
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnmasking the constitution and bonding of the proposed lithium nickelate “Li3NiPh3(solv)3”: revealing the hidden C6H4 ligand†
Rosie J.
Somerville‡
 a,
Andryj M.
Borys‡
b,
Marina
Perez-Jimenez
a,
Ainara
Nova
c,
David
Balcells
c,
Lorraine A.
Malaspina
b,
Simon
Grabowsky
b,
Ernesto
Carmona
a,
Eva
Hevia
*b and
Jesús
Campos
*a
a,
Andryj M.
Borys‡
b,
Marina
Perez-Jimenez
a,
Ainara
Nova
c,
David
Balcells
c,
Lorraine A.
Malaspina
b,
Simon
Grabowsky
b,
Ernesto
Carmona
a,
Eva
Hevia
*b and
Jesús
Campos
*a
aInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica, Centro de Innovación en Química Avanzada (ORFEO-CINQA), Consejo Superior de Investigaciones Científicas (CSIC), University of Sevilla, Avenida Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: jesus.campos@iiq.csic.es
bDepartement für Chemie, Biochemie und Pharmazie, Universität Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: eva.hevia@unibe.ch
cHylleraas Centre for Quantum Molecular Sciences, Centre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, 0315 Oslo, Norway
First published on 11th April 2022
Abstract
More than four decades ago, a complex identified as the planar homoleptic lithium nickelate “Li3NiPh3(solv)3” was reported by Taube and co-workers. This and subsequent reports involving this complex have lain dormant since; however, the absence of an X-ray diffraction structure leaves questions as to the nature of the Ni–PhLi bonding and the coordination geometry at Ni. By systematically evaluating the reactivity of Ni(COD)2 with PhLi under different conditions, we have found that this classical molecule is instead a unique octanuclear complex, [{Li3(solv)2Ph3Ni}2(μ-η2:η2-C6H4)] (5). This is supported by X-ray crystallography and solution-state NMR studies. A theoretical bonding analysis from NBO, QTAIM, and ELI perspectives reveals extreme back-bonding to the bridging C6H4 ligand resulting in dimetallabicyclobutane character, the lack of a Ni–Ni bond, and pronounced σ-bonding between the phenyl carbanions and nickel, including a weak σC–Li → sNi interaction with the C–Li bond acting as a σ-donor. Employing PhNa led to the isolation of [Na2(solv)3Ph2NiCOD]2 (7) and [Na2(solv)3Ph2(NaC8H11)Ni(COD)]2 (8), which lack the benzyne-derived ligand. These findings provide new insights into the synthesis, structure, bonding and reactivity of heterobimetallic nickelates, whose prevalence in organonickel chemistry and catalysis is likely greater than previously believed.
Introduction
During the last decade, organonickel chemistry has undergone a period of rapid growth, particularly when it comes to the development of new reactions that take advantage of nickel's accessible one-electron mechanistic steps, the often favorable oxidative addition of strong bonds to nickel, and its lower cost compared to platinum-group metals.1,2 As this chemistry progresses, it remains based on fundamental knowledge, be it in understanding mechanistic steps, ligand choices, or the nature of the bonding interactions. A key aspect of the latter is the work that was carried out around half a century ago involving low-valent and highly reduced Ni species.3 This continues to be relevant today; for example, early work involving the isolation of nickelate complexes with Ni–E interactions (E = Li, Mg) has recently been implicated in catalytic cross-coupling reactions by Hevia and Cornella.4–6 Other early work led to the isolation of zerovalent trigonal species derived from organosodium or organolithium reagents.7–11 In parallel, σ-organyl complexes of first row transition metals (Ni, Fe and Co) were synthesised by Taube and others and transformed into intriguing complexes again bearing organolithium ligands.12–19 Finally, there is also the finding that organolithium reagents may behave as oxidants in certain cases.20Amongst these complexes, our attention was drawn to “Li3NiPh3(solv)3”, which was proposed to adopt a planar geometry based exclusively on 13C NMR spectroscopy (Scheme 1).13,16,19,21 Although it seems to have been almost completely overlooked since its publication in 1979, the implications of this purported homoleptic phenyllithium complex are numerous. First, without crystallographic studies it is not clear whether the “Li3NiPh3(solv)3” complex contains the extremely rare hexagonal planar coordination geometry recently reported by the Crimmin group for a Pd complex.22 Furthermore, the coordination of Li–C bonds to transition metals is considerably less explored than that of E–H bonds, where E is a main-group element such as Li, Al, Mg or Zn.23–25 The binding of three phenyllithium molecules to a Ni(0) centre might imply multi-centre-multi-electron interactions, suggested by Jonas and co-workers,26–28 that can now be investigated by computational methods. With the above in mind, we aimed to investigate the existence of “Li3NiPh3(solv)3” and to explore its structure, bonding and reactivity. Overall, our results do not support the existence of “Li3NiPh3(solv)3” and instead reveal a unique dinickel complex [{Li3(solv)2Ph3Ni}2(μ-η2:η2-C6H4)] containing a bridging benzyne-type moiety (Scheme 1). Experimental and computational studies have been performed to understand the nature of the bonding and the structural features of this and related alkali-metal nickelates. Preliminary investigations into the reactivity and catalytic activity of this exotic complex are also provided.
