
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntitumour imidazotetrazines: past, present… and future?
Malcolm F. G.
Stevens
*a and
Richard T.
Wheelhouse
 *b
*b
aBiodiscovery Institute, School of Pharmacy, University of Nottingham, NG7 2RD, UK
bInstitute of Cancer Therapeutics, School of Pharmacy and Medical Sciences, University of Bradford, BD7 1DP, UK. E-mail: r.t.wheelhouse@bradford.ac.uk
First published on 29th August 2023
Abstract
It is 40 years since the publication of the patent that announced the imidazotetrazines temozolomide and mitozolomide to the world and 30 since the discovery that they function as prodrugs of alkyldiazonium reactive intermediates. Temozolomide combined with radiation is established as the first-line treatment for glioma but despite the attentions of the inventors and others, further examples of this intriguing ring system have yet to enter the clinic.
 Malcolm F. G. Stevens | Malcolm F. G. Stevens is Emeritus Professor at the University of Nottingham. So far, 5 new molecules from his laboratory have entered clinical trials. His achievements have been recognised by awards including: Royal Society of Chemistry (RSC) Interdisciplinary Research Award (1991); RSC George and Christine Sosnovsky Award for chemical contributions to cancer research (2002); British Association of Cancer Research Tom Connors Award Lecture (2002) and the American Association of Cancer Research Bruce F. Cain Memorial Award (2003). He was appointed OBE in the 1999 New Year Honours and elected Fellow of the Royal Society in 2009. |
 Richard T. Wheelhouse | Richard T. Wheelhouse is Reader in Medicinal Chemistry at the University of Bradford. A graduate of the University of Leicester, he spent 6 years in his early career in the Cancer Research Campaign Experimental Cancer Chemotherapy Research Group, led by Malcolm Stevens, at Aston and Nottingham Universities. He discovered the intermediacy of alkyldiazonium ions in the prodrug activation of the imidazotetrazines and established the ground rules that enabled the design of the first new, biologically-active 3-substituents. Work in his lab has focused on alkyldiazonium ions as aziridinium ion precursors and defining substituent effects on the kinetics of imidazotetrazine prodrug activation. |
There were no specific biological insights that motivated the synthesis of imidazotetrazines: rather a desire to conduct inquisitive Chemistry on nitrogen-rich molecules. The first examples were synthesised in 1979 by research student Robert Stone at Aston University in Birmingham UK. The Stone Synthesis involved the interaction of 5-diazoimidazole-4-carboxamide (Diazo-IC), prepared efficiently and safely from 5-aminoimidazole-4-carboxamide (AIC), and an isocyanate in a mixed solvent system (Fig. 1A). The original synthesis, which is notable for being an exemplar of atom efficiency, was limited by the availability of commercially available isocyanates at the time; substrates available included 2-chloroethyl isocyanate and a range of aryl isocyanates. Sponsorship by May & Baker, Dagenham UK, led by Dr Eddy Lunt, resulted in further isocyanates becoming available, including methyl isocyanate. Stone's lab book dated 29th April, 1980 (Fig. 1B) records the first synthesis of 8-carbamoyl-3-methylimidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one, which subsequently became known to medicine as temozolomide (1).
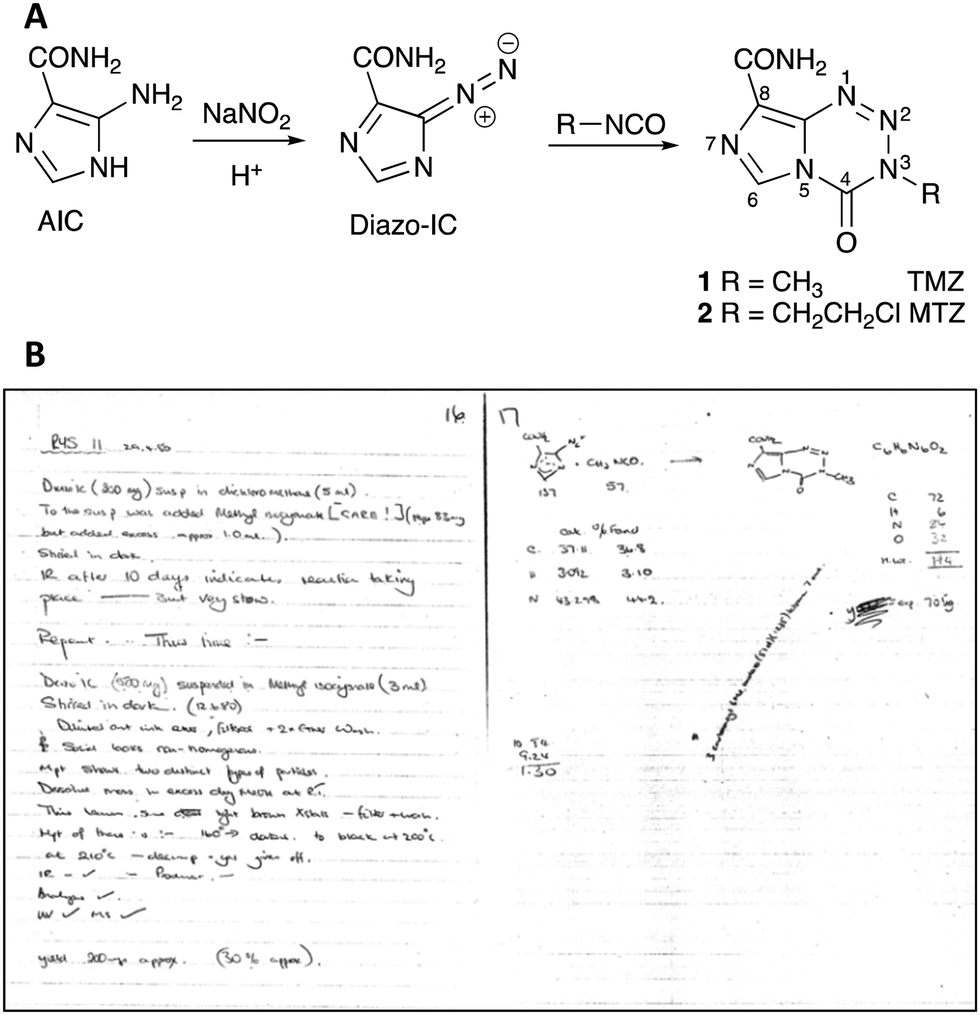 | ||
| Fig. 1 (A) Scheme for the synthesis of temozolomide 1 and mitozolomide 2; (B) entry for 29th April, 1980 in Robert Stone's laboratory book. | ||
U.S. Patent 5260291 (1983) revealed the structures of the first cohort of imidazotetrazines, a new ring-system, and taught that 1 and the 3-(2-chloroethyl)-derivative, subsequently known as mitozolomide (2, MTZ), were of “particular importance.” The initial publication on the synthesis and antitumour properties of imidazotetrazines appeared in 1984.1 It soon became apparent that MTZ and temozolomide were prodrugs, being converted in vivo to triazenes (3, MTIC) and 4, respectively that, in turn, generated electrophilic cations,2Scheme 1.
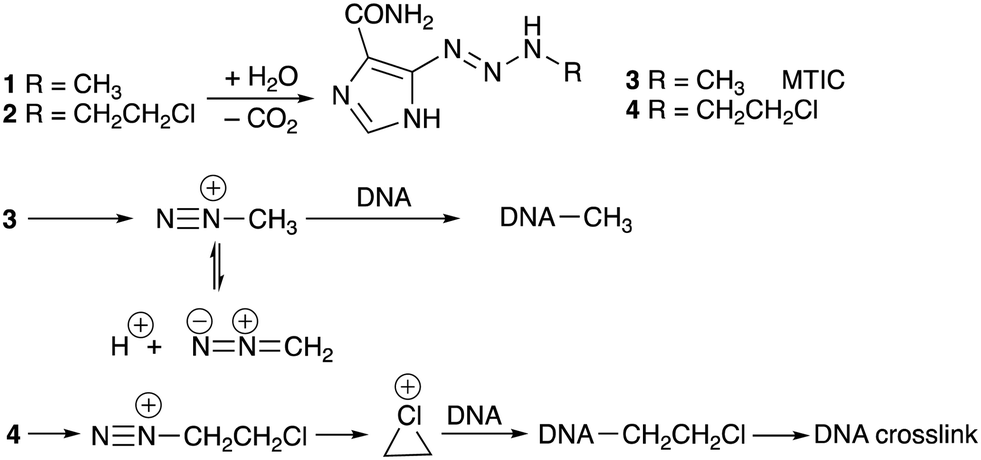 | ||
| Scheme 1 Conversion of temozolomide (1) and mitozolomide (2) to triazenes and alkyl diazonium ions and their subsequent reactions with DNA. | ||
The first compound studied in detail was MTZ because of its spectacular experimental antitumor activity. In several cases curative activity was recorded from a single dose of agent against a range of mouse tumour models employed in the 1970s.1,3 Initial reports of the effects of MTZ on the DNA of mouse L1210 cells suggested that the drug elicited DNA cross-links from interaction of reactive species generated from triazene 4. In this respect MTZ behaved like a 2-chloroethylnitrosourea.4 More detailed examination of the cross-linking process in many laboratories subsequently identified the eventual formation of an etheno linkage between a guanine residue on one strand and a cytosine N(3) locus on an adfacent DNA strand (Scheme 2). It is interesting to note in retrospect that telomeres present a substantial region of the molecular target of the imidazotetrazines – G-rich DNA sequences. Surviving 'cured' mice died prematurely with shrunken bodies and white fur, possibly indicative of some effect against telomere function.
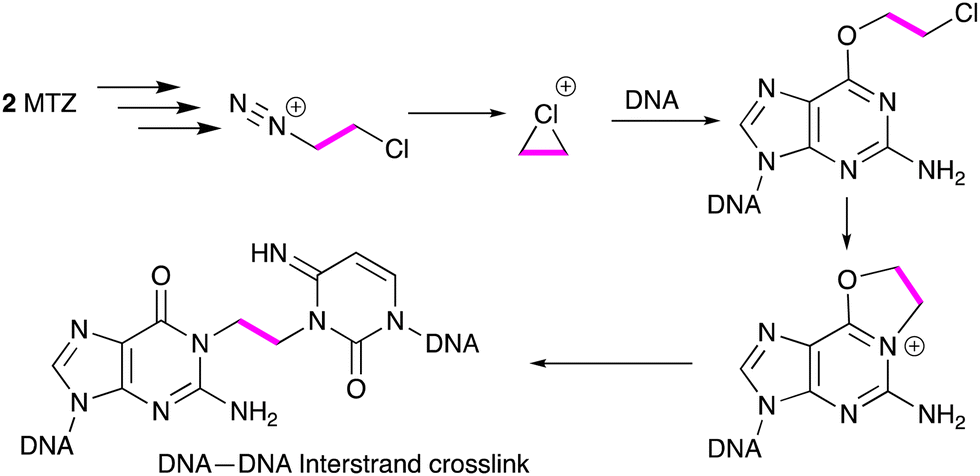 | ||
| Scheme 2 Crosslinking of DNA by MTZ. | ||
MTZ was fast-tracked into clinical trial at hospitals in Birmingham and London, UK in 1983. Two operational problems were encountered with the drug formulated for i.v. injection in DMSO. Patients excreted unchanged and (presumably) mutagenic drug in urine and for safety reasons this was conveniently oxidised in bedpans by bleach to the corresponding non-toxic carboxylic acid. Complaints from nursing staff that patients excreted nauseous methyl mercaptan (a metabolite of DMSO) through their lungs and skin were overcome by positioning beds adjacent to open windows and blowing a current of air across the patient with a fan!
Despite the high hopes of inventors that they had discovered ‘the magic bullet’, severe unpredictable and irreversible thrombocytopenia was elicited by the drug in Phase 1 evaluation.5 One study even concluded that the drug was ineffective and its use “dangerous.”6 These reports spelt the end of MTZ and, despite efforts to identify a more tolerable 8-substituted 3-(2-choroethyl)imidazotetrazine,7 further work on this structural type was reluctantly abandoned by its sponsor (May & Baker) in 1985. To anti-cancer drug discoverers, there was a clear lesson: activity against mouse tumour models was irrelevant as a filter to select molecules for clinical use.
Academic researchers are stubborn types and even when their favourite projects are clearly doomed, find some excuse to persist with them. Thus, from a box of assorted imidazotetrazines a sample of CCRG 81045, M&B 39831, NSC 362856, methazolastone, malkazole (aka temozolomide 1) was selected for further study. As a prodrug of a mono-functional methylating agent, temozolomide could not, unlike the forlorn mitozolomide, act as a DNA cross-linking agent and therefore would be predicted to have a better safety profile. The orphaned agent was to have an important role in the treatment of glioblastoma.
In the mid-1980s Cancer Research Campaign UK initiated a novel strategy to select compounds for clinical trial. Essentially candidates had to satisfy four criteria: they had to challenge a biological process in cancer cells not hitherto adequately studied (methylation of DNA); they had to be synthetically accessible; their physical properties should be amenable for the development of a simple pharmaceutical formulation; and physicians should be interested in conducting clinical trials. Temozolomide was adjudged to have fulfilled these criteria.8 Despite several alternative synthetic routes being devised to obviate the use of volatile and toxic methyl isocyanate,9 bulk drug was most efficiently manufactured by the ‘one-pot’ Stone Synthesis. It was stable in acid and suitable for the development of an oral formulation; it was free from metabolic complications; and it proved substantially less toxic to rodents than mitozolomide.10
Temozolomide was introduced into Phase 1 clinical trial in 1987. Two formulations were developed by Pharmacists at Aston University: an oral dosage form consisting of drug plus citric acid in capsules; and ampoules containing drug in DMSO. When published in 199210 the results of the Phase 1 trial attracted wide attention. The study confirmed that the drug was well tolerated over a 5 day schedule and the oral medication was fully bioavailable. Remarkably, in Phase 1, two complete responses were observed in mycosis fungoides, and several partial responses in melanoma and glioblastoma. Subsequent Phase 2 studies focussed on confirming the observed activity against glioma tumours.
Over a period of 20 years a large inventory of work, including syntheses of a range of imidazotetrazine analogues (including temozolomide labelled with 2H, 11C, 13C and 14C in the methyl group and 15N in the tetrazine ring), physical chemistry, pharmacology, pharmaceutical and clinical development, was completed and has been reviewed on multiple occasions (see ref. 5, 11 and 12 and references therein), including a personal account of the discovery and development of temozolomide by one of the authors.13
Several instrumental methods have been employed to clarify aspects of the structure and mechanism of action of temozolomide. An X-ray structure determination showed two distinct rotamers of the carboxamide group in the asymmetric unit. In one case there is a H-bond between the carboxamide NH and N(1) of the tetrazine ring: in the other there is a H-bond between NH and N(7) of the imidazole ring.14
The decomposition of the drug in deuteriated buffer and measuring the fate of the imidazole proton by NMR, showed only two species – the intact drug and AIC indicating that the decomposition of the triazene intermediate is faster than that of the prodrug molecule and that the methyl group is transferred intact to nucleophilic sites in DNA. Methyl group transfer was accompanied by deuterium incorporation from the solvent, an observation that implicated alkyldiazonium ions in the reaction process.2,15
The biological fates of all atoms in the molecule have been determined. N(2) and N(3) atoms in the tetrazine ring are eliminated as dinitrogen gas; the carbonyl fragment at C(4) is liberated as CO2; the imidazole carboxamide moiety and N(1) of the tetrazine ring are excreted as 5-aminoimidazole-4-carboxamide (Fig. 2). All these sub-components combined act as a delivery system to transfer a methyl group to nucleophilic sites on DNA, especially O(6) residues of guanine bases in runs of guanines.16
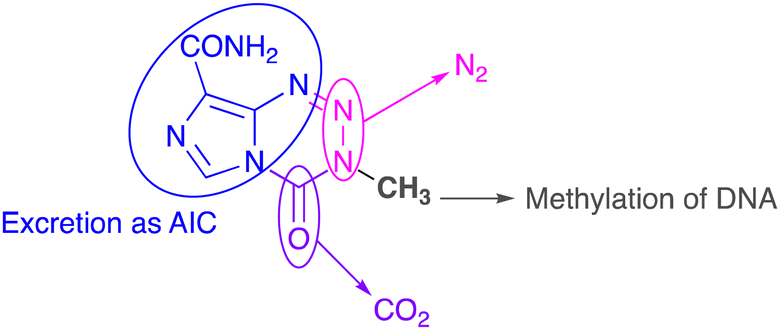 | ||
| Fig. 2 Metabolic fates of all atoms in the temozolomide molecule. | ||
Radiosynthesis of the 11C-Me variant of temozolomide17 allowed the metabolic activation of temozolomide to be studied in vivo in a patient bearing a glioma tumour by positron emission tomography (PET). This work enabled generation of remarkable images of the 11C label located in tumour, Fig. 3, and triggered interest in PET during pre-clinical drug development programmes.
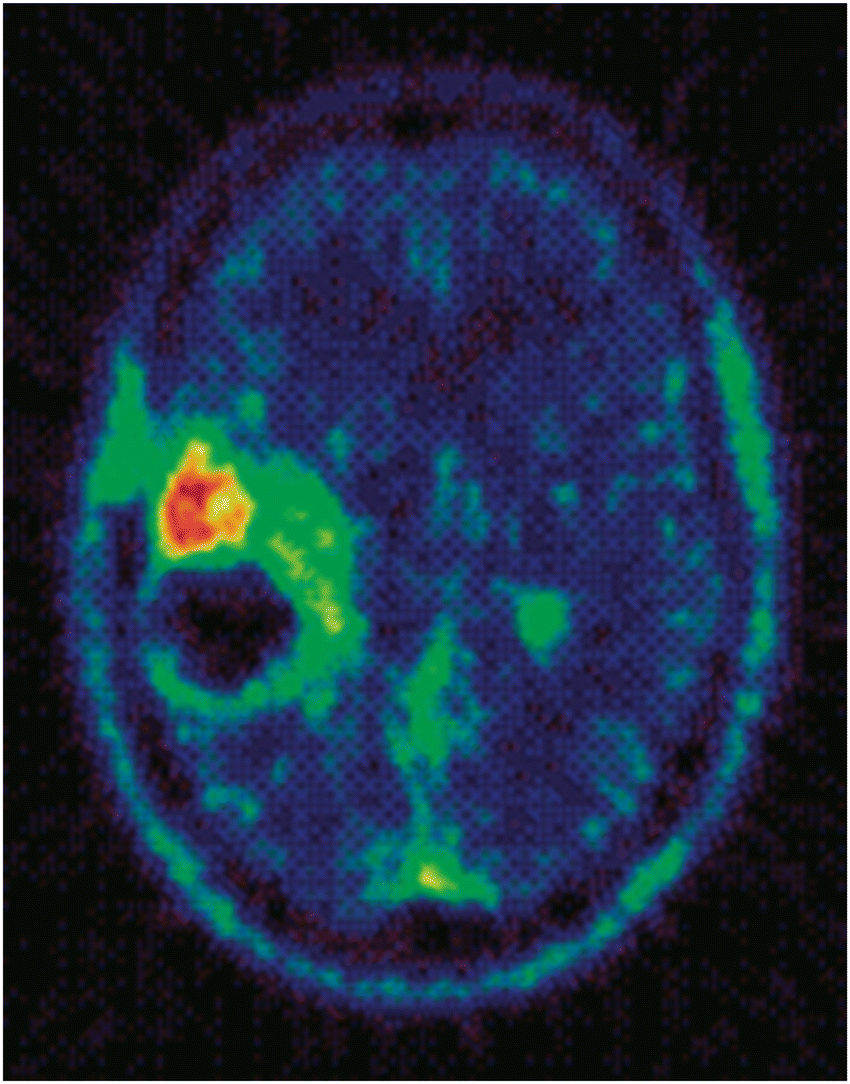 | ||
| Fig. 3 PET image of mean radioactivity concentration during the first 90 min after injection of [N3-11C-methyl]-temozolomide (the image represents a transverse section of brain with a right parietal lesion). Reprinted with permission from Brown et al, J. Med. Chem., 2002, 45, 5448–5457. Copyright 2002 American Chemical Society. | ||
