
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResistance is futile: targeting multidrug-resistant bacteria with de novo Cys-rich cyclic polypeptides
Alvaro
Mourenza†
a,
Rajasekaran
Ganesan†
a and
Julio A.
Camarero
 *ab
*ab
aDepartment of Pharmacology and Pharmaceutical Sciences, Alfred E. Mann School of Pharmacy, Los Angeles, CA90033, USA. E-mail: jcamarer@usc.edu; Tel: +1-(323) 442-1417
bNorris Comprehensive Cancer Center, Keck School of Medicine, University of Southern California, Los Angeles, CA90033, USA
First published on 21st August 2023
Abstract
The search for novel antimicrobial agents to combat microbial pathogens is intensifying in response to rapid drug resistance development to current antibiotic therapeutics. The use of disulfide-rich head-to-tail cyclized polypeptides as molecular frameworks for designing a new type of peptide antibiotics is gaining increasing attention among the scientific community and the pharmaceutical industry. The use of macrocyclic peptides, further constrained by the presence of several disulfide bonds, makes these peptide frameworks remarkably more stable to thermal, biological, and chemical degradation showing better activities when compared to their linear analogs. Many of these novel peptide scaffolds have been shown to have a high tolerance to sequence variability in those residues not involved in disulfide bonds, able to cross biological membranes, and efficiently target complex biomolecular interactions. Hence, these unique properties make the use of these scaffolds ideal for many biotechnological applications, including the design of novel peptide antibiotics. This article provides an overview of the new developments in the use of several disulfide-rich cyclic polypeptides, including cyclotides, θ-defensins, and sunflower trypsin inhibitor peptides, among others, in the development of novel antimicrobial peptides against multidrug-resistant bacteria.
Introduction
According to the Centers for Disease Control and Prevention, the six ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanii, Pseudomonas aeruginosa, and Enterobacter species) bacterial species cause around two-thirds of healthcare-associated infections (e.g., pneumonia, septicemia), leading to close to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually in the United States.1 A trait of these emerging difficult-to-treat clinical superbugs is their ability to “escape” the action of multiple traditional antibiotics, in part due to biofilm formation as well as mechanisms of drug resistance. It is estimated that globally, at least 1.2 million people died in 2019 because of bacterial infections caused by multidrug-resistant (MDR) bacterial strains.2 It is expected that the rapid spread of MDR bacterial strains may cause about 10 million deaths per year globally by 2050.2 These terrifying facts seriously highlight the urgent need to develop novel antibiotic therapeutic agents.
000 deaths annually in the United States.1 A trait of these emerging difficult-to-treat clinical superbugs is their ability to “escape” the action of multiple traditional antibiotics, in part due to biofilm formation as well as mechanisms of drug resistance. It is estimated that globally, at least 1.2 million people died in 2019 because of bacterial infections caused by multidrug-resistant (MDR) bacterial strains.2 It is expected that the rapid spread of MDR bacterial strains may cause about 10 million deaths per year globally by 2050.2 These terrifying facts seriously highlight the urgent need to develop novel antibiotic therapeutic agents.
Several approaches have been proposed for the development of novel antimicrobial therapeutics. For example, the use of antibody protein pseudo capsids, probiotics, metal chelation, CRISPR-Cas9, bioengineered toxins, bacteriocins, vaccines and antibodies are also currently being explored to overcome antibiotic resistance (see ref. 3 and references cited there).
Among the different approaches this review will focus on the use of highly constrained peptides, specifically on highly constrained disulfide-rich backbone-cyclized antimicrobial peptides. Antimicrobial peptides are essential host defense molecules found in a wide variety of species and have been proposed as promising antibacterial therapeutic candidates.4 To date, several hundreds of antimicrobial peptides have been identified in a variety of life forms ranging from bacteria, fungi, plants, amphibians, and mammals, including humans.5,6 In mammals, cathelicidins, protegrins, and defensins are the three major types of host defense peptides.7,8
However, the utility of these peptides as antimicrobial therapeutic agents has been typically hampered by their generally poor stability and limited bioavailability.9 To overcome these limitations, the use of highly constrained peptides has recently received special attention, mostly through backbone cyclization, incorporation of non-natural amino acids, and Cys-based disulfide bridges for the design of novel peptide-based antimicrobial agents with better stability.10–13
In this review, we will present recent developments in the use of Cys-rich cyclic polypeptides for designing novel peptide antibiotics. Cyclic Cys-rich peptides are widely distributed natural product polypeptides among different species, including animals and plants (Scheme 1). They have attractive features, including thermal, chemical, and biological stability against proteases. Peptides that will be discussed in this review include the use of cyclotides, mammalian defensins, and sunflower protease inhibitor 1, among others, as molecular frameworks to produce novel peptide-based antimicrobial agents.
 | ||
| Scheme 1 Schematic representation of the structural complexity of naturally occurring disulfide-rich cyclic peptides that are commonly used as molecular scaffolds for the development of novel antibacterial therapeutic leads. Techniques used to improve or introduce antimicrobial activities include the use of SAR introducing charged/hydrophobic or non-natural amino acids, backbone cyclization, molecular grafting, and evolution techniques. As indicated in the test, some of these engineered constructs have shown to exhibit high activity against multi-drug resistant (MDR) pathogenic bacteria. Molecular structures shown on the scheme include θ-defensins (RTD-1, pdb: 1HVZ),156 BBI peptides (sunflower peptide trypsin inhibitor 1, SFTI-1, pdb: 1JBL),157 α-defensins (cryptidin 4, pdb: 2GW9),158 cyclotides (cycloviolacin O2, pdb: 1NBJ),159 and β-hairpin antimicrobial peptides gomesin (pdb: 1KFP)137 and porcine PG-1 (pdb: 1PG1).67 | ||
Defensins
Defensins are Cys-rich peptides containing three disulfide bridges that belong to the β-sheet class of antimicrobial peptides in vertebrates.14 Defensins are small (29 to 42 residues long), mostly cationic and amphipathic peptides showing broad-spectrum and direct and indirect antimicrobial activity against Gram-positive and Gram-negative bacteria, fungi, and viruses.15 Defensins can be divided into three subfamilies: α-, β-, and θ-defensins, depending on the position of the Cys residues forming the corresponding disulfide bridges (Fig. 1 and Table 1).14 While α- and β-defensins are linear polypeptides that are widely distributed in vertebrate species, θ-defensins are backbone cyclized or circular cyclic polypeptides found only in certain non-human primates (Fig. 1).16,17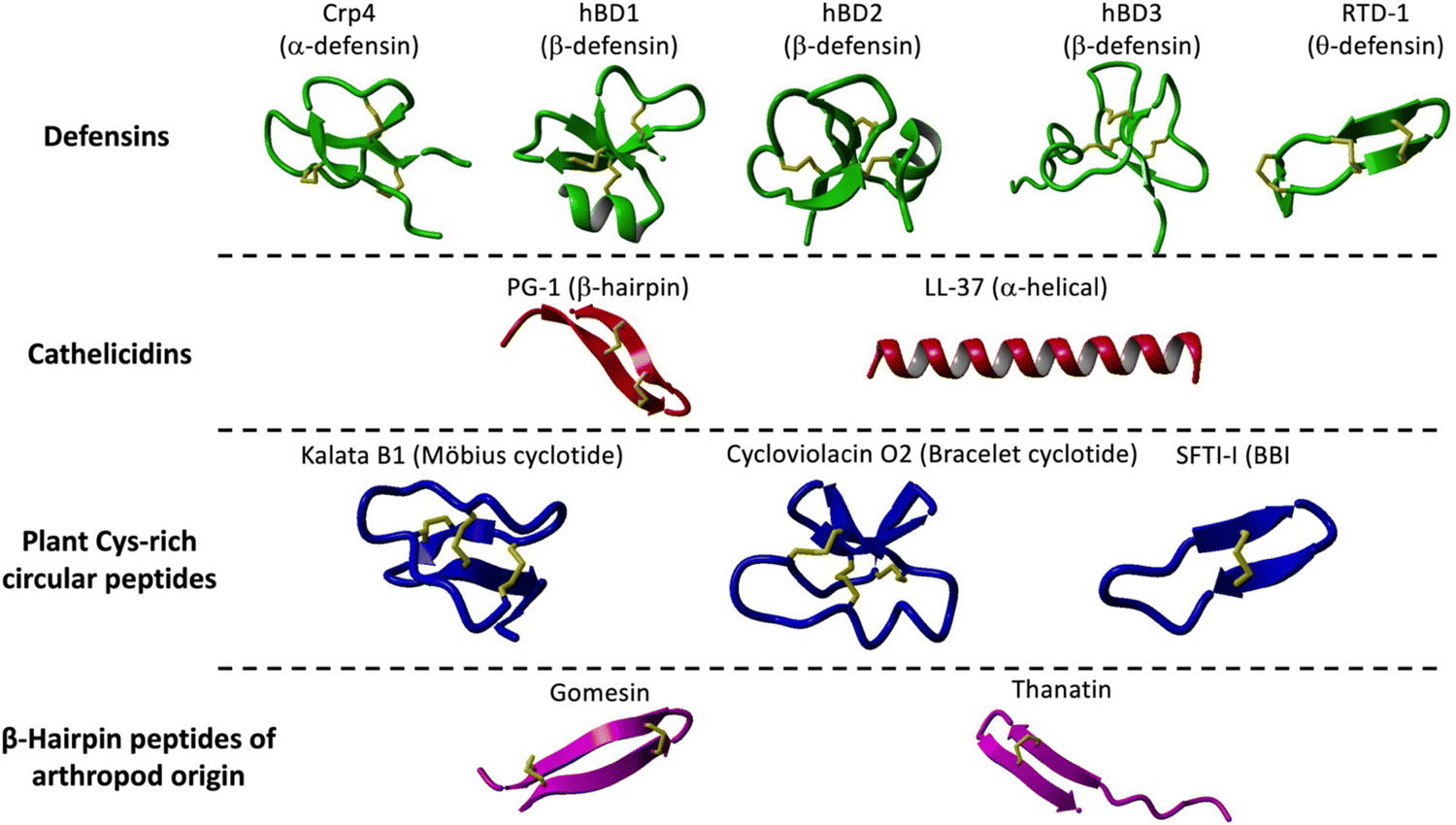 | ||
| Fig. 1 Families and structures of different Cys-rich cyclic peptides used as molecular frameworks for the design of novel antimicrobial peptides as described in the manuscript. These include α-defensins: Crp4 (PDB: 2GW9),158 β-defensins: hBD1 (PDB: 1IJV),160 hBD2 (PDB: 1FD3),161 hBD3 (PDB: 1KJ6)162 and RTD1 (PDB: 1HVZ);163 cathelicidins: PG-1 (PDB:1PG1)67 and LL-37 (pdb: 7PDC);164 Cys-rich backbone cyclized peptides: kalata B1 (pdb: 1NB1),99 cycloviolacin O2 (pdb: 1DF6)159 and SFTI-I (pdb: 4TTK);137 and β-hairpin peptides: gomesin (pdb: 1KFP)137 and thanatin (pdb: 5XO4). Their primary structures and antimicrobial properties are summarized in Table 1. | ||
| Group | Peptide name | Sequence and disulfide structure | MIC (μM) | Ref. |
|---|---|---|---|---|
| α-Defensin | CRP4 |

|
<5 | 38 and 39 |
| β-Defensins |
|

|
N.A. oxidized | 49 |

|
2.3–23 | 50 | ||
|
0.7–1.2 | 51 | ||
| θ-Defensin | RTD-1 |

|
0.5–5 | 61 and 62 |
| Cathelicidins | PG-1 |

|
0.03–0.4 | 72 and 76 |
| LL-37 |

|
1–10 | 88 | |
| Cyclotides | Kalata B1 |
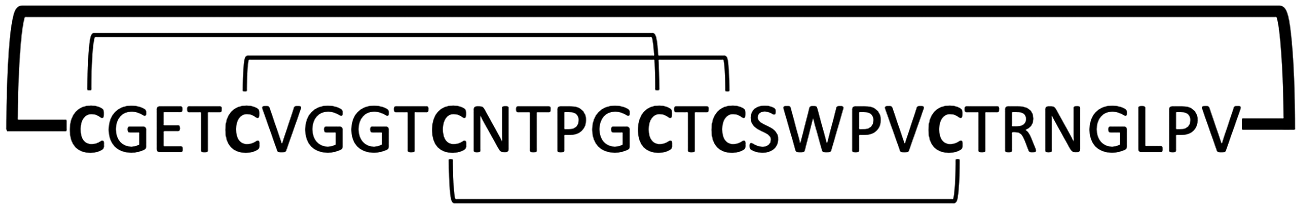
|
0.29–14 | 122 |
| MCo-PG2 |
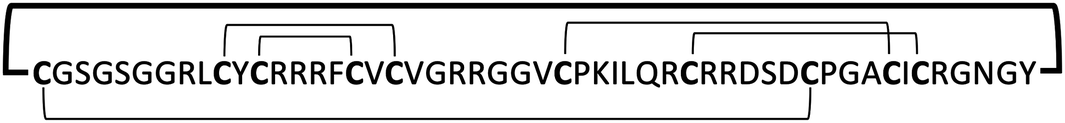
|
0.8–12.5 | 77 | |
| SFTI-I derived | RV3 |

|
0.6–4 | 134 and 135 |
| β-Hairpin | Gomesin |
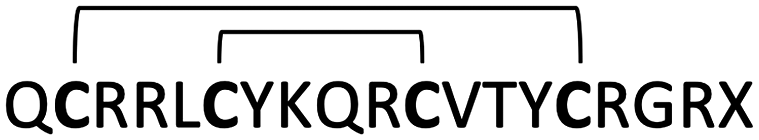
|
0.2–6 | 136 |
| Thanatin |

|
0.5–2 | 142 and 143 |
α- and β-Defensins