 | ||
| Scheme 1 Investigation of Taube's “Li3NiPh3(solv)3”. | ||
Results and discussion
Taube's initial report presented us with two synthetic routes to “Li3NiPh3(THF)3”: (i) ligand substitution from Ni(COD)2 (COD = 1,5-cyclooctadiene) via “Li2Ph2Ni(COD)” and; (ii) reduction of Li2NiPh4(THF)4 with excess PhLi.13,16,18 However, the exact experimental conditions necessary for the synthesis and isolation of “Li3NiPh3(THF)3” from either route were not specified, so each route was investigated in turn. The first route involves the formal displacement of COD by PhLi. Following pioneering work from Jonas,3,9 Borys and Hevia treated Ni(COD)2 with PhLi in THF and obtained three isolable COD complexes (1–3) depending on the stoichiometry and crystallisation conditions (Fig. 1a).6 When a similar reaction is performed with PhLi prepared from PhBr and tBuLi, which gives [(PhLi·Et2O)3LiBr],29 the lithium nickelate obtained, [Li2(Et2O)4(LiBr)Ph2Ni]2COD (4), contains co-complexed LiBr (Fig. 1b; see ESI† for full details and solid-state structure). No spectroscopic evidence for Taube's proposed planar complex was observed in these experiments. Following Taube's ligand substitution route, we envisioned that we could displace the COD ligands by treating intermediate complexes 1, 2 or 4 with additional PhLi, or, more conveniently, by treating Ni(COD)2 directly with excess PhLi. Thus, Ni(COD)2 was treated with donor-free PhLi (5 equiv.) and refluxed in a mixture of THF and hexane as described by Taube. A deep red solution was obtained as indicated in Taube's reports. However, signals originally attributed to “Li3NiPh3(THF)3” were not detected by 1H and 13C NMR spectroscopy.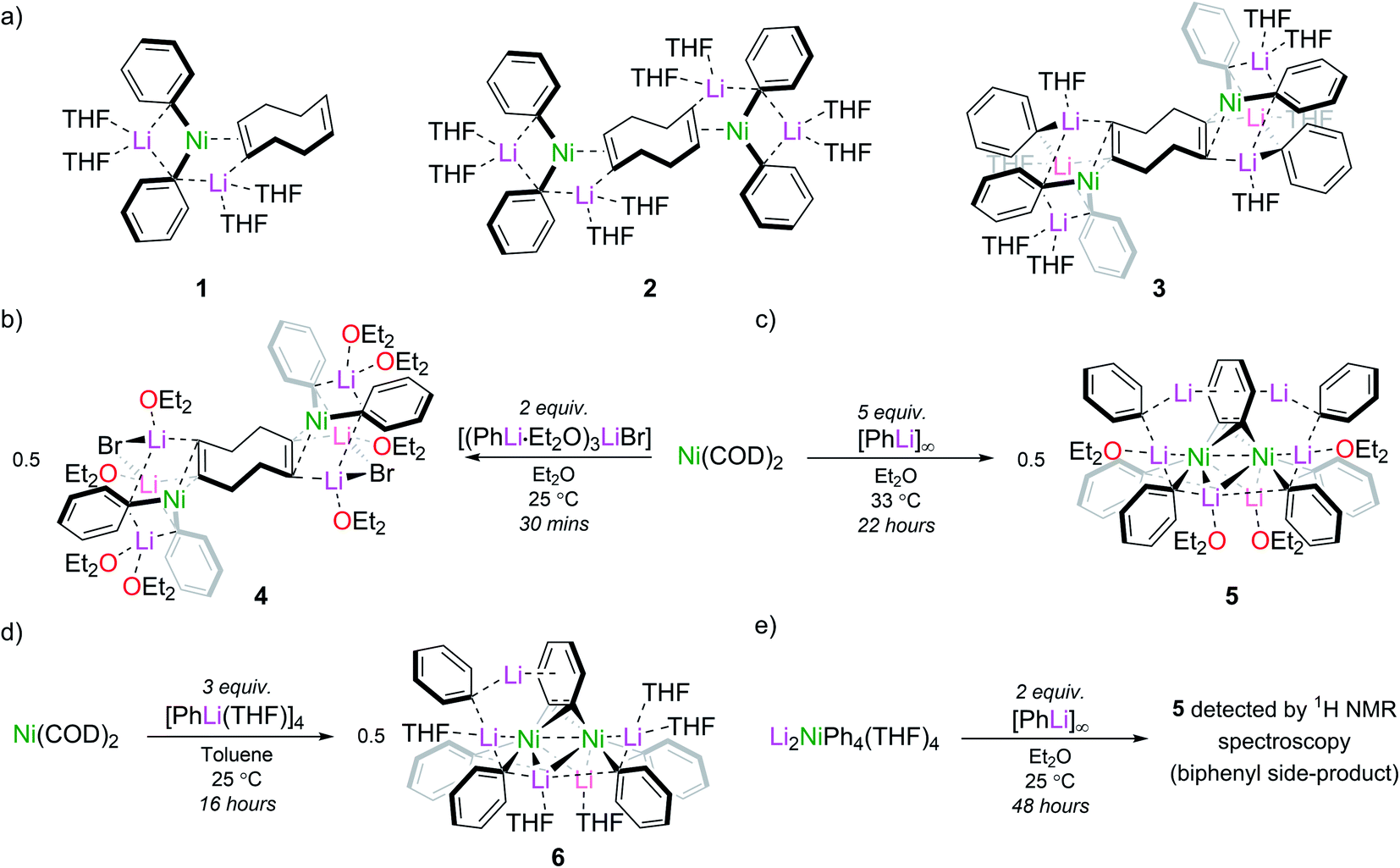 | ||
| Fig. 1 (a) Lithium nickelates (1–3) derived from Ni(COD)2 and PhLi; (b) synthesis of [Li2(Et2O)4(LiBr)Ph2Ni]2COD (4); (c) synthesis of [{Li3(Et2O)2Ph3Ni}2(μ-η2:η2-C6H4)] (5); (d) synthesis of heptametallic lithium nickelate 6; (e) observation of 5 by reduction of Li2NiPh4(THF)4. | ||
Nickelate intermediate 2 was the major product of these reactions. We found that the choice of solvent was crucial for converting 2 to a COD-free nickelate. Employing Et2O with gentle heating for 22 hours also gave a characteristic deep red solution; however, in this case a crystalline solid identified as [{Li3(Et2O)2Ph3Ni}2(μ-η2:η2-C6H4)] (5) could be isolated in low but reliable yields (10–25%) (Fig. 1c). These are lowered as removing the excess PhLi was challenging due to its similar solubility to the reaction product. Although the well-resolved 1H NMR spectrum of this material indicated a diamagnetic complex as anticipated for “Li3NiPh3(solv)3”, our attention was drawn to an unexpected doublet-of-doublets at 5.77 ppm (JHH = 5.5 Hz, 2.2 Hz, 20 °C, THF-d8) correlated to a signal at 6.70 ppm. This indicated the presence of a disubstituted arene, which would be inconsistent with Taube's original structure. Also inconsistent were two sets of Ni–Ph signals and a complex, broad, 7Li{1H} resonance from −0.40 to 1.0 ppm that upon cooling resolved into three signals at 1.28, 0.85, and −0.68 ppm (−80 °C, THF-d8, Fig. S6†). A discussion of the available NMR data (13C only) of Taube's “Li3NiPh3(THF)3” indicates the presence of just four aromatic signals at 111.6, 142.2, 125.0 and 120.3 ppm.13 Notably, the ipso-carbon was assigned as having the lowest chemical shift (111.6 ppm)30 which is inconsistent with the downfield signals reported for other phenyl-nickelate complexes (cf. 192.0 ppm for 1).6 Additional signals at 115, 125, and 127 ppm are noted and were attributed to a side product, “Li2NiPh2(η2-C6H6)”.31 Our experiments instead showed that the signal found at 115.5 ppm is correlated to the previously mentioned doublet-of-doublets at 5.77 ppm. This experiment was unavailable to Taube and demonstrates that we have isolated the same complex that was misidentified as “Li3NiPh3(THF)3”, and that this formula is inconsistent with its true composition.
We observed identical spectroscopic signatures following Taube's second synthetic route, the treatment of Li2NiPh4(THF)4 with two equivalents of PhLi. These were accompanied by a large amount of biphenyl and minor unidentified species due to the competing thermal decomposition of Li2NiPh4(THF)4 (Fig. 1e and S1, S2†). A final attempt was made to target Taube's proposed “Li3NiPh3(THF)3” complex. Treating Ni(COD)2 with 3 equivalents of [PhLi(THF)]46 in toluene at room temperature afforded the heptametallic lithium nickelate, [{Li3(THF)2Ph3Ni}(μ-η2:η2-C6H4){Li2(THF)3Ph2Ni}] (6) in 50% yield (Fig. 1d). It is important to note that in all attempted syntheses of COD-free nickelates the exclusion of dinitrogen is required. During our studies we crystallographically characterised a tetranickel dinitrogen structure (Fig. S22†) related to “side-on” dinitrogen complexes reported by Jonas, Krüger and co-workers, obtained by treating Ni(CDT) (CDT = trans,trans,trans-cyclododecatriene) with excess PhLi or PhNa under a N2 atmosphere.26–28
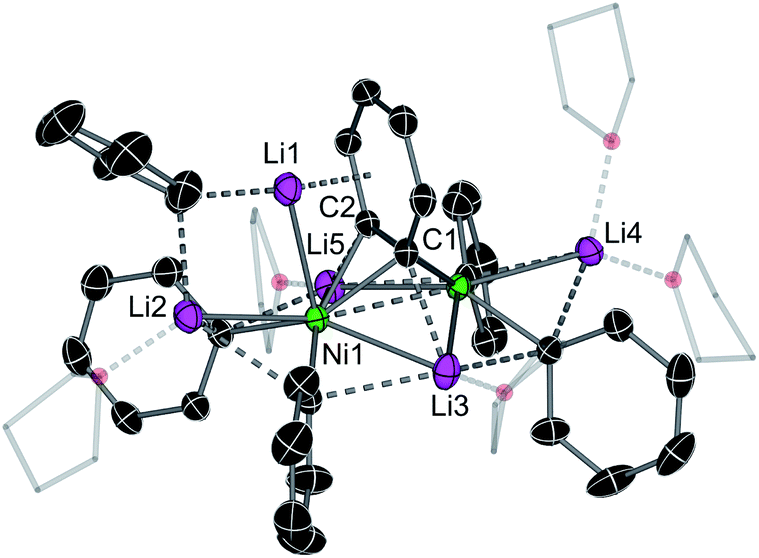
The inconsistencies between the original formulation and our complete set of NMR data were resolved with the aid of single-crystal X-ray diffraction studies. Dark red crystals of 5 and 6 were obtained from Et2O and revealed an unusual polynuclear cluster containing numerous interactions between C, Li and Ni atoms (Fig. 2 and 3). As these complexes are structurally very similar, we focus our discussion on complex 5. The complex contains an 8-membered ring (Li2–C19–Li4–C25–Li3–C13–Li1–C7) of alternating Li and C atoms surrounding a dinickel core that is bridged by a perpendicular C6H4 moiety sandwiched between two PhLi molecules. The PhLi molecules interact with the C6H4 moiety though the Li atoms, with Li⋯centroid distances of 1.958 and 1.968 Å for complex 5. The distances between the Ni centres and the two closest C atoms of the C6H4 moiety have an average value of 1.96 Å, indicating the presence of Ni–C bonds. Compared to COD complexes 2 and 4, the Li–Cipso and Ni–Li distances found in 5 are longer (ca. 0.12 Å).
 | ||
| Fig. 2 (a) Solid-state structure of 5 – top-down view. Thermal ellipsoids at 30% probability. Hydrogen atoms omitted and Et groups drawn as wireframes for clarity. Selected bond lengths (Å); Ni1–Ni2 2.7117(8); Ni1–C7 1.944(6); Ni1–C13 1.955(6); Ni2–C19 1.940(4); Ni2–C25 1.945(5); Ni2–Li2 2.65(1); Ni2–Li3 2.65(1); Ni2–Li4 2.564(9); Ni1–Li1 2.560(8); Ni1–Li2 2.664(9); Ni1–Li3 2.610(9); (b) side-on view. Selected bond lengths (Å); Ni1–C53 1.938(5); Ni1–C54 1.945(5); Ni2–C53 1.937(4); Ni2–C54 1.925(3); C53–C54 1.449(6); Ni2–Li6 2.55(1); Ni1–Li5 2.556(9). | ||
 | ||
| Fig. 3 Solid-state structure of 6. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and THF rings drawn as wireframes for clarity. Selected bond lengths (Å); C1–C2 1.436(3); Ni1–C1 1.951(1); Ni1–C2 1.958(2); Ni2–C1 1.954(2); Ni2–C2 1.945(2); Ni1–Ni2 2.7308(4); Ni1–Li1 2.552(3); Ni1–Li2 2.553(3); Ni1–Li3 2.583(3); Ni1–Li5 2.561(3); Ni2–Li3 2.592(3); Ni2–Li4 2.538(3); Ni2–Li5 2.569(3). | ||
Importantly, the bridging C–C bond in the C6H4 moiety (1.449(6) Å) is significantly longer than the analogous bond in a previously reported 4,5-difluorobenzyne complex [{(PEt3)2Ni}2(μ-η2:η2-C6H2-4,5-F2)] (1.390(3) Å)32 and the mononuclear Ni–benzyne complex [(Cy2PCH2CH2PCy2)Ni(η2-C6H2)] (1.331(3) Å)33 (Scheme 2). Compared to dinickel complexes, where Ni–Ni bond distances tend to be around 2.5 Å, and in a rare case 2.6027(17) Å,34 the Ni⋯Ni distance of 2.7117(8) Å is very long and is similar to the 2.7242(3) Å observed in [{(PEt3)2Ni}2(μ-η2:η2-C6H2-4,5-F2)].32 In that case, Ni⋯Ni interactions were not studied.
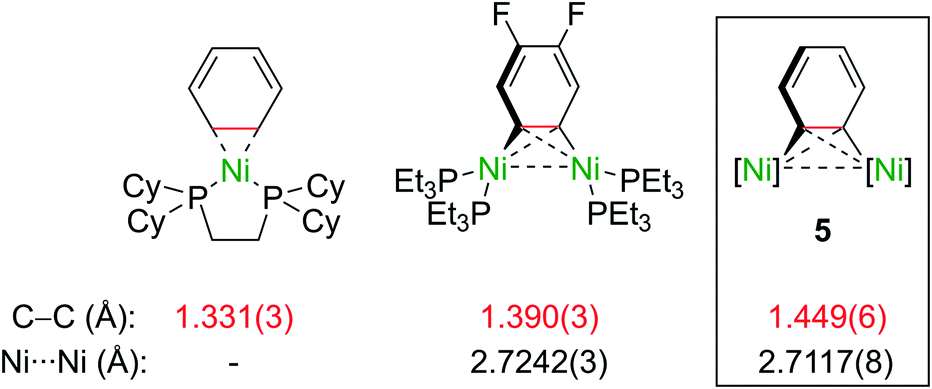 | ||
| Scheme 2 Comparison of selected bond lengths in mono- and dinuclear Ni–aryne complexes.32,33 | ||
Computational studies on the entire complex 5 were performed to analyse the bonding. First, we aimed to understand the Ni⋯Ni interaction (if any), then we investigated the nature of the C6H4 moiety bridging the Ni atoms and the bonding of the phenyl ligands. We also compared the bonding in 5 to that in the related COD complex 2 (see ESI† for further details).
The electronic structure of 5 was investigated by means of DFT calculations at the MN15//MN15L/def2SVP level. Three different electron configurations, namely the closed-shell singlet, the open-shell singlet and open-shell triplet were computed. The triplet state converged at an energy level +25.6 kcal mol−1 above the closed-shell singlet, whereas the open-shell singlet was unstable. Electronic states with localized unpaired electrons, including Ni(↑)Ni(↓) and Ni(↑)Ni(↑) configurations, were thus excluded. The fully optimised geometry of the lowest-energy state, i.e. the closed-shell singlet, agreed with the crystal structure (RMSD = 0.023 Å, for all Ni–C distances). In contrast, the triplet state yielded a larger RMSD of 0.084 Å. These results are consistent with the diamagnetic nature of the complex as observed by NMR spectroscopy.
Natural bond orbital (NBO) calculations,35 including second-order perturbation analysis, were carried out to explore the electronic structure of the complex, with a particular focus on the nature of the bimetallic core. First, the presence of a Ni–Ni bond was not reflected in the molecular orbitals. Any relevant d-orbital combinations that we found were either non-bonding or antibonding. There is a weak dNi → sNi interaction between the two nickel atoms, however the stabilisation energy (SE) of this interaction, which measures its strength, is only 1.9 kcal mol−1. The absence of a Ni–Ni bond is consistent with the comparatively long Ni⋯Ni distance discussed above.
Subsequent topological analyses of the real-space functions electron density (according to the quantum theory of atoms in molecules, QTAIM)36 and electron localisability indicator (ELI),37 add a complementary view of the bonding.38 These were conducted at the B3LYP/def2-TZVP level including empirical GD3BJ dispersion corrections. The same potential energy minimum was found as for the MN15//MN15L/def2SVP calculations (confirmed by frequency analyses). In the electron density, neither a bond path for the Ni⋯Ni contact, nor an ELI basin was found. This confirmed that no bonding interaction is present. However, the delocalisation index (related to QTAIM, measure of the bond order) between the two Ni atomic basins is 0.20, which is consistent with the very weak charge transfer between these two atoms as also found in the NBO analysis. This is the basis of our choice to depict a Ni⋯Ni interaction as a dashed line in schemes and figures.
The strongest interaction found in the NBO analysis of 5 was the backdonation from a Ni d orbital to a π* orbital of the C6H4 bridging fragment. This is consistent with the considerable elongation of the C–C bond in the C6H4 moiety and its low natural bond order (1.12). The stabilisation energy of this interaction is 474.1 kcal mol−1, and the shape and symmetry of the orbitals involved can be seen in the associated natural localised molecular orbital (NLMO) and schematic (Fig. 4a and b).
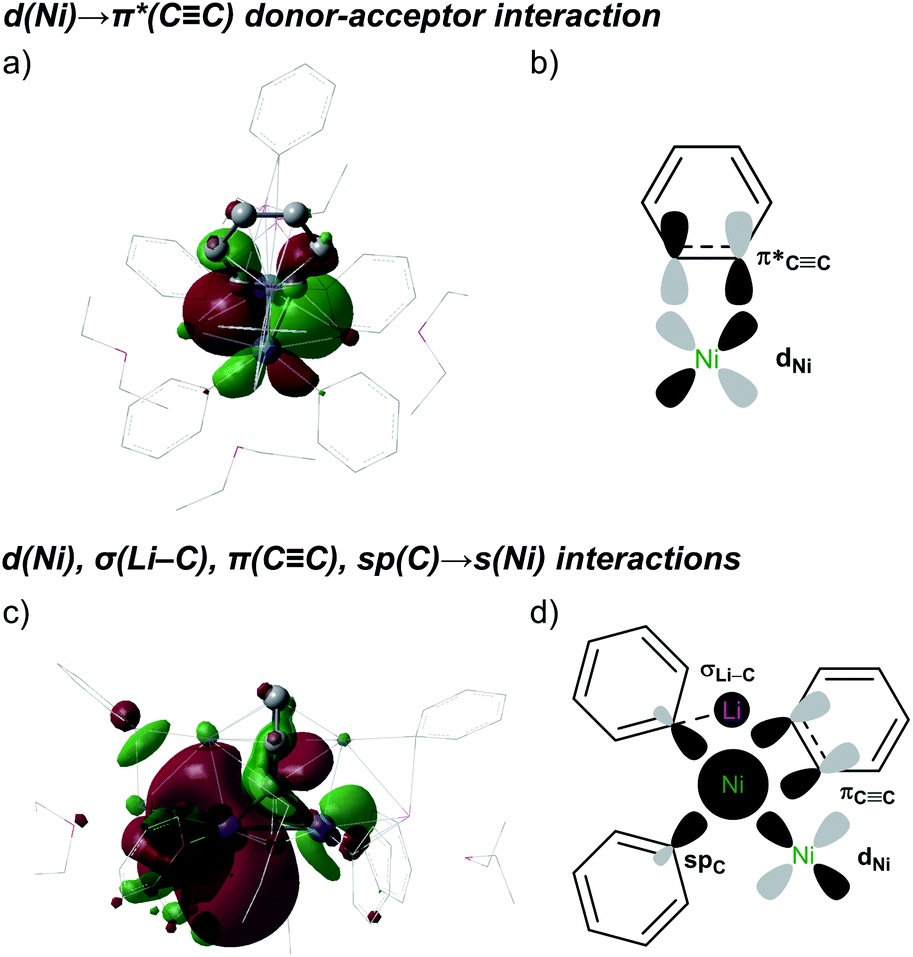 | ||
Fig. 4 NLMO (a; isovalue = 0.02 a.u.) and schematic representation (b) of the d(Ni) → π*(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) donor–acceptor interaction. NLMO (c; isovalue = 0.01 a.u.) and schematic representation (d) of the d(Ni), σ(Li–C), π(CC), sp(C) → s(Ni) donor–acceptor interactions. For clarity, H atoms were excluded. Representations: ball-and-stick for the Ni2(μ-C6H4) core and wireframe for the remainder. Calculations were performed on the entire complex 5. C) donor–acceptor interaction. NLMO (c; isovalue = 0.01 a.u.) and schematic representation (d) of the d(Ni), σ(Li–C), π(CC), sp(C) → s(Ni) donor–acceptor interactions. For clarity, H atoms were excluded. Representations: ball-and-stick for the Ni2(μ-C6H4) core and wireframe for the remainder. Calculations were performed on the entire complex 5. | ||
Electron donation to the empty s orbital of nickel is also relevant, with the spC orbitals of the phenyl ligands making the strongest contribution (SE = 50.4 kcal mol−1, on average). The πCC system also makes a notable contribution (SE = 29.3 kcal mol−1), whereas that of the σCC orbital is weaker (SE = 3.4 kcal mol−1). The shapes and symmetries of these different contributions can be observed in the NLMO plot of the empty sNi orbital (Fig. 4c and d). Interestingly, when this NLMO was augmented to visualise the weakest interactions, as shown in Fig. 4c, we found a σC–Li→ sNi interaction (SE = 2.1 kcal mol−1) in which the Ph–Li moiety acts as an electron donor to the nickel centre. The HOMO and LUMO orbitals did not show any other additional interactions (Fig. S26†).
The overall electron distribution was quantified with a combined ELI and QTAIM analysis. Fig. 5 shows ELI localisation domains, electron populations of the respective bond basins and QTAIM delocalization indices for these bonds. We start from an isolated benzyne molecule (Fig. 5a) to better understand the impressive backdonation upon bonding to nickel (Fig. 5b). In the benzyne molecule there are 3.56 electrons in the putative triple bond with a bond order of 2.25. There are also two additional 0.62 π-electrons that are not attributed to any bond, but instead fused to the localisation domain of the adjacent C–C bond (strained acetylenes become biradicals and as such electron donors).39 Aside from this, the aromatic π-system is intact with the other C–C bonds having uniformly 2.74–2.80 electrons with a bond order of 1.37/1.38.
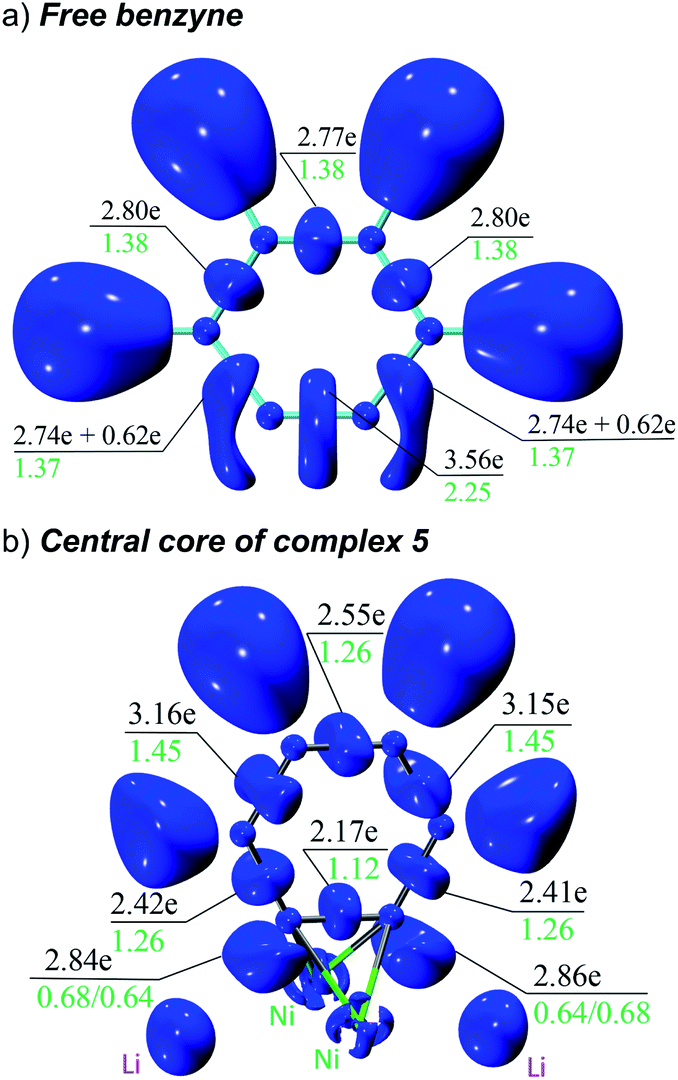 | ||
| Fig. 5 ELI localisation domain representations (isovalue = 1.40) of: (a) isolated benzyne, and; (b) close-up view of the Ni–benzyne bonding in complex 5. There is one ELI attractor and corresponding bond basin attributed to each localisation domain, except for the C–C bonds adjacent to the putative triple bond in (a) where the domains are reducible to give rise to two basins each. Electron populations of the ELI basins are given in black, QTAIM delocalisation indices in green. | ||
Upon complexation to nickel, the situation changes drastically (Fig. 5b): the former triple bond is now the most depleted bond of the six-membered ring with only 2.17 electrons remaining and a bond order of 1.12. Bond orders and electron populations of the ELI bond basins are no longer uniform, but alternate around the ring. This disruption of the aromatic system is caused by both πCC → s(Ni) bonding and 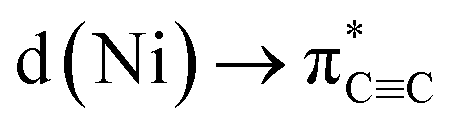 backbonding, as discussed in the NBO analysis. These two contributions cannot be separated in the real-space analyses. However, the consequence of this is that a trisynaptic valence basin V(C, Ni1, Ni2) forms on each side of the former triple bond which is populated with approximately 2.85 electrons on each side. In the QTAIM picture, this coincides with two C–Ni bond paths on each side with a formal bond order of about 0.65 each. Consistent with the NBO analysis and decreased bond order of the bridging C–C bond, the QTAIM analysis and ELI bond basins build a picture of strong backbonding such that the central core has metallabicyclobutane character. In terms of assigning a formal oxidation state to the Ni atoms in 5 and 6, the data therefore suggest considering them Ni(I) with a reduced C6H42− ligand and the two unpaired electrons located in a Ni–(μ-η2:η2-C6H4)–Ni orbital (HOMO in Fig. S26†).
backbonding, as discussed in the NBO analysis. These two contributions cannot be separated in the real-space analyses. However, the consequence of this is that a trisynaptic valence basin V(C, Ni1, Ni2) forms on each side of the former triple bond which is populated with approximately 2.85 electrons on each side. In the QTAIM picture, this coincides with two C–Ni bond paths on each side with a formal bond order of about 0.65 each. Consistent with the NBO analysis and decreased bond order of the bridging C–C bond, the QTAIM analysis and ELI bond basins build a picture of strong backbonding such that the central core has metallabicyclobutane character. In terms of assigning a formal oxidation state to the Ni atoms in 5 and 6, the data therefore suggest considering them Ni(I) with a reduced C6H42− ligand and the two unpaired electrons located in a Ni–(μ-η2:η2-C6H4)–Ni orbital (HOMO in Fig. S26†).
Finally (not shown in Fig. 5b, see ESI Fig. S27†), there is strong directed σ-bonding from the six phenyl groups to Ni, each with between 3.8 and 4.0 electrons in the ELI bond basin and only a minor contribution from the Li atoms (less than 0.2 electrons charge transfer between each Ni and Li pair). This is consistent with the NLMO depicted in Fig. 4c and d.
Given that we showed that Taube's proposed “Li3NiPh3(solv)3” complex instead corresponds to a more complicated dinickel structure, we naturally had questions about why the simpler complex does not form. We attribute this to the carbanionic phenyl ligands being such strong donor ligands that the Ni(0) centre cannot accept the electron density that would be required in “Li3NiPh3(solv)3”. An indication of the donor properties of the phenyl ligands is already seen in the elongation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond distance of the coordinated COD ligand in 1 (1.448(3) Å)6 and 4 (1.445(6) Å) (c.f. 1.439(5) Å) in [{Ni(dcpp)}2(COD)]40 (dcpp = 1,2-bis(dicyclohexylphosphino)propane) and 1.425(4) Å in [{Ni(NHC)2}2(COD)]41 (NHC = 1,3-diisopropylimidazole-2-ylidene). We postulate that the approach of the third PhLi molecule to the Ni centre might induce the insertion of extremely electron-rich Ni into a C–H bond. Subsequently, release of LiH may occur to form benzyne. This reaction might involve a bimetallic mechanism with participation of two Ni centres.42
C bond distance of the coordinated COD ligand in 1 (1.448(3) Å)6 and 4 (1.445(6) Å) (c.f. 1.439(5) Å) in [{Ni(dcpp)}2(COD)]40 (dcpp = 1,2-bis(dicyclohexylphosphino)propane) and 1.425(4) Å in [{Ni(NHC)2}2(COD)]41 (NHC = 1,3-diisopropylimidazole-2-ylidene). We postulate that the approach of the third PhLi molecule to the Ni centre might induce the insertion of extremely electron-rich Ni into a C–H bond. Subsequently, release of LiH may occur to form benzyne. This reaction might involve a bimetallic mechanism with participation of two Ni centres.42
We attempted to trap the plausible LiH byproduct by the addition of benzophenone to the reaction mixture (see ESI† for further details). The product of hydrolithiation, Ph2CHOLi, was not observed. Instead, the reaction is accompanied by a dramatic colour change to deep blue which suggested the formation of the benzophenone ketyl radical anion. This can be attributed to complex redox processes that, alongside the expected reactivity of the PhLi groups with benzophenone, made it difficult to rule out or confirm the formation of LiH. Experiments with [(13C6-PhLi·Et2O)3·LiBr] and unlabelled COD-bearing intermediate 4 showed that, as expected, the 13C-labelled PhLi ends up in the C6H4 moiety. However, the PhLi-13C6 was also observed to be in exchange with the other phenyl ligands (vide infra) making it difficult to determine the mechanism by experimental means. Although small amounts of C6H6 were detected due to decomposition of the nickelate species, likely via hydrolysis from trace water, an alternative mechanism via deprotonation was discarded. It does not account for the overall charge balance and Li(0) was not observed as a precipitate during the course of the reaction.
Beyond the mechanism by which compound 5 is formed, we were also interested in understanding its dynamic behaviour in solution. NMR spectroscopy experiments reveal that although the pseudo-C2v symmetry of 5 appears to be retained when it is dissolved in THF-d8, the PhLi molecules that are coordinated to the C6H4 moiety in the solid state dissociate. This is supported by 1H DOSY NMR studies which reveal two major independent species that do not co-diffuse, with an estimated molecular weight suggesting a composition of “{Li2(THF)3Ph2Ni}2(μ-η2:η2-C6H4)” (Fig. 6). 1H{1H} NOE and 1H–1H EXSY NMR studies (Fig. S2 and S3†) also demonstrate that the free PhLi is in rapid exchange with the Ni-bound phenyl ligands, which is consistent with our observations reacting 4 with 13C-labelled PhLi (see ESI† for further details). Variable temperature NMR studies of 5 in a mixture of toluene-d8 and Et2O suggest that the dissociation of PhLi is hampered at lower temperatures, since at −80 °C the 7Li NMR spectrum contains five unique signals in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:1:1 ratio (Fig. S9†).
:2:1:1:1 ratio (Fig. S9†).
 | ||
| Fig. 6 1H DOSY NMR spectrum showing the dissociation of 5 in THF-d8. | ||
Compound 6 displays similar solution-state behaviour to 5 as illustrated by its apparent symmetric structure in THF-d8 due to dissociation of PhLi. As for 5, variable temperature NMR studies also revealed that rotation about the Cipso–Ni bonds could be frozen out upon cooling to −80 °C (Fig. S5†).
To further explore the nature of compound 5 we performed reactivity studies. Taube reported the formation of biphenyl when the purported “Li3NiPh3(THF)3” was reacted with I2.13 Upon addition of excess I2 to 5, we observed the complete disappearance of signals related to 5 and the appearance of biphenyl and ortho-terphenyl in a 3:1 ratio after quenching the reaction (Scheme 3a). The formation of ortho-terphenyl is interesting as it suggests sequential reductive elimination reactions involving the C6H4 moiety. Moreover, when 5 was reacted with MeI, we observed the complete disappearance of 5 and the appearance of toluene and methyl-biphenyl in a 2:1 ratio by 1H NMR alongside biphenyl (Scheme 3b). Since the former triple bond in the bridging C6H4 moiety has been diminished by the significant Ni → π* back-bonding to merely a single bond (bond order 1.12), typical benzyne reactivity was not observed. We did not detect cycloaddition products upon reaction of 5 with either anthracene or furan. These exploratory tests, along with the previously discussed computational studies, suggest that the nature of the bridging C6H4 fragment is rather that of a dimetallabicyclobutane.
 | ||
| Scheme 3 Stoichiometric reactivity of 5: (a) reaction of 5 with I2; (b) reaction of 5 with MeI. | ||
Studies using compounds 2 and 5 as pre-catalysts for Ni-catalysed cross-coupling reactions were also conducted (Scheme 4). The ability of 5 to act as a pre-catalyst for the Csp2–Csp3 Kumada coupling shown in Scheme 4a is significant as this reaction was investigated in the context of Ni(0)-ate complexes such as Li2(THF)4Ni(COD)2.4 Highly electron-rich complexes 2 and 5 gave improved yields compared to the <6% yield reported when Ni(COD)2 was used as the pre-catalyst (74% and 43%, respectively). A Buchwald–Hartwig coupling reaction that involves a Ni(I)/Ni(III) mechanism was also explored (Scheme 4b).43 The desired amine was obtained in rather lower yields than previously reported pre-catalysts (28% with 2, 22% with 5) or with Ni(COD)2 alone (52%).43,44 This is likely due to the nickelates lacking the robust NHC or bidentate phosphine ligands necessary for nickel intermediates to withstand the harsh reaction conditions. Compound 5 is also a competent pre-catalyst in the Ni-catalysed cross-coupling of 2-methoxynaphthalene with PhLi (46% yield of 2-phenylnaphthalene), albeit with a slight reduction in yield compared to 2 (Scheme 4c).6
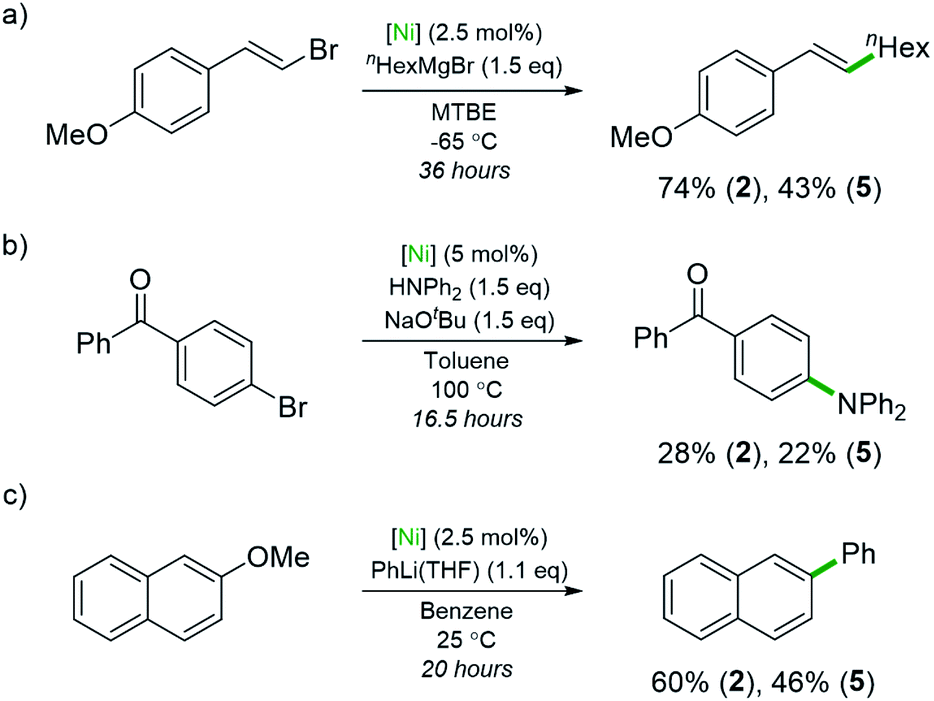 | ||
| Scheme 4 Use of lithium nickelates 2 and 5 as pre-catalysts in various cross-coupling reactions: (a) Csp²–Csp³ Kumada cross-coupling; (b) C–N Buchwald–Hartwig cross-coupling; (c) cross-coupling of aryl ethers. | ||
Having explored the structure, bonding, and reactivity of 5, we next investigated whether this chemistry could be extended to PhNa. Although we did not observe any analogous sodium nickelates containing the C6H4 ligand, the species that were obtained do provide some insight into how the polynuclear lithium nickelates 5 and 6 may be constructed. Treating Ni(COD)2 with 2 equivalents of PhNa in Et2O at −30 °C followed by crystallisation from THF/hexane afforded the 2:1 sodium nickelate, [Na2(THF)3Ph2Ni(COD)]2 (7) (Fig. 7a). Although the structure of compound 7 was unambiguously determined by single crystal X-ray diffraction, 1H and 13C NMR spectroscopic analysis of the isolated material indicated a bulk constitution of [Na2(THF)nPh2Ni(C2H4)]2, a previously reported compound.7,8
 | ||
| Fig. 7 (a) Reactions of Ni(COD)2 with PhNa for the synthesis of [Na2(THF)3Ph2Ni(olefin)]2 (7) and identification of [Na2(Et2O)3Ph2(NaC8H11)Ni(COD)]2 (8); (b) solid-state structure of 7. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and THF rings drawn as wireframes for clarity; (c) solid-state structure of 8. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and Et groups drawn as wireframes for clarity. | ||
The ethene ligands of this compound are proposed to originate from cleavage of the ethereal solvent (Et2O or THF),45 a process that is also observed when PhNa is dissolved in THF. Compound 7 exists as a discrete dimeric structure that contains an 8-membered ring of alternating carbon and sodium atoms akin to that found in lithium nickelates 5 and 6 (Fig. 7b).
Since organosodium compounds tend to form higher aggregates compared to their lithium congeners,46 we propose that during the synthesis of 5 in Et2O, a lithium-containing analogue of 7 may form. This electron-rich dinickel intermediate could then react with additional PhLi via a bimetallic mechanism as discussed previously to give the corresponding C6H4 complex. When Ni(COD)2 is treated with a greater excess of PhNa (3–5 equiv.) significant decomposition is observed due to competing deprotonative sodiation of 1,5-cyclooctadiene. In one case, an octanuclear sodium nickelate [Na2(Et2O)3Ph2(NaC8H11)NiCOD]2 (8) was identified from the reaction mixture by single-crystal X-ray diffraction (Fig. 7a and c). Attempts to isolate this in pure form however were unsuccessful. Compound 8 can be viewed as a co-complex of 7 and NaC8H11, with the Na3 atoms indicated in Fig. 7c solvated solely by numerous interactions to allyl, aryl and alkenyl carbons (Na⋯C range = 2.506(3)–2.952(3) Å).
Conclusions
In conclusion, we have demonstrated that the proposed hexagonal planar, homoleptic complex “Li3NiPh3(solv)3” is instead a unique dinickel complex [{Li3(solv)2Ph3Ni}2(μ-η2:η2-C6H4)] containing a C6H4 unit that is formally derived from a molecule of benzyne. By analysing the bonding in 5 by NBO, QTAIM, and ELI techniques, we have shown that remarkably strong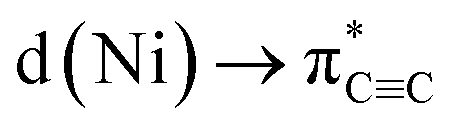 backbonding supported by πCC→ s(Ni) charge transfer depletes the triple bond to only a single bond with a bond order of 1.12. Strong σ-bonding of the remaining phenyl carbanionic ligands to the Ni atoms was also detected with very weak contributions from the Li atoms. Preliminary reactivity studies revealed the formation of 1,2-disubstituted benzene compounds that formally derive from PhLi. We have also uncovered an interesting alkali-metal effect, where using PhNa gives new nickelate species that do not contain the C6H4 ligand. Looking at the broader implications of these findings, since many Ni(0)-catalysed transformations involve the use of Ni(COD)2 with a large excess of ArLi or other polar organometallic reagents, it cannot be discarded that compounds similar to 4–8 can also be formed or even be involved in some of these processes.
backbonding supported by πCC→ s(Ni) charge transfer depletes the triple bond to only a single bond with a bond order of 1.12. Strong σ-bonding of the remaining phenyl carbanionic ligands to the Ni atoms was also detected with very weak contributions from the Li atoms. Preliminary reactivity studies revealed the formation of 1,2-disubstituted benzene compounds that formally derive from PhLi. We have also uncovered an interesting alkali-metal effect, where using PhNa gives new nickelate species that do not contain the C6H4 ligand. Looking at the broader implications of these findings, since many Ni(0)-catalysed transformations involve the use of Ni(COD)2 with a large excess of ArLi or other polar organometallic reagents, it cannot be discarded that compounds similar to 4–8 can also be formed or even be involved in some of these processes.
Data availability
Synthetic procedures, NMR spectra, and supplementary computational details are included as ESI.†Author contributions
R. J. S., A. M. B. and M. P.-J. synthesised and characterised all new complexes and carried out reactivity studies. A. N., D. B., L. A. M. and S. G. carried out computational studies. E. C., E. H. and J. C. supervised the overall project. R. J. S. and A. M. B. wrote the original draft and all authors contributed to review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, 756575), the Spanish Ministry of Science and Innovation (Project PID2019-110856GA-I00) and Junta de Andalucía (P18-FR-4688). The authors gratefully acknowledge the financial support provided by the Swiss National Science Foundation (SNSF) (Award 188573 to EH). A. N. and D. B. acknowledge the support from the Norwegian Research Council through the Hylleraas Centre for Quantum Molecular Sciences (Project 262695) and the Norwegian Metacenter for Computational Science (NOTUR; Grant nn4654k). The Rigaku Synergy-S instrument used for X-ray diffraction studies was purchased through the SNSF R'Equip grant 206021_177033.Notes and references
- J. B. Diccianni and T. Diao, Trends Chem., 2019, 1, 830–844 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS.
- K. Jonas and C. Krüger, Angew. Chem., Int. Ed., 1980, 19, 520–537 CrossRef.
- L. Nattmann, S. Lutz, P. Ortsack, R. Goddard and J. Cornella, J. Am. Chem. Soc., 2018, 140, 13628–13633 CrossRef CAS PubMed.
- S. Lutz, L. Nattmann, N. Nöthling and J. Cornella, Organometallics, 2021, 40, 2220–2230 CrossRef CAS.
- A. M. Borys and E. Hevia, Angew. Chem., Int. Ed., 2021, 60, 24659–24667 CrossRef CAS PubMed.
- B. D. J. Brauer, C. Krüger, P. J. Roberts and Y.-H. Tsay, Angew. Chem., Int. Ed., 1976, 15, 48–49 CrossRef.
- K. Jonas, Angew. Chem., Int. Ed., 1976, 15, 47 CrossRef.
- K. Jonas, K. R. Pörschke, C. Krüger and Y.-H. Tsay, Angew. Chem., Int. Ed., 1976, 15, 621–922 CrossRef.
- K.-R. Pörschke, K. Jonas, G. Wilke, R. Benn, R. Mynott and R. Goddard, Chem. Ber., 1985, 118, 275–297 CrossRef.
- K.-R. Pörschke, K. Jonas and G. Wilke, Chem. Ber., 1988, 121, 1913–1919 CrossRef.
- R. Taube and G. Honymus, Angew. Chem., Int. Ed., 1975, 14, 261–262 CrossRef.
- R. Taube and N. Stransky, Z. Chem., 1979, 19, 412–413 CrossRef CAS.
- T. A. Bagenova, A. K. Shilova, E. Deschamps, M. Gruselle, G. Leny and B. Tchoubar, J. Organomet. Chem., 1981, 222, 2–5 CrossRef.
- R. Taube and N. Stransky, Z. Anorg. Allg. Chem., 1982, 490, 91–100 CrossRef CAS.
- R. Taube, Pure Appl. Chem., 1983, 55, 165–176 CAS.
- T. A. Bazhenova, R. M. Lobkovskaya, R. P. Shibaeva, A. E. Shilov, A. K. Shilova, M. Gruselle, G. Leny and B. Tchoubar, J. Organomet. Chem., 1983, 244, 265–272 CrossRef CAS.
- R. Taube, Comments Inorg. Chem., 1984, 3, 69–81 CrossRef CAS.
- R. Taube, D. Steinborn, H. Drevs, P. Nguyen Chuong, N. Stransky and J. Langlotz, Z. Chem., 1988, 11, 381–396 Search PubMed.
- J. Wei, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2015, 54, 5999–6002 CrossRef CAS PubMed.
- S. U. Koschmieder and G. Wilkinson, Polyhedron, 1991, 10, 135–173 CrossRef CAS.
- M. Garçon, C. Bakewell, G. A. Sackman, J. P. Andrew, R. I. Cooper, A. J. Edwards and M. R. Crimmin, Nature, 2019, 574, 390–396 CrossRef PubMed.
- I. M. Riddlestone, J. A. B. Abdalla and S. Aldridge, Adv. Organomet. Chem., 2015, 63, 1–38 CrossRef CAS.
- M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348–1365 RSC.
- M. Perez-Jimenez, N. Curado, C. Maya, J. Campos, J. Jover, S. Alvarez and E. Carmona, J. Am. Chem. Soc., 2021, 143, 5222–5230 CrossRef CAS PubMed.
- K. Jonas, Angew. Chem., Int. Ed., 1973, 12, 997–998 CrossRef.
- C. Krüger and Y.-H. Tsay, Angew. Chem., Int. Ed., 1973, 12, 998–999 CrossRef.
- K. Jonas, D. J. Brauer, C. Krüger, P. J. Roberts and Y.-H. Tsay, J. Am. Chem. Soc., 1976, 98, 74–81 CrossRef CAS.
- H. Hope and P. P. Power, J. Am. Chem. Soc., 1983, 105, 5320–5324 CrossRef CAS.
- Taube compared the chemical shifts of “Li3NiPh3(THF)3” to that of PhLi which was assigned as having a chemical shift of 121.7 ppm for the ipso-carbon.13 H. J. Reich, D. P. Green, M. A. Medina, W. S. Goldenberg, B. Ö. Gudmundsson, R. R. Dykstra and N. H. Phillips, J. Am. Chem. Soc., 1998, 120, 7201–7210 CrossRef CAS This is inconsistent with reports by Reich which shows that for PhLi in Et2O, the ipso-carbon has a chemical shift of 187.0 ppm (dimer) or 174.0 ppm (tetramer). , ref. 31 also mentions this incorrect assignment..
- J. Langlotz, PhD thesis, Technische Hochschule Carl Schorlemmer, Merseburg, 1989.
- A. L. Keen and S. A. Johnson, J. Am. Chem. Soc., 2006, 128, 1806–1807 CrossRef CAS.
- M. A. Bennett, T. W. Hambley, N. K. Roberts and G. B. Robertson, Organometallics, 1985, 4, 1992–2000 CrossRef CAS.
- P. A. Cleaves, A. J. Ayres, L. Vondung, J. C. Stewart, P. J. Cobb, A. J. Wooles and S. T. Liddle, Organometallics, 2020, 39, 4735–4746 CrossRef CAS.
- F. Weinhold and C. R. Landis, Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2012 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, 1995 Search PubMed.
- M. Kohout, Int. J. Quantum Chem., 2004, 97, 651–658 CrossRef CAS.
- S. Grabowsky, Complementary Bonding Analysis, De Gruyter, 2021 Search PubMed.
- P. Prieto, A. De La Hoz, I. Alkorta, I. Rozas and J. Elguero, Chem. Phys. Lett., 2001, 350, 325–330 CrossRef CAS.
- F. D'Accriscio, A. Ohleier, E. Nicolas, M. Demange, O. Thillaye Du Boullay, N. Saffon-Merceron, M. Fustier-Boutignon, E. Rezabal, G. Frison, N. Nebra and N. Mézailles, Organometallics, 2020, 39, 1688–1699 CrossRef.
- T. Schaub and U. Radius, Chem.–Eur. J., 2005, 11, 5024–5030 CrossRef CAS.
- B. J. Graziano, M. V. Vollmer and C. C. Lu, Angew. Chem., Int. Ed., 2021, 60, 15087–15094 CrossRef CAS PubMed.
- T. Inatomi, Y. Fukahori, Y. Yamada, R. Ishikawa, S. Kanegawa, Y. Koga and K. Matsubara, Catal. Sci. Technol., 2019, 9, 1784–1793 RSC.
- A. Bismuto, P. Müller, P. Finkelstein, N. Trapp, G. Jeschke and B. Morandi, J. Am. Chem. Soc., 2021, 143, 10642–10648 CrossRef CAS PubMed.
- R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588–591 CrossRef CAS.
- A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470–7562 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2127441–2127443 and 2127151–2127153. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01244h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |