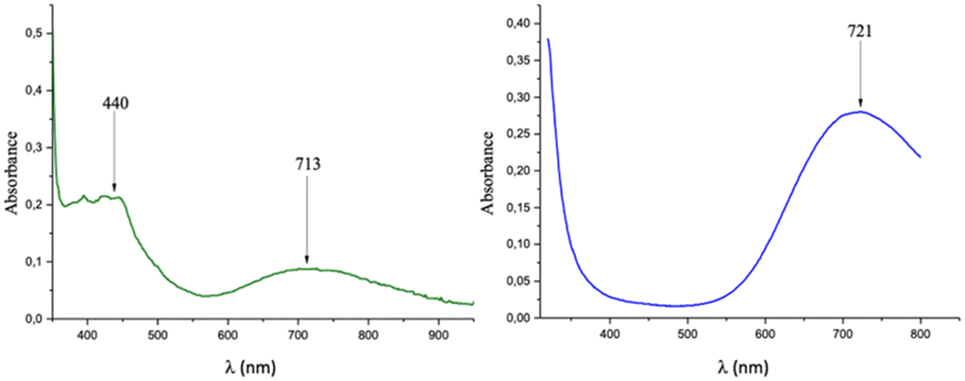 | ||
| Fig. 5 UV-vis spectra of 1 (left) and 2 (right). | ||
3.5 PXRD analysis and SEM
The powder X-ray diffraction (PXRD) pattern was used to verify the crystallinity and phase purity of 1 and 2. The experimental and the simulated powder XRD patterns are presented in Fig. S4.† The experimental diffraction peak patterns of each complex are well defined, which demonstrates the crystallization of both complexes. We can also observe that the diffraction peaks of both experimental and simulated patterns match well in the key positions besides some intensity differences, which indicates a good phase purity for both compound and down to 100 K for 1.The surface morphologies of the as-prepared samples were investigated by scanning electron microscopy (SEM) and are shown in Fig. S9,† also indicating phase homogeneity. It can be noted that 1 is formed as rod-like crystals with numerous leaves. In contrast to 1, the surface morphology of 2 appears as a block with a lot of fractures. Energy-dispersive X-ray spectroscopy (EDS) analysis was also carried out to detect the composition of the as-fabricated compounds, as shown in Fig. S5.† Cu, O, N and C were all detected, which are in agreement with the crystal structure analysis.
3.6 Thermogravimetric analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) for compounds 1 and 2 were carried out in a flowing air atmosphere with a heating rate of 10 °C min−1 from room temperature to 900 °C (Fig. S6†). Each of these compounds depicts no mass loss until 180 °C, thus confirming the absence of crystallization water molecules. Thereafter, both of them exhibit two steps of mass loss in the temperature range of ∼180–270 °C and ∼270–500 °C respectively. The first mass loss of 1, supported by an endothermic effect, corresponds to the removal of the two 2-amino-3-hydroxypyridine moieties 38% (calc. 40%). The second process, which is also associated with endothermic effect, proceeds with a 32% (calc. 35%) and involves the formation of a CuO residue (Fig. S8a†).In contrast to the case of 1, the first mass loss of 78% (calc. 81%) in 2 immediately involves the formation of the CuO residue. It deserves to be noted that during the first stage, an unstable product was formed at 195 °C, which was suddenly decomposed to yield the residue. The formation of this product is associated with the exothermic effect, and that of the residue is with the endothermic effect. After that, 2 shows a little mass loss 2% (calc. 3%), supported by an endothermic effect, which can be due to the release of CO2 (Fig. S8b†).
To confirm the nature of the residue obtained in each compound, they were characterized by X-ray powder diffraction analysis. The powder XRD patterns for both residues were in agreement with the reported pattern of CuO (Fig. S8†).