Defensins were initially discovered in rabbit lung macrophages being later also reported in other vertebrate species.14 Defensin-like peptides have also been isolated from plants, where they were initially classified as γ-thionins10 and later classified as defensins based on sequence homology, structure, and functional similarity with animal defensins.18 Despite the fact that plant defensins have a quite similar amphiphilic β-sheet structure to that of animal defensins, their evolutionary relationship is unclear (see ref. 10 for an extensive review on plant defensins).Defensins can kill bacteria or inhibit their growth through a multiplicity of antimicrobial mechanisms such as direct membrane disruption10,19,20 and inhibition of bacterial cell wall synthesis.21–23 Although defensins are classically known for their antimicrobial activities, they also possess many other defense-involved activities, including wound healing,24,25 immune modulation,26–29 neutralization of endotoxins,30,31 and anti-cancer activities.32
Mammalian α-defensins are, in general, less cationic and more hydrophobic than β-defensins (hBDs) (Table 1), and they use different mechanisms to kill bacteria.19 For example, while hBD-1 and hBD-2 show better activity against Gram-negative bacteria,33 hBD-3 displays potent bactericidal activity against both Gram-positive and Gram-negative bacteria.34,35
The antibacterial activity of α- and β-defensins is highly dependent on the ionic strength of the media, and salt-dependent inactivation of defensins in patients with cystic fibrosis has been proposed as the potential cause of chronic pulmonary infections in these patients.36
The antimicrobial activity of the naturally cyclic θ-defensins (Fig. 1 and Table 1), in contrast, has been shown to be less sensitive to salt concentration than α- and β-defensins.37 This difference has been attributed to the circular structure of θ-defensins since the acyclic forms are more salt sensitive.37 In agreement with this hypothesis, the replacement of the characteristic Cys–Cys disulfide bridge end-to-end, Cys3,31 (Cys I:Cys VI) in the rabbit α-defensin neutrophil peptide 1 (NP-1) by a backbone-cyclization, provided biologically active defensin analogs that were less sensitive to salt.38 No studies, however, were provided in this work on the stability of the resulting engineered circular α-defensins lacking the characteristic C- to N-termini Cys–Cys disulfide bridge. In another independent study, the backbone-cyclization of the native α-defensin cryptidin-4 (Crp4) from murine Paneth cells demonstrated that the backbone-cyclized version of Crp4 was able to adopt a natively-folded α-defensin structure displaying equivalent or better microbicidal activities against several Gram-positive and Gram-negative bacteria when compared to the native linear Crp4.39 Both forms of Crp4, linear and circular, showed no detectable hemolytic activity against human red blood cells. In addition, the circular version of Crp4 displayed significantly higher stability against proteolytical degradation when incubated with human serum.39 The presence of the disulfide array in Crp4 has previously been shown to be a necessary component in resistance to proteolytic degradation but does not affect antimicrobial activity.40 These properties make cyclized Crp4 α-defensins promising scaffolds for drug development of novel antibiotics, although further studies may be required to evaluate their metabolic stability and bioavailability.
The hydrophobic and highly cationic nature of defensins favors the accumulation of these peptides on the mostly negatively charged bacterial membranes in both Gram-positive and Gram-negative bacteria independently of their actual target of action.41 For many years, it was believed that the mechanism of action of defensins was the permeabilization of the bacterial membranes. Different models have been proposed for the membrane permeabilization of antimicrobial peptides (Fig. 2). These include: (i) the barrel-stave pore model, where the peptide molecules can use their amphipathic nature to form dimers or multimers that cross the membrane forming barrel-like channels,42 (ii) the toroidal pore model where the defensin creates a monolayer connecting the outer and the inner lipid layers in the pore;43 (iii) the carpet model, where antimicrobial peptides produce a carpet-like structure that covers the outer surface of the membrane and disrupts it in a detergent-like mechanism of action;44 (iv) other less frequent models involve the sinking-raft and the molecular electroporation models, where the peptide molecules can either bind or sink into the bacterial membrane or just disrupt the electrostatic potential across the membrane to generate pores.45
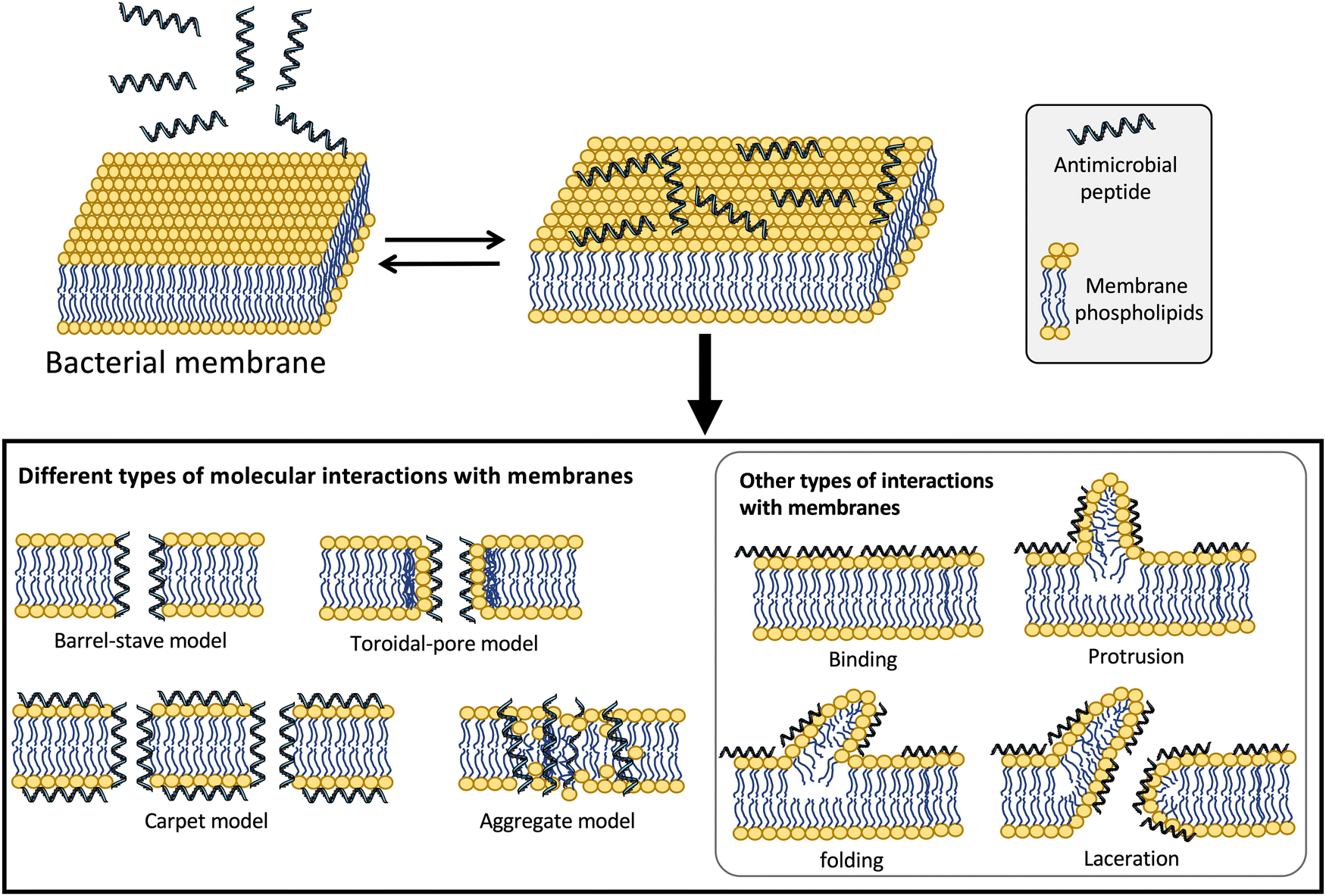 | ||
| Fig. 2 Different types of models proposed for the molecular mechanism of action of antimicrobial peptides.41,165 | ||
More recent studies have suggested that some defensins can exert their antibacterial activity using alternative modes of action (Fig. 2).41 For example, human defensins can also interact with additional bacterial targets to kill bacteria. α-Defensin HNP1 has been shown to interact mostly with the bacterial membrane in Escherichia coli but instead employs a different bactericidal mechanism on Staphylococcus aureus by interacting and inhibiting the cell-wall precursor lipid II.23 The bactericidal activity of α-defensins HNP2 and HNP3 also involve lysing the bacterial membrane, although HNP2 has also been reported to produce aggregation and fusion of vesicles as alternative mechanisms of action for its bactericidal activity.46 Interestingly, HNP4, the less abundant of the α-defensins found in the azurophilic granules of neutrophils, is about 100 times more active against E. coli than its homologs HNP1-3 and also targets the bacterial membrane.47 Other α-defensins like HD5 and HD6 also have been shown to permeabilize the bacterial cell membrane as the main mechanism for their bactericidal activity.48 HD5 has, in addition, the ability to bind strongly to DNA, suggesting that this interaction may be key in dysregulating essential processes associated with DNA replication, transcription, or translation of genes involved in bacterial survival.49
The proposed role for β-defensins like hBD1 is acting like a protective barrier for epithelial cells, therefore, preventing infection by commensal bacteria.50 Although the antimicrobial activity of hBD1 has been found to be lower than other defensins, after the complete reduction of its three disulfide bridges, its antimicrobial properties become more potent but also more labile to proteolytic degradation.50 In human skin, hBD1 is found colocalized with the redox protein thioredoxin, which supports the hypothesis that thioredoxin may act as an in situ physiological mediator catalyzing the reduction of the less active folded hBD1 into the most active, fully reduced form of hBD1 in human epithelia.50 Other β-defensins, like hBD2-4, also employ the common mechanism of action by binding to the negatively charged bacterial membrane and causing membrane permeabilization and cellular death.51,52 In addition, hBD3 has also been shown to bind and inhibit lipid II, perturbing the bacterial cell wall biosynthesis.52
θ-Defensins
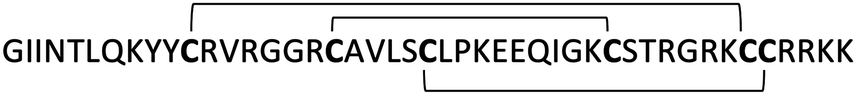
In contrast with α- and β-defensins, θ-defensins are naturally backbone cyclized antimicrobial peptides which are formed by the head-to-tail covalent assembly of two nonapeptides derived from α-defensin related precursors37,53 (see ref. 17 for an extended review on the chemistry and biology of θ-defensins). As mentioned earlier, θ-defensins are, to date, the only known backbone-cyclized polypeptides expressed in animals37 and show antimicrobial activities in the 0.5–5 μM range. The antimicrobial activity of θ-defensins is less sensitive to physiological salt content when compared to α- and β-defensins, which can be attributed to their backbone circular topology as the linear analog of the Rhesus defensin 1 (RTD-1) is about three times less active than the cyclic RTD-1.37 Interestingly, it has been reported that the disulfides of θ-defensins are not essential for antimicrobial activity.54 θ-Defensins have also been shown to bind and neutralize bacterial toxins such as anthrax lethal factor.55–57The recent development of efficient approaches for the chemical and recombinant production of θ-defensins53,56–59 has allowed the rapid and efficient production of many θ-defensin analogs to study their chemical and biological properties.17 The recent use of a ‘tea-bag’ approach60 in combination with a one-pot cyclization method involving native chemical ligation and oxidative folding allowed the development of more potent analogs of θ-defensin RTD-1 that can inhibit anthrax lethal factor (LF) as well as the TNF-α converting enzyme (TACE) with Ki values ≈40 nM and ≈157 nM, respectively.56
Natively folded and bioactive θ-defensin RTD-1 has also been produced in high yield (0.7 mg of RTD-1 per gram of wet cells) inside E. coli cells by making use of intracellular protein trans-splicing in combination with a highly efficient split-intein.57 This approach was employed to produce a genetically-encoded RTD-1-based peptide library in E. coli cells encoding ≈2 × 107 different RTD-1-based sequences.57 This result is intriguing as θ-defensins possess antimicrobial activities against several microbial pathogens, including P. aeruginosa, S. aureus and E. coli, pathogenic yeast Candida albicans, and HIV.61,62 θ-Defensins also display potent anti-inflammatory properties in vitro and in vivo mediated by the suppression of numerous pro-inflammatory cytokines and blockade of TNF-α release by inhibition of TACE.63 A potent TACE inhibitor was recently developed using the Rhesus θ-defensin as a molecular scaffold.56
Cathelicidins
Cathelicidins are antimicrobial peptide precursors widely distributed in mammalian and some fish granulocytes and certain epithelia. Their name indicates the presence of a cathelin domain, a well-conserved structural element of about 100 amino acids, that is followed by a C-terminal antimicrobial peptide domain. The C-terminal antimicrobial domain can adopt α-helical, β-sheet, or other structures once released by proteolytic cleavage. Cathelicidins are considered part of the innate immune system in many vertebrates and show in general a broad-spectrum antimicrobial activity against bacteria, some enveloped viruses, and fungi. In addition to their antimicrobial activity, cathelicidins are also able to activate host defense responses (see ref. 64 for a detailed review of cathelicidins).Protegrins
Mature protegrins are Arg and Cys-rich cationic polypeptides 16 to 18 residues long with an amidated C-terminus.65 Protegrins adopt a β-hairpin structure stabilized by two disulfide bridges (Fig. 1 and Table 1). Protegrins were first discovered in porcine leukocytes,65 and their structure resembles that of the θ-defensins53 and tachyplesin/polyphemusin peptides found in the hemocytes of horseshoe crabs.66Five different porcine protegrins (PGs) have been isolated and characterized, PG-1 through PG-5.65 Protegrins show broad-spectrum antimicrobial activity against Gram-positive and Gram-negative bacteria, including multi-drug resistant bacterial strains.67 They can bind important components of microbial membranes, including lipopolysaccharides (LPS), lipid A and lipoteichoic acid (LTA),68 which help them to insert into the bacterial membrane and permeabilize it.69 Protegrins have also shown antiviral activity against HIV-170 and HSV-2,71 enveloped virus responsible for AIDS and genital herpes.
In contrast with defensins, protegrins have a broader antimicrobial pH range, being effective against acid-tolerant microorganisms even at pH 4.5.68 Another interesting feature of protegrins is that their antimicrobial activity is not affected by physiological NaCl concentration, also remaining largely unaffected by the presence of divalent cations.72 The salt tolerance observed in protegrins is imparted by the disulfide bridges, which stabilize the β-hairpin even in the presence of high salt content.68,72 For example, a disulfide-free protegrin analog of PG-1, obtained by replacing each Cys residue with Ala, displayed similar antimicrobial activity to that of the native disulfide-containing protegrin but was practically inactive in media containing 100 mM NaCl.72
The presence of serum proteins also has a negligible effect on the antimicrobial activity of protegrins, and in some cases, it has been described to have a positive effect enhancing their antimicrobial activity, likely through the additive effects of complement or other serum constituents.68 This is completely the opposite effect that human serum has on α-defensins, where several serum components (e.g., α2-macroglobulin and serpins) avidly bind human α-defensins, significantly reducing their antimicrobial activity.73
On the other hand, native protegrins also display high cytotoxicity and hemolytic properties against eukaryotic and red blood cells.74 Cytotoxicity to eukaryotic cells has also been described for other β-sheet antimicrobial peptides like tachyplesin and polyphemusin peptides.75
Extensive structure–activity studies have been carried out in porcine protegrins to define the importance of sequence length, residue chirality, C-terminal amidation state, and disulfide-bridge structure required for the antimicrobial and cytotoxic activity of protegrins in order to improve their biological activity and reduce cytotoxicity.76 Several studies have shown that the introduction of structural alterations on protegrins can greatly reduce their cytotoxic properties while keeping intact their broad-spectrum antibacterial activity.72,76,77
Backbone cyclization and introduction of an extra disulfide in the β-sheet region to produce a cyclic tri-cystine analog of protegrin PG-1 has been shown to reduce about 10 times the hemolytic activity of PG-1 while improving 6 to 30 times the membranolytic selectivity against several pathogenic microorganisms.72 Introduction of the V14T mutation on PG-1 also significantly reduced the cytotoxicity against mammalian cells while keeping similar antibacterial activity to that of the parent PG-1 peptide.68
The porcine PG-1 was used as starting point to generate the analog IB-367, also known as iseganan.78 IB-367 has shown promising results in the prevention of chemotherapy- and radiation-induced oral mucositis, and the drug has been evaluated in phase III clinical trials for this application.79,80
Murepavadin, also known as POL7080, is a recently developed synthetic cyclic β-hairpin peptidomimetic based on protegrin PG-1.81,82 This antibacterial peptide was designed by using a D-Pro-L-Pro template to mimic and stabilize the β-hairpin conformation of PG-1 generating a diverse peptide library that was then screened for antibacterial activity.83 Murepavadin is a highly active Pseudomonas spp-specific peptide antibiotic that targets the outer membrane protein transporter LptD of P. aeruginosa.82 This is the first example of a peptide antibiotic with a novel nonlytic mechanism of action. This peptide has shown very potent bactericidal antimicrobial activity against P. aeruginosa in vitro, including over 1000multi-drug-resistant (MDR) clinical isolates of P. aeruginosa.84–86
These results, together with the good safety profile observed in a phase 1 study,87 and combined with results from ongoing clinical studies, are highly encouraging to continue further clinical development of murepavadin for treating serious P. aeruginosa infections.84
More recently, a topologically modified version of protegrin PG-1 was also used to generate novel antibacterial cyclotides with effective broad-spectrum antibacterial activity against several ESKAPE bacterial strains and a panel of MDR clinical isolates of P. aeruginosa and S. aureus.77 The most active antibacterial cyclotide showed similar activity to that of PG-1 while displaying little hemolytic activity and being extremely stable in serum.77 This novel cyclotide was also able to provide in vivo protection in a murine model of P. aeruginosa peritonitis.77
LL-37
Despite the great abundance of β-defensin-encoding genes in the human body, LL-37 is the only cathelicidin identified so far in humans.64 LL-37 is expressed as the protein precursor hCAP-18 that is stored in cytoplasmic granules and lamellar bodies.64 The C-terminal segment of hCAP18 is then proteolytically cleaved by proteinase 3, providing the fully active 37-residue long LL-37 peptide.64 Expression and secretion of LL-37 is mostly carried out by epithelial cells, although immune cells are also known to express and secrete peptide LL-37.64 LL-37 shows good antimicrobial activity against Gram-negative and Gram-positive bacteria with MIC values ranging from 1–10 μM depending on the method used to calculate it and the bacteria strain.88 The antibacterial activity of LL-37 is through direct interaction and disruption of the bacterial membrane, although it also shows strong immunomodulatory properties.64Mature LL-37 is a linear peptide with a high α-helical content in physiological buffers. The α-helical content has been estimated to be around 35% at pH 7.4.89 The helicity of the LL-37 increases upon binding to the bacterial membrane, which is a common feature observed in lipid-binding peptides.89 Synthetic LL-37 peptide analogs have been evaluated in clinical trials for treating venous leg ulcers.90
Despite the interesting bioactivities of LL-37, its clinical potential has been mainly hampered by its proteolytic instability and cytotoxicity.91 To overcome these issues, different approaches have been employed for the stabilization of LL-37.
The N-terminal fragment of LL-37, peptide LL-12, completely lacks any antibacterial activity.92 The C-terminal region of LL-37, in particular the segment corresponding to LL-37 (18–29), also known as peptide KR12, retains most of the antimicrobial activity of LL-37 while also showing reduced cytotoxicity.93,94 The activity of KR12 can be further improved by introducing specific mutations, Q5A and D9K, in the peptide sequence.95 KR12-derived linear peptides, however, are highly susceptible to proteolytic degradation. A recent report used a backbone cyclized homodimer of the improved KR12 Q5A, D9K analog sequence to improve its stability to proteolytic degradation (Fig. 3).96 The most active peptide, CD4-PP, displayed 16-fold higher antibacterial activity compared to KR-12 against P. aeruginosa and S. aureus, and 8-fold increased fungicidal activity against C. albicans.96 Unfortunately, CD4-PP also displayed increased hemolytic and cytotoxic activity.96 This interesting work on peptide KR-12 suggests that backbone cyclo-dimerization can be used as an effective strategy to improve both the potency and stability of linear antimicrobial peptides, although more studies may be required to decrease the hemolytic and cytotoxic activity of CD4-PP.
 | ||
| Fig. 3 Design of peptide CD4-PP, a cyclo-homodimer based on an improved KR12 peptide (LL37 218-29 fragment) containing activating mutations Q5A and D9K (shown in green). The optimized KR12 Q5A, D9K was used to produce a backbone cyclized anti-parallel homodimer. The flexible linker G/CPGG (shown in blue) was used to join the identical α-helical segments.96 The resulting cyclic peptide CD4-PP displayed 16-fold higher antibacterial activity compared to KR-12 against P. aeruginosa and S. aureus, and 8-fold increased fungicidal activity against C. albicans.96 | ||
Cyclotides
Cyclotides are fascinating circular Cys-rich micro-proteins containing ≈30 residues and three disulfides that are found in plants.97 Cyclotides display various biological properties such as protease inhibitory, antimicrobial, insecticidal, cytotoxic, anti-HIV, and hormone-like activities (see ref. 12, 13 and 98 for recent reviews of the properties and applications of cyclotides). They share a unique head-to-tail cyclic cystine knot (CCK) scaffold of three disulfide bridges, with one disulfide penetrating through a macrocycle formed by the two other disulfides and inter-connecting peptide backbones, forming what is called a cystine knot topology (Fig. 1 and 4).99 The main features of cyclotides are remarkable stability due to the cystine knot, a small size making them readily accessible to chemical synthesis, and an excellent tolerance to sequence variations. Cyclotides have also been shown to be orally bioavailable,100,101 and capable of crossing cell membranes102,103 to efficiently target extracellular104–106 and intracellular molecular targets in vivo.107 Cyclotides also display poor immunogenicity due to their highly constrained nature.108,109 All these properties make them an ideal scaffold for peptide drug design.13,98,110 | ||
| Fig. 4 Design of antimicrobial cyclotide MCo-PG2 using molecular grafting. A topologically modified version of protegrin 1 (PG-1) was grafted into loop 6 of trypsin inhibitor cyclotide MCoTI-II. The resulting engineered cyclotide precursor was readily produced by solid-phase peptide synthesis. The crude linear thioester was then cyclized and oxidatively folded in a “one-pot reaction”.77 The resulting cyclotide, MCo-PG2, displayed broad-spectrum antimicrobial activity in vitro against different ESKAPE pathogens (P. aeruginosa, S. aureus, K. pneumoniae, and E. coli), including 20 MDR clinical isolates for the human pathogens S. aureus and P. aeruginosa. Cyclotide MCo-PG2 also improved the survival rate in a peritonitis mice model using the clinical isolate of P. aeruginosa (ATCC 27853) from 0% for the untreated mice group to 90% for the mice group treated with 25 mg kg−1 of cyclotide.77 | ||
Naturally-occurring cyclotides from the Möbius and bracelet subfamilies display strong insecticidal activity and are thought to work mainly as host-defense agents.111–116 Other biological activities reported for cyclotides from these two subfamilies also include inhibiting the growth of parasitic worms.117–119
Folded cyclotides from these two subfamilies display well-defined hydrophobic and hydrophilic patches on the molecular surface, conferring them an amphipathic character.120 These amphipathic properties are also typically found in classical antimicrobial peptides. This molecular characteristic has been used to explain their antibacterial activity.121 For example, the Möbius cyclotide kalata B1 has been described to possess antimicrobial activity against Gram-positive and Gram-negative bacteria.122 Other cyclotides isolated from plants have also shown similar antimicrobial activities.123–125 The more potent naturally-occurring antimicrobial cyclotide tested so far is the bracelet cyclotide cycloviolacin O2.126 This cyclotide also showed activity against S. aureus in a mouse infection model.127 As with many other antimicrobial peptides, the in vitro antimicrobial activity of these types of cyclotides strongly depends on the buffer composition, showing promising antimicrobial activity only in low salt content buffers. This may suggest that the in vivo antimicrobial activity of cycloviolacin O2 could also be due to an indirect effect.
Cyclotides can also be used as molecular frameworks to introduce other biological activities. For example, a recent study reported the first design and synthesis of a novel cyclotide with broad-spectrum antimicrobial activity in vitro against different ESKAPE pathogens (P. aeruginosa, S. aureus, K. pneumoniae, and E. coli), including 20 MDR clinical isolates for the human pathogens S. aureus and P. aeruginosa (Fig. 4). The median minimal inhibitory concentration (MIC) 50% (MIC50) and MIC 90% (MIC90) values for several MDR clinical strains of P. aeruginosa were 1.5 μM and 3.1 μM, respectively; while for clinical isolates of MDR S. aureus the MIC50 and MIC90 values were 6.25 μM and 12.5 μM, respectively.77 More importantly, the most active cyclotide, MCo-PG2, showed strong antimicrobial activity in vivo using a murine model of acute P. aeruginosa peritonitis.77 Cyclotide MCo-PG2 was shown to improve the survival rate in a peritonitis mice model using the clinical isolate of P. aeruginosa (ATCC 27853) from 0%, for the untreated mice group, to 90% for the mice group treated with 25 mg kg−1 of cyclotide.77
The results reported with cyclotide-MCo-PG2 are very encouraging, demonstrating for the first time the design of a novel cyclotide exhibiting potent antimicrobial activity under physiological-like conditions and showing also strong in vivo antibacterial efficacy in a murine P. aeruginosa-induced peritonitis animal model.77
The trypsin inhibitor cyclotide MCoTI-II has also recently been used to graft potent antimicrobial peptide optP7 (H-KRRVRWIIW-NH2)128 to produce antimicrobial cyclotide [L6-Opt7]-Mco.129 Some of the fractions obtained during the folding of this cyclotide exhibited promising antimicrobial activity against a panel of ESKAPE pathogens, with MIC values in the low μM range.129 MIC values were, however, obtained under optimized buffer conditions, and the antimicrobial activity was shown to be strongly dependent on the ionic strength of the assay buffer.129 Unfortunately, this is a feature commonly found in many antimicrobial peptides. The activity dependence on ionic strength of cyclotide [L6-Opt7]-Mco, however, could be attributed to the intrinsic properties of the grafted sequence, peptide optP7.128 In contrast, the activity of cyclotide MCo-PG-2, based on cyclotide MCoTI-I and protegrin PG-1, was not affected by the buffer composition and exhibited full activity in vivo using a peritonitis animal model.77
Sunflower trypsin inhibitor 1 (SFTI-1)
SFTI-1 is a 14 amino acid backbone-cyclized peptide containing a single disulfide bond that is naturally found in the seeds of the sunflower plant (Helianthus annuus).130 SFTI-1 belongs to the Bowman–Birk inhibitor (BBI) family, whose members are found in many plants and are potent serine protease inhibitors.131 Structural analysis of SFTI-1 shows a well-defined double β-hairpin loop linked by two short anti-parallel β-strands (Fig. 1).132 The backbone-cyclized SFTI-1 is the smallest and the most potent protease member of the family with a Ki against trypsin in the low nM range.133The relatively rigid backbone of SFTI-1 makes it extremely resistant to proteolytic degradation and a good molecular framework to introduce novel biological activities).130 A recent report used the trypsin-binding loop (CTKSIPPIC) of SFTI-1 as a scaffold to design a novel type of peptide antibiotic. In this work, several peptides designed using an alternate arrangement of hydrophobic and cationic amino acids ((RX)nW(RX)n, where n = 2 or 3, and X represents a hydrophobic residue) were fused to the N-terminal of the oxidized trypsin-binding loop (CTKSIPPIC) of SFTI-1.134 The most active peptide, RV3 (n = 3 and X = Val), displayed broad antimicrobial activity against Gram-positive and Gram-negative bacteria, with MIC values ranging from 1 to 4 μM, and showed antimicrobial efficacy in a mouse skin inflammation model established by P. aeruginosa infection.134 Peptide RV3 was able to effectively kill the pathogen, promote wound healing, inhibit inflammatory cell infiltration, and inhibit mRNA and protein expression of TNF-α, IL-6, and IL-1β inflammatory factors.134 This study concluded the mechanism of action of RV3 involved binding to lipopolysaccharides in the bacterial membrane, increasing the cell membrane permeability and finally leading to cell membrane rupture and death.134
Another study recently reported the design of a hybrid antimicrobial peptide using the trypsin binding loop (loop 1) of SFTI-1 and the sequence corresponding to loop 2 of peptide HVBBI.135 HVBBI is a β-hairpin BBI peptide found in the skin secretions of the Chinese bamboo odorous frog.135 The resulting hybrid peptide, HSEP-1, displayed only modest antimicrobial activities against Micrococcus luteus (MIC ≈40 μM). Further optimization of HSEP-1 by increasing the hydrophobicity of loop 2 and charge of the peptide yielded peptide HSEP-3. This peptide displayed a MIC value of 0.6 μM against M. luteus, although antimicrobial activity against other bacterial pathogens was rather modest.135 These designed peptides elicit their antibacterial action via both membrane destabilization and inhibition of intracellular trypsin, which are attributable to the two separate peptide segments, loop 1 (trypsin binding loop) and loop 2 (antimicrobial segment). The toxicity studies and food preservation assays in this work indicated good safety and efficacy profiles for its potential use as a food preservative, although more optimization may be required before its commercial application.135
β-Hairpin-containing peptides of arthropod origin
Gomesin
Gomesin is an 18-residue long Cys-rich cationic antimicrobial peptide originally isolated from the hemocytes of the Brazilian tarantula Acanthoscurria gomesiana (see ref. 136 for a recent review on the structural and biological properties of gomesin) (Fig. 1 and Table 1). The peptide shows potent cytotoxic activity against clinical isolates of bacteria, including pathogenic Gram-positive and Gram-negative, fungi and yeast with MIC values ranging in all cases from 0.2 μM to 6 μM.136The solution structure of gomesin shows that the peptide adopts a β-hairpin-like structure, with two-stranded anti-parallel β-strands connected by a 4-residue non-canonical β-turn (Y7-K8-Q9-R10) (Fig. 1).137 The β-strands are stabilized by two inter-strand disulfide bridges (C2–C15 and C6–C11) and six inter-strand backbone–backbone hydrogen bonds.137
Numerous studies have been reported on the chemical modification of gomesin to increase its antibacterial activity while reducing its hemolytic properties.138–140 For example, a report showed that backbone cyclization produced a circular analog of gomesin that was more resistant to serum proteases and able to retain the potent anticancer and antimicrobial activities of native gomesin.139 Cyclization of gomesin usually produces a 2- to 3-fold increase in its antimicrobial activity while significantly increasing its biological resistance to proteolytic degradation.139 More recently, the same group also reported the introduction of mutations in the cyclic analog of gomesin to reduce the cytotoxicity to eukaryotic cells while keeping the biological activities of gomesin.138,141
Thanatin
Thanatin is a 21-residue pathogen-inducible antimicrobial peptide that, as gomesin, also adopts in solution a β-hairpin structure stabilized by a single-disulfide bond (Fig. 1 and Table 1).142 This antimicrobial peptide was originally isolated from the insect Podisus maculiventris and exhibits broad-spectrum activity against both Gram-positive and Gram-negative bacteria as well as against various species of fungi [see ref. 143 for a recent review on the antimicrobial properties of thanatin]. Thanatin presents low hemolytic activity and low toxicity against mammalian cells. MIC values range from ≈0.5 μM (E. coli) to ≈30 μM (P. aeruginosa). Thanatin is less active against Gram-positive bacteria, with MIC values ranging from ≈2 μM (M. luteus) to no activity (S. aureus).142 A comparison study across different disulfide-containing β-hairpin antimicrobial peptides indicates that thanatin had the lowest hemolytic and cytotoxic activity hence highlighting its potential for therapeutic applications.143 Accordingly, the antimicrobial activity of thanatin and its analogs have been extensively studied in vivo with very promising results. For example, S-thanatin (thanatin with the T15S mutation) was shown to improve the survival rate in a septicemic mice model using the MDR clinical isolate of K. pneumoniae (CI120204205, resistant to carbapenems) from 0%, for the untreated mice group, to 100% for the mice group treated with 15 mg kg−1 of the peptide.144 A C-terminal amidated analog of thanatin (thanatin A) also increased the survival rate in an extended-spectrum β-lactamase-producing E. coli (ESBL-EC)-infected mice model from 0%, for the control group, to 92% for the mice treated with 10 mg kg−1 of A-thanatin.145A recent report indicated that the binding of thanatin with periplasmic proteins involved in LPS transport to the outer membrane is the principal antimicrobial mode of action against Gram-negative bacteria.146
The broad-spectrum antimicrobial activity of thanatin, in conjunction with its high in vivo stability and low cytotoxicity, are quite promising, although further research may be required before thanatin-based peptides can be developed into effective treatments against bacterial infection caused by MDR pathogenic bacteria. These studies should be focused on the development of more potent thanatin-based antimicrobial peptides. For example, more SAR studies on thanatin are still required to understand better how the sequence and structural modifications in the peptide composition can be translated into more active analogs.143 In addition, more detailed studies are required on the mode of action of this peptide. The antibacterial activity of thanatin against Gram-positive bacteria and fungi remains largely unexplained as these microorganisms do not contain LPS or LPS-translocating protein complexes.
Concluding remarks
The spreading of antimicrobial-resistant genes in new bacterial strains is a matter of course and inevitable but this has been accelerated by the consequence of antibiotics abuse.147 The search for new treatments should be urged to efficiently treat infections mediated by established and new MDR pathogenic bacterial strains.Antimicrobial peptides are a highly diverse group of compounds that are being investigated as potentially attractive alternatives to classic antibiotics.148 Among them, Cys-rich cyclic peptides have gained popularity holding many advantageous characteristics as therapeutic compounds in contrast to classical small molecule-based antibiotics. Among the different examples reviewed in this manuscript, cyclotide-based antimicrobials are emerging as one of the most attractive alternatives for the design of novel antimicrobial peptides with broad-spectrum and effective in MDR pathogenic bacteria.
Cyclotides present extraordinary stability to thermal/chemical denaturation as well as to proteolytic degradation.149 This high stability to proteolytic degradation and potential low cytotoxicity allows them to be used for the treatment of systemic infections.77 In fact, proteolysis stability has been one of the main limitations associated with the development of antimicrobial peptides, which has limited their use mostly to topical applications.150 Cyclotides can in some cases cross mammalian cellular membranes,103,151 which should also allow them to target intracellular bacteria. Intracellular bacteria remain one of the most challenging targets when designing novel antimicrobial peptide-based therapeutics.150 Intracellular bacteria are also known to interfere with the host immune system to exacerbate the bacterial infection process.150 Cyclotides and the circularized version of the antimicrobial peptide gomesin have been shown to cross mammalian cellular membranes103,141,151 and they have been shown to have potent antimicrobial activities in animal models of bacterial infection,77 which makes them ideal candidates for further development into effective therapeutic agents able to target intracellular pathogens.
The small size of these peptides allows their chemical synthesis by using standard solid-phase peptide synthesis methods facilitating the use of structure–activity relationship (SAR) studies to improve their biological properties. This allows the introduction of chemical modifications such as PEGylation and/or non-natural amino acids to improve their pharmacological profiles, e.g., half-life extension and proteolytic stability.105,152
In the case of the cyclotide scaffold, its high plasticity, tolerance to sequence variation, and the presence of up to five hypervariable loops make it an ideal substrate for molecular grafting and molecular evolution techniques to target a specific set of bacteria or bacterial targets.98,153 A recent study showcased the use of the cyclotide scaffold in the design of de-novo cyclotides with potent broad-spectrum antimicrobial activity.77 The most active cyclotide (MCo-PG2) displayed better activity than colistin in a peritonitis infection model in mice.77 Colistin is presently considered the last line of defense against human infections caused by multidrug-resistant Gram-negative organisms such as carbapenemase-producer E. coli, A. baumanni, and P. aeruginosa.154 Cyclotide MCo-PG2 was also highly active against a panel (N = 20) of different MDR clinical isolates of S. aureus and P. aeruginosa indicating its high translational potential as an antimicrobial therapeutic lead.77
Another exciting feature of cyclotides is the potential to be orally active. In fact, several bioactive cyclotides have already been reported as orally active,100,155 although more detailed pharmacological studies in this regard may be required in the future.
It is anticipated that some of the challenges still affect antimicrobial peptides before they can move into the clinic. These affect mostly immunogenicity and oral bioavailability, which should be soon addressed as novel and more potent antimicrobial peptides are reported.
Conflicts of interest
The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.Acknowledgements
This work was supported by National Institutes of Health Research Grants R35-GM132072 (JAC). AM is partially supported by a postdoctoral fellowship “Ayudas Margarita Salas para la formación de jóvenes doctores” by University of León, Spain.References
- J. M. Pogue, K. S. Kaye, D. A. Cohen and D. Marchaim, Clin. Microbiol. Infect., 2015, 21, 302–312 CrossRef CAS PubMed.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castaneda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiu, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. VictoriaRobotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek, M. Naghavi and A. R. Collabora, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- I. E. Kepiro, I. Marzuoli, K. Hammond, X. Ba, H. Lewis, M. Shaw, S. B. Gunnoo, E. De Santis, U. Lapinska, S. Pagliara, M. A. Holmes, C. D. Lorenz, B. W. Hoogenboom, F. Fraternali and M. G. Ryadnov, ACS Nano, 2020, 14, 1609–1622 CrossRef CAS PubMed.
- G. Wang, B. Mishra, K. Lau, T. Lushnikova, R. Golla and X. Wang, Pharmaceuticals, 2015, 8, 123–150 CrossRef CAS PubMed.
- R. Sher Khan, A. Iqbal, R. Malak, K. Shehryar, S. Attia, T. Ahmed, M. Ali Khan, M. Arif and M. Mii, 3 Biotech, 2019, 9, 192 CrossRef PubMed.
- P. M. Silva, S. Goncalves and N. C. Santos, Front. Microbiol., 2014, 5, 97 Search PubMed.
- T. Tecle, S. Tripathi and K. L. Hartshorn, Innate Immun., 2010, 16, 151–159 CrossRef CAS PubMed.
- K. A. Brogden, M. Ackermann, P. B. McCray and B. F. Tack, Int. J. Antimicrob. Agents, 2003, 22, 465–478 CrossRef CAS PubMed.
- D. P. McGregor, Curr. Opin. Pharmacol., 2008, 8, 616–619 CrossRef CAS PubMed.
- S. Srivastava, K. Dashora, K. L. Ameta, N. P. Singh, H. A. El-Enshasy, M. C. Pagano, A. E. Hesham, G. D. Sharma, M. Sharma and A. Bhargava, Phytother. Res., 2021, 35, 256–277 CrossRef CAS PubMed.
- A. Falanga, E. Nigro, M. G. De Biasi, A. Daniele, G. Morelli, S. Galdiero and O. Scudiero, Molecules, 2017, 22, 1217 CrossRef PubMed.
- C. K. Wang and D. J. Craik, Nat. Chem. Biol., 2018, 14, 417–427 CrossRef CAS PubMed.
- A. Gould and J. A. Camarero, ChemBioChem, 2017, 18, 1350–1363 CrossRef CAS PubMed.
- M. E. Selsted and A. J. Ouellette, Nat. Immunol., 2005, 6, 551–557 CrossRef CAS PubMed.
- X. Gao, J. Ding, C. Liao, J. Xu, X. Liu and W. Lu, Adv. Drug Delivery Rev., 2021, 179, 114008 CrossRef CAS PubMed.
- M. E. Selsted, Curr. Protein Pept. Sci., 2004, 5, 365–371 CrossRef CAS PubMed.
- A. C. Conibear and D. J. Craik, Angew. Chem., Int. Ed., 2014, 53, 10612–10623 CrossRef CAS PubMed.
- J. P. Tam, S. Wang, K. H. Wong and W. L. Tan, Pharmaceuticals, 2015, 8, 711–757 CrossRef CAS PubMed.
- J. Koehbach and D. J. Craik, Trends Pharmacol. Sci., 2019, 40, 517–528 CrossRef CAS PubMed.
- R. I. Lehrer and W. Lu, Immunol. Rev., 2012, 245, 84–112 CrossRef CAS PubMed.
- B. L. Kagan, M. E. Selsted, T. Ganz and R. I. Lehrer, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 210–214 CrossRef CAS PubMed.
- R. Lehrer, A. Barton, K. A. Daher, S. S. L. Harwig, T. Ganz and M. E. Selsted, J. Clin. Invest., 1989, 84, 553–561 CrossRef CAS PubMed.
- E. de Leeuw, C. Li, P. Zeng, C. Li, M. Diepeveen-de Buin, W. Y. Lu, E. Breukink and W. Lu, FEBS Lett., 2010, 584, 1543–1548 CrossRef CAS PubMed.
- F. Niyonsaba, H. Ushio, N. Nakano, W. Ng, K. Sayama, K. Hashimoto, I. Nagaoka, K. Okumura and H. Ogawa, J. Invest. Dermatol., 2007, 127, 594–604 CrossRef CAS PubMed.
- L. Steinstraesser, T. Koehler, F. Jacobsen, A. Daigeler, O. Goertz, S. Langer, M. Kesting, H. Steinau, E. Eriksson and T. Hirsch, Mol. Med., 2008, 14, 528–537 CAS.
- D. Yang, Q. Chen, O. Chertov and J. J. Oppenheim, J. Leukocyte Biol., 2000, 68, 9–14 CrossRef CAS PubMed.
- M. C. Territo, T. Ganz, M. E. Selsted and R. Lehrer, J. Clin. Invest., 1989, 84, 2017–2020 CrossRef CAS PubMed.
- D. Yang, Z. H. Liu, P. Tewary, Q. Chen, G. de la Rosa and J. J. Oppenheim, Curr. Pharm. Des., 2007, 13, 3131–3139 CrossRef CAS PubMed.
- L. M. Rehaume and R. E. Hancock, Crit. Rev. Immunol., 2008, 28, 185–200 CrossRef CAS PubMed.
- M. G. Scott, A. C. Vreugdenhil, W. A. Buurman, R. E. Hancock and M. R. Gold, J. Immunol., 2000, 164, 549–553 CrossRef CAS PubMed.
- D. Motzkus, S. Schulz-Maronde, A. Heitland, A. Schulz, W. G. Forssmann, M. Jubner and E. Maronde, FASEB J., 2006, 20, 1701–1702 CrossRef CAS PubMed.
- N. Droin, J. B. Hendra, P. Ducoroy and E. Solary, J. Proteomics, 2009, 72, 918–927 CrossRef CAS PubMed.
- E. Kudryashova, R. Quintyn, S. Seveau, W. Lu, V. H. Wysocki and D. S. Kudryashov, Immunity, 2014, 41, 709–721 CrossRef CAS PubMed.
- R. I. Lehrer, G. Jung, P. Ruchala, W. Wang, E. D. Micewicz, A. J. Waring, E. J. Gillespie, K. A. Bradley, A. J. Ratner, R. F. Rest and W. Lu, Infect. Immun., 2009, 77, 4028–4040 CrossRef CAS PubMed.
- G. A. D. Blyth, L. Connors, C. Fodor and E. R. Cobo, Front. Immunol., 2020, 11, 965 CrossRef CAS PubMed.
- R. Bals, M. J. Goldman and J. M. Wilson, Infect. Immun., 1998, 66, 1225–1232 CrossRef CAS PubMed.
- Y. Q. Tang, J. Yuan, G. Osapay, K. Osapay, D. Tran, C. J. Miller, A. J. Ouellette and M. E. Selsted, Science, 1999, 286, 498–502 CrossRef CAS PubMed.
- Q. Yu, R. I. Lehrer and J. P. Tam, J. Biol. Chem., 2000, 275, 3943–3949 CrossRef CAS PubMed.
- A. E. Garcia, K. P. Tai, S. S. Puttamadappa, A. Shekhtman, A. J. Ouellette and J. A. Camarero, Biochemistry, 2011, 50, 10508–10519 CrossRef CAS PubMed.
- A. Maemoto, X. Qu, K. J. Rosengren, H. Tanabe, A. Henschen-Edman, D. J. Craik and A. J. Ouellette, J. Biol. Chem., 2004, 279, 44188–44196 CrossRef CAS PubMed.
- M. E. Pachon-Ibanez, Y. Smani, J. Pachon and J. Sanchez-Cespedes, FEMS Microbiol. Rev., 2017, 41, 323–342 CrossRef CAS PubMed.
- I. Ben-Efraim and Y. Shai, Biophys. J., 1997, 72, 85–96 CrossRef CAS PubMed.
- A. Mor and P. Nicolas, J. Biol. Chem., 1994, 269, 1934–1939 CrossRef CAS PubMed.
- Z. Oren and Y. Shai, Biopolymers, 1998, 47, 451–463 CrossRef CAS PubMed.
- R. M. Dawson and C. Q. Liu, Crit. Rev. Microbiol., 2008, 34, 89–107 CrossRef CAS PubMed.
- C. J. Pridmore, A. Rodger and J. M. Sanderson, Biochim. Biophys. Acta, 2016, 1858, 892–903 CrossRef CAS PubMed.
- B. Ericksen, Z. Wu, W. Lu and R. I. Lehrer, Antimicrob. Agents Chemother., 2005, 49, 269–275 CrossRef CAS PubMed.
- B. O. Schroeder, D. Ehmann, J. C. Precht, P. A. Castillo, R. Kuchler, J. Berger, M. Schaller, E. F. Stange and J. Wehkamp, Mucosal Immunol., 2015, 8, 661–671 CrossRef CAS PubMed.
- B. Mathew and R. Nagaraj, Peptides, 2015, 71, 128–140 CrossRef CAS PubMed.
- B. O. Schroeder, Z. Wu, S. Nuding, S. Groscurth, M. Marcinowski, J. Beisner, J. Buchner, M. Schaller, E. F. Stange and J. Wehkamp, Nature, 2011, 469, 419–423 CrossRef CAS PubMed.
- K. R. Parducho, B. Beadell, T. K. Ybarra, M. Bush, E. Escalera, A. T. Trejos, A. Chieng, M. Mendez, C. Anderson, H. Park, Y. Wang, W. Lu and E. Porter, Front. Immunol., 2020, 11, 805 CrossRef CAS PubMed.
- V. Sass, T. Schneider, M. Wilmes, C. Korner, A. Tossi, N. Novikova, O. Shamova and H. G. Sahl, Infect. Immun., 2010, 78, 2793–2800 CrossRef CAS PubMed.
- A. C. Conibear, C. K. Wang, T. Bi, K. J. Rosengren, J. A. Camarero and D. J. Craik, J. Phys. Chem. B, 2014, 118, 14257–14266 CrossRef CAS PubMed.
- A. C. Conibear, K. J. Rosengren, N. L. Daly, S. T. Henriques and D. J. Craik, J. Biol. Chem., 2013, 288, 10830–10840 CrossRef CAS PubMed.
- W. Wang, C. Mulakala, S. C. Ward, G. Jung, H. Luong, D. Pham, A. J. Waring, Y. Kaznessis, W. Lu, K. A. Bradley and R. I. Lehrer, J. Biol. Chem., 2006, 281, 32755–32764 CrossRef CAS PubMed.
- Y. Li, A. Gould, T. Aboye, T. Bi, L. Breindel, A. Shekhtman and J. A. Camarero, J. Med. Chem., 2017, 60, 1916–1927 CrossRef CAS PubMed.
- T. Bi, Y. Li, A. Shekhtman and J. A. Camarero, Bioorg. Med. Chem., 2018, 26, 1212–1219 CrossRef CAS PubMed.
- A. Gould, Y. Li, S. Majumder, A. E. Garcia, P. Carlsson, A. Shekhtman and J. A. Camarero, Mol. BioSyst., 2012, 8, 1359–1365 RSC.
- T. L. Aboye, Y. Li, S. Majumder, J. Hao, A. Shekhtman and J. A. Camarero, Bioorg. Med. Chem. Lett., 2012, 22, 2823–2826 CrossRef CAS PubMed.
- R. A. Houghten, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5131–5135 CrossRef CAS PubMed.
- T. J. Bensman, J. G. Jayne, M. Sun, E. Kimura, J. Meinert, J. C. Wang, J. B. Schaal, D. Tran, A. P. Rao, O. Akbari, M. E. Selsted and P. M. Beringer, Antimicrob. Agents Chemother., 2017, 61, e00154-17 CrossRef PubMed.
- A. Seidel, Y. Ye, L. R. de Armas, M. Soto, W. Yarosh, R. A. Marcsisin, D. Tran, M. E. Selsted and D. Camerini, PLoS One, 2010, 5, e9737 CrossRef PubMed.
- J. B. Schaal, D. Tran, P. Tran, G. Osapay, K. Trinh, K. D. Roberts, K. M. Brasky, P. Tongaonkar, A. J. Ouellette and M. E. Selsted, PLoS One, 2012, 7, e51337 CrossRef CAS PubMed.
- E. M. Kosciuczuk, P. Lisowski, J. Jarczak, N. Strzalkowska, A. Jozwik, J. Horbanczuk, J. Krzyzewski, L. Zwierzchowski and E. Bagnicka, Mol. Biol. Rep., 2012, 39, 10957–10970 CrossRef CAS PubMed.
- V. N. Kokryakov, S. S. L. Harwig, E. A. Panyutich, A. A. Shevchenko, G. M. Aleshina, O. V. Shamova, H. A. Korneva and R. I. Lehrer, FEBS Lett., 1993, 327, 231–236 CrossRef CAS PubMed.
- T. Nakamura, H. Furunaka, T. Miyata, F. Tokunaga, T. Muta, S. Iwanaga, M. Niwa, T. Takao and Y. Shimonishi, J. Biol. Chem., 1988, 263, 16709–16713 CrossRef CAS PubMed.
- R. L. Fahrner, T. Dieckmann, S. S. Harwig, R. I. Lehrer, D. Eisenberg and J. Feigon, Chem. Biol., 1996, 3, 543–550 CrossRef CAS PubMed.
- L. Bellm, R. I. Lehrer and T. Ganz, Expert Opin. Invest. Drugs, 2000, 9, 1731–1742 CrossRef CAS PubMed.
- B. Yasin, S. S. Harwig, R. I. Lehrer and E. A. Wagar, Infect. Immun., 1996, 64, 709–713 CrossRef CAS PubMed.
- H. Tamamura, A. Otaka, W. Takada, Y. Terakawa, H. Yoshizawa, M. Masuda, T. Ibuka, T. Murakami, H. Nakashima and M. Waki, et al. , Chem. Pharm. Bull., 1995, 43, 12–18 CrossRef CAS PubMed.
- B. Yasin, W. Wang, M. Pang, N. Cheshenko, T. Hong, A. J. Waring, B. C. Herold, E. A. Wagar and R. I. Lehrer, J. Virol., 2004, 78, 5147–5156 CrossRef CAS PubMed.
- J. P. Tam, C. Wu and J. L. Yang, Eur. J. Biochem., 2000, 267, 3289–3300 CrossRef CAS PubMed.
- E. Kudryashova, S. M. Seveau and D. S. Kudryashov, Biol. Chem., 2017, 398, 1069–1085 CAS.
- N. Soundrarajan, S. Park, Q. L. V. Chanh, H. S. Cho, G. Raghunathan, B. Ahn, H. Song, J. H. Kim and C. Park, Sci. Rep., 2019, 9, 11569 CrossRef PubMed.
- M. B. Marggraf, P. V. Panteleev, A. A. Emelianova, M. I. Sorokin, I. A. Bolosov, A. A. Buzdin, D. V. Kuzmin and T. V. Ovchinnikova, Mar. Drugs, 2018, 16, 466 CrossRef CAS PubMed.
- J. Chen, T. J. Falla, H. Liu, M. A. Hurst, C. A. Fujii, D. A. Mosca, J. R. Embree, D. J. Loury, P. A. Radel, C. Cheng Chang, L. Gu and J. C. Fiddes, Biopolymers, 2000, 55, 88–98 CrossRef CAS PubMed.
- R. Ganesan, M. A. Dughbaj, L. Ramirez, S. Beringer, T. L. Aboye, A. Shekhtman, P. M. Beringer and J. A. Camarero, Chemistry, 2021, 27, 12702–12708 CrossRef CAS PubMed.
- D. A. Mosca, M. A. Hurst, W. So, B. S. Viajar, C. A. Fujii and T. J. Falla, Antimicrob. Agents Chemother., 2000, 44, 1803–1808 CrossRef CAS PubMed.
- O. Simonetti, O. Cirioni, R. Ghiselli, F. Orlando, C. Silvestri, S. Mazzocato, W. Kamysz, E. Kamysz, M. Provinciali, A. Giacometti, M. Guerrieri and A. Offidani, Peptides, 2014, 55, 17–22 CrossRef CAS PubMed.
- B. Chaveli-Lopez and J. V. Bagan-Sebastian, J. Clin. Exp. Dent., 2016, 8, e201–209 Search PubMed.
- I. Martin-Loeches, G. E. Dale and A. Torres, Expert Rev. Anti-Infect. Ther., 2018, 16, 259–268 CrossRef CAS PubMed.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, B. Van der Meijden, F. Bernardini, A. Lederer, R. L. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Kach, L. Eberl, K. Riedel, S. J. DeMarco and J. A. Robinson, Science, 2010, 327, 1010–1013 CrossRef CAS PubMed.
- A. Luther, K. Moehle, E. Chevalier, G. Dale and D. Obrecht, Curr. Opin. Chem. Biol., 2017, 38, 45–51 CrossRef CAS PubMed.
- H. S. Sader, G. E. Dale, P. R. Rhomberg and R. K. Flamm, Antimicrob. Agents Chemother., 2018, 62, e00311-18 CrossRef PubMed.
- H. S. Sader, R. K. Flamm, G. E. Dale, P. R. Rhomberg and M. Castanheira, J. Antimicrob. Chemother., 2018, 73, 2400–2404 CrossRef CAS PubMed.
- D. Chaudhuri, R. Ganesan, A. Vogelaar, M. A. Dughbaj, P. M. Beringer and J. A. Camarero, J. Org. Chem., 2021, 86, 15242–15246 CrossRef CAS PubMed.
- A. Wach, K. Dembowsky and G. E. Dale, Antimicrob. Agents Chemother., 2018, 62, e02355-17 CrossRef PubMed.
- J. Turner, Y. Cho, N. N. Dinh, A. J. Waring and R. I. Lehrer, Antimicrob. Agents Chemother., 1998, 42, 2206–2214 CrossRef CAS PubMed.
- M. Shahmiri, M. Enciso, C. G. Adda, B. J. Smith, M. A. Perugini and A. Mechler, Sci. Rep., 2016, 6, 38184 CrossRef CAS PubMed.
- M. Mahlapuu, A. Sidorowicz, J. Mikosinski, M. Krzyzanowski, J. Orleanski, K. Twardowska-Saucha, A. Nykaza, M. Dyaczynski, B. Belz-Lagoda, G. Dziwiszek, M. Kujawiak, M. Karczewski, F. Sjoberg, T. Grzela, A. Wegrzynowski, F. Thunarf, J. Bjork, J. Ekblom, A. Jawien and J. Apelqvist, Wound Repair Regen., 2021, 29, 938–950 CrossRef PubMed.
- J. K. White, T. Muhammad, E. Alsheim, S. Mohanty, A. Blasi-Romero, S. Gunasekera, A. A. Stromstedt, N. Ferraz, U. Goransson and A. Brauner, Cell. Mol. Life Sci., 2022, 79, 411 CrossRef CAS PubMed.
- X. Li, Y. Li, H. Han, D. W. Miller and G. Wang, J. Am. Chem. Soc., 2006, 128, 5776–5785 CrossRef CAS PubMed.
- G. Wang, J. Biol. Chem., 2008, 283, 32637–32643 CrossRef CAS PubMed.
- S. X. Ren, J. Shen, A. S. Cheng, L. Lu, R. L. Chan, Z. J. Li, X. J. Wang, C. C. Wong, L. Zhang, S. S. Ng, F. L. Chan, F. K. Chan, J. Yu, J. J. Sung, W. K. Wu and C. H. Cho, PLoS One, 2013, 8, e63641 CrossRef CAS PubMed.
- S. Gunasekera, T. Muhammad, A. A. Stromstedt, K. J. Rosengren and U. Goransson, ChemBioChem, 2018, 19, 931–939 CrossRef CAS PubMed.
- S. Gunasekera, T. Muhammad, A. A. Stromstedt, K. J. Rosengren and U. Goransson, Front. Microbiol., 2020, 11, 168 CrossRef PubMed.
- J. Weidmann and D. J. Craik, J. Exp. Bot., 2016, 67, 4801–4812 CrossRef CAS PubMed.
- J. A. Camarero and M. J. Campbell, Biomedicines, 2019, 7, 31 CrossRef CAS PubMed.
- K. J. Rosengren, N. L. Daly, M. R. Plan, C. Waine and D. J. Craik, J. Biol. Chem., 2003, 278, 8606–8616 CrossRef CAS PubMed.
- C. T. Wong, D. K. Rowlands, C. H. Wong, T. W. Lo, G. K. Nguyen, H. Y. Li and J. P. Tam, Angew. Chem., Int. Ed., 2012, 51, 5620–5624 CrossRef CAS PubMed.
- K. Thell, R. Hellinger, G. Schabbauer and C. W. Gruber, Drug Discovery Today, 2014, 19, 645–653 CrossRef CAS PubMed.
- L. Cascales, S. T. Henriques, M. C. Kerr, Y. H. Huang, M. J. Sweet, N. L. Daly and D. J. Craik, J. Biol. Chem., 2011, 286, 36932–36943 CrossRef CAS PubMed.
- J. Contreras, A. Y. Elnagar, S. F. Hamm-Alvarez and J. A. Camarero, J. Controlled Release, 2011, 155, 134–143 CrossRef CAS PubMed.
- W. G. Lesniak, T. Aboye, S. Chatterjee, J. A. Camarero and S. Nimmagadda, Chemistry, 2017, 23, 14469–14475 CrossRef CAS PubMed.
- T. L. Aboye, H. Ha, S. Majumder, F. Christ, Z. Debyser, A. Shekhtman, N. Neamati and J. A. Camarero, J. Med. Chem., 2012, 55, 10729–10734 CrossRef CAS PubMed.
- T. Aboye, Y. Kuang, N. Neamati and J. A. Camarero, ChemBioChem, 2015, 16, 827–833 CrossRef CAS PubMed.
- Y. Ji, S. Majumder, M. Millard, R. Borra, T. Bi, A. Y. Elnagar, N. Neamati, A. Shekhtman and J. A. Camarero, J. Am. Chem. Soc., 2013, 135, 11623–11633 CrossRef CAS PubMed.
- D. J. Craik, R. J. Clark and N. L. Daly, Expert Opin. Invest. Drugs, 2007, 16, 595–604 CrossRef CAS PubMed.
- B. Slazak, M. Kapusta, S. Malik, J. Bohdanowicz, E. Kuta, P. Malec and U. Goransson, Planta, 2016, 244, 1029–1040 CrossRef CAS PubMed.
- J. A. Camarero, Bioorg. Med. Chem. Lett., 2017, 27, 5089–5099 CrossRef CAS PubMed.
- C. Jennings, J. West, C. Waine, D. Craik and M. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10614–10619 CrossRef CAS PubMed.
- C. V. Jennings, K. J. Rosengren, N. L. Daly, M. Plan, J. Stevens, M. J. Scanlon, C. Waine, D. G. Norman, M. A. Anderson and D. J. Craik, Biochemistry, 2005, 44, 851–860 CrossRef CAS PubMed.
- A. G. Poth, M. L. Colgrave, R. E. Lyons, N. L. Daly and D. J. Craik, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10127–10132 CrossRef CAS PubMed.
- M. F. Pinto, I. C. Fensterseifer, L. Migliolo, D. A. Sousa, G. de Capdville, J. W. Arboleda-Valencia, M. L. Colgrave, D. J. Craik, B. S. Magalhaes, S. C. Dias and O. L. Franco, J. Biol. Chem., 2012, 287, 134–147 CrossRef CAS PubMed.
- D. J. Craik, Toxins, 2012, 4, 139–156 CrossRef CAS PubMed.
- E. K. Gilding, M. A. Jackson, A. G. Poth, S. T. Henriques, P. J. Prentis, T. Mahatmanto and D. J. Craik, New Phytol., 2016, 210, 717–730 CrossRef CAS PubMed.
- M. L. Colgrave, A. C. Kotze, Y. H. Huang, J. O'Grady, S. M. Simonsen and D. J. Craik, Biochemistry, 2008, 47, 5581–5589 CrossRef CAS PubMed.
- M. L. Colgrave, A. C. Kotze, D. C. Ireland, C. K. Wang and D. J. Craik, ChemBioChem, 2008, 9, 1939–1945 CrossRef CAS PubMed.
- D. Malagon, B. Botterill, D. J. Gray, E. Lovas, M. Duke, C. Gray, S. R. Kopp, L. M. Knott, D. P. McManus, N. L. Daly, J. Mulvenna, D. J. Craik and M. K. Jones, Biopolymers, 2013, 100, 461–470 CrossRef CAS PubMed.
- S. Troeira Henriques and D. J. Craik, Biochemistry, 2017, 56, 669–682 CrossRef CAS PubMed.
- A. A. Stromstedt, S. Park, R. Burman and U. Goransson, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1986–2000 CrossRef PubMed.
- J. P. Tam, Y. A. Lu, J. L. Yang and K. W. Chiu, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8913–8918 CrossRef CAS PubMed.
- G. K. Nguyen, S. Zhang, W. Wang, C. T. Wong, N. T. Nguyen and J. P. Tam, J. Biol. Chem., 2011, 286, 44833–44844 CrossRef CAS PubMed.
- C. T. Wong, M. Taichi, H. Nishio, Y. Nishiuchi and J. P. Tam, Biochemistry, 2011, 50, 7275–7283 CrossRef CAS PubMed.
- G. K. Nguyen, S. Zhang, N. T. Nguyen, P. Q. Nguyen, M. S. Chiu, A. Hardjojo and J. P. Tam, J. Biol. Chem., 2011, 286, 24275–24287 CrossRef CAS PubMed.
- M. Pranting, C. Loov, R. Burman, U. Goransson and D. I. Andersson, J. Antimicrob. Chemother., 2010, 65, 1964–1971 CrossRef PubMed.
- I. C. Fensterseifer, O. N. Silva, U. Malik, A. S. Ravipati, N. R. Novaes, P. R. Miranda, E. A. Rodrigues, S. E. Moreno, D. J. Craik and O. L. Franco, Peptides, 2015, 63, 38–42 CrossRef CAS PubMed.
- E. Grimsey, D. W. P. Collis, R. Mikut and K. Hilpert, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183195 CrossRef CAS PubMed.
- J. Koehbach, J. Gani, K. Hilpert and D. J. Craik, Microorganisms, 2021, 9, 1249 CrossRef CAS PubMed.
- S. J. de Veer, A. M. White and D. J. Craik, Angew. Chem., Int. Ed., 2021, 60, 8050–8071 CrossRef CAS PubMed.
- M. L. Korsinczky, H. J. Schirra and D. J. Craik, Curr. Protein Pept. Sci., 2004, 5, 351–364 CrossRef CAS PubMed.
- M. L. Korsinczky, H. J. Schirra, K. J. Rosengren, J. West, B. A. Condie, L. Otvos, M. A. Anderson and D. J. Craik, J. Mol. Biol., 2001, 311, 579–591 CrossRef CAS PubMed.
- J. Austin, R. H. Kimura, Y. H. Woo and J. A. Camarero, Amino Acids, 2010, 38, 1313–1322 CrossRef CAS PubMed.
- C. Wang, C. Shao, Y. Fang, J. Wang, N. Dong and A. Shan, Acta Biomater., 2021, 124, 254–269 CrossRef CAS PubMed.
- Y. L. Vishweshwaraiah, A. Acharya, V. Hegde and B. Prakash, NPJ Sci. Food, 2021, 5, 26 CrossRef PubMed.
- J. D. Tanner, E. Deplazes and R. L. Mancera, Molecules, 2018, 23, 1733 CrossRef PubMed.
- N. Mandard, P. Bulet, A. Caille, S. Daffre and F. Vovelle, Eur. J. Biochem., 2002, 269, 1190–1198 CrossRef CAS PubMed.
- S. Troeira Henriques, N. Lawrence, S. Chaousis, A. S. Ravipati, O. Cheneval, A. H. Benfield, A. G. Elliott, A. M. Kavanagh, M. A. Cooper, L. Y. Chan, Y. H. Huang and D. J. Craik, ACS Chem. Biol., 2017, 12, 2324–2334 CrossRef CAS PubMed.
- L. Y. Chan, V. M. Zhang, Y. H. Huang, N. C. Waters, P. S. Bansal, D. J. Craik and N. L. Daly, ChemBioChem, 2013, 14, 617–624 CrossRef CAS PubMed.
- M. A. Fazio, V. X. Oliveira, Jr., P. Bulet, M. T. Miranda, S. Daffre and A. Miranda, Biopolymers, 2006, 84, 205–218 CrossRef CAS PubMed.
- A. H. Benfield, S. Defaus, N. Lawrence, S. Chaousis, N. Condon, O. Cheneval, Y. H. Huang, L. Y. Chan, D. Andreu, D. J. Craik and S. T. Henriques, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183480 CrossRef CAS PubMed.
- P. Fehlbaum, P. Bulet, S. Chernysh, J. P. Briand, J. P. Roussel, L. Letellier, C. Hetru and J. A. Hoffmann, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1221–1225 CrossRef CAS PubMed.
- R. Dash and S. Bhattacharjya, Int. J. Mol. Sci., 2021, 22, 1522 CrossRef CAS PubMed.
- G. Wu, P. Wu, X. Xue, X. Yan, S. Liu, C. Zhang, Z. Shen and T. Xi, Peptides, 2013, 45, 73–77 CrossRef CAS PubMed.
- Z. Hou, J. Lu, C. Fang, Y. Zhou, H. Bai, X. Zhang, X. Xue, Y. Chen and X. Luo, J. Infect. Dis., 2011, 203, 273–282 CrossRef CAS PubMed.
- S. U. Vetterli, K. Zerbe, M. Muller, M. Urfer, M. Mondal, S. Y. Wang, K. Moehle, O. Zerbe, A. Vitale, G. Pessi, L. Eberl, B. Wollscheid and J. A. Robinson, Sci. Adv., 2018, 4, eaau2634 CrossRef CAS PubMed.
- F. Akram, M. Imtiaz and I. U. Haq, Microb. Pathog., 2023, 174, 105923 CrossRef CAS PubMed.
- A. Hollmann, M. Martinez, P. Maturana, L. C. Semorile and P. C. Maffia, Front. Chem., 2018, 6, 204 CrossRef PubMed.
- D. Chaudhuri, T. Aboye and J. A. Camarero, Biochem. J., 2019, 476, 67–83 CrossRef CAS PubMed.
- A. Pfalzgraff, K. Brandenburg and G. Weindl, Front. Pharmacol., 2018, 9, 281 CrossRef PubMed.
- Y. Ji, S. Majumder, M. Millard, R. Borra, T. Bi, A. Y. Elnagar, N. Neamati, A. Shekhtman and J. A. Camarero, J. Am. Chem. Soc., 2013, 135, 11623–11633 CrossRef CAS PubMed.
- T. Aboye, C. J. Meeks, S. Majumder, A. Shekhtman, K. Rodgers and J. A. Camarero, Molecules, 2016, 21, 152 CrossRef PubMed.
- B. Jacob, A. Vogelaar, E. Cadenas and J. A. Camarero, Molecules, 2022, 27, 6430 CrossRef CAS PubMed.
- F. F. Andrade, D. Silva, A. Rodrigues and C. Pina-Vaz, Microorganisms, 2020, 8, 1716 CrossRef CAS PubMed.
- K. Thell, R. Hellinger, E. Sahin, P. Michenthaler, M. Gold-Binder, T. Haider, M. Kuttke, Z. Liutkeviciute, U. Goransson, C. Grundemann, G. Schabbauer and C. W. Gruber, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3960–3965 CrossRef CAS PubMed.
- M. Trabi, H. J. Schirra and D. J. Craik, Biochemistry, 2001, 40, 4211–4221 CrossRef CAS PubMed.
- M. L. J. Korsinczky, H. J. Schirra, K. J. Rosengren, J. West, B. A. Condie, L. Otvos, M. A. Anderson and D. J. Craik, J. Mol. Biol., 2001, 311, 579–591 CrossRef CAS PubMed.
- K. J. Rosengren, N. L. Daly, L. M. Fornander, L. M. Jonsson, Y. Shirafuji, X. Qu, H. J. Vogel, A. J. Ouellette and D. J. Craik, J. Biol. Chem., 2006, 281, 28068–28078 CrossRef CAS PubMed.
- K. J. Rosengren, N. L. Daly, M. R. Plan, C. Waine and D. J. Craik, J. Biol. Chem., 2003, 278, 8606–8616 CrossRef CAS PubMed.
- D. M. Hoover, O. Chertov and J. Lubkowski, J. Biol. Chem., 2001, 276, 39021–39026 CrossRef CAS PubMed.
- D. M. Hoover, K. R. Rajashankar, R. Blumenthal, A. Puri, J. J. Oppenheim, O. Chertov and J. Lubkowski, J. Biol. Chem., 2000, 275, 32911–32918 CrossRef CAS PubMed.
- D. J. Schibli, H. N. Hunter, V. Aseyev, T. D. Starner, J. M. Wiencek, P. B. McCray, Jr., B. F. Tack and H. J. Vogel, J. Biol. Chem., 2002, 277, 8279–8289 CrossRef CAS PubMed.
- M. Trabi, H. J. Schirra and D. J. Craik, Biochemistry, 2001, 4211–4221 CrossRef CAS PubMed.
- E. Sancho-Vaello, D. Gil-Carton, P. Francois, E. J. Bonetti, M. Kreir, K. R. Pothula, U. Kleinekathofer and K. Zeth, Sci. Rep., 2020, 10, 17356 CrossRef CAS PubMed.
- Q. Y. Zhang, Z. B. Yan, Y. M. Meng, X. Y. Hong, G. Shao, J. J. Ma, X. R. Cheng, J. Liu, J. Kang and C. Y. Fu, Mil. Med. Res., 2021, 8, 48 CAS.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |