
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and hybridizing properties of P-stereodefined chimeric [PS]-{DNA:RNA} and [PS]-{DNA:(2′-OMe)-RNA} oligomers†
Katarzyna Jastrzębska *,
Anna Maciaszek,
Rafał Dolot,
Agnieszka Tomaszewska-Antczak,
Barbara Mikołajczyk and
Piotr Guga
*,
Anna Maciaszek,
Rafał Dolot,
Agnieszka Tomaszewska-Antczak,
Barbara Mikołajczyk and
Piotr Guga
Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Department of Bioorganic Chemistry, Sienkiewicza 112. 90-363 Łódź, Poland. E-mail: kjastrz@cbmm.lodz.pl; Fax: +48-42-6803261; Tel: +48-42-6803248
First published on 21st September 2022
Abstract
Oxathiaphospholane derivatives of 2′-OMe-ribonucleosides and 2′-O-TBDMS-ribonucleosides (MN-OTP and TN-OTP, respectively; nucleobase protected) were synthesized and separated into pure P-diastereomers. X-ray analysis showed the RP absolute configuration of the phosphorus atom in the fast-eluting diastereomer of TA-OTP. The fast- and slow-eluting P-diastereomers of MN-OTP and TN-OTP were used in the solid-phase synthesis of phosphorothioate dinucleotides (MNPST and NPST, respectively), which were subsequently hydrolyzed with RP-selective phosphodiesterase svPDE and SP-selective nuclease P1 to determine the absolute configuration of the phosphorus atoms. P-Stereodefined phosphorothioate ([PS]) 10-mer chimeric oligomers [PS]-{DNA:(2′-OMe)-RNA} and isosequential [PS]-{DNA:RNA} containing two MNPS or NPS units were synthesized. Melting experiments performed for their complexes with Watson–Crick paired DNA matrix showed that MNPS or NPS units decrease the thermal stability of the duplexes (ΔTm = −0.5 ÷ −5.5 °C per modification) regardless of the absolute configuration of the P-atoms. When the (2′-OMe)-RNA matrix was used an increase in Tm was noted in all cases (ΔTm = +1 ÷ +7 °C per modification). The changes in thermal stability of the duplexes formed by [PS]-chimeras with DNA and (2′-OMe)-RNA matrices do not correlate with the absolute configuration of the phosphorus atoms.
Introduction
In the last five decades, many DNA analogs have been chemically synthesized, mainly to improve their stability against nucleases and in this way to ensure long-lasting recognition of the target nucleic acid. This increased stability was expected to produce new probes useful for example for controlling gene expression or biochemical manipulations. To some extent, this goal has been achieved by the introduction of phosphorothioate analogs of DNA ([PS]-DNA),1 although nonspecific binding to proteins2 and immunological side effects3,4 initially hindered their application in vivo. [PS]-RNA oligomers were obtained shortly after using phosphoramidite or H-phosphonate monomers with sulfurization. Note: interestingly, it has been shown that [PS]-DNA probes in the absence of transfecting agents enter mammalian cells more efficiently than DNA.5 An explanation based on the dynamic covalent exchange of phosphorothioates with cellular thiols and disulfides has recently been published.6 Some drugs containing [PS]-DNA and [PS]-RNA-derived units have been approved by the FDA and other agencies, and several others are in advanced clinical trials.7Noteworthy, in 2007 phosphorothioation of DNA was discovered in bacteria,8 and in 2019, a DNA phosphorothioation-based antiviral system was discovered in archaea.9 The identified chimeric phosphate:phosphorothioate compounds ([PO]:[PS]) contained the phosphorothioate phosphorus atoms only of RP absolute configuration.10 In 2020, the RP phosphorothioate modification was detected in RNA isolated from prokaryotes and eukaryotes.11
Although the phosphorothioate internucleotide bond is isoelectronic with the natural phosphate moiety, the properties of [PS]-DNA differ from natural DNA ([PO]-DNA) in several aspects, mainly due to the different steric requirements of the sulfur atom, the altered affinity for metal ions (“soft” sulfur vs. “hard” oxygen), and unsymmetrical distribution of the negative charge in the diester phosphorothioate anion.12 Depending on the intended application, these altered properties may be considered either disadvantageous or beneficial, but in the vast majority of biology-related experiments, one should consider the stereochemistry of the internucleotide phosphorothioate moieties. Since the substitution of sulfur for a nonbridging oxygen atom creates a stereogenic center, for example, a [PS]-oligonucleotide decamer synthesized by a non-stereocontrolled method exists as a mixture of 29 = 512 P-diastereomers, and the content of a single diastereomer is less than 0.2%. Since each P-diastereomer can interact with a biological target (usually with fixed chirality) in slightly different ways, the mixtures of hundreds of P-diastereomers may be useless for detecting the interactions of interest. To overcome this problem, stereocontrolled synthetic methods have been developed.13–15 Among them, the approach utilizing so called oxathiaphospholane monomers (OTP; developed in this laboratory)13,16 has yielded the most P-stereodefined oligomers so far, and some of them showed interesting P-stereodependent properties in biochemical studies.17–21 So far, the OTP method has mainly used separated P-diastereomers of 3′-O-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) derivatives of 2′-deoxyribonucleosides (1, X = H, Y = S, R,R = –(CH2)5–, Chart 1), although 2-selena monomers (1, X = H, Y = Se, R,R = –(CH2)5–) have been used in the synthesis of P-stereodefined phosphoroselenoate analogs of DNA.22 In 1996, a set of four monomers 3 (X = O-TBDMS, R = H) was synthesized and chromatographically separated into P-diastereomers.23 They were used in the solid-phase synthesis of dinucleoside phosphorothioates NPSN, which were obtained in 66–98% yields. However, the separation of fast- and slow-eluting monomers was very difficult and inefficient. In other attempts to synthesize P-stereodefined [PS]-RNA, the synthesis of up to 10-mers was achieved using a method based on nucleoside-3′-O-oxazaphospholidine monomers with the 2′-O-TBDMS protecting group.24 Good coupling yields (97–99%) were observed, but stereoselectivity was not perfect (≥96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4). A diastereoselectivity ≥98:2 was observed when oxazaphospholidine monomers with 2′-O-(2-cyanoethoxymethyl) group were used.25
:4). A diastereoselectivity ≥98:2 was observed when oxazaphospholidine monomers with 2′-O-(2-cyanoethoxymethyl) group were used.25
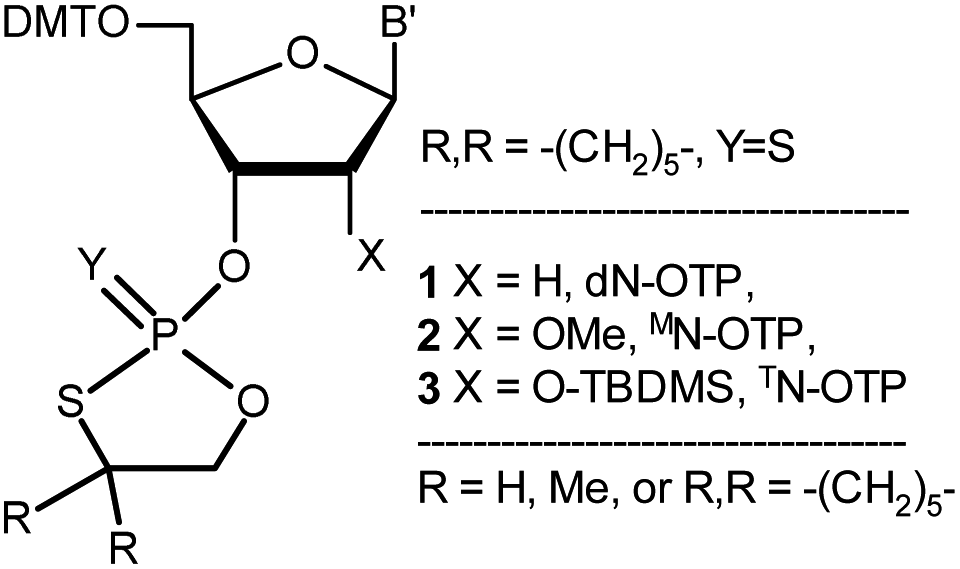 | ||
| Chart 1 Classes of the OTP derivatives of 2′-deoxyribo- and ribonucleosides. B′ = AdeBz, CytBz, GuaiBu, or Ura. For those with R = H or Me, see ref. 13 and 16, respectively. | ||
This discovery prompted our attempt to develop a fully stereospecific method for the synthesis of [PS]-(2′-OMe)-RNA ([PS]-MRNA) and [PS]-RNA oligonucleotides with P-stereodefined phosphorothioate internucleotide linkages. (For simplicity the (2′-OMe)-, (2′-O-TBDMS)-, 2′–OH– and 2′-deoxyribonucleosides will be referred to as MN, TN, N, and dN, respectively). In a next step we will attempt to develop a method for the synthesis of {[PO]-DNA:[PS]-MRNA},‡ {[PO]-DNA:[PS]-RNA}, and {[PO]-RNA:[PS]-RNA} chimeras that could be used as model compounds in biochemical studies.
In this report, we present the synthesis of OTP monomers 2 (X = OMe, MN-OTP, Chart 1) and 3 (X = O-TBDMS, TN-OTP) and their separation into pure P-diastereomers, as well as the result of the successful crystallographic analysis of detritylated 3a (B′ = AdeBz). The P-diastereomerically pure monomers 2 and 3 were used for the synthesis of chimeric dinucleotides 8a and 8b, respectively (Scheme 1), and the absolute configuration of the P atoms was determined enzymatically. Melting curves were recorded to determine the thermal stability of duplexes formed by P-stereodefined chimeric [PS]-{DNA:MRNA} and [PS]-{DNA:RNA} oligonucleotides (10 nt in length) with Watson–Crick paired DNA and MRNA matrices.
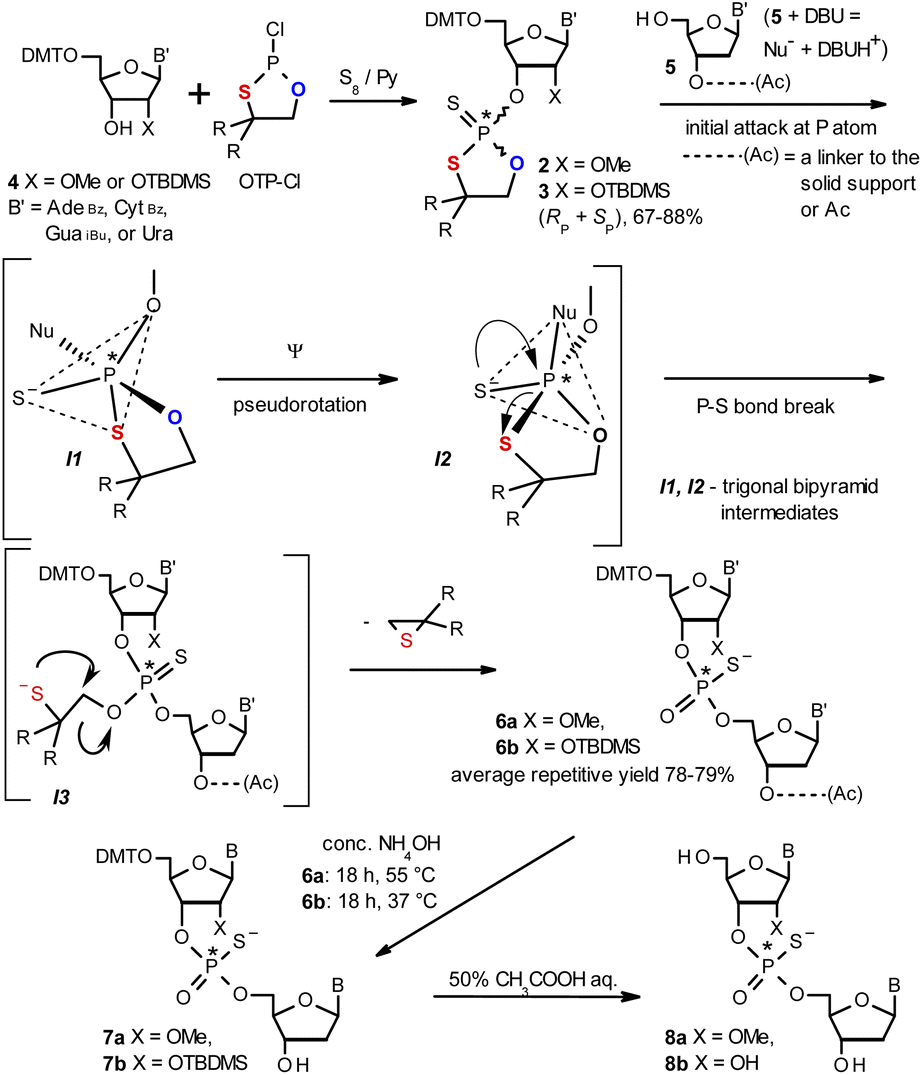 | ||
| Scheme 1 Synthesis of OTP monomers 2 and 3 (R,R = –(CH2)5–) and the mechanistic principle of the coupling step. | ||
Results and discussion
The mechanistic principle of the OTP method
The synthesis of OTP monomers 2 and 3 and the mechanistic principle of the coupling step are shown in Scheme 1. It has been documented26 that the condensation does not follow a stereoinvertive SN2P mechanism but is a stereoretentive process. The initial attack of a nucleoside 5 occurs from the side opposite to the most electronegative atom bonded to the phosphorus center (the oxygen atom in the oxathiaphospholane ring of 2 or 3, marked in blue; Scheme 1) to form a trigonal bipyramidal intermediate I1. Subsequently, a pseudorotation process furnishes the intermediate I2, in which the thioalkyl leaving group occupies the axial position, followed by cleavage of the P–S bond to form the triester intermediate I3. The condensation process concludes with the elimination of episulfide. Finally, deprotection of the resulting intermediates 6 and 7 and purification steps yield [PS]-oligomers 8. The range of P-stereodefined phosphorothioate oligonucleotides was extended by the synthesis of [PS]-LNA27 and [PS]-GNA28 oligomers as well as oligomers containing 3′-amino-2′,3′-dideoxy nucleosides ([NPS]-DNA).29 Unfortunately, oligomers of the latter two series were obtained in low yields due to unexpected side reactions and unfavorable conformational factors.Preparation of P-diastereomerically pure MN-OTP and TN-OTP monomers
Protected ribonucleosides 4 (Scheme 1, B′ = AdeBz, CytBz, GuaiBu, or Ura; X = OMe or O-TBDMS), were phosphitylated with 2-chloro-4,4-pentamethylene-1,3,2-oxathiaphospholane (OTP-Cl, 1.2 equiv.) and then sulfurized with elemental sulfur.30 The resultant monomers 2 and 3 were isolated in 67–88% yields. The P-diastereomers (formed in nearly equimolar amounts) were separated by preparative HPLC on a silica gel column (Fig. S1a and b, ESI†), and their structures were confirmed by HR MS (Fig. S2a–h and S3a–h, ESI†). (For 1H, 13C and 31P NMR spectra see ESI, Fig. S4–S6,† respectively). Interestingly, 31P NMR analysis (Tables 1 and 2) showed that all fast-eluting isomers of 3 had lower chemical shifts (δ) than their slow-eluting counterparts (as observed for dN-OTP monomers16 and LNA-OTP27) whereas this relationship was reverse for compounds 2.| MA-OTP (2a) | MC-OTP (2b) | MG-OTP (2c) | MU-OTP (2d) | |
|---|---|---|---|---|
| a Yield of the isolated mixture of P-diastereomers.b Eluent CHCl3:MeOH (9:1, v/v).c Separation of P-diastereomers at an AcOEt:hexane ratio (v/v).d A Phenomenex Luna 5u Silica gel column (100 Å; 250 × 10 mm; flow rate 5 mL min−1, isocratically).e Recorded with a SYNAPT G2-Si High Definition Mass Spectrometer.f In anhydrous CDCl3. |
||||
| Yielda (%) | 78 | 88 | 69 | 83 |
| Rfb (TLC) | 0.72 | 0.83 | 0.70 | 0.81 |
| Eluent for TLC/HPLCc | 80:20 |
70:30 |
100 | 40:60 |
| MM calc. (Da) | 893 | 869 | 875 | 766 |
|
||||
| For fast-2/slow-2 | ||||
| Rtd (min) | 16/24 | 13/18 | 12/17 | 19/26 |
| HR MSe (m/z) | 892.2630/892.2625 | 868.2510/868.2510 | 874.2706/874.2715 | 765.2079/765.2082 |
| δ 31P NMRf (ppm) | 106.555/106.132 | 106.690/106.579 | 107.042/106.688 | 106.719/106.571 |
| TA-OTP (3a) | TC-OTP (3b) | TG-OTP (3c) | TU-OTP (3d) | |
|---|---|---|---|---|
| a Yield of the isolated mixture of P-diastereomers.b Eluent CHCl3:MeOH (9:1, v/v).c Separation of P-diastereomers at an AcOEt:hexane ratio (v/v).d A Phenomenex Luna 5u Silica gel column (100 Å; 250 × 10 mm; flow rate 5 mL min−1, isocratically).e Recorded with a SYNAPT G2-Si High Definition Mass Spectrometer.f In anhydrous CDCl3. |
||||
| Yielda (%) | 70 | 72 | 67 | 73 |
| Rfb (TLC) | 0.64 | 0.74 | 0.62 | 0.71 |
| Eluent for TLC/HPLCc | 50:50 |
70:30 |
100 | 40:60 |
| MM calc. (Da) | 993 | 969 | 975 | 866 |
|
||||
| For fast-3/slow-3 | ||||
| Rtd (min) | 15/17 | 35/40 | 10/15 | 25/35 |
| HR MSe (m/z) | 992.3309/992.3311 | 968.3198/968.3192 | 974.3427/974.3418 | 865.2776/865.2775 |
| δ 31P NMRf (ppm) | 105.880/107.519 | 106.966/107.022 | 106.244/107.342 | 106.500/107.369 |
X-ray crystallography analysis of the detritylated oxathiaphospholane monomer 3a (B′ = AdeBz)
Previously published results on the hydrolysis of dinucleotides d(NPSN) with snake venom phosphodiesterase (svPDE) and nuclease P1 (nP1) (RP- and SP-specific phosphodiesterase, respectively) showed that the compounds obtained from fast- and slow-eluting monomers dN-OTP (1, X = H, Y = S, R = Me or R,R = –(CH2)5–) contain the internucleotide bonds with the P atoms of the absolute configuration RP and SP, respectively.16 The same correlation was found for the analogous OTP derivatives of LNA nucleosides.27 X-ray analysis of LNA-thymidine-3′-O-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) confirmed the stereoretentive course of the condensation step. The “locked” structure naturally resulted in the C3′-endo conformation of the sugar ring.In the present studies, a crystal suitable for X-ray analysis was obtained from the 5′-OH deprotected fast-eluting monomer 3a (B′ = AdeBz). Detritylation was carried out in anhydrous acetonitrile using a sodium hydrogen sulfate suspension deposited on silica gel.31 The crystal structure was resolved (Fig. 1) with the refinement parameter R = 0.0563 and ESI.† The crystal data and the data acquisition and refinement parameters are listed in Table S1 (ESI†). It should be noted that despite the presence of a 2′-oxygen atom, the (normally strong) anomeric effect is not decisive in this case and the sugar ring exists in the C2′-endo conformation (Fig. 2), perhaps due to the steric requirements of the bulky TBDMS group.
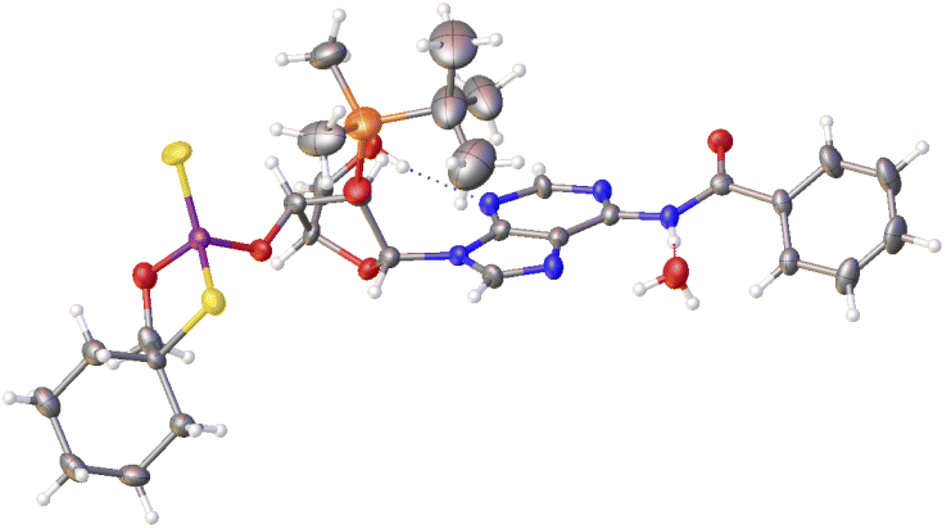 | ||
| Fig. 1 X-ray ORTEP diagram of the detritylated fast-eluting P-diastereomer of 3a showing the RP absolute configuration. The displacement ellipsoids are shown at the 50% probability level.32 Colors: red – oxygen, blue – nitrogen, pink – phosphorus, yellow – sulfur, orange – silicon, black – carbon, white – hydrogen. | ||
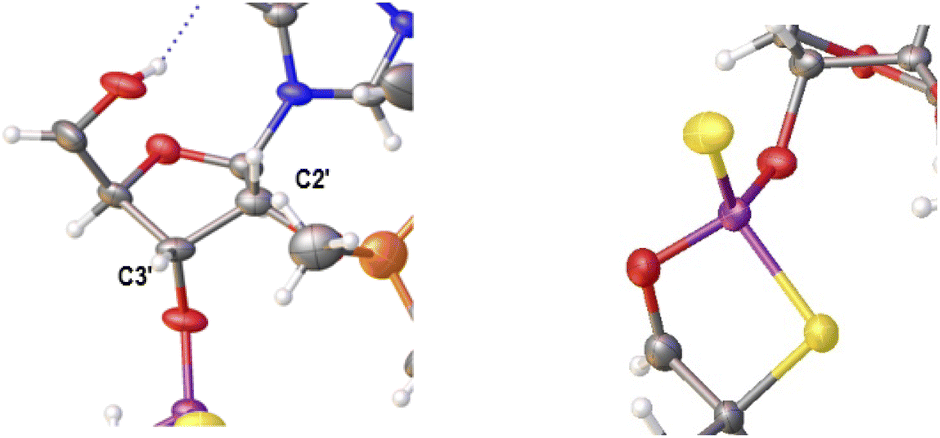 | ||
| Fig. 2 Parts of the X-ray ORTEP diagram show: left image–a DNA-like C2′-endo conformation of detritylated fast-eluting 3a; right image – spatial orientation of substituents on the phosphorus atom. Colors as in Fig. 1. | ||
Crystallization experiments with all other DMT-labeled as well as detritylated OTP derivatives were unsuccessful.
Synthesis and enzymatic analysis of MNPST and NPST dinucleotides
Since X-ray analysis was successful only for the derivative of fast-eluting 3a, enzymatic analysis was performed to complete the stereochemical assignment in the remaining 2 and 3. For this purpose, 4 pairs of P-diastereomeric diesters MNPST (6a, X = OMe) and 4 pairs of P-diastereomeric diesters NPST (6b, X = OH) were prepared from 3′-O-acetyl-thymidine (5, B′ = Thy, Scheme 1) and diastereomerically pure (fast- or slow-eluting) MN-OTP 2a–d or TN-OTP 3a–d, respectively. The base-labile protecting groups in intermediates 6 were removed with concentrated ammonia. Subsequently, products 7a (X = OMe; derived from 2) were routinely detritylated with 50% AcOHaq. The DMT and TBDMS moieties in the freeze-dried products 7b (X = O-TBDMS; derived from 3) were removed by treatment with 3HF × NEt3. The MALDI TOF MS data for the resulting dinucleotides 8a and 8b are given in Table 3.| MN-OTP (2a–d) or TN-OTP (3a–d) | MALDI TOF MS m/z [m/zcalc] | ||
|---|---|---|---|
| B′ | Isomer | MNPST (8a, X = OMe) | NPST (8b, X = OH) |
| AdeBz | Fast | 600.1283 [601] | 586.1066 [587] |
| Slow | 600.1283 [601] | 586.1064 [587] | |
| CytBz | Fast | 576.1171 [577] | 562.1013 [563] |
| Slow | 576.1174 [577] | 562.1011 [563] | |
| GuaiBu | Fast | 616.1229 [616] | 602.1076 [602] |
| Slow | 616.1232 [616] | 602.1074 [602] | |
| Ura | Fast | 577.1006 [578] | 563.0858 [564] |
| Slow | 577.1007 [578] | 563.0848 [564] | |
Small amounts (2–3 OD254 units) of all 16 P-diastereomeric dinucleotides 8 were isolated using RP-HPLC and further treated with the svPDE and nP1 enzymes, which are RP- and SP-stereoselective, respectively. Subsequent RP-HPLC analysis revealed that all dinucleotides 8 obtained from the fast-eluting 2a–d and 3a–d were hydrolyzed with svPDE and not with nP1. Their counterparts obtained from the slow-eluting monomers were hydrolyzed with nP1 and not with svPDE. These results, together with the X-ray data collected for 3a, confirm the stereoretentive course of the condensation step.
Relative reactivity of MN-OTP and TN-OTP compared with dN-OTP
It has been reported previously that when monomers 1 were used, 92–94% yields were observed with single coupling, allowing 18–23 nt long PS-DNA oligomers to be assembled.18 Previous experiments with the OTP derivatives of LNA nucleosides showed that double coupling was necessary to achieve an acceptable yield of the condensation step.27 Using this modified protocol, chimeric [PS]-{DNA:LNA} oligomers were obtained with LNA units in every third position.§ This lower reactivity was tentatively attributed to steric hindrance due to the C2′–O–CH2–C4′ bridge near the reacting phosphorus center. However, unsatisfactory attempts to synthesize oligomers with 3′-amino-2′,3′-dideoxy nucleosides ([NPS]-DNA)29 and subsequent molecular modeling experiments indicated the importance of the deoxyribose ring conformation in the OTP monomers. Importantly, only the OTP derivative of 3′-amino-3′-deoxy-thymidine allowed the synthesis of [NPS]-DNA 8-mer, while none of the monomers with the other three nucleobases allowed more than four coupling steps. Therefore, competition experiments were performed to determine the relative reactivity of selected purine and pyrimidine monomers 2 and 3 compared to the corresponding dN-OTP. For this purpose, the approximately equimolar mixtures 1/2 and 1/3 (measured by 31P NMR) were reacted with thymidine bound to a solid support (10 mg of each OTP monomer was used, i.e., a 14–18-fold excess). The resultant MNPST/dNPST and TNPST/dNPST mixtures were detritylated, detached from the solid support, deprotected with concentrated ammonia (the TBDMS group in the TNPST dinucleotides was not removed), and analyzed using RP-HPLC. The required standards were prepared independently. The results (Table 4) generally show that the MN-OTP monomers studied were two to three times less reactive compared with the corresponding dN-OTP, as the selectivity ratio values (calculated by dividing the percent composition of the dinucleotide products by the percent composition of the OTP substrates) ranged from 0.36 to 0.55. Only for the slow-eluting MA-OTP the difference was almost negligible (Table 4, selectivity ratio 0.91, double underlined). The reactivity of TN-OTP was 5–12 times lower, with the lowest selectivity ratio value of 0.08 (Table 4, underlined). Reactivity comparable to MN-OTP was found only for the fast-eluting TA-OTP (selectivity ratio 0.39).| OTP substrates, dinucleotide productsab | N = A | N = U | ||
|---|---|---|---|---|
| Fast | Slow | Fast | Slow | |
| a The dinucleotide products were detritylated before cleavage from the solid support.b In the adenosine containing products, the benzoyl protecting group was removed with concentrated ammonia.c The selectivity ratios are calculated by dividing % composition of products by % composition of substrates.d The TBDMS group in TNPST dinucleotides was not removed. | ||||
| MN-OTP (2):dN-OTP (1) |
49:51 |
44:56 |
53:47 |
46:54 |
| MNPST:dNPST |
26:74 |
42:58 |
31:69 |
32:68 |
| Selectivity ratioc | 0.36 | 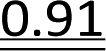 |
0.40 | 0.55 |
| TN-OTP (3):dN-OTP (1) |
42:58 |
44:56 |
53:47 |
55:45 |
| TNPSTd:dNPST |
22:78 |
6:94 |
19:81 |
15:85 |
| Selectivity ratioc | 0.39 | 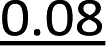 |
0.21 | 0.14 |
The generally low reactivity observed for 2 and 3 suggested that the synthesis protocol optimized for [PS]-DNA34 and later modified for the synthesis of [PS]-LNA oligomers27 would be inadequate. In addition, the widely varying selectivity ratios indicated that several factors might influence reactivity, so that optimization of the protocol for all 16 diastereomers of MN-OTP and TN-OTP would be a lengthy process with uncertain prospects for success. An attempt to synthesize uniformly modified [PS]-(MA)4T, in which the most reactive slow-eluting MA-OTP (2a, see Table 4) was double-coupled (thymidine was bound to the support), yielded only a small amount of product. The results of the DMT cation assay, shown in Fig. 3 (green line), clearly indicate that although the first coupling was carried out with 86% yield, the next three coupling steps were much less efficient with 74, 67 and 49% yield, respectively. The synthesis of [PS]-(MA)4T showed that the synthesis of [PS]-MRNA and [PS]-RNA oligomers with mixed sequences and longer than penta- or hexamers will be very difficult.
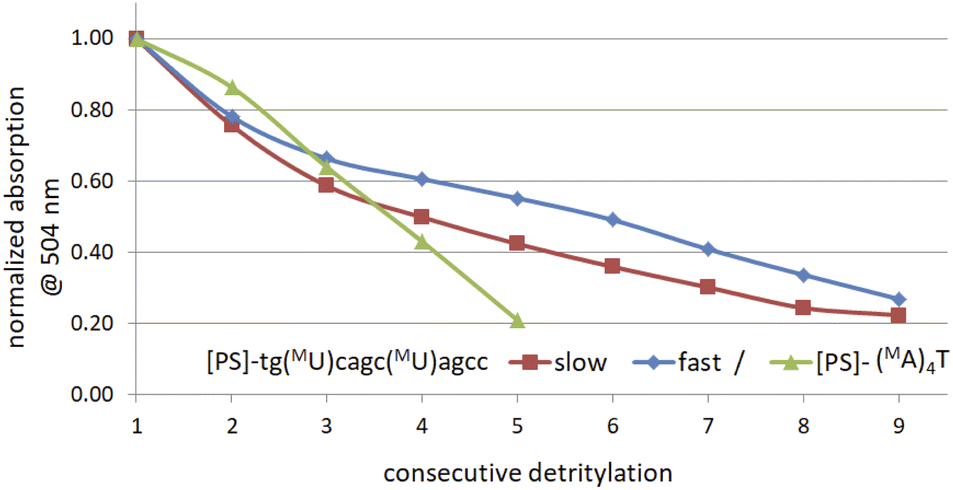 | ||
| Fig. 3 Decay of absorption (at 504 nm) of the DMT-cation released in consecutive detritylation steps. | ||
Synthesis and properties of chimeric [PS]-{DNA:MRNA} and [PS]-{DNA:RNA} oligonucleotides
Because of the relatively low repetitive yield of coupling steps in syntheses with monomers 2 or 3, we focused on the synthesis of P-stereodefined chimeric [PS]-{DNA:MRNA} and [PS]-{DNA:RNA} oligonucleotide decamers (collectively referred to as [PS]-chimeras or PSCh) with base sequence 5′-tgtcagctag-3′, where lowercase letters represent DNA units. In the sequences of analogous chimeric oligomers, the introduced NPS are indicated in uppercase letters (e.g. A or U), whereas the MNPS units are indicated in uppercase letters with a superscript M prefix (e.g. MA or MU).In the synthesis of each oligomer, the dN-OTP monomer was replaced in two selected cycles by MN-OTP or TN-OTP carrying the same nucleobase, except for the T-OTP monomer, which was replaced by MU-OTP or TU-OTP congeners. The codes, specific sequences, and HR MS data for two synthesized reference [PS]-DNA and sixteen chimeric [PS]-oligomers are listed in Table 5. [Note: in codes such as UR or MUS, the capital letters R or S denote [PS]-oligomers with phosphorus atoms with either RP or SP absolute configuration, respectively].
| Code | [PS]-oligomera sequence (5′ → 3′) | MS ES (m/z) | |
|---|---|---|---|
| Calc. | Found | ||
| a In the sequences, the units containing 2′-deoxyribo- and ribonucleosides are indicated in lowercase and uppercase letters, respectively. The MNPS units are marked with a superscript M prefix (MN). | |||
| [PS]-DNA from 1a–d | |||
| BR | tgtcagctag | 3185.34 | 3185.3401 |
| BS | tgtcagctag | 3185.34 | 3185.3301 |
|
|||
| [PS]-{DNA:MRNA} from 1a–d and 2a–d | |||
| MAR | tgtcMAgctMAg | 3245.36 | 3245.3501 |
| MAS | 3245.3501 | ||
| MCR | tgtMCagMCtag | 3245.36 | 3245.3401 |
| MCS | 3245.3500 | ||
| MGR | tMGtcaMGctag | 3245.36 | 3245.4001 |
| MGS | 3245.3401 | ||
| MUR | tgMUcagcMUag | 3217.33 | 3217.3000 |
| MUS | 3217.3000 | ||
|
|||
| [PS]-{DNA:RNA} from 1a–d and 3a–d | |||
| AR | tgtcAgctAg | 3217.33 | 3217.3181 |
| AS | 3217.3101 | ||
| CR | tgtCagCtag | 3217.33 | 3217.3101 |
| CS | 3217.3000 | ||
| GR | tGtcaGctag | 3217.33 | 3217.3101 |
| GS | 3217.3101 | ||
| UR | tgUcagcUag | 3189.30 | 3189.3000 |
| US | 3189.3000 | ||
The DMT cation decay assay was performed for each synthesis, and calculations for the steps using MN-OTP or TN-OTP (double coupling was performed) gave the average coupling yields of 78 and 79%, respectively (Table S2, ESI†). After cleavage from the support and removal of the base-labile protecting groups, the DMT-labeled oligomers were purified by RP-HPLC, and the collected samples were detritylated and isolated by RP-HPLC. Fig. 4 shows typical chromatograms recorded during the DMT-ON and DMT-OFF purification steps; here those for MGS (SP). MALDI TOF MS spectra for PSCh are shown in ESI.†
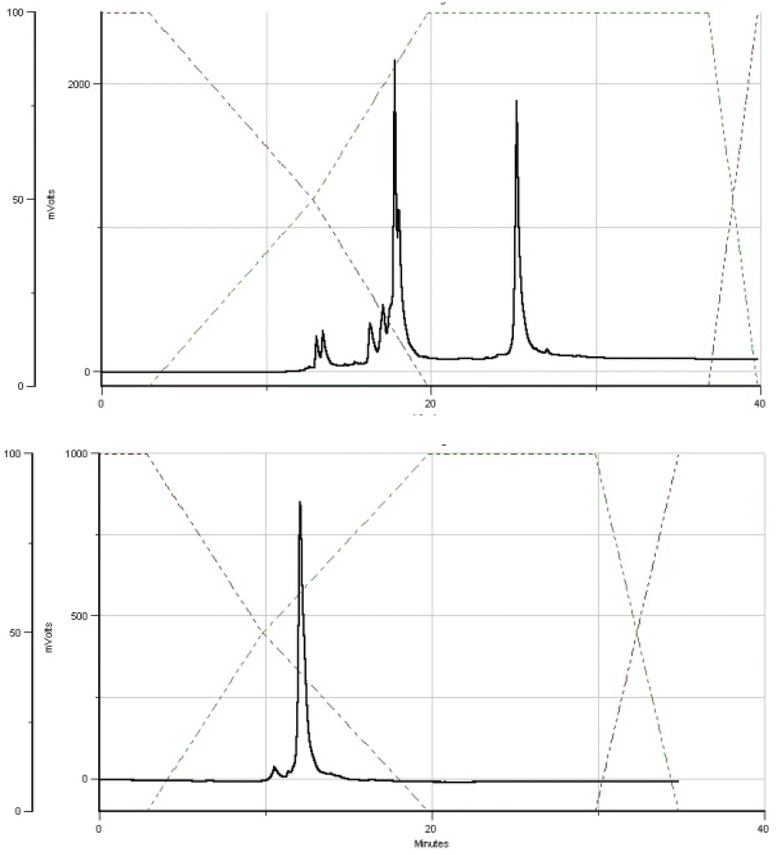 | ||
| Fig. 4 HPLC profiles recorded for MGS during DMT-ON (the upper panel) and DMT-OFF (the lower panel) purification steps. | ||
Melting experiments for canonical and hybrid duplexes
For the melting experiments, the oligomers were mixed with the equimolar amount of Watson–Crick-paired DNA matrix (Md) or mRNA matrix (Mm) (2.0 μM concentration each) in pH 7.2 buffer. The observed changes in UV absorbance followed the two-state mechanism and the recorded curves had the expected S-shape. An example set of melting curves is shown in Fig. 5. Melting temperatures (compiled in Table 6) were calculated using the numerical fitting method provided by the UV spectrophotometer manufacturer. The ΔTm values were calculated by subtracting the control Tm from the values recorded for the duplexes tested.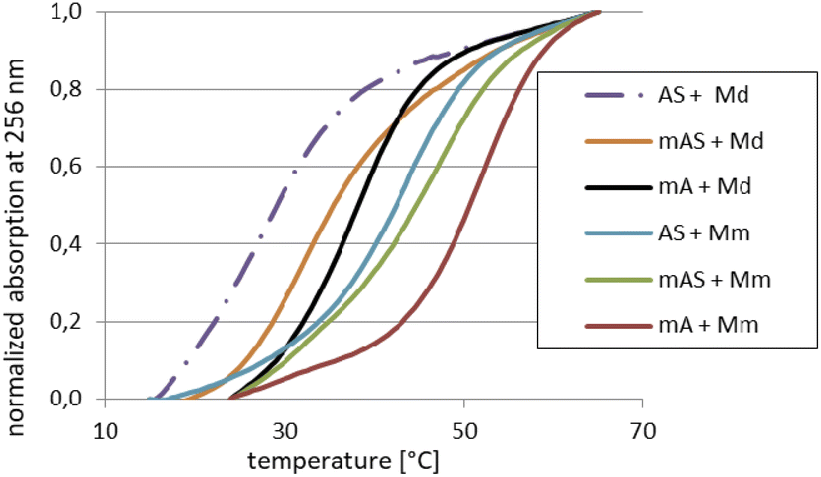 | ||
| Fig. 5 Normalized melting curves for selected duplexes (see legend) dissolved in pH 7.2 buffer (10 mM Tris–HCl, 100 mM NaCl, and 10 mM MgCl2). The curves designations mA and mAS (in the legend) correspond to MA and MAS, respectively. | ||
:1 molar ratio
| Code | Sequence (5′ → 3′)a | Md | Mm | ||
|---|---|---|---|---|---|
| Tm [°C] | ΔTm [°C] | Tm [°C] | ΔTm [°C] | ||
| a In the sequences, the DNA and RNA units are written in lowercase and uppercase letters, respectively. The MNPS units are marked with a superscript M prefix (MN).b The ΔTm values were calculated using the Tm values for the corresponding duplexes Bd/Md or Bd/Mm.c The ΔTm values were calculated using the Tm values for the corresponding duplexes B(R,S)/Md or B(R,S)/Mm. | |||||
| [PO]-DNA reference oligomer | |||||
| Bd | tgtcagctag | 41 | — | 51 | — |
|
|||||
| [PO]-{DNA:MRNA} chimerasb | |||||
| MA | tgtcMAgctMAg | 38 | −3 | 52 | +1 |
| MC | tgtMCagMCtag | 36 | −5 | 48 | −3 |
| MG | tMGtcaMGctag | 40 | −1 | 55 | +4 |
| MU | tgMUcagcMUag | 32 | −9 | 46 | −5 |
|
|||||
| [PS]-DNA reference oligomers | |||||
| BR | tgtcagctag | 35 | — | 32 | — |
| BS | 35 | — | 35 | — | |
|
|||||
| [PS]-{DNA:RNA} chimerasc | |||||
| AR | tgtcAgctAg | 30 | −5 | 40 | +8 |
| AS | 27 | −8 | 43 | +8 | |
| CR | tgtCagCtag | 30 | −5 | 38 | +6 |
| CS | 34 | −1 | 39 | +4 | |
| GR | tGtcaGctag | 31 | −4 | 39 | +7 |
| GS | 30 | −5 | 37 | +2 | |
| UR | tgUcagcUag | 27 | −8 | 40 | +8 |
| US | 31 | −4 | 40 | +5 | |
|
|||||
| [PS]-{DNA:MRNA} chimerasc | |||||
| MAR | tgtcMAgctMAg | 30 | −5 | 44 | +12 |
| MAS | 32 | −2 | 47 | +12 | |
| MCR | tgtMCagMCtag | 31 | −4 | 41 | +9 |
| MCS | 32 | −4 | 39 | +4 | |
| MGR | tMGtcaMGctag | 30 | −5 | 46 | +14 |
| MGS | 31 | −4 | 48 | +13 | |
| MUR | tgMUcagcMUag | 29 | −6 | 40 | +8 |
| MUS | 24 | −11 | 41 | +6 | |
Melting experiments for duplexes formed by [PO]-chimeras
When analyzing the results of melting experiments with duplexes formed by [PO]-{DNA:MRNA} chimeras (MA, MC, MG, and MU), an oligonucleotide with the sequence [PO]-5′-tgtcagctag-3′ (Basal DNA oligomer, Bd) was used as a reference. Compared to the reference Bd/Md duplex (Tm = 41 °C, Table 6), the [PO]-{DNA:MRNA}/Md duplexes were 1 ÷ 9 °C less thermally stable (Tm = 32 ÷ 40 °C, Fig. 6, green bars), with the pyrimidine units (MCPO, MUPO) destabilizing the duplexes more than the purine units.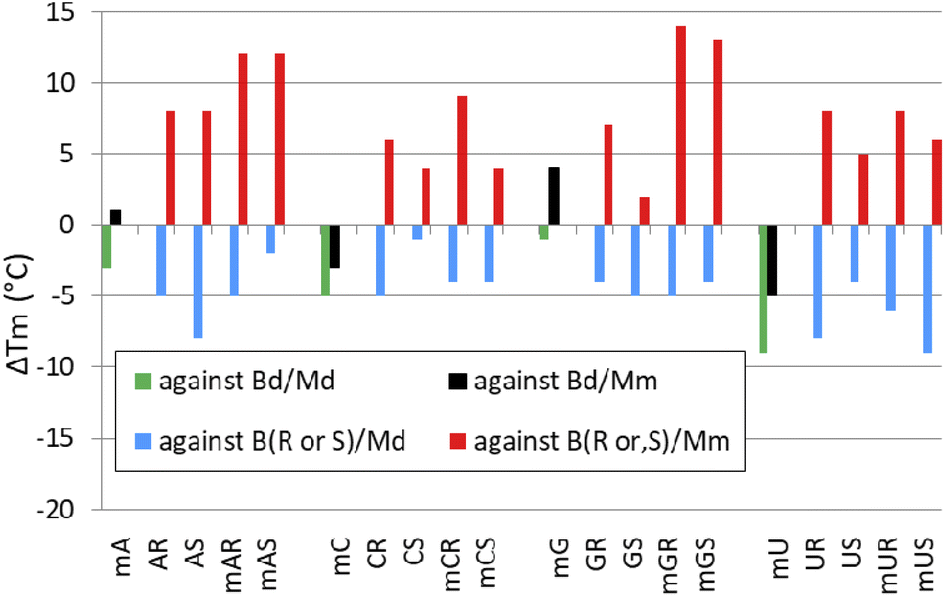 | ||
| Fig. 6 Changes in melting temperature of duplexes (ΔTm; °C) formed by [PO]-{DNA:MRNA} chimeras with Md (green bars) or Mm (black bars), and PSCh oligomers with Md (blue bars) and Mm (red bars). The ΔTm values were calculated using the Tm values for the corresponding duplexes Bd/Md, Bd/Mm, B(R,S)/Md and B(R,S)/Mm. Designations along the horizontal axis such as mAR or mGS correspond to MAR and MGS, respectively. | ||
Melting experiments with the Mm matrix showed that the purine units (MAPO, MGPO) slightly stabilized the duplexes (ΔTm = 1 ÷ 4 °C, Fig. 6, black bars), compared to the reference Bd/Mm duplex (Tm = 51 °C). The duplexes containing the MCPO or MUPO nucleotides were less stable than Bd/Mm and the destabilizing effect was strongest for MU (ΔTm = −5 °C). These results differ from those of Wengel and coworkers, who showed that one to three LNA nucleotides introduced into the DNA strand at either the adenine (5′-AGCACCAG) or thymine (5′-TGCTCCTG) residues increased the thermal stability of the corresponding {DNA:LNA}/RNA heteroduplexes in 110 mM Na+ buffer.33 For two introduced A-LNA or T-LNA units, ΔTm = 10 and 16 °C, respectively, were found. It can be argued that RNA matrices were used in these experiments, but Dolot and coworkers showed that crystals of DNA/RNA and DNA/MRNA duplexes have very similar geometries.34
Conclusions
The 2-thio-1,3,2-oxathiaphospholane monomers of 2′-OMe and 2′-O-TBDMS series (MN-OTP and TN-OTP, 2 and 3, respectively) were synthesized in good yield and effectively separated into pure P-diastereomers. The X-ray crystallographic analysis revealed the RP absolute configuration of the phosphorus atom in the fast-eluting diastereomer 3a (B′ = AdeBz). Using the RP-selective phosphodiesterase svPDE and the SP-selective Nuclease P1 it was found that in both monomer series the fast- and slow-eluting P-diastereomers are precursors of dinucleotides with RP and SP absolute configuration of the phosphorus atoms, respectively. Importantly, no traces of P-epimerization were observed during coupling steps. P-Stereodefined chimeric [PS]-{DNA:MRNA} and isosequential [PS]-{DNA:RNA} 10-mer oligomers with two MNPS or NPS units were synthesized. Melting experiments using complexes with Watson–Crick paired DNA matrix showed that MNPS or NPS units decrease the thermal stability of the duplexes (ΔTm = −0.5 ÷ −5.5 °C per modification), regardless of the absolute configuration of the P-atoms. When the (2′-OMe)-RNA matrix was used an increase in Tm was observed in all cases (ΔTm = +1 ÷ +7 °C per modification). The changes in thermal stability of duplexes formed by [PS]-chimeras with DNA and (2′-OMe)-RNA matrices did not correlate with the absolute configuration of phosphorus atoms.Despite of double-coupling, the average repetitive yields of the condensation steps with either 2 or 3 were rather moderate (78–79%). Nevertheless, even these moderate yields should allow the synthesis of {[PO]-DNA:[PS]-MRNA} and {[PO]-RNA:[PS]-RNA} chimeras that could be used as model compounds in biochemical studies. To achieve this goal, the conditions for an effective combination of phosphoramidite and OTP synthesis methods need to be worked out. This work is in progress.
Experimental section
1H NMR and 31P NMR spectra were acquired using Bruker instruments (AV-200, DRX-500, or Avance Neo 400; operating for 1H at 200, 500, and 400 MHz, respectively). Chemical shift (δ) values are in ppm, referenced to internal tetramethylsilane (TMS) or residual solvent protons for 1H NMR and external 85% H3PO4 for 31P NMR.High-resolution mass spectra (HR MS) were acquired using a Synapt G2 Si mass spectrometer (Waters Corporation, Milford, MA) equipped with an ESI source and a quadrupole time-of-flight mass analyzer. Measurements were performed in negative ion mode with the capillary and sampling cone voltages set at 2.7 kV and 20 V, respectively. The temperature of the source was 110 °C. To ensure satisfactory accuracy, data were acquired in a centroid mode and readings were corrected during acquisition using leucine-enkephalin as an external reference (Lock-SprayTM) generating reference ions at m/z 554.2615 Da ([M–H]−) in negative ESI mode. Data sets were processed using MassLynx 4.1 software (Waters).
MALDI-TOF MS analyses of oligonucleotides were performed with negative ion detection using a Voyager Elite instrument (PerSeptive Biosystems Inc, Framingham, MA) operating in reflector mode or an Axima Performance instrument (Shimadzu Biotech Corp., Japan) operating in linear mode.
TLC silica gel 60 plates F254 developed in CHCl3/MeOH, 9:1 (v/v), were used for routine analyses. HP TLC Silica gel 60 plates with a UV-F254 indicator were used to evaluate the separability of the P-diastereomers of 2 and 3.
A Varian binary HPLC system (two PrepStar 210 pumps, 25 mL pump heads, one ProStar 320 UV/vis detector at 275 nm) was used for HPLC separation of the P-diastereomers of monomers 2 and 3. A Phenomenex Luna 5 μm silica column (100 Å; 250 × 10 mm; flow rate 5 mL min−1) was used in search of the appropriate conditions. Preparative separation was performed with a Pursuit XRs silica gel column (10 μm, 250 × 21.2 mm) eluted at a flow rate of 25 mL min−1. Gradient-grade HPLC solvents from Sigma-Aldrich, Baker, or ChemPur were used.
Crystallographic data were collected using an XtaLAB Synergy, Dualflex, HyPix diffractometer at T = 100.00(10) K.
Routine UV spectra were recorded with a CINTRA 4040 spectrophotometer (GBC, Dandenong, Australia). UV-monitored melting experiments were performed at 260 nm in cuvettes with 1 cm path length using a UV-vis NIR spectrophotometer V-770 (Jasco, Japan) equipped with a 6 × 1 Peltier thermal cell.
For thermal dissociation experiments, oligonucleotide samples were dissolved in pH 7.2 buffer containing 10 mM Tris–HCl, 100 mM NaCl, and 10 mM MgCl2. Annealing was performed from 85 °C to 15 °C with a temperature gradient of 1 °C min−1. Melting profiles were recorded over a range of 15→85 °C (0.5 °C min−1). Melting temperatures (Tm) were calculated using the software provided by Jasco.
P-Stereodefined PSCh oligonucleotides were synthesized manually at a 1 μmol scale, using the slightly modified previously published protocols.16,35 The first nucleoside unit was anchored to the solid support by a DBU-resistant  –succinoyl linker [–C(O)CH2CH2C(O)–
–succinoyl linker [–C(O)CH2CH2C(O)– –LCAA-CPG].36 Routine coupling steps were performed using 20 mg of the dN-OTP monomers 1. In the cycles where the monomers 2 or 3 were incorporated, double coupling was executed (20 mg + 20 mg) and in both steps the coupling time was extended to 20 minutes. The assembled DMT-labeled oligomers were cleaved from the support (25% NH4OH, r.t., 3 h) and the protecting groups from nucleobases were removed with 25% NH4OHaq over 18 h ([PS]-{DNA:MRNA}, 55 °C; [PS]-{DNA:TRNA}, 37 °C). The samples were concentrated under reduced pressure and the DMT-labeled oligonucleotides were isolated using RP-HPLC. The collected fractions were concentrated under reduced pressure. The [PS]-{DNA:MRNA} oligomers were detritylated with 50% AcOHaq for 20 minutes and the volatile components were evaporated. The TBDMS and DMT groups in [PS]-{DNA:TRNA} oligomers were simultaneously removed by treatment with 3HF × NEt3 at room temperature for 20 hours. The reagent was quenched with sterile water and all chimeric [PS]-oligomers were isolated by means of RP-HPLC. The identity of oligonucleotides was confirmed by MALDI-TOF MS (for the relevant spectra see ESI†).
–LCAA-CPG].36 Routine coupling steps were performed using 20 mg of the dN-OTP monomers 1. In the cycles where the monomers 2 or 3 were incorporated, double coupling was executed (20 mg + 20 mg) and in both steps the coupling time was extended to 20 minutes. The assembled DMT-labeled oligomers were cleaved from the support (25% NH4OH, r.t., 3 h) and the protecting groups from nucleobases were removed with 25% NH4OHaq over 18 h ([PS]-{DNA:MRNA}, 55 °C; [PS]-{DNA:TRNA}, 37 °C). The samples were concentrated under reduced pressure and the DMT-labeled oligonucleotides were isolated using RP-HPLC. The collected fractions were concentrated under reduced pressure. The [PS]-{DNA:MRNA} oligomers were detritylated with 50% AcOHaq for 20 minutes and the volatile components were evaporated. The TBDMS and DMT groups in [PS]-{DNA:TRNA} oligomers were simultaneously removed by treatment with 3HF × NEt3 at room temperature for 20 hours. The reagent was quenched with sterile water and all chimeric [PS]-oligomers were isolated by means of RP-HPLC. The identity of oligonucleotides was confirmed by MALDI-TOF MS (for the relevant spectra see ESI†).
Unmodified DNA and (2′-OMe)-RNA oligonucleotide matrices (Md and Mm, respectively) were synthesized on a Gene-World synthesizer (K&A Laborgeraete GbR, Schaafheim, Germany) and routinely isolated by a two-step RP-HPLC method.
Synthesis of oxathiaphospholane monomers 2 and 3 – a general procedure
Oxathiaphospholane monomers 2 and 3 were synthesized analogously to a general procedure published for the synthesis of standard OTP monomers.34 Briefly, a suitable nucleoside 4 (2 mmol, N-protected if necessary; Scheme 1) was dried overnight in a 25 mL round bottom flask with two necks (with a magnetic stirrer inside) at high vacuum (0.01 mm Hg). To the flask purged with dry argon, 10 mL of anhydrous pyridine was added using a gas-tight syringe. To the stirred solution, 0.36 mL of 2-chloro-4,4-pentamethylene-1,3,2-oxathiaphospholane (OTP-Cl, 0.51 g, 2.4 mmol) was added at room temperature for 5 minutes using a gas-tight syringe. After approximately 5 minutes, 0.2 g of dry elemental sulfur (approximately 6 mmol) was added, and stirring was continued at room temperature for 12 hours. Excess sulfur was filtered off, the solvent evaporated, and the residue dissolved in 2 mL of acid-free chloroform (distilled with pyridine). The crude product was applied to a silica gel column (glass tube 15 × 3 cm, height of gel suspension layer – 5 cm, 230–400 mesh) and the column was eluted with acid-free chloroform. Appropriate fractions (8–10 mL each, analyzed by TLC on silica gel 60 plates, Rf's are given in Table 1) were combined, and the solvent was evaporated under reduced pressure (15–20 mm Hg) at a water bath temperature not exceeding 30 °C. Anhydrous toluene (5–6 mL) was then added and the solution was evaporated to dryness excluding moisture. Finally, the samples were dried under high vacuum (oil pump) for 12 hours and stored at −15 °C. The yields of the isolated mixtures of P-diastereomers are given in Tables 1 and 2.Detritylation of the fast-eluting diastereomer of 3a
To a solution of 120 mg (0.12 mmol) of fast-eluting isomer of 5′-O-DMT-2′-O-TBDMS-N6-Bz-adenosine-3′-O-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) in 2 mL of anhydrous acetonitrile a sodium hydrogen sulfate/silica gel reagent (50 mg) was added and the suspension was stirred at room temperature. The reaction progress was monitored by TLC using a 9:1 (v/v) chloroform:methanol mixture as an eluent (Rf of the substrate 0.57; Rf of the product 0.31). After 26 hours the reaction went to complete. The silica gel was filtered off and the solvent was evaporated in vacuo. The product was isolated by semipreparative HPLC (a silica gel column eluted with ethyl acetate) and evaporation at reduced pressure furnished white solid material (52 mg, 62%).
Details of X-ray data collection and reduction
Single colorless transparent plate-shaped crystals of the detritylated fast-eluting P-diastereomer of 3a were obtained by re-crystallization from a mixture of ethyl acetate and methanol (4:1 v/v). A suitable crystal of 0.11 × 0.08 × 0.02 mm dimension was selected and mounted on a suitable support. During data collection the crystal was kept at a steady temperature T = 100.00(10) K. The structure was solved with the XT structure solution program37 using an intrinsic phasing solution method and Olex2 (ref. 38) as a graphical interface. The model was refined with ShelXL (version 2018/3; ref. 39), using the least squares minimization.
In solution synthesis of dinucleoside 3′,5′-phosphorothioates MNPST (8a) and NPST (8b)
To a sample of a P-diastereomerically pure (fast- or slow-eluting) oxathiaphospholane monomer 2 or 3 (5 mg, ca. 5 μmol) and 3′-O-Ac-thymidine (3 mg, ca. 11 μmol), 600 μL of dry acetonitrile and 5 μL of 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU, 1.1 molar equivalent over the OTP monomers) were added under dry argon. After 2 h the reaction was complete. The acetonitrile was removed under reduced pressure and the products 6 were treated (in a tightly closed vessel) with concentrated ammonia solution (1 mL) for 18 h either at 55 °C (6a) or at 37 °C (6b), except for B = Ura (2 h at room temperature). The products 7a were routinely detritylated using 50% AcOHaq. In case of 7b, approximately 10% of the ammoniacal solution was evaporated to dryness and the DMT and TBDMS moieties were removed with 3HFxNEt3 (150 μL, 17 h, room temperature) followed by evaporation under reduced pressure. The dimers 8 were isolated using RP-HPLC. Conditions: a C18 column, 250 × 4.6 mm, 5 μm; elution at 1.0 mL min−1 with a gradient of 0.1 M TEAB, pH 7.3 to 40% CH3CN in 0.1 M TEAB over 20 minutes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Centre of Science, Poland, grants UMO-2015/19/B/ST5/03116 (to P. G.) and UMO-2021/43/D/ST4/02433 (to K. J.). An Avance Neo 400 NMR spectrometer was purchased using funds provided by the EU Regional Operational Program of the Lodz Region, RPLD.01.01.00-10-0008/18.Notes and references
- F. Eckstein, Angew. Chem., Int. Ed. Engl., 1983, 22, 423 CrossRef CAS
; W. J. Stec, G. Zon, W. Egan and B. Stec, J. Am. C. Soc., 1984, 106, 6077 CrossRef
- C. A. Stein, Biochim. Biophys. Acta, 1999, 1489, 45 CrossRef CAS
- A. M. Krieg, Antisense Nucleic Acid Drug Dev., 2001, 11, 181 CrossRef CAS PubMed
- A. M. Krieg, P. Guga and W. J. Stec, Oligonucleotides, 2003, 13, 491 CrossRef CAS PubMed
- S. T. Crooke, S. Wang, T. A. Vickers, W. Shen and X. Liang, Nat. Biotechnol., 2017, 35, 230 CrossRef CAS
- Q. Laurent, R. Martinent, D. Moreau, N. Winssinger, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2021, 60, 19102 CrossRef CAS PubMed
- S. T. Crooke, P. P. Seth, T. A. Vickers and X. Liang, J. Am. Chem. Soc., 2020, 142, 14754 CrossRef CAS PubMed
- L. Wang, S. Chen S, T. Xu, K. Taghizadeh, J. S. Wishnok, X. Zhou, D. You, Z. Deng and P. C. Dedon, Nat. Chem. Biol., 2007, 3, 709 CrossRef CAS
- L. Xiong, S. Liu, S. Chen, Y. Xiao, B. Zhu, Y. Gao, Y. Zhang, B. Chen, J. Luo, Z. Deng, X. Chen, L. Wang and S. Chen, Nat. Commun., 2019, 10, 1 CrossRef PubMed
- P. Guga and M. Koziołkiewicz, Chem. Biodiversity, 2011, 8, 1642 CrossRef CAS PubMed
- Y. Wu, Y. Tang, X. Dong, Y. Y. Zheng, P. Haruehanroengra, S. Mao, Q. Lin and J. Sheng, ACS Chem. Biol., 2020, 15, 1301 CrossRef CAS
- P. A. Frey and R. D. Sammons, Science, 1985, 228, 541 CrossRef CAS PubMed
- W. J. Stec, A. Grajkowski, M. Koziołkiewicz and B. Uznański, Nucleic Acids Res., 1991, 19, 5883 CrossRef CAS
- A. Wilk, A. Grajkowski, L. R. Phillips and S. L. Beaucage, J. Am. Chem. Soc., 2000, 122, 2149 CrossRef CAS
- N. Oka, M. Yamamoto, T. Sato and T. Wada, J. Am. Chem. Soc., 2008, 130, 16031 CrossRef CAS
- W. J. Stec, B. Karwowski, M. Boczkowska, P. Guga, M. Koziołkiewicz, M. Sochacki, M. Wieczorek and J. Błaszczyk, J. Am. Chem. Soc., 1998, 120, 7156 CrossRef CAS
- M. Boczkowska, P. Guga and W. J. Stec, J. Biomol. Struct. Dyn., 1999, 16, 1291 Search PubMed
- T. Inagawa, H. Nakashima, B. Karwowski, P. Guga, W. J. Stec, H. Takeuchi and H. Takaku, FEBS Lett., 2002, 528, 48 CrossRef CAS
- P. Guga, Curr. Top. Med. Chem., 2007, 7, 695 CrossRef CAS PubMed
- P. Guga, M. Boczkowska, M. Janicka, A. Maciaszek, S. Kuberski and W. J. Stec, Biophys. J., 2007, 92, 2507 CrossRef CAS PubMed
- P. Guga, M. Janicka, A. Maciaszek, B. Rębowska and G. Nowak, Biophys. J., 2007, 93, 3567 CrossRef CAS PubMed
- P. Guga, A. Maciaszek and W. J. Stec, Org. Lett., 2005, 7, 3901 CrossRef CAS PubMed
- A. Sierzchała, A. Okruszek and W. J. Stec, J. Org. Chem., 1996, 61, 6713 CrossRef PubMed
- N. Oka, T. Kondo, S. Fujiwara, Y. Maizuru and T. Wada, Org. Lett., 2009, 11, 967 CrossRef CAS PubMed
- Y. Nukaga, K. Yamada, T. Ogata, N. Oka and T. Wada, J. Org. Chem., 2012, 77, 7913 CrossRef CAS PubMed
- W. J. Stec, A. Grajkowski, A. Kobylańska, B. Karwowski, M. Koziołkiewicz, K. Misiura, A. Okruszek, A. Wilk, P. Guga and M. Boczkowska, J. Am. Chem. Soc., 1995, 117, 12019 CrossRef CAS
- K. Jastrzębska, A. Maciaszek, R. Dolot, G. Bujacz and P. Guga, Org. Biomol. Chem., 2015, 13, 10032 RSC
- A. Tomaszewska-Antczak, K. Jastrzębska, A. Maciaszek, B. Mikołajczyk and P. Guga, RSC Adv., 2018, 8, 24942 RSC
- E. Radzikowska, R. Kaczmarek, D. Korczyński, A. Krakowiak, B. Mikołajczyk, J. Baraniak, P. Guga, K. A. Wheeler, T. Pawlak and B. Nawrot, RSC Adv., 2020, 10, 35185 RSC
- R. Wallin, M. Kalek, A. Bartoszewicz, M. Thelin and J. Stawinski, Phosphorus, Sulfur, and Silicon, 2009, 184, 908 CrossRef CAS
- R. K. Kannasani, V. V. S. Peruri and S. R. Battula, Chem. Cent. J., 2012, 6, 136 CrossRef CAS PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS
- H. Kaur, J. Wengel and S. Maiti, Biochemistry, 2008, 47, 1218–1227 CrossRef CAS PubMed
- R. Dolot, A. Maciaszek, B. Mikołajczyk and B. Nawrot, Crystals, 2022, 12, 760 CrossRef CAS
- P. Guga and W. J. Stec, in Current Protocols in Nucleic Acid Chemistry, ed. S. L. Beaucage, D. E. Bergstrom, G. D. Glick and R. A. Jones, John Wiley and Sons, Hoboken, N. J., 2003, p. 4.17.1 Search PubMed
- T. Brown, C. E. Pritchard, G. Turner and S. A. Salisbury, J. Chem. Soc., Chem. Commun., 1989, 891 RSC
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC profiles for the separation of P-diastereomers of 2 and 3; HR MS, 1H, 13C and 31P NMR spectra for P-diastereomers of 2 and 3; the crystal data and data collection and refinement parameters for the fast-eluting detritylated 3a (B′ = AdeBz); MALDI-TOF mass spectra recorded for chimeric PS-oligonucleotides. CCDC 2063388. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra04855h |
| ‡ A colon in structures such as {[PO]-RNA:[PS]-RNA} or [PS]-{DNA:MRNA} indicates that the nucleotides of both types are contained in the same oligonucleotide strand. |
| § Uniformly modified [PS]-LNA oligomers were not synthesized because of expected too strong rigidity of the resultant strand. |
| This journal is © The Royal Society of Chemistry 2022 |