3.7 Magnetic properties
The temperature dependence of the χT product (χ is the magnetic susceptibility) of 1 and 2 is shown in Fig. 6a and b, respectively. The field dependence of the magnetization per formula unit M1 = Mmol/NAμB at a constant temperature is shown in Fig. 10c and 10d for 1 and 2, respectively.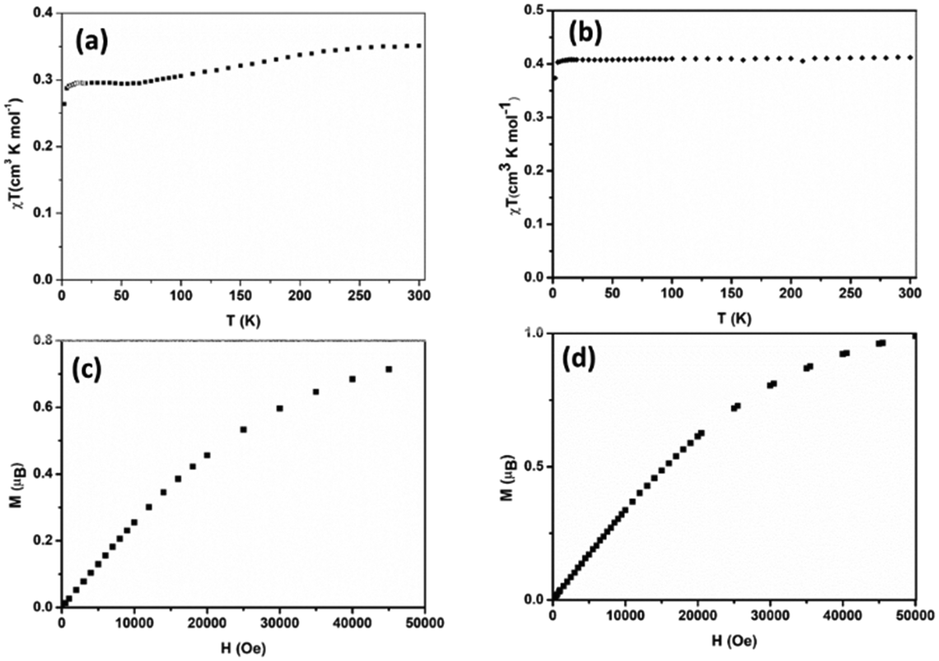 | ||
| Fig. 6 Temperature dependence of the χT product (top) and field dependence of the magnetization (bottom) of 1 (a and c) and 2 (b and d). | ||
For complex 1, the value of χT at room temperature is 0.36 cm3 K mol−1, which perfectly matches the calculated value of one isolated Cu2+ ion (0.36 cm3 K mol−1, S = 1/2). Upon cooling, χT decreases slowly down to 65 K where it reaches almost a plateau (0.30 cm3 K mol−1) down to 10 K. Below this temperature, χT decreases again. The decrease of χT in the high temperature regime together with the plateau could suggest a system involving an odd number of antiferromagnetically coupled spin S = 1/2. However, it was not possible to fit the experimental data with such systems. Furthermore, this is not realistic due to the crystal structure as described above. It shows a network of hydrogen bonds that link the (C5H7N2O)2[Cu(C2O4)2] entities in the form of a 1D chain but are in no way compatible with an odd spin system. Fitting the experimental by considering a 1D chain of S = 1/2 was also unsuccessful. In any case, in this compound, the intermolecular bonding network consisting mainly of hydrogen bonds, even reinforced by the π–π stacking between the pyridine rings, is not expected to transmit such strong magnetic exchange interactions.36 A possible explanation may be a phase transition occurring below 65 K, which is below the reach of our X-ray diffraction setups.
For complex 2, the value of χT is 0.4 cm3 K mol−1 at room temperature, a value which is close to the value expected for one isolated Cu(II) (0.36 cm3 K mol−1). Upon cooling, χT remains constant for almost all temperature domain and decreases only below 10 K. This feature indicates that Cu(II) are almost magnetically independent of the occurrence of very weak antiferromagnetic interactions between the Cu(II) centers operating only below 10 K. This is in agreement with axial coordination and the bridge angle (Cu–O–Cu) of 110.81° of the single oxygen-bridged Cu(II) ions within one-dimensional chain. This does not allow any overlap with the 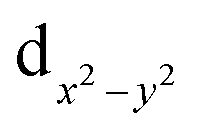 magnetic orbital and does not promote any strong magnetic exchange interaction between the Cu(II) centers.37 It has also been demonstrated by Oshio and Nagashima38 through theoretical calculations on 1D chains of copper(II) complexes using the Heisenberg linear chain model, showing that the weak antiferromagnetic interaction in the chain is due to the long axial Cu–O bond length (ca. 2.54 Å), which causes a small induction of the spin on the
magnetic orbital and does not promote any strong magnetic exchange interaction between the Cu(II) centers.37 It has also been demonstrated by Oshio and Nagashima38 through theoretical calculations on 1D chains of copper(II) complexes using the Heisenberg linear chain model, showing that the weak antiferromagnetic interaction in the chain is due to the long axial Cu–O bond length (ca. 2.54 Å), which causes a small induction of the spin on the 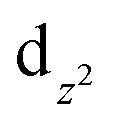 orbital of the nearby complex. Therefore, the weak antiferromagnetism observed is in agreement with the value of the axial Cu–O bond length, 2.454(2) Å, in this compound.
orbital of the nearby complex. Therefore, the weak antiferromagnetism observed is in agreement with the value of the axial Cu–O bond length, 2.454(2) Å, in this compound.
In order to confirm the magnetic coupling observed in 1 and 2, the magnetic field dependence of the magnetization M(μB) (Fig. 6c and d) has been measured. From the obtained curve, it is well observed that for both compounds, the magnetization M has reached B = 5 T, a saturation plateau. It is important to recall at this stage that it is well known for Cu(II) complexes with ground state S = 1/2 that the magnetization M(μB) should be saturated to the value of Msat = 1.0 μB. In the case of 1, the magnetization (M = 0.73 μB at B = 5 T) was saturated below the value expected for one isolated Cu(II) (Msat = 1.0 μB) (Fig. 6c). This indicates rather sizable (negative) J-values and may be due to the paramagnetic impurities, which could have an influence on the interaction between the paramagnetic centers.36 In contrast to what has been observed in 1, the magnetization in 2 was saturated to the value expected for one isolated Cu(II) (Fig. 6d).
4. Conclusions
We report two pyridinium copper(II) oxalate organic–inorganic salts, namely, (C5H7N2O)2[Cu(C2O4)2] 1 and C13H16N2[Cu(C2O4)2] 2, exhibiting differences in i) the coordination mode of oxalate ligands, ii) the coordination geometry around Cu(II) and iii) the supramolecular association (C5H7N2O = 2-amino-3-hydroxypyridinium; C13H16N2 = 4,4′-trimethylenedipyridinum). One remarkable feature of 2 is that the oxalate(2-) ligand has adopted the unusual μ-oxalato-κ3O1,O2:O1′ coordination mode. Moreover, in 2 [Cu(C2O4)2]2− anions polymerize via the axial Cu–O bond, leading to a uniform zigzag Cu(II) chain with a Cu⋯Cu separation of 5.32 Å, whereas in 1 they do not polymerize. These differences between 1 and 2 reveal that the cationic species play key roles in tuning the architectures of the target compounds. Regarding the thermal stability of these compounds, both were found to be stable up to 180 °C. Magnetic studies reveal the weak antiferromagnetic interactions in compounds 1 and 2. This work not only expands the family of copper(II) oxalate salts involving pyridinium type counterions, but also further provides insight into the influence of these cations on the crystal architecture. However, existing correlations which could make the structural architecture of such salts controllable are to be discovered yet.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CLDF, LÖ and JN are thankful to the Swedish Research Council for funding, and the Berkeley Global Science Institute (BGSI) for a supporting network. AKI acknowledges support from the Swedish Foundation for Strategic Research (SSF). We gratefully acknowledge Mr Erik Svensson Grape (Stockholm University) for the SEM analysis, Mr David Kiefer (Chalmers University of Technology) for the TGA measurements and Chalmers Materials Analysis Laboratory, CMAL, and the Olle Enqvist Foundation for funding the single crystal diffractometer.Notes and references
- L. Öhrström and J. Covès, The Rhubarb Connection and Other Revelations: The Everyday World of Metal Ions, https://pubs.rsc.org/en/content/ebook/978-1-78801-094-8, Royal Society of Chemistry, London, 2018.
- C. L. F. Dazem, F. M. A. Noa, J. Nenwa and L. R. Öhrström, CrystEngComm, 2019, 21, 6156–6164 RSC
.
- J. Y. Lee, M. G. Hur, Y. B. Kong, E. J. Lee, S. D. Yang, P. S. Choi and J. H. Park, J. Radioanal. Nucl. Chem., 2021, 330, 455 CrossRef CAS
- H.-M. Li, G.-M. Zhong, S.-Q. Wu, O. Sato, X.-Y. Zheng, Z.-S. Yao and J. Tao, Inorg. Chem., 2021, 60, 8042–8048 CrossRef CAS PubMed
- Z.-L. Xie, L. Deng, C. Yuan, W.-Z. Weng and Z.-H. Zhou, Inorg. Chem. Commun., 2021, 129, 108661 CrossRef CAS
- N. Marino, M. L. Calatayud, M. Orts-Arroyo, A. Pascual-Álvarez, N. Moliner, M. Julve, F. Lloret, G. De Munno, R. Ruiz-García and I. Castro, Magnetochemistry, 2021, 7, 65 CrossRef CAS
- L. Wang, W. Wang, D. Guo, A. Zhang, Y. Song, Y. Zhang and K. Huang, CrystEngComm, 2014, 16, 5437–5449 RSC
- Y. Y. Sun, Y. X. Zong, H. R. Ma, A. Zhang, K. Liu, D. B. Wang, W. Q. Wang and L. Wang, J. Solid State Chem., 2016, 237, 225–233 CrossRef CAS
- C. J. Kepert, Chem. Commun., 2006, 695–700, 10.1039/B515713G
- X. Chen, S. Han and R. Wang, CrystEngComm, 2012, 14, 6400–6403 RSC
- M.-H. Zeng, W.-X. Zhang, X.-Z. Sun and X.-M. Chen, Angew. Chem., Int. Ed., 2005, 44, 3079–3082 CrossRef CAS PubMed
- O. Castillo, A. Luque, P. Román, F. Lloret and M. Julve, Inorg. Chem., 2001, 40, 5526–5535 CrossRef CAS PubMed
- O. Castillo, A. Luque, F. Lloret and P. Román, Inorg. Chim. Acta, 2001, 324, 141–149 CrossRef CAS
- C. F. N. Nguemdzi, F. Capet, J. Ngouné, G. Bebga, M. Foulon and J. Nenwa, J. Coord. Chem., 2018, 71, 1484–1496 CrossRef CAS
-
F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999 Search PubMed
- C. L. F. Dazem, B. N. Ndosiri, E. N. Nfor, R. Köferstein, P. Shankhari, B. P. T. Fokwa and J. Nenwa, J. Mol. Struct., 2020, 1203, 127399 CrossRef CAS
- J. Nenwa, E. D. Djomo, E. N. Nfor, P. L. Djonwouo, M. Mbarki and B. P. T. Fokwa, Polyhedron, 2015, 99, 26–33 CrossRef CAS
- I. N. Kamga, B. N. Ndosiri, A. N. Nana, T. Roman, L. S. Pouamo and J. Nenwa, Polyhedron, 2021, 205, 115291 CrossRef CAS
-
A. Earnshaw, Introduction to Magnetochemistry, Academic Press, London, 2nd edn, 1968 Search PubMed
- M. A. Viswamitra, J. Chem. Phys., 1962, 37, 1408–1414 CrossRef CAS
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- P. A. Gaye, A. D. Sarr, M. Gaye, M. Sanselme and P. V. Agasse, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1046 CrossRef CAS PubMed
- Z.-C. Pan, K.-L. Zhang and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m221 CrossRef CAS PubMed
- Y.-Q. Sun, J. Zhang, J.-L. Chen and G.-Y. Yang, Eur. J. Inorg. Chem., 2004, 2004, 3837–3841 CrossRef
- R. Selvakumar, S. J. Geib, T. Premkumar and S. Govindarajan, J. Therm. Anal. Calorim., 2016, 124, 375–385 CrossRef CAS
- K. Matelková, L. Kucková, A. Mašlejová, J. Moncoľ, V. Jorík and J. Kožíšek, Chem. Pap., 2016, 70, 82–92 Search PubMed
- F. Klongdee, J. Boonmak and S. Youngme, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 300–306 CrossRef CAS PubMed
- W. Li, H.-P. Jia, Z.-F. Ju and J. Zhang, Inorg. Chem. Commun., 2008, 11, 591–594 CrossRef CAS
- K. Muraleedharan and S. Kripa, J. Anal. Appl. Pyrolysis, 2014, 107, 298–305 CrossRef CAS
- Y.-H. Xiao, Z.-P. Deng, Z.-B. Zhu, L.-H. Huo and S. Gao, Dalton Trans., 2017, 46, 16493–16504 RSC
- N. K. Singh, M. K. Bharty, R. Dulare and R. J. Butcher, Polyhedron, 2009, 28, 2443–2449 CrossRef CAS
- W. E. Hatfield and T. S. Piper, Inorg. Chem., 1964, 3, 841–843 CrossRef CAS
- M. Zhou, L. Song, F. Niu, K. Shu and W. Chai, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 463–466 CrossRef CAS PubMed
- S. Roy, P. Mitra and A. K. Patra, Inorg. Chim. Acta, 2011, 370, 247–253 CrossRef CAS
- M. Barwiolek, E. Szlyk, A. Kozakiewicz, T. Muziol, A. Bieńko and J. Jezierska, Dalton Trans., 2018, 47, 13902–13912 RSC
-
D. J. Hodgson, in Prog. Inorg. Chem., 1975, ch. 4, pp. 173–241, DOI:10.1002/9780470166208
- H. Oshio and U. Nagashima, Inorg. Chem., 1992, 31, 3295–3301 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1848472, 1848473, 2217520, and 2217521. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01513g |
| This journal is © The Royal Society of Chemistry 2023 |