Even before temozolomide entered Phase 1 trial it was anticipated that resistance to the drug in glioma tumours might be mediated through the agency of O6-methylguanine-DNA methyltransferase (MGMT), a protein whose reactive thiolate group will remove the methyl residue on guanine bases by an SN2 reaction (Scheme 3). Hegi and her colleagues18,19 showed that patients with tumours where the MGMT gene was silenced by methylation within its promoter sequence experienced more robust responses to temozolomide than those bearing activated MGMT and hence levels of MGMT that compromised the robustness of response. Other mechanisms of resistance, both constitutive and acquired, including DNA mismatch repair (MMR) status of the tumour, have been identified.20,21
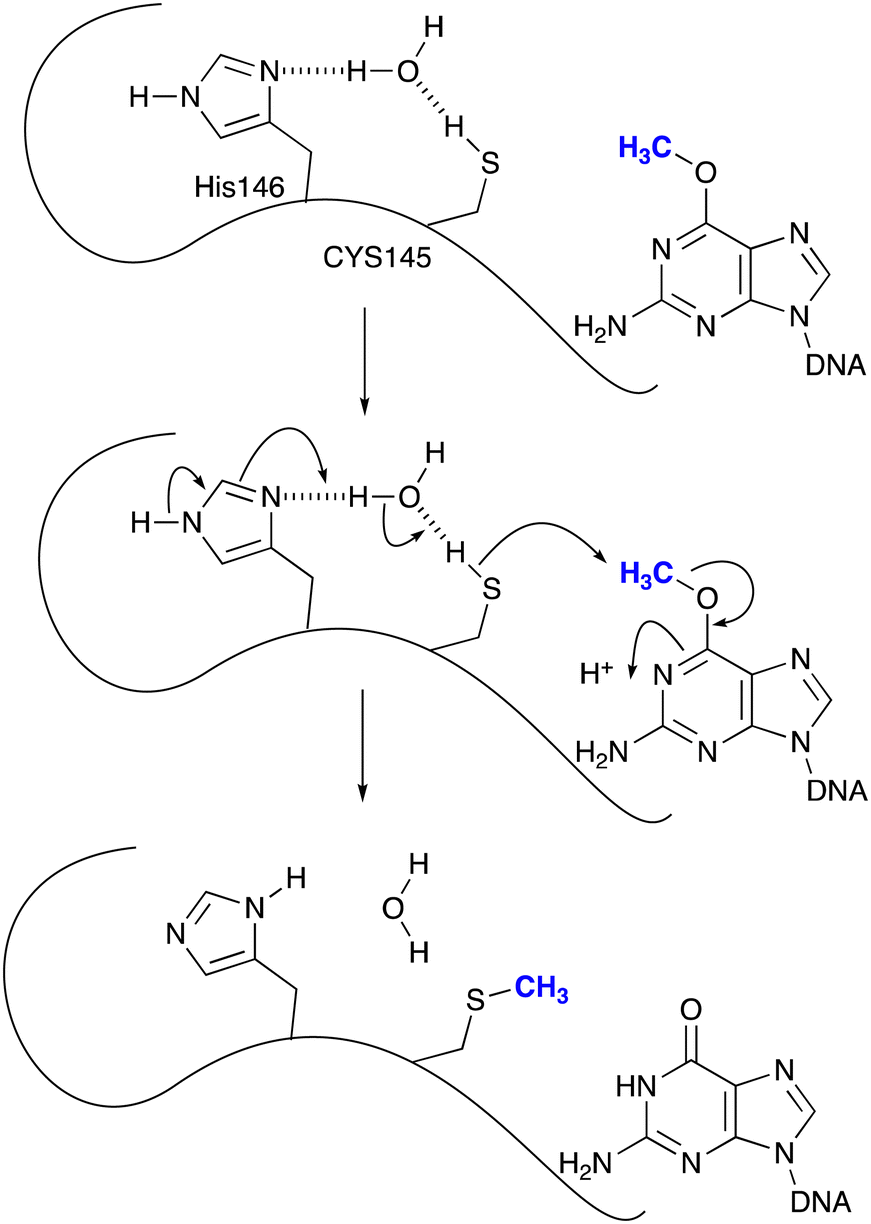 | ||
| Scheme 3 Repair of O(6)-methylguanines by MGMT. | ||
Temozolomide was licensed to Schering-Plough Corporation in 1993 by Cancer Research Campaign UK and marketed, firstly in the US in 1999 (as Temodar), followed thereafter worldwide (as Temodal). It first achieved ‘block buster’ status (annual sales > $1 billion) in 2008 and is still the agent of choice, combined with radiotherapy, for the treatment of glioma, 45 years after the initiation of the imidazotetrazines project. Temozolomide, of molecular weight only 194 Daltons (Da), occupies a rewarding niche in the cancer therapeutics market. As only the methyl group (15 Da) is responsible for biological activity, one might argue that the drug is the most lucrative anticancer agent in terms of dollars earned per Da of working molecular mass. There have been considerable efforts since 2000 to develop a ‘neo-temozolomide’ that might overcome the resistance inevitably mounted to the original drug by glioma tumours. Essentially the question to be answered is a simple one: is it possible to develop a monofunctional agent which elicits a cytotoxic O(6) lesion on DNA that cannot be repaired by MGMT and is not compromised by other resistance mechanisms? Because of the mitozolomide clinical tragedy, it was deemed undesirable to consider DNA cross-linking agents. Despite this imperative, a recent publication in Science claims excellent pre-clinical properties for the 3-(2-fluoroethyl)-imidazotetrazine analogue of mitozolomide.22 This publication is flawed on two counts: because the fluoro compound is a ‘mee-too’ of MTZ rather than temozolomide, the former agent should have been used as a reference compound in the thorough biological studies conducted; and secondly, and unaccountably, no reference was made to the bone marrow toxicity encountered in the Phase 1 study of mitozolomide.6
Only two sites on the imidazotetrazine scaffold are readily amenable to modification: N(3) and C(8) (Fig. 4) and many variants at these two positions have been synthesised.23,24 Understanding the significance of alkyldiazonium ions allowed the first rationalisation of the inactivity of the many early 3-substituent variants on MTZ and temozolomide and established ground rules for the rational design of active new agents. The scope for competing side reactions of released alkyldiazonium ions, for example elimination to alkenes,25 had to be controlled and some form of stabilisation mechanism was needed to ensure the reactive intermediates once free, have an adequate aqueous lifetime in which to locate their DNA target. This insight enabled the rational design of, inter alia, the 3-aminoethyl26,27 and 3-propargyl28 substituted imidazotetrazines.
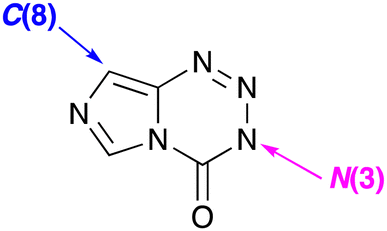 | ||
| Fig. 4 Accessible sites of modification of the imidazotetrazine scaffold. | ||
Unlike the mouse tumours used in anti-cancer drug discovery in the 1980s, molecularly-characterised, paired human glioma cell lines (MGMT+ and MGMT− and companion MMR lines) were available as screening tools in the search for a new agent. Of the large range of alkyl variations at the N(3) position synthesised28 the 3-propargyl-imidazotetrazine 5 was particularly active against a range of tumour cell lines in vitro that are resistant to temozolomide.29–32 This agent could be synthesised from diazo-IC and propargyl isocyanate28 or from nor-temozolomide 6 and propargyl iodide.33 Replacing the 8-substituent by a thiazol-2-yl group, a carboxamide surrogate, afforded an agent 7 with modified DMPK properties when compared with 1 and 5 and could be considered a new clinical candidate from the imidazotetrazine stable (Scheme 4).34 Recent research by the Nottingham group has explored encapsulation of temozolomide and propargyl analogues in apoferritin nanocages as a strategy to improve delivery of drugs to brain tumours.35
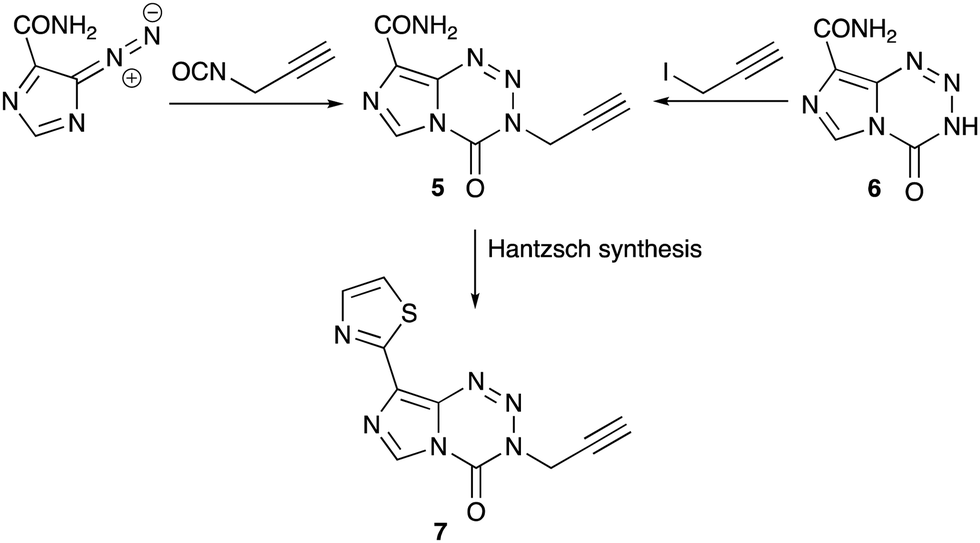 | ||
| Scheme 4 Synthesis of 3-propargyl-imidazotetrazines. | ||
Conclusions
What does the future hold? In the early 1980s it was possible for a small academic group of pharmacists to discover and develop two imidazotetrazines which underwent clinical trial under the aegis of a small charity – Cancer Research Campaign: one of them (temozolomide) was a major commercial success, albeit of limited efficacy in the clinic. However, new regulations require expensive investment to bridge the chasm between bench and clinic that is unaffordable to small companies. Big Pharma is focussing its investment in biologics to address the brain cancer problem but their efforts are thwarted by the blood–brain-barrier that denies large molecules access to the brain. Perversely, there appears to be little current interest in small molecules, despite the clear potential of compounds such as 7.Recent communications to national newspapers have highlighted the paucity of funding for brain cancer research and reasonably criticised the drug development community for the lack of progress against the diseases and the reliance on a drug first synthesised over 40 years ago. Unless Big Pharma changes its priorities to re-embrace small molecules, temozolomide may still be the drug of choice against glioma in another 40 years.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. F. G. Stevens, J. A. Hickman, R. Stone, N. W. Gibson, G. U. Baig, E. Lunt and C. G. Newton, J. Med. Chem., 1984, 27, 196–201 CrossRef CAS PubMed.
- R. T. Wheelhouse and M. F. G. Stevens, J. Chem. Soc., Chem. Commun., 1993, 1177–1179 RSC.
- J. A. Hickman, M. F. G. Stevens, N. W. Gibson, S. P. Langdon, C. Fizames, F. Lavelle, G. Atassi, E. Lunt and R. M. Tilson, Cancer Res., 1985, 45, 3008–3013 CAS.
- N. W. Gibson, L. C. Erickson and J. A. Hickman, Cancer Res., 1984, 44, 1767–1771 CAS.
- E. S. Newlands, M. F. G. Stevens, S. R. Wedge, R. T. Wheelhouse and C. Brock, Cancer Treat. Rev., 1997, 23, 35–61 CrossRef CAS PubMed.
- J. H. Schornagel, G. Simonetti, R. Dubbelman, W. W. ten Bokkel Huinink and J. G. McVie, Cancer Chemother. Pharmacol., 1990, 26, 237–238 CrossRef CAS PubMed.
- K. R. Horspool, M. F. G. Stevens, C. G. Newton, E. Lunt, R. J. A. Walsh, B. L. Pedgrift, G. U. Baig, F. Lavelle and C. Fizames, J. Med. Chem., 1990, 33, 1393–1399 CrossRef CAS PubMed.
- M. F. G. Stevens, J. A. Hickman, S. P. Langdon, D. Chubb, L. Vickers, R. Stone, G. Baig, C. Goddard, N. W. Gibson, J. A. Slack, C. Newton, E. Lunt, C. Fizames and F. Lavelle, Cancer Res., 1987, 47, 5846–5852 CAS.
- Y. Wang, M. F. G. Stevens, T. M. Chan, D. DiBenedetto, Z. X. Ding, D. Gala, D. Hou, M. Kugelman, W. Leong and S. C. Kuo, J. Org. Chem., 1997, 62, 7288–7294 CrossRef CAS PubMed.
- E. S. Newlands, G. R. P. Blackledge, J. A. Slack, G. J. S. Rustin, D. B. Smith, N. S. A. Stuart, C. P. Quarterman, R. Hoffman, M. F. G. Stevens, M. H. Brampton and A. C. Gibson, Br. J. Cancer, 1992, 65, 287–291 CrossRef CAS PubMed.
- C. L. Moody and R. T. Wheelhouse, Pharmaceuticals, 2014, 7, 797–838 CrossRef CAS.
- M. F. G. Stevens, Cancer Drug Design and Discovery, Elsevier Science, San Diego, 2nd edn, 2014, pp. 145–164 Search PubMed.
- M. F. G. Stevens, The Kestrels of Gosta Green, Carnegie Book Production, Lancaster, UK, 2018 Search PubMed.
- P. R. Lowe, C. E. Sansom, C. H. Schwalbe, M. F. G. Stevens and A. S. Clark, J. Med. Chem., 1992, 35, 3377–3382 CrossRef CAS.
- B. J. Denny, R. T. Wheelhouse, M. F. G. Stevens and L. L. H. Tsang, Biochemistry, 1994, 33, 9045–9051 CrossRef CAS PubMed.
- A. S. Clark, B. Deans, M. F. G. Stevens, M. J. Tisdale, R. T. Wheelhouse, B. J. Denny and J. A. Hartley, J. Med. Chem., 1995, 38, 1493–1504 CrossRef CAS PubMed.
- G. D. Brown, S. K. Luthra, C. S. Brock, M. F. G. Stevens, P. M. Price and F. Brady, J. Med. Chem., 2002, 45, 5448–5457 CrossRef CAS PubMed.
- M. E. Hegi, A. C. Diserens, S. Godard, P. Y. Dietrich, L. Regli, S. Ostermann, P. Otten, G. Van Melle, N. de Tribolet and R. Stupp, Clin. Cancer Res., 2004, 10, 1871–1874 CrossRef CAS PubMed.
- M. E. Hegi, A. Diserens, T. Gorlia, M. Hamou, N. de Tribolet, M. Weller, J. M. Kros, J. A. Hainfellner, W. Mason, L. Mariani, J. E. C. Bromberg, P. Hau, R. O. Mirimanoff, J. G. Cairncross, R. C. Janzer and R. Stupp, N. Engl. J. Med., 2005, 352, 997–1003 CrossRef CAS PubMed.
- J. Zhang, M. F. G. Stevens and T. D. Bradshaw, Curr. Mol. Pharmacol., 2012, 5, 102–114 CrossRef CAS PubMed.
- J. J. Zhang, M. F. G. Stevens, C. A. Laughton, S. Madhusudan and T. D. Bradshaw, Clin. Transl. Res., 2010, 78, 103–114 CAS.
- K. Lin, S. E. Gueble, R. K. Sundaram, E. D. Huseman, R. S. Bindra and S. B. Herzon, Science, 2022, 377, 502–511 CrossRef CAS PubMed.
- M. C. Hummersone and D. Cousin, WO 2011/107726 A1, 2011.
- M. G. Hummersone, M. F. G. Stevens and D. Cousin, US Pat., 9024018 B2, 2015 Search PubMed.
- Y. Wang, R. T. Wheelhouse, L. Zhao, D. A. F. Langnel and M. F. G. Stevens, J. Chem. Soc., Perkin Trans. 1, 1998, 1669–1676 RSC.
- E. A. E. Garelnabi, D. Pletsas, L. Li, K. Kiakos, N. Karodia, J. A. Hartley, R. M. Phillips and R. T. Wheelhouse, ACS Med. Chem. Lett., 2012, 3, 965–968 CrossRef CAS PubMed.
- D. Pletsas, E. A. E. Garelnabi, L. Li, R. M. Phillips and R. T. Wheelhouse, J. Med. Chem., 2013, 56, 7120–7132 CrossRef CAS PubMed.
- D. Cousin, J. H. Zhang, M. G. Hummersone, C. S. Matthews, M. Frigerio, T. D. Bradshaw and M. F. G. Stevens, MedChemComm, 2016, 7, 2332–2343 RSC.
- J. H. Zhang, M. F. G. Stevens, M. Hummersone, S. Madhusudan, C. A. Laughton and T. D. Bradshaw, Oncology, 2011, 80, 195–207 CrossRef CAS PubMed.
- R. T. Othman, I. Kimishi, T. D. Bradshaw, L. C. D. Storer, A. Korshunov, S. M. Pfister, R. G. Grundy, I. D. Kerr and B. Coyle, Acta Neuropathol. Commun., 2014, 2, 57 CrossRef PubMed.
- J. H. Zhang, M. Hummersone, C. S. Matthews, M. F. G. Stevens and T. D. Bradshaw, Oncology, 2015, 88, 28–48 CrossRef CAS PubMed.
- R. L. Svec, S. A. McKee, M. R. Berry, A. M. Kelly, T. M. Fan and P. J. Hergenrother, ACS Chem. Biol., 2022, 17, 299–313 CrossRef CAS PubMed.
- D. Cousin, M. F. G. Stevens and M. G. Hummersone, MedChemComm, 2012, 3, 1419–1422 RSC.
- H. S. Summers, W. Lewis, H. E. L. Williams, S. Shnyder, P. Cooper, T. D. Bradshaw, C. J. Moody and M. F. G. Stevens, Eur. J. Med. Chem., 2023, 257, 115507 CrossRef CAS PubMed.
- K. Bouzinab, H. S. Summers, M. F. G. Stevens, C. J. Moody, N. R. Thomas, P. Gershkovich, N. Weston, M. B. Ashford, T. D. Bradshaw and L. Turyanska, ACS Appl. Mater. Interfaces, 2020, 12, 12609–12617 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |