
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater oxidation at low potential exploiting a nitroxide/oxoammonium ion redox couple as mediator†
Antonio
Barbon
 *a,
Abdirisak Ahmed
Isse
a,
Armando
Gennaro‡
a,
Raanan
Carmieli
b,
Itzhak
Bilkis
*c and
Lev
Weiner
*bd
*a,
Abdirisak Ahmed
Isse
a,
Armando
Gennaro‡
a,
Raanan
Carmieli
b,
Itzhak
Bilkis
*c and
Lev
Weiner
*bd
aDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: antonio.barbon@unipd.it
bDepartment of Chemical Research Support, Weizmann Institute of Science, Rehovot 7610001, Israel. E-mail: leweiner1950@gmail.com
cFaculty of Agricultural, Food and Environmental Sciences, Hebrew University, Rehovot 7610001, Israel. E-mail: Itzhak.bilkis@mail.huji.ac.il
dDepartment of Brain Sciences, Weizmann Institute of Science, Rehovot 7610001, Israel
First published on 27th September 2022
Abstract
The oxoammonium cation (OAC) of 3-carboxy proxyl, a nitroxide radical (NitR), could be produced either by chemical or by electrochemical oxidation (0.8–1.0 V vs. NHE) of the radical. We have determined that in dilute aqueous basic solutions (pH ≥ 9), OAC is reduced quantitatively to the original radical with concomitant formation of molecular oxygen in a ratio ca. 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (4 moles of OAC reduced per 1 mole of O2), and the redox cycle can be repeated. The low electrolysis potential (0.8 V) contrasts with the high redox potential of the bare OH− anion (2–2.6 V vs. NHE for the first outer-sphere electron transfer). This apparent thermodynamic paradox was solved by a careful study of its possible mechanism. In our opinion, OAC/NitR's may represent a new class of redox mediators for a novel approach to water oxidation (and generation of molecular oxygen) at a low potential.
:1 (4 moles of OAC reduced per 1 mole of O2), and the redox cycle can be repeated. The low electrolysis potential (0.8 V) contrasts with the high redox potential of the bare OH− anion (2–2.6 V vs. NHE for the first outer-sphere electron transfer). This apparent thermodynamic paradox was solved by a careful study of its possible mechanism. In our opinion, OAC/NitR's may represent a new class of redox mediators for a novel approach to water oxidation (and generation of molecular oxygen) at a low potential.
Introduction
Stable nitroxide radicals (NitRs) and oxoammonium cations (OACs) form redox couples with redox potentials in the range of 0.8–1.0 V vs. NHE in water. Both species have attracted attention of academia and industry because of their possible applications. The NitRs have been used as spin labels and spin probes, but at the same time they are exploited for their reactivity, for example they are well known to act as antioxidants.1–4 Nitroxides are formed as spin-adducts in the spin-trapping technique,5 the final products between a precursor and short-lived radicals, bearing structural information on these transient species.The OACs have been extensively studied in the past 20 years due to their ability to perform specific oxidations of various organic compounds, e.g., primary alcohols to aldehydes and carboxylic acids,6–8 and of N-alkyl anilines to quinolones.9
Earlier, we used some OAC/NitR redox couples to study electron transfer processes in aqueous solutions or between different compartments of biomolecules, e.g., proteins, phospholipid membranes.3,10,11 These investigations revealed, unexpectedly, that the OACs are reduced back to the corresponding NitRs at alkaline pH values.
We here propose that in the back reaction from the OAC to the NitR in aqueous basic solutions, the reduction process involves the oxidation of HO− ions, finally resulting in oxidation of water. This reaction is tremendously important for its role in water splitting by electrolysis, which is an intensely investigated topic for a low-cost production of hydrogen as a clean and sustainable energy carrier.12 Both heterogeneous and homogeneous molecular electrocatalysts based on transition metals are widely used for water oxidation to O2.13,14 Also, metal-free catalysts based on carbon materials have been widely investigated for water oxidation.15 In general, oxidation of water on these catalysts requires potentials that are significantly higher than the equilibrium potential of the H2O/O2 redox couple. Typically, overpotentials in the range of 0.3–0.7 V are reported for electrocatalysts based on various metals such as Mn, Ru, Ir, Fe, Co, Cu.16–19
A few examples of organic catalysts for the electrochemical oxidation of H2O to O2 have also been reported, but at potentials substantially higher than equilibrium potentials.20,21
In biological systems, like in the photosynthetic P680 system, able to drive the oxidation of water at pH 7,22 the redox potential is of 1.25 V vs. NHE.
In the present study we provide experimental evidence for the oxidation of HO− by the OAC in diluted solutions with concomitant formation of O2 during its conversion back to the original NitR. Paradoxically, the process should suffer a high overpotential relative to the oxidation of the hydroxyl ion to form the hydroxyl radical HO˙, (E0 = 2.73 V vs. NHE),23 which renders the process thermodynamically and kinetically highly demanding. We investigate the mode of action of the oxoammonium cation of 3-carboxyproxyl (3CP) as an oxidant with a relatively low redox potential (0.8–1.0 V vs. NHE) and propose a plausible mechanism for the process.
Results and discussion
Kinetics of recovery of the initial NitR from the photo- and electrochemically-generated OAC
OAC was obtained from a water-soluble NitR (3-carboxy-proxyl, 3-CP, see inset in Fig. 1a), as already reported,10,11 by using Ru(III). This last species was formed by oxidation of [Ru(II)(bpy)3]2+ in the presence of ammonium persulphate, (NH4)2S2O8, and laser pulse excitation at 436 nm (blue lightning in Fig. 1). Fig. 1a shows the initial continuous-wave cw-EPR spectrum of 3-CP (blue line). It is typical of an NitR, with three resolved lines due to the hyperfine splitting of the 14N atom (I = 1) of the nitroxide. It is also possible to distinguish a further splitting of the lines due to 13C (I = 1/2) in natural abundance.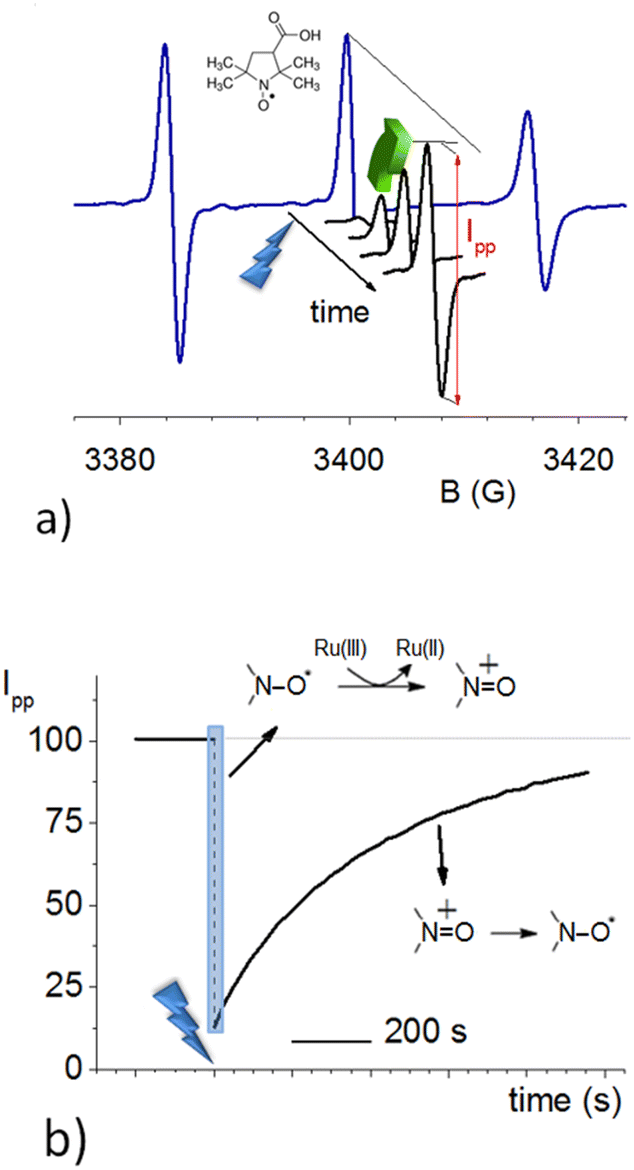 | ||
| Fig. 1 Typical profiles of EPR signals as a function of time obtained using a diluted aqueous solution of 3-CP (0.1 mM ca) at pH 9.0 containing [Ru(II)(bpy)3]2+ and (NH4)2S2O8. (a) The initial spectrum of the NitR is displayed as a blue trace; the central line intensity (peak-to-peak, Ipp) is representative of the NitR concentration. (b) Time-dependence of Ipp. | ||
Upon pulsed laser illumination, the radical is 70–90% oxidized within a few seconds, as seen by the decrease in the EPR signal. After the laser pulses are terminated, the EPR signal recovers almost to its initial intensity (green arrow) with pseudo-first-order kinetics (Fig. 1b). The characteristic recovery time (τrec) of the exponential curve (Fig. 1b) was found to be sensitive to the pH of the solution. A further investigation revealed that the recovery time decreases with increasing pH and the trend is not affected by the type of buffer; Fig. 2 displays the kinetic constant for recovery, krec = 1/τrec, as a function of pH. The data clearly establish that OH− ions are implicated in the recovery process.
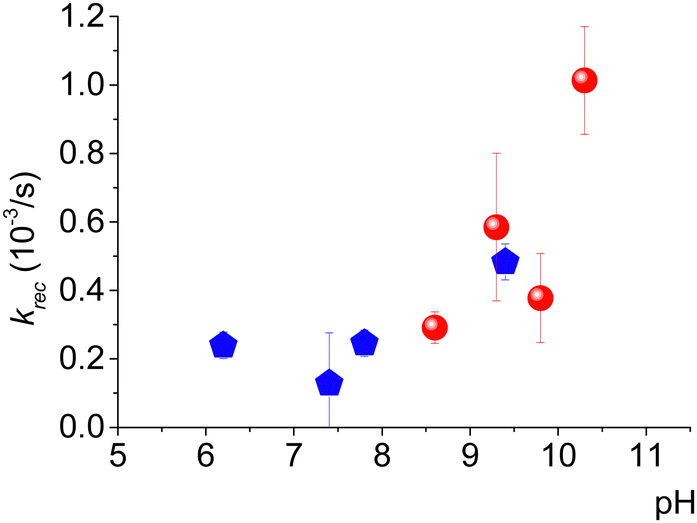 | ||
| Fig. 2 pH-dependence of the rate constant, krec, for regeneration of 3-CP from 3-CP+. Values of krec were obtained from exponential fits of few recovery profiles. Measurements were performed in either carbonate buffer (red circles) or phosphate buffer (blue pentagons). | ||
With the aim of excluding possible reduction pathways that would involve the Ru(II) complex, and/or other compounds initially present in the solution, and confirming that OH− acts as the reducing agent, we further investigated the process by electrolytic generation of the OAC. Cyclic voltammetry (CV) of 3-CP shows a reversible peak couple attributed to the reversible one-electron oxidation of the radical to 3-CP+ (Fig. S1, ESI†). The standard potential of the 3-CP+/3-CP redox couple was obtained as E0 = (Epa + Epc)/2, where Epa and Epc are the anodic and cathodic peak potentials, respectively. Cyclic voltammetry (CV) measurements at pH 6 and pH 9 showed that the redox properties of the couple are not affected by the pH in this range. The value of the standard potential obtained was E0 = 0.56 V vs. SCE (SCE = saturated calomel electrode). 3-CP can be converted to 3-CP+ (the OAC) by controlled-potential electrolysis at a potential (0.8 V vs. SCE) higher than the oxidation peak potential, and the recovery reaction can be easily monitored either by linear sweep voltammetry (LSV) at a rotating disk electrode (RDE), or by EPR. Fig. 3a shows voltammograms recorded before and after electrolysis of 3-CP in a phosphate buffer at pH 6.0.
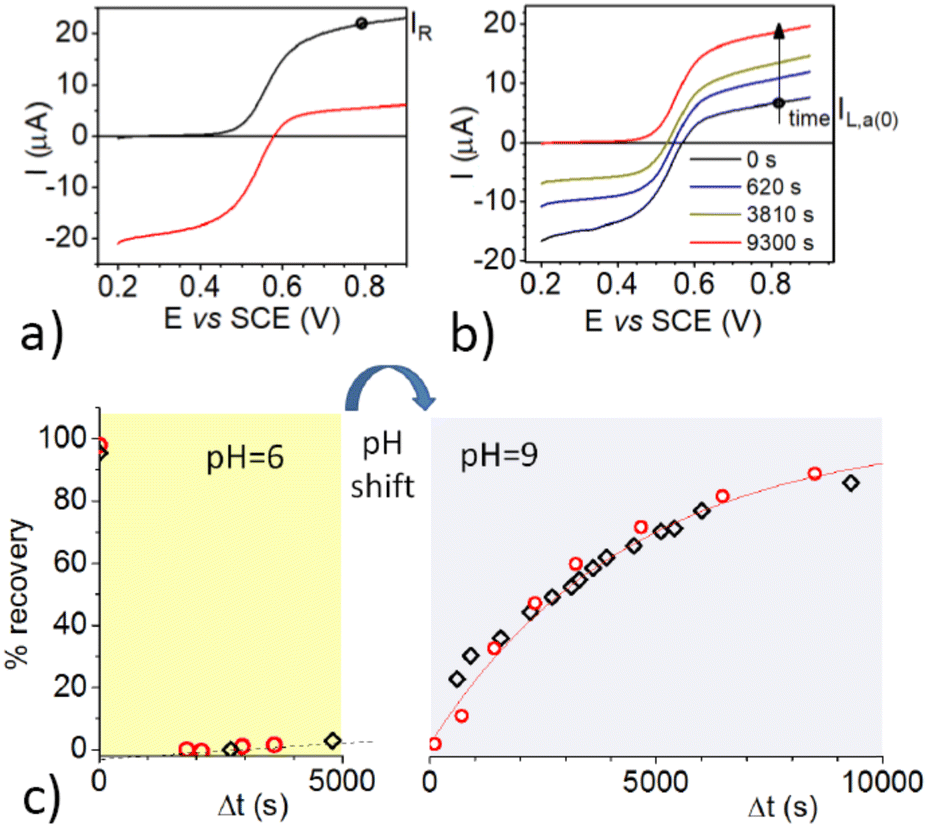 | ||
| Fig. 3 (a) LSV of 0.8 mM 3-CP in a phosphate buffer at pH 6.0, recorded on a glassy carbon RDE (ω = 1000 rpm) before and after electrolysis at 0.8 V; scan rate = 0.01 V s−1; (b) LSV of the same solution, performed as in (a), as a function of time after adjustment of the pH to 9.0; (c) percentage recovery of 3-CP as function of time, as obtained from the anodic current (relative to the 3-CP concentration, red circles) and from EPR intensity measurements (related to the 3-CP concentration, black tetragons) at pH 6.0, and subsequently after the pH had been shifted to pH 9.0. The OAC was generated during the initial times at pH 6.0 by electrochemical oxidation. | ||
Prior to electrolysis, a well-defined oxidation wave with a half-wave potential, E1/2, of 0.56 V vs. SCE was observed for the one-electron oxidation of 3-CP to 3-CP+ as already described. After electrolysis, the anodic wave was replaced by a cathodic one with the same E1/2, due to the one-electron reduction of 3-CP+ to 3-CP. No other waves were observed, indicating that oxidation of 3-CP yields exclusively 3-CP+. The intensity of the reduction wave of 3-CP+ at pH 6.0 remained constant for more than 1 h. Thus, the OAC cation is stable at this pH.
The pH of the electrolyzed solution was then raised to 9.0 by a small addition of concentrated NaOH solution, and LSV measurements were taken as a function of time. In parallel, the reaction was followed also by EPR measurements. Some representative LSV curves, taken at suitable time intervals after the pH change, are shown in Fig. 3b.
It is worth noting that while the cathodic limiting current, IL,c, for the reduction of 3-CP+ decreases, the anodic limiting current, IL,a, for the oxidation of 3-CP (see Fig. 3b) increases. Nearly full recovery of the radical was achieved in less than 3 h, as testified by the complete disappearance of the cathodic branch of the wave, and by the increase of the anodic branch to a limiting current comparable to that of the starting 3-CP. No other signals appeared in the LSV trace, confirming that, at high pH and at low concentration, the oxoammonium cation is quantitatively reduced back to the starting nitroxide radical.
The limiting currents measured at the RDE can be used to calculate the concentrations of 3-CP and 3-CP+, since IL is proportional to the concentration of the redox species according to the Levich equation:24
| IL = 0.62nFAD2/3ν−1/6ω1/2C | (1) |
The data of the recovery of the NitR are reported in Fig. 3c. Both EPR and LSV show that reduction of 3-CP+ at pH 6.0 is so slow that only ∼5% of the radical is regenerated after 1 h. At pH = 9.0, the reaction becomes much faster, with a recovery of ∼50% within 1 h. This result emphasizes the involvement of OH− in the reduction of the OAC to the nitroxide radical. It is important to note that the EPR and LSV display nicely overlapping plots, and clearly show that almost full regeneration of the NitR is achieved within a few hours under basic conditions.
In order to provide further evidence of the involvement of OH−, the recovery reaction was conducted in an initially basic and unbuffered solution after electrolysis. The initial pH of the solution was adjusted to 9.4, and a constant potential of 0.8 V vs. SCE was applied to selectively oxidize the radical. After oxidation to 3-CP+ had proceeded almost to completion (see Fig. 4), the electrolysis was stopped. During electrolysis the solution pH dropped to 7.2. The system was then left to evolve freely overnight, and it was found that only ∼25% of the OAC reverted to the NitR, since the pH of the unbuffered solution had further dropped steeply, to pH 5.2, thus greatly slowing the back reaction. Thus, the OH− ion is both involved and consumed in the recovery reaction (more details are supplied under ESI†).
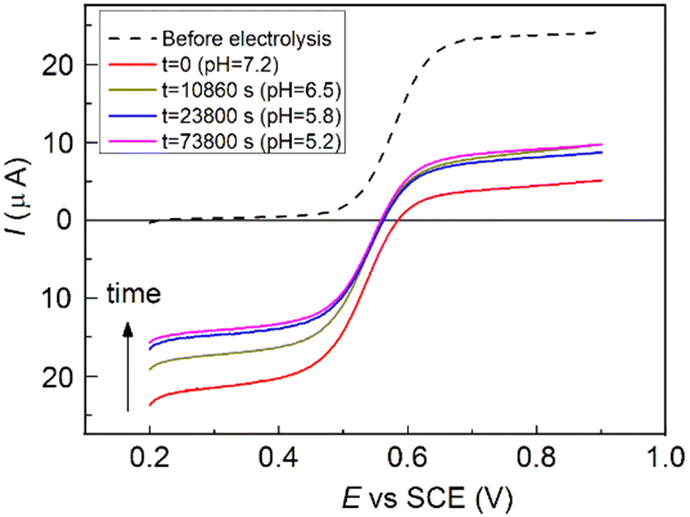 | ||
| Fig. 4 LSV of 0.8 mM 3-CP in 0.1 M NaClO4 (initial pH 7.2) recorded on a glassy carbon RDE (ω = 1000 rpm), scan rate = 0.01 V s−1. The relative variation of the anodic and cathodic currents is different from that shown in Fig. 3 because of the variation toward acidic pH values which might affect the system with the disproportion reaction typical of nitroxides. | ||
Oxidation products
The identification and determination of the products, and in particular of the main oxidation product (OP), is fundamental for the full understanding of the recovery mechanism. The very high percentage of nitroxide radical recovery (nearly 100% both, in photo- and in electrochemical experiments) indicates that: (1) neither the nitroxide nor the oxoammonium cation contribute to the OP; (2) oxidation–reduction cycles can be repeated with accumulation of increasing amounts of the OP; (3) when several such cycles were performed in the same sealed EPR cell by using the photochemical oxidation of NitR, we observed formation of gas bubbles, as can be seen in Fig. 5. In the absence of the NitR/OAC couple the bubbles were not seen. Formation of gas bubbles during the recovery process was also observed when the OAC was generated by electrolysis.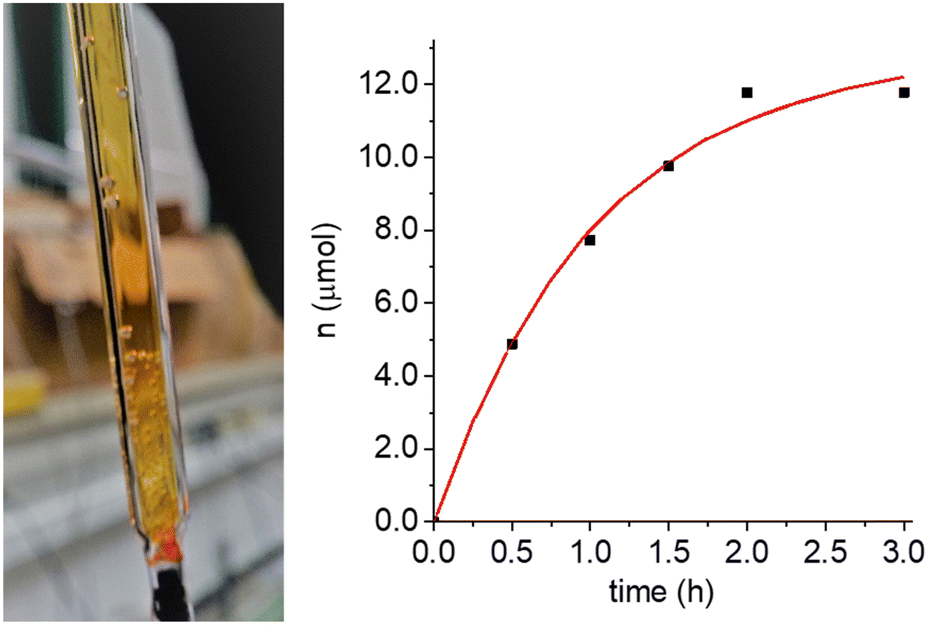 | ||
| Fig. 5 Appearance of gas bubbles inside the flat cell (left) in which the EPR experiment was performed, after oxidation of 3-CP by Ru(III), and subsequent recovery of the initial EPR signal. (right) quantitative determination of the number of moles (n) of the produced gas by volumetric method as a function of time, after electrolysis of a 3-CP solution at pH = 9 ca. It should be noted that the solution was fluxed with argon before sealing the cell. In red the exponential-growth fitting curve with a typical krec value of 0.28 ± 0.04 ·10−3 s−1. | ||
Identification of the gas as oxygen and quantification of the gas production was made by use of well-established methods, namely by oximetric EPR procedure,25,26 Gas Chromatography-Mass Spectrometry (GC-MS),27 and by volumetric measurement.
Oximetry is a classical method to measure oxygen content,25,26,28,29 in particular in biological environments. It was initially chosen because the recovery process conducted in the EPR cell was accompanied by an unusual progressive increase in the linewidth of the recovered NitR EPR spectrum. The formation of paramagnetic gas molecules (NOx or O2 gasses), unobserved by EPR, is the only possible source of line broadening of NitR; since electrochemical measurements showed that NOx gasses were not formed in appreciable quantities in the reverse reaction, oxygen is the only able to induce the increase in the linewidth. In homogenous solutions the line broadening is linearly dependent on oxygen content.28
In the experiment, a 0.2 mM radical solution containing Ru(bpy)32+ and persulfate buffered at pH = 10 was purged with an argon flow to remove the dissolved oxygen. The cell was then sealed and irradiated with visible light to convert NitR to OAC quantitatively. The line broadening analysis allowed to determine that the formed oxygen was at a concentration comparable to what expected from a quantitative conversion of OAC to NitR (a molecular 1:4 ratio of oxygen per converted OAC was considered, see further).
Determination of oxygen was also conducted by GC-MS using a similar chemical system. A solution of the radical at pH = 9 containing Ru(bpy)32+ and persulfate was carefully purged with a nitrogen flow inside a reactor and sealed. After irradiation of the system and the complete recovery of the radical, the gas accumulated in the headspace of the reactor was analysed quantitatively by GC-MS. The experiment was conducted in duplicate. The experiment was designed to obtain an oxygen content in the headspace of 1.5% (a molecular 1:4 ratio of produced oxygen per converted OAC was used, see further). In the headspace we measured an oxygen content of 1.7 ± 0.4%, in excellent agreement with the estimate. Contamination from air oxygen, leakage of the reactor, direct oxidation of water from Ru(III) were checked to be small, and in any case subtracted. More details on the experiments are provided in the ESI.†
For a further quantitative analysis of the recovery reaction by volumetric method, 56 μmoles of 3-CP were oxidized on a Pt gauze working electrode at 0.8 V vs. Ag/AgCl (pH 10.0). After complete oxidation, the solution was transferred to a volumetric apparatus, from which the number of moles of evolved gas could be measured. 12 μmoles of gas (0.29 mL) were produced within 2 h with an asymptotic value of 13 μmoles of evolved gas (0.31 mL) obtained from the exponential-growth fitting (red curve in Fig. 5). The asymptotic value is rather close to that expected for a 4:1 stoichiometry (0.34 mL), therefore, we can conclude that the recovery of 4 molecules of 3-CP+ to 4 molecules of 3-CP was accompanied by the formation of ca. 1 molecule of gas (O2).
Thus, the reduction of 3-CP+ to 3-CP in an alkaline aqueous solution can be described by Scheme 1.
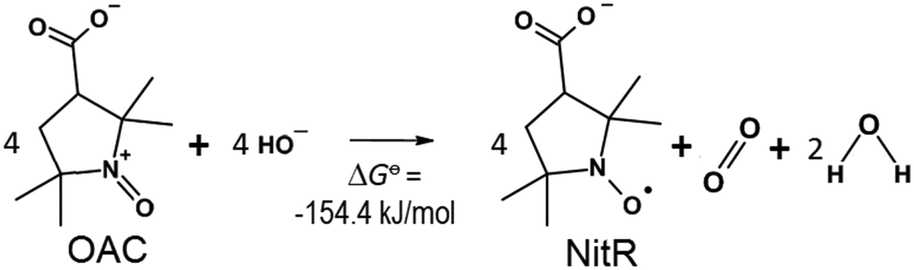 | ||
| Scheme 1 Reverse reaction in which the OAC was transformed to the NitR with concomitant formation of oxygen. The thermodynamic calculation of the free energy accounts for a favourable reaction. The detail of the calculation is supplied in the ESI.† | ||
The change in the Gibbs free energy of the reaction in Scheme 1 could be easily calculated using the data for the corresponding half-reactions, and was found to be thermodynamically favourable, with ΔG0 = −154.4 kJ mol−1; the detail of the calculation is supplied in the ESI.†
The data presented allow us to conclude that the OAC plays the role of a mediator in electrochemical or photochemical oxidation of water at relatively low potentials, viz., 0.8 V vs. SCE. Previously, only a few organic mediators had been described, but they were characterized by much higher potentials, i.e., 1.7–1.9 V.30
We will discuss a plausible mechanism for the reaction in the following section.
Reaction mechanism and reaction scheme
Scheme 1 has shown the global reaction which involves four molecules of OAC and four molecules of OH−. As it is very improbable that it occurs in a single step, we have to consider a multistep scheme.In a multistep scheme, in principle we can prefigure two different possible reactions for the initial step. The first is a simple outer-sphere electron transfer between the OAC and the hydroxyl anion with the formation of the hydroxyl radical HO• together with the first molecule of NitR. Alternatively, we can consider the formation of a complex between the opposite-charged partners, and eventually an inner-sphere electron transfer within the complex.
We have proven the formation of the complex between the hydroxyl anion OH− and the OAC by conducting the reaction in 17O-labelled water. The experiment started with a solution of the 3-CP in 70% 17O-labeled water, in the presence of the Ru complex and the ammonium persulfate.
The initial EPR spectrum (Fig. 6, top) was a typical nitroxide triplet. After photo-oxidation of the radical stimulated by the laser flashes (and almost complete disappearance of the initial EPR spectrum), the EPR signal was allowed to recover in the dark, producing a new spectrum (Fig. 6, middle). This spectrum is a superposition of two contributions: the initial 3-CP spectrum (N16O species), and a spectrum with 8 resolved lines compatible with a 3-CP radical containing the N17O group, with I(17O) = 5/2, and comparable hyperfine splitting for both 14N and 17O.
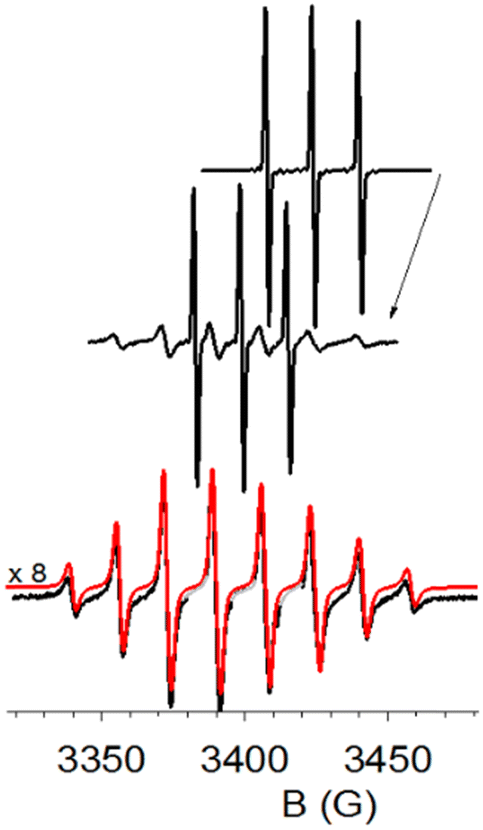 | ||
| Fig. 6 Oxidation and regeneration of 3-CP at pH 9.0 in 70% H217O. Top: The cw-EPR spectrum of 3-CP before oxidation; Middle: after regeneration of the signal; Bottom: the black trace, attributed to the N17O 3-CP radical, was calculated as the weighted difference of the two experimental spectra. The red trace overlaid on it is a simulation spectrum (see Table 1 for the parameters of the N17O species), with a small offset in order to make it readily distinguishable. | ||
The characteristic hyperfine structure can be better observed after a weighted subtraction of the initial triplet spectrum (see Fig. 6, bottom). The spectrum thus obtained is in perfect agreement with the nitroxide EPR spectrum observed by Luz et al.31 for the 17O-labeled Fremy salt.
The spectrum was simulated using the Easyspin package32 for an isotropically tumbling radical (correlation time of the motion τc = 1.5 × 10−11 s) coupled with two paramagnetic nuclei: 14N (I = 1) and 17O (I = 5/2). Table S1 (ESI†) displays the best fit parameters for the simulation. The hyperfine coupling constant of the nitrogen is identical in the unlabeled and labeled species, thus confirming a straightforward isotopic substitution, not a structural change. The reduced number of lines is due to overlapping of the transitions for comparable hyperfine coupling constants of 14N and 17O nuclei. Further details are provided under ESI.†
The percentage of 3-CP labeling does not change with increase in the concentration of 3-CP; it remains equal to the 17O enrichment of the water (70%). This means that we can rule out the outer-sphere mechanism, as the first NitR molecule is certainly formed without isotopic substitution.
An inner-sphere mechanism is also ruled out on the base of the % of substitution, as detailed in the ESI.†
Therefore, we have evidence that the recovery reaction starts with the formation of a complex between 3-CP+ and the hydroxyl anion OH−: an azinic acid (Scheme 2a). The formation of an azinic acid by interaction of an OH− ion with an OAC is also documented in the literature.33–35
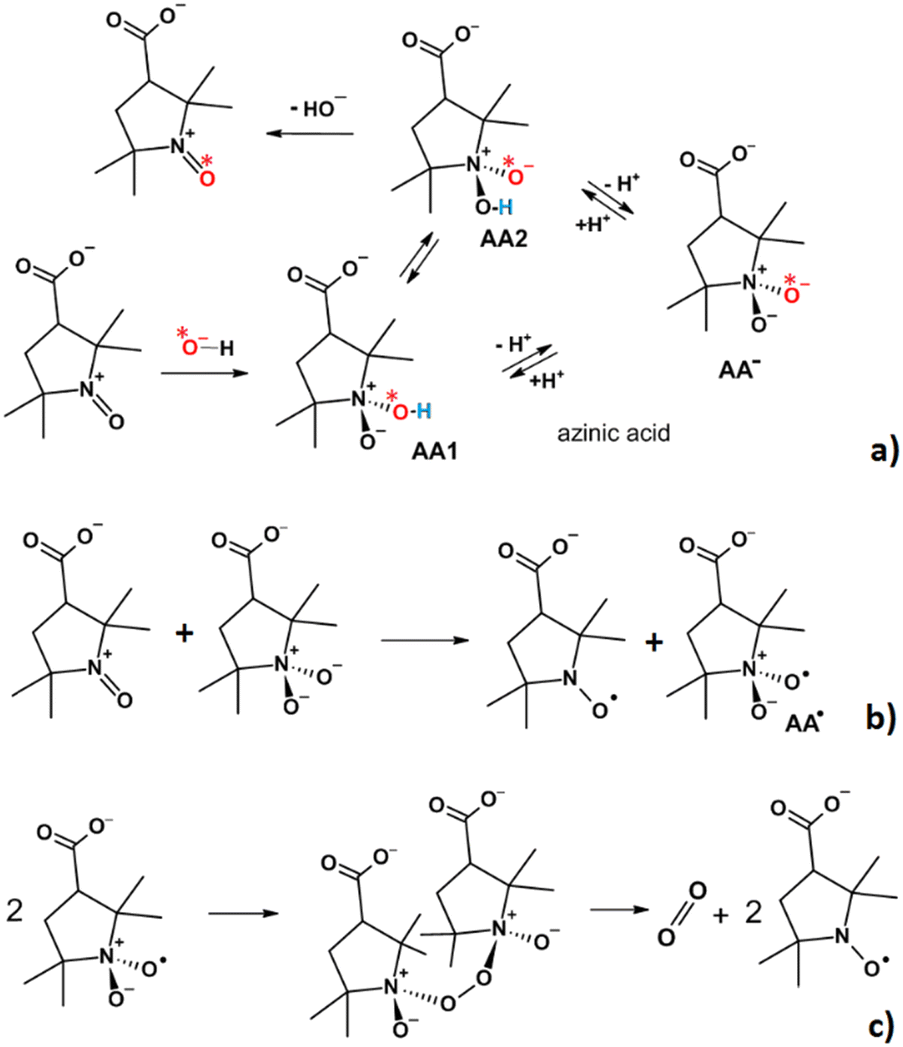 | ||
| Scheme 2 Proposed mechanism for reformation of 3-CP from the OAC. (a) Interaction of the OAC with OH−, to form azinic acid, whose anionic form AA−, exists in two tautomeric forms, AA1 and AA2, in which a proton can be bound to one or the other of the two oxygen atoms attached to the nitrogen. The star near the red oxygen labels the oxygen atom deriving from the solvent (OH−), whereas the hydrogen undergoing tautomerization is colored in blue; (b) the azinic acid anion is oxidized by a second OAC molecule to generate two radicals, 3-CP and AA˙; (c) Two AA˙ radicals dimerize to form an unstable peroxide that decomposes to 3-CP and molecular oxygen. The star indicates the oxygen which can be incorporated from the solvent water due to acid/base equilibrium, thus resulting the presence of 17O in the 3-CP when the reaction is performed in 17O-enriched water. | ||
We propose that the reaction continues with a proton exchange between the two oxygens bound to the nitrogen, competing with the deprotonation of the azinic acid. The oxidation step involves electron transfer from the formed azinic anion to another free OAC to form two radical species (Scheme 2b), 3-CP and AA˙. Finally, the two AA˙ radicals can dimerize to an unstable peroxo intermediate that breaks down to yield two 3-CP molecules and one O2 molecule (Scheme 2c). Overall, the mechanism is consistent with the stoichiometry of the process that we have established experimentally (Scheme 1) and correctly accounts for the isotopic substitution.
We used quantum chemical calculations to support the tautomerization reaction and the deprotonation of the azinic acid. The kinetic parameters for the proton exchange process between the two nitrogen-bound oxygen atoms were obtained from the Gibbs free energies of the two diastereoisomeric azinic acids, AA1 and AA2, and of the transition state (TS) in which the proton bridges the two oxygen atoms, were optimized by the DFT method (B3LYP and 6-311+G** basis set with water as solvent). The Gibbs free energies of AA1 and AA2 are slightly different. Consequently, the free energy of the TS for the forward (AA1 to AA2) reaction is 86.40 kJ mol−1 higher than AA1, and that for the TS of the reverse reaction is 82.08 kJ mol−1 higher than AA2. Using these values, the rate constant for the tautomerization reaction, estimated using the Eyring equation,36k = (kBT/h)exp(−ΔG#/RT) (assuming a transmission coefficient of (1) is ca. two orders of magnitude higher than the experimental rate of 3-CP recovery observed.
The next issue is the chemical structure of the species that is oxidized. Assuming it to be the azinic acid, its redox potential is expected to be pH-dependent because it becomes deprotonated with increasing pH.
According to the quantum chemical calculations, AA1 and AA2 are weak acids, with proton transfer to CO32− being thermodynamically favored: the ΔG0 values for deprotonation of AA1 and AA2 are −17.42 kJ mol−1 and −13.10 kJ mol−1, respectively. From these values, and from the known pKa of bicarbonate,37 10.33, we have calculated that the pKa of AA1 is ca. 7.3, and that of AA2 is ca. 8.0. It is plausible that these pKa values account for the decrease in the rate of the recovery reaction at neutral pH and at lower values. Furthermore, it appears that the deprotonated, anionic forms of the azinic acids are the true reducing agents. According to classical work of Eigen,38 cases in which tautomerism occurs between two groups with close pKa values the rate of tautomerism is predicted to be very high, and is surely higher than the rate of recovery that we are studying.
To further support the mechanism that we have proposed, we calculated the Gibbs free energies of the initial and oxidized states of AA1, AA2 and of the common anion, AA−, by DFT, and were thus able to obtain the corresponding redox potentials. The E0 values thus obtained are presented in Table 1. The E0 value for 3-CP was calculated for comparison and is in good agreement with the experimental value (see above and Table 1). The calculated E0 values show that the azinic anion has by far the lowest value, and is even ca. 0.5 V lower than that of 3-CP. We can thus conclude that this species is able to reduce 3-CP+ due to the reaction being thermodynamically favorable.
| Compound | E 0 (V) | E 0calc (V) |
|---|---|---|
| 3-CP | 0.802 | 0.674 |
| AA1 | — | 1.522 |
| AA2 | — | 1.545 |
| AA− | — | 0.146 |
According to the scheme that we propose, electron transfer between AA− and 3-CP+ generates two radicals, 3-CP, which is stable, and AA˙, which is reactive (Scheme 2b). Two molecules of AA˙ then react to form a peroxide bond (for the structure and further comments refer to the ESI†). We propose that this dimer decomposes to 3-CP and molecular oxygen (Scheme 2c). The overall mechanism described explains both the almost complete regeneration of 3-CP, and the observed stoichiometric ratio of 4:1 for molecular oxygen generated concomitantly with the 3-CP.
Regarding the stability of the system, we point out that the nitroxide in diluted condition is rather stable and the cycle can be repeated with very small loss of the initial concentration. We just observed a slow dark reaction in the systems containing persulfate, which is more evident in more concentrated solution, possibly due to a ring opening reaction of the NitR.39
From our experimental data and calculations, taken together, we can conclude that the NitR/OAC couple is a novel type of mediator that can facilitate both photochemical and electrochemical oxidation of water. The active OAC form is obtained at relatively low oxidation potentials, 0.8–1.0 V vs. NHE, and the NitR is recovered almost quantitatively. The N(5)-ethylflavinium cation/N(5)-ethylflavinium dication radical couple that was earlier described,40 is less effective, since its active dication radical form is obtained at much higher potentials (ca. 1.7–1.9 V vs. NHE).
Conclusions
OAC produced by either photochemical or electrochemical oxidation of a diluted solution of NitR, was shown to undergo quantitative reduction back to the initial NitR at alkaline pH values the reaction involves formal oxidation of water with concomitant generation of molecular oxygen.We have established the stoichiometry of the reaction: four OAC molecules are reduced while one molecule of oxygen is formed. The reaction is thermodynamically favored (ΔG0 = −154.4 kJ mol−1) and we have obtained quantitative production of oxygen by conducting an electrolysis of 3-CP at 0.8–1.0 V vs. NHE, that is a potential much lower than that typical for a direct oxidation of the hydroxyl ion to hydroxyl radical (E0 = 2.226 V vs. NHE).41 For 3-CP, the timescale of the reaction of the relative OAC in a buffered mildly basic solution is around 103 min; from the data in Fig. 3 we can derive that the Turnover frequency (TOF) is around 2 × 10−3 s−1.
We have proposed a plausible mechanism which accounts for the experimental evidence, and we expect that this mechanism is general for nitroxides. Therefore, in our opinion we have discovered an original approach to water oxidation at a low potential having molecular oxygen as product.
We think that it is worth to investigate this class of molecules as possible catalysts for the oxidation of water. The ability to conduct such a reaction might be exploited in technological contexts, for example for energy production, but we envisage a range of possible applications for NitR/OAC couple. It may provide a novel approach to studying OH−/H2O penetration (diffusion) into buried regions of proteins and other biomacromolecules that have been labeled with nitroxides at specific sites.33
Author contributions
Antonio Barbon (investigation, data curation, conceptualization, writing – original draft), Abdirisak Ahmed Isse (investigation, data curation, writing – review & editing), Armando Gennaro (writing – review & editing), Raanan Carmieli (investigation), Itzhak Bilkis (methodology, formal analysis, conceptualization), Lev Weiner (conceptualization, writing – review & editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. B and L. W. are grateful to the Dept. of Chemical Sciences and the Internationalization Programs of the University of Padova (IT) for financial support. A. B. acknowledges the support of the Kimmel Center for Molecular Design, and L.W. that of the Kimmelman Center for Biomolecular Structure and Assembly, both at the Weizmann Institute of Science (IL). The authors thank Prof. Israel Silman (Weizmann Inst.) for constructive suggestions and critical reading of the manuscript, Dr Alex Khenkin (Weizmann Inst.) for valuable assistance in the volumetric experiments and prof. Andrea Sartorel (University of Padova) for helping with GC-MS experiments. L. W. acknowledges profs. Yuri Molin and Kolya Bazin from the University of Novosibirsk (RU) and prof. Vasilii Sen' from the Russian Academy of Sciences (RU) for constructive discussions of possible mechanisms for the recovery reaction.References
- A. Kumar and A. Singh, Front. Pharmacol., 2015, 6, 206 CrossRef PubMed
.
- E. A. Haidasz, D. Meng, R. Amorati, A. Baschieri, K. U. Ingold, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2016, 138, 5290–5298 CrossRef CAS
- G. Mobbili, E. Crucianelli, A. Barbon, M. Marcaccio, M. Pisani, A. Dalzini, E. Ussano, M. Bortolus, P. Stipa and P. Astolfi, RSC Adv., 2015, 5, 98955–98966 RSC
- M. P. Murphy and R. A. J. Smith, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 629–656 CrossRef CAS
- Edward G. Janzen, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS
- M. A. Mercadante, C. B. Kelly, J. M. Bobbitt, L. J. Tilley and N. E. Leadbeater, Nat. Protoc., 2013, 8, 666–676 CrossRef CAS PubMed
- S. Weik, G. Nicholson, G. Jung and J. Rademann, Angew. Chem., Int. Ed., 2001, 40, 1436–1439 CrossRef CAS
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed
- H. Richter and O. García Mancheño, Org. Lett., 2011, 13, 6066–6069 CrossRef CAS PubMed
- T. Eliash, A. Barbon, M. Brustolon, M. Sheves, I. Bilkis and L. Weiner, Angew. Chem., Int. Ed., 2013, 52, 8689–8692 CrossRef CAS PubMed
- M. Schmallegger, A. Barbon, M. Bortolus, A. Chemelli, I. Bilkis, G. Gescheidt and L. Weiner, Langmuir, 2020, 36, 10429–10437 CrossRef CAS
- S. Wang, A. Lu and C. J. Zhong, Nano Convergence, 2021, 8, 4 CrossRef CAS PubMed
- J. Li, C. A. Triana, W. Wan, D. P. Adiyeri Saseendran, Y. Zhao, S. E. Balaghi, S. Heidari and G. R. Patzke, Chem. Soc. Rev., 2021, 50, 2444–2485 RSC
- M. Sutradhar, A. J. L. Pombeiro and J. A. L. da Silva, Coord. Chem. Rev., 2021, 439, 213911 CrossRef CAS
- J. Wang, H. Kong, J. Zhang, Y. Hao, Z. Shao and F. Ciucci, Prog. Mater. Sci., 2021, 116, 100717 CrossRef CAS
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 RSC
- H. A. Younus, Y. Zhang, M. Vandichel, N. Ahmad, K. Laasonen, F. Verpoort, C. Zhang and S. Zhang, ChemSusChem, 2020, 13, 5088–5099 CrossRef CAS PubMed
- S. Watabe, Y. Tanahashi, M. Hirahara, H. Yamazaki, K. Takahashi, E. A. Mohamed, Y. Tsubonouchi, Z. N. Zahran, K. Saito, T. Yui and M. Yagi, Inorg. Chem., 2019, 58, 12716–12723 CrossRef CAS PubMed
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC
- X. Yang, J. Walpita, E. Mirzakulova, S. Oottikkal, C. M. Hadad and K. D. Glusac, ACS Catal., 2014, 4, 2635–2644 CrossRef CAS
- J. Meng, P. Bi, J. Jia, X. Sun and R. Chen, ChemistrySelect, 2017, 2, 4882–4888 CrossRef CAS
- F. Rappaport, M. Guergova-Kuras, P. J. Nixon, B. A. Diner and J. Lavergne, Biochemistry, 2002, 41, 8518–8527 CrossRef CAS PubMed
- D. A. Armstrong, R. E. Huie, W. H. Koppenol, S. V. Lymar, G. Merényi, P. Neta, B. Ruscic, D. M. Stanbury, S. Steenken and P. Wardman, Pure Appl. Chem., 2015, 87, 1139–1150 CrossRef CAS
-
A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2nd edn, 2001 Search PubMed
-
V. V. Khramtsov, in Nitroxides – Theory, Experiment and Applications, ed. A. Kokorin, InTech, 2012 Search PubMed
- J. Weaver, S. R. Burks, K. J. Liu, J. P. Y. Kao and G. M. Rosen, J. Magn. Reson., 2016, 271, 68–74 CrossRef CAS PubMed
- A. Volpe, C. Tubaro, M. Natali, A. Sartorel, G. W. Brudvig and M. Bonchio, Inorg. Chem., 2019, 58, 16537–16545 CrossRef CAS PubMed
- V. V. Khramtsov, A. A. Bobko, M. Tseytlin and B. Driesschaert, Anal. Chem., 2017, 89, 4758–4771 CrossRef CAS PubMed
- J. Shen, N. Khan, L. D. Lewis, R. Armand, O. Grinberg, E. Demidenko and H. Swartz, Biophys. J., 2003, 84, 1291–1298 CrossRef CAS
- K. Fominykh, G. C. Tok, P. Zeller, H. Hajiyani, T. Miller, M. Döblinger, R. Pentcheva, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2017, 27, 1605121 CrossRef
- Z. Luz, B. L. Silver and C. Eden, J. Chem. Phys., 1966, 44, 4421–4426 CrossRef CAS
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed
- W. L. Hubbell, A. Gross, R. Langen and M. A. Lietzow, Curr. Opin. Struct. Biol., 1998, 8, 649–656 CrossRef CAS
- V. A. Golubev, E. G. Rozantsev and M. B. Neiman, Izv. Akad. Nauk SSSR, Ser. Khim., 1965, 11, 1927–1936 Search PubMed
- J. B. Gerken, Y. Q. Pang, M. B. Lauber and S. S. Stahl, J. Org. Chem., 2018, 83, 7323–7330 CrossRef CAS
-
P. W. Atkins, Physical chemistry, Freeman, New York, 4th edn, 1990 Search PubMed
-
Nagra/PSI chemical thermodynamic data base 01/01, ed. W. Hummel, U. Berner and Paul Scherrer Institut, Universal Publishers, Parkland, FL, 2002 Search PubMed
- M. Eigen, Angew. Chem., Int. Ed. Engl., 1964, 3, 1–19 CrossRef
-
Nitroxides: Synthesis, Properties and Applications, ed. O. Ouari and D. Gigmes, Royal Society of Chemistry, Cambridge, 2021 Search PubMed
- V. Sichula, P. Kucheryavy, R. Khatmullin, Y. Hu, E. Mirzakulova, S. Vyas, S. F. Manzer, C. M. Hadad and K. D. Glusac, J. Phys. Chem. A, 2010, 114, 12138–12147 CrossRef CAS PubMed
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimentals, CV of 3-CP, electrolysis of an unbuffered solution of 3-CP, interpretation of the EPR spectrum of 3-CP with 17O, comments on the mechanism of the redox process. See DOI: https://doi.org/10.1039/d2ma00668e |
| ‡ Deceased on April 2022. |
| This journal is © The Royal Society of Chemistry 2022 |