
Electrocatalytic reduction of N2 and nitrogen-incorporation process on dopant-free defect graphene†
Yanqiu
Du
a,
Cheng
Jiang
a,
Wei
Xia
a,
Li
Song
a,
Peng
Li
 a,
Bin
Gao
a,
Chao
Wu
a,
Lei
Sheng
a,
Jinhua
Ye
b,
Tao
Wang
*a and
Jianping
He
*a
a,
Bin
Gao
a,
Chao
Wu
a,
Lei
Sheng
a,
Jinhua
Ye
b,
Tao
Wang
*a and
Jianping
He
*a
aCollege of Materials Science and Technology, Jiangsu Key Laboratory of Electrochemical Energy Storage Technologies, Nanjing University of Aeronautics and Astronautics, 29 Yudao Street, Nanjing, 210016, P. R. China. E-mail: wangtao0729@nuaa.edu.cn; jianph@nuaa.edu.cn; Fax: +86 25 52112626; Tel: +86 25 52112900
bInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 13th November 2019
Abstract
The electrochemical reduction of N2 to NH3 under ambient conditions is a promising N2 fixation method, which provides a new technical solution to remedy the limitations of the Haber–Bosch process. Defect engineering is considered an inspiring strategy for strong N2 activation. Herein, we demonstrate the reduction of N2 on dopant-free defect graphene, which was prepared via the molten salt method. Systematic experiments and density functional theory calculation revealed that the defect sites are the unique active sites for nitrogen adsorption and activation. The phenomenon of N incorporation into graphene using the product NH3 from the NRR as the N source has never been reported before. This was thoroughly studied in this study, and thus serves as a unique perspective to illustrate the significance of defect sites in activating N2.
The process of converting free nitrogen into nitrogenous compounds is known as N2 fixation, which provides a crucial element for the survival of living organisms. As the most widely used process for N2 fixation, the Haber–Bosch method can be performed only under high temperature and high pressure,1 which is extremely energy-dependent and responsible for massive CO2 emissions. Considering the urgent demand for more sustainable alternatives, many efforts have been devoted to finding cleaner ways to remedy the defects of traditional processes. The N2 reduction reaction (NRR) is the most promising candidate, during which N2 fixation is driven by renewable electricity.2 Unfortunately, the low efficiency of the NRR is still far from satisfactory for its widespread use because it is limited by the ultrahigh stability of the N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond and the competitive hydrogen evolution reaction.3 Consequently, the development of high-performance electrocatalysts with the ability to significantly reduce the N2 activation energy is essential for the NRR.
N bond and the competitive hydrogen evolution reaction.3 Consequently, the development of high-performance electrocatalysts with the ability to significantly reduce the N2 activation energy is essential for the NRR.
Recently, numerous electrocatalysts based on noble metals and transition metals showed high activity, such as Ru4 and MoS2.5 The unsaturated d orbitals, which synergistically accept electron density from and back donate to N2,6 endow transition metal catalysts with prominent performances. Notably, many transition metal catalysts are anchored on carbon nanomaterials, such as porous carbon7 and CNT.8 However, the signification contribution of dopant-free carbon substrates for the NRR has rarely been investigated.
Graphene, as an ideal platform to load electrocatalysts,9 is always modified via the introduction of heteroatoms such as nitrogen and boron to improve its application. It has been long accepted that graphene can be activated by lone-pair electrons from electron-rich dopants or vacant orbitals from electron-deficient dopants by breaking the integrity of π conjugation.10,11 This activation process is usually advantageous for improving catalytic activity. However, intrinsic carbon defects can produce the same effects, breaking the original electron arrangement and improving the electron transfer from the electrocatalyst to chemisorbed molecules.12 Fortunately, a previous study proved that the edge-rich graphene is more active than the basal plane in the oxygen reduction reaction.13 Besides, theoretical calculations and experimental works confirmed that defective graphene is also effective for other catalytic reactions such as the OER and HER.14 This inspired us to consider whether the defects in graphene are functional for the N2 reduction reaction.
Herein, we designed dopant-free few-layer graphene with rich defects, which was obtained by controlling the synthetic conditions, and investigated its NRR activity. The synthesized ultrathin porous graphene achieved a faradaic efficiency (FE) of 8.51% at −0.4 V vs. RHE for NH3 electrosynthesis. This remarkable performance was enabled by the enhancement in defect density, higher surface area and larger pore volume. This study not only shows the feasibility of pure carbon nanomaterials as highly efficient NRR catalysts, but also serves as a model system to investigate the origin of NRR activity in carbon-based electrocatalysts.
The process to create defects is illustrated in Fig. 1a, where freeze-dried oxidized graphite was annealed in molten salts. Molten salt-based exfoliation has been successfully applied to synthesize thin BN nanosheets from bulk materials.15,16 Inspired by this, we adopted this method to synthesize few-layer graphene from bulk oxidized graphite (Fig. S1†). The scanning electron microscopy (SEM) images reveal that the morphology of graphene after annealing at 800 °C, labelled as DG-800, is wrinkled and crimped (Fig. 1b and S2†), suggesting that the exfoliation process is based on its attachment to salts and subsequent peeling off (Fig. S3†).17 The morphology of graphene is sensitively dependent on the annealing temperature. When the temperature was up to 900 °C, the product DG-900 with distinct stacking was not favorable for effective utilization of its active area (Fig. S2e†). The graphene sheet was investigated by atomic force microscopy (AFM), showing a thickness of 3 nm (Fig. 1c). Furthermore, numerous nanopores with a size in the range of 1–5 nm were observed on the surface of DG-800 in the TEM images (Fig. 1d and e) and aberration-corrected HAADF-STEM image (Fig. S4†).18 Concomitant with numerous nanopores, a higher edge exposure will offer more active sites for N2 adsorption and activation. Contrary to that treated with molten salt, obvious defects were not found in the pristine graphene (denoted as PG), and its graphene sheet was multi-layer (Fig. S5†) and unbroken (Fig. 1f and g). According to the N2-physisorption measurements, the Brunauer–Emmett–Teller (BET) specific surface area of DG-800 is 420.20 m2 g−1, which is much higher than that of PG (78.33 m2 g−1). The inset pore size distribution curves indicate that the size of the mesopores is centered at 3.4 nm and the mesopore volume was significantly boosted, which is mainly attributed to the etching effect of the molten salt (Fig. 2a and Table S1†). With the aid of the molten salt, the multi-layer bulk graphene was exfoliated into few-layer ultrathin porous nanosheets with abundant mesopores and exposed edges. These features revealed by microscopy are beneficial for the maximum inherent activity.
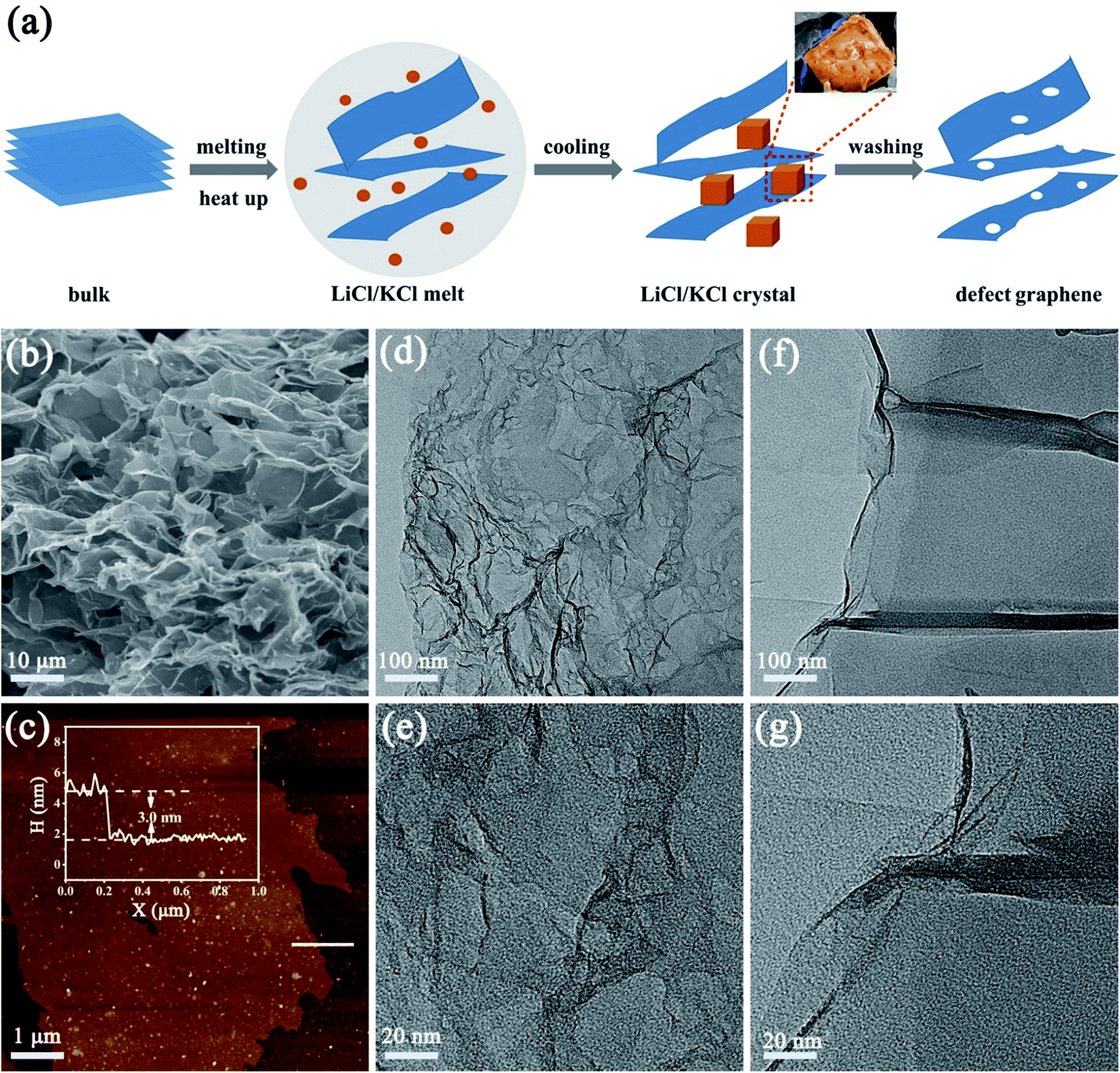 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of defect graphene. (b) SEM image of defect graphene DG-800, where DG-800 is the sample treated with molten salt at 800 °C. (c) AFM image of DG-800. (d and e) TEM images of DG-800 at different magnifications. (f and g) TEM images of PG at different magnifications, where PG is the sample treated without molten salt. | ||
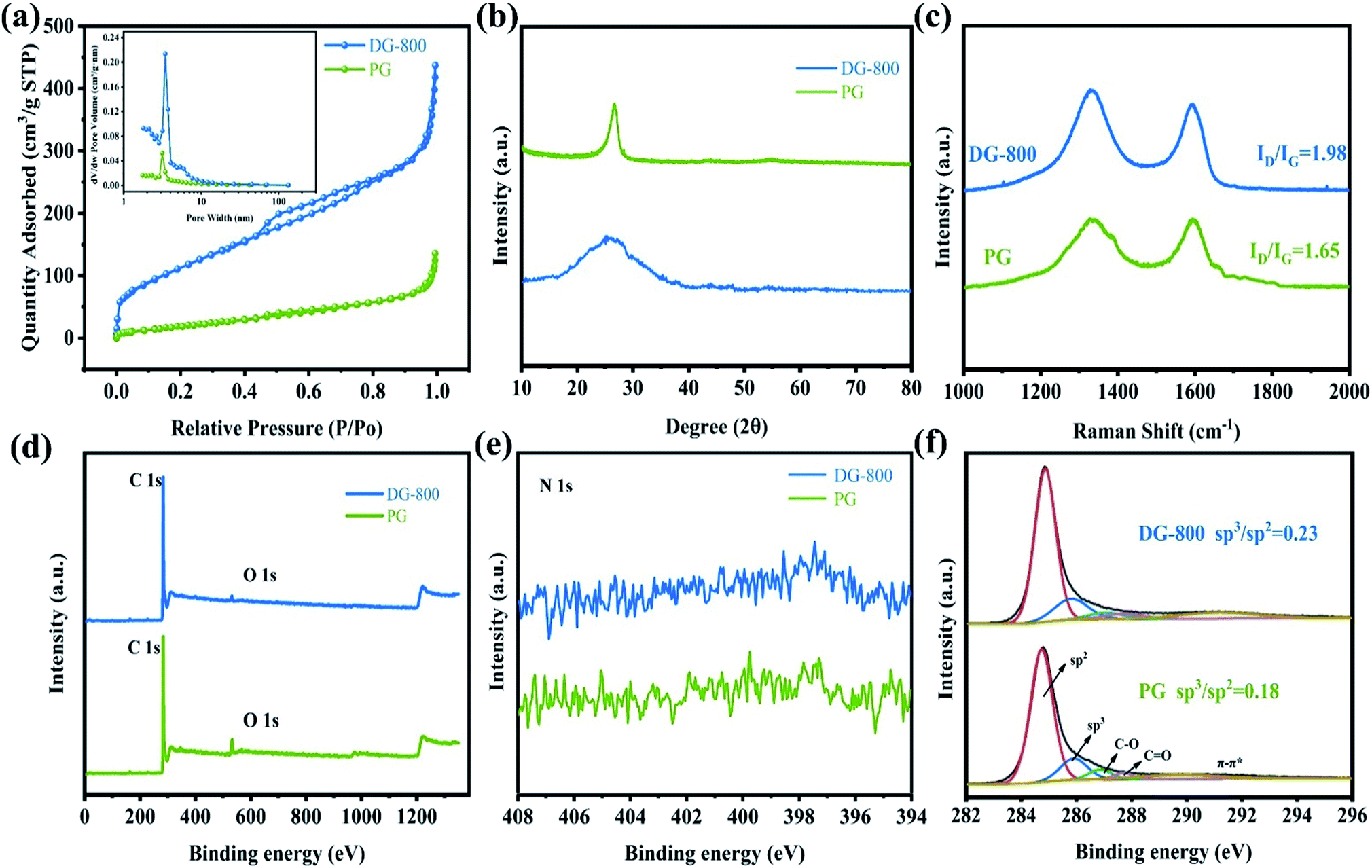 | ||
| Fig. 2 (a) Nitrogen adsorption–desorption isotherms and the inset corresponding pore size distribution of the catalyst DG-800 and PG. (b) XRD spectra of DG-800 and PG. (c) Raman spectra of PG and DG-800. (d) Full XPS spectra of DG-800 and PG. (e) N 1s XPS spectrum of DG-800. (f) C 1s XPS spectra of DG-800 and PG. | ||
The defect-induced alteration in local structure was further characterized. Primarily, the X-ray diffraction (XRD) characterization of PG and representative defect graphene is presented in Fig. 2b. The diffraction peak at 26.6° changes from a sharp peak to a broader peak due to the destruction of the long-range order in the atomic arrangements after annealing.19 The ID/IG ratio of PG and DG-800 in the Raman spectra, which is a powerful tool to quantify disorder, increased from 1.65 to 1.98, implying a higher defect concentration and coinciding well with the XRD measurements and TEM images (Fig. 2c).20 X-ray photoelectron spectroscopy (XPS) (Fig. 2d) indicated that only carbon and oxygen were present in the sample. Since no peaks for K+ and Li+ were initially detected by XPS (as shown in Fig. 2d), we then proceeded to use ICP, a sensitive technique suitable for trace elements, to detect the metallic impurities of K+ and Li+. In DG-800, the amount of K and Li was found to be 0.048 wt% and 0.0086 wt%, respectively, which are too low to be electroactive. Considering permanganates are used in the Hummers' method, the analysis of residual Mn was also conducted, which was determined to be only 0.043 wt%. More specifically, no obvious peak can be detected on the N 1s XPS spectra (Fig. 2e). Thus, the problem of nitrogen species causing false positive results can be avoided.21 The asymmetric high-resolution C 1s spectra were deconvoluted into five peaks, where the peaks at 284.75 and 285.9 eV correspond to sp2 and sp3 carbon,22,23 respectively (Fig. 2f). The ratio of sp3 to sp2 carbons in DG-800 was determined to be approximately 0.23, which is higher than that in PG and another quantitative description of more defects in DG-800.13 Thus, the above spectroscopic characterization fully confirms the successful preparation of defect graphene.
The NRR activity of defect graphene was preliminarily probed via the noticeable difference between Ar- and N2-saturated electrolytes in LSV tests (Fig. 3a). Considering the increase in current density and the existence of N2, the N2 reduction reaction is the most convincing reason for the above difference. Inspired by this, further NRR tests were performed to confirm the electrocatalytic activity of the defect graphene. All the NRR electrochemical tests were performed in an H-type cell with a continuous N2 flow, which was separated by a Nafion membrane. To clean ammonia from the membrane in between tests, the Nafion membrane was retreated in 3% H2O2 and 0.5 M H2SO4 for 1 h at 80 °C after the test. The NH3 produced in the chronoamperometry tests were analyzed by Nessler's reagent and indophenol blue together, thus ensuring the accuracy of the results (the calibration curves are shown in Fig. S6 and S7†). Furthermore, carefully controlling the conditions ensured that the calculated NH4+ had an acceptable error, with a standard deviation of <2% (Table S2†). The absorbance of the electrolyte catalyzed by DG-800 was overwhelmingly enhanced compared with that of PG, indicating the better performance of DG-800 toward NH3 synthesis (Fig. 3b and S8†). The introduction of mesoporosity and accessible defect sites in DG-800, acting as channels for the reaction medium,24 greatly favored the electrocatalytic activity. By contrast, PG was a poor catalyst towards the NRR. More specific performances are presented in Fig. 3c and d, where it can be seen that the faradaic efficiency (FE) and NH3 yield both increased with an increase in potential up to −0.4 V vs. RHE and both have peak values, and then decreased for potentials <−0.4 V vs. RHE. The degradation of the performances at a more negative potential can be explained by the competing hydrogen evolution reaction (HER), in which more protons occupy the active sites.25 Subsequently, we studied the effect of the defect density on NRR activity by comparing the properties of the graphene synthesized at different temperatures (Fig. S9†). DG-800 achieved the best performance, NH3 yield rate (4.31 μg h−1 mgcat−1) and FE (8.51%). Consistent with Fig. S10,† we found that the defect density positively affected the NRR process. Besides, we further performed the method reported by Watt and Chrisp to detect the side product of N2H4 (the calibration curve is shown in Fig. S11†). As illustrated in Fig. S12,† even a negligible gap between the pristine electrolyte and electrolytes after NRR could not be detected. Therefore, it is reasonable to conclude that no N2H4 was produced, indicating the high selectivity of the electrocatalyst. Considering the possible existence of NOx residue such as NO3− in the electrolyte after the chronoamperometry tests at −0.4 V for 2 h, DG-800 as the catalyst was analyzed using an ion chromatography (IC) to verify the presence of nitrate. The IC spectra of the electrolyte after being tested and NO3− at different concentrations are provided in Fig. S13.† The peak area of the electrolyte was barely visibly, indicating negligible NO3− was present, thus excluding NH3 oxidation and the influence of NO3− on the ammonia yield. To unambiguously confirm the source of ammonia, a series of control experiments were implemented. Blank carbon paper was used as the electrode and the N2 flow was replaced by an Ar flow, and the absorption spectra showed no obvious difference with the pristine electrolyte (Fig. 3e), that is, no NH4+ was generated. The former experiment made it clear that the synthesized graphene, rather than carbon paper, was the catalyst for the NRR. In the latter case, we did not detect NH4+ in the Ar-saturated electrolyte, which lacked a nitrogen source for synthesizing ammonia since the electrocatalyst was nitrogen-free.
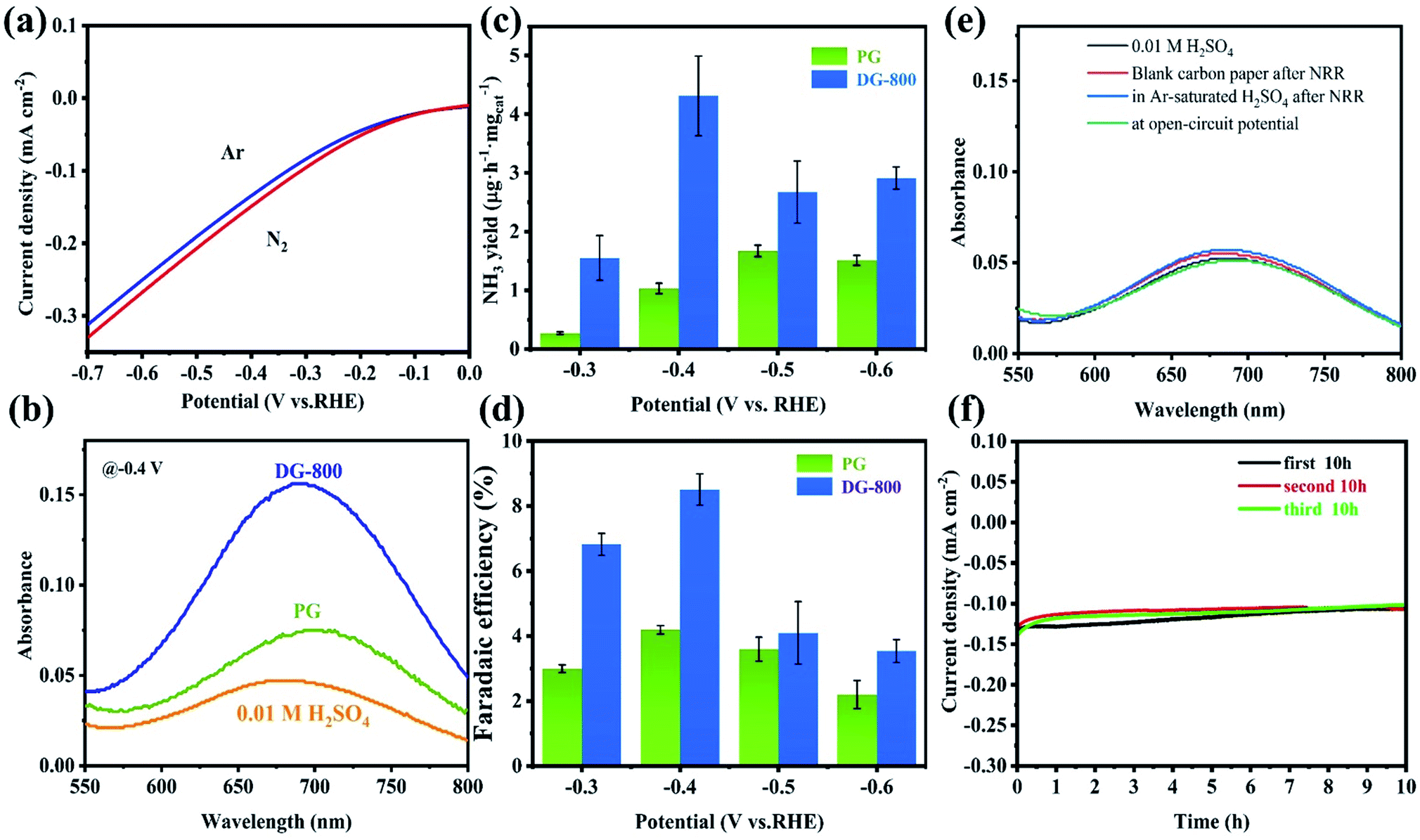 | ||
| Fig. 3 (a) Linear sweep voltammetry curves of DG-800 in Ar- and N2-saturated electrolyte. (b) UV-vis spectra of the electrolytes after NRR at −0.4 V vs. RHE for 2 h, where pristine electrolyte with indophenol indicator was used as a reference (yellow line). (c) NH3 yield rate at various potentials. (d) Faradaic efficiency at various potentials. (e) UV-vis absorption spectra of the electrolytes in the control experiment stained with indophenol indicator after 2 h electrolysis. (f) Stability test of the DG-800 catalyst in N2-saturated 0.01 M H2SO4 at −0.4 V vs. RHE under long-time electrolysis. | ||
To evaluate the stability, a durability test consisting of three cycles of 10 h chronoamperometric tests at −0.4 V vs. RHE was conducted (Fig. 3f). The current density was well maintained at around 0.1 mA cm−2 at the of end each cycle, showing little degradation. The key performance metrics after three cycles of FE (7.24%) and NH3 yield rate (1.84 μg h−1 mgcat−1) were still at a high level for a carbon-based material without any heteroatoms or transition metals. This remarkable performance clearly demonstrates the superiority of DG-800 for the NRR.
The FT-IR spectra of the catalysts after the NRR allowed us to directly trace the protonation of N2 on the electrocatalyst surface (Fig. 4a). As reported in the literature, the two sharp peaks at 2930 cm−1 and 2850 cm−1 are attributed to the characteristic H–N–H stretching of NH4+.26 The intensities of these two bands increased with time, suggesting an increase in the production of NH3.26 Contrary to the blank carbon paper, a new band at 1459 cm−1 was observed, which is related to the bending vibration of H–N–H in the intermediates and NH3 species.27 The appearance of H–N–H bending and stretching bands implies that proton–electron pairs transferred to the adsorbed N2 and reaction intermediates. When PG was used as the electrocatalyst, the characteristic stretching bands of H–N–H at 2930 cm−1 and 2850 cm−1 on the electrode surface were not evident until the reaction time was 10 h (Fig. S14†). This result is due to the low NRR activity of PG.
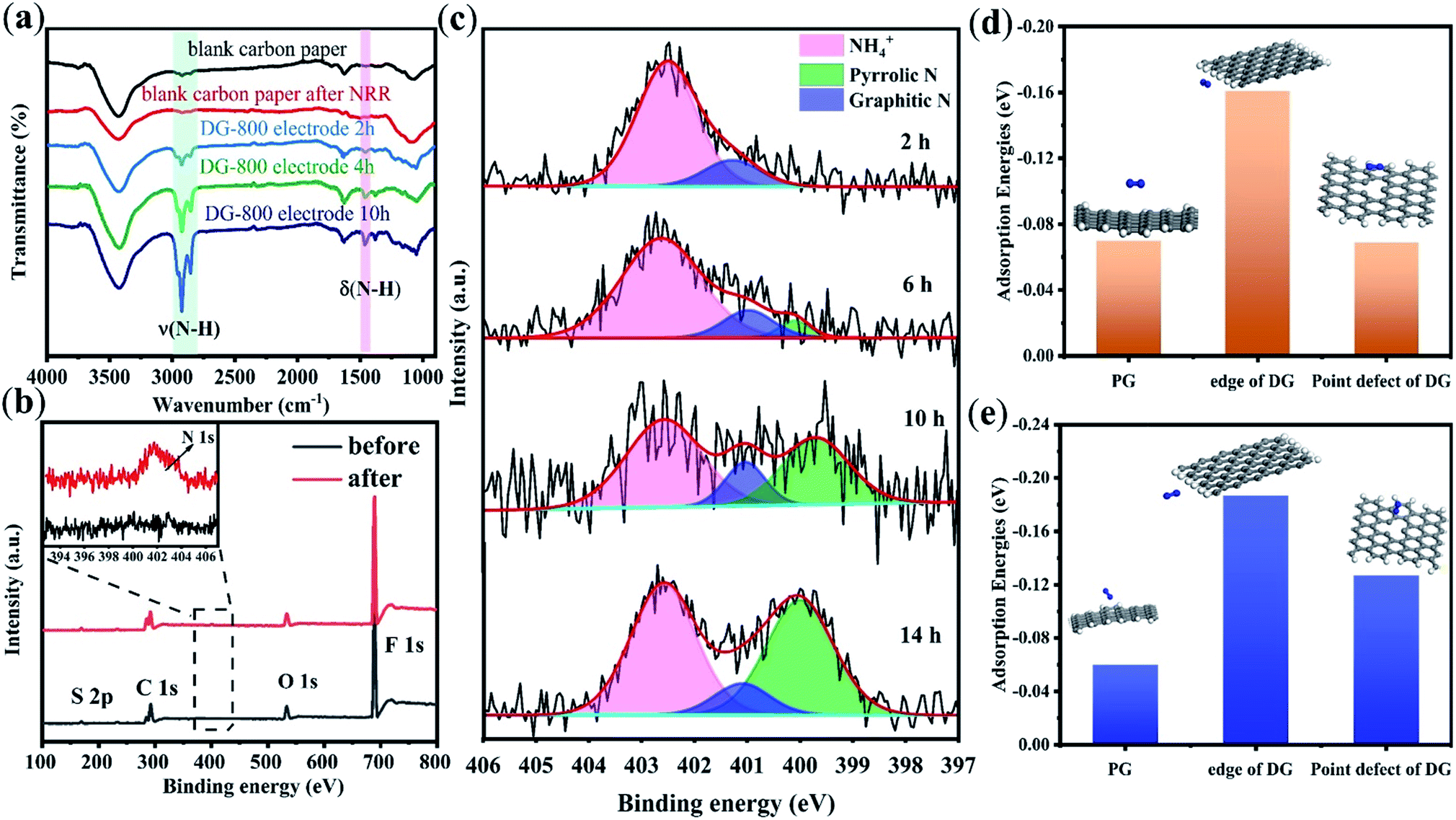 | ||
| Fig. 4 (a) FT-IR spectra of blank electrode and DG-800 electrode after different reaction times. (b) Full XPS spectra and N 1s spectra (the inset) of the DG-800 electrode before and after the NRR. (c) N 1s spectra of the DG-800 electrode after different reaction times. (d) Adsorption energies (Eads, eV) of N2 molecule with the side-on configuration. (e) Adsorption energies (Eads, eV) of the N2 molecule with the end-on configuration. | ||
XPS measurements, which give fresh insight into N2 fixation, were conducted on the carbon paper with DG-800 after the NRR test, and as a control, the same film sample before the NRR test. As shown in Fig. 4b, the XPS spectra on the film sample before the test revealed C, O, F, and S signals without N, where F and S originate from the Nafion binder (C9HF17O5S). Apparently, a clear N signal on the sample after the NRR test was detected, whereas there was no N signal on the control sample. The XPS data for nitrogen content (Table S3†) distinctly changed from 0 to 1.4%, providing solid spectroscopic evidence of the successful fixation of N2 on DG-800. Further, the N 1s spectrum was deconvoluted into three peaks (Fig. 4c) corresponding to NH4+ (402.5 eV),28–30 graphitic N (401.2 eV),31,32 and pyrrolic N (400.1 eV).33 Thus, the reduction of N2 to ammonia on DG-800 was verified again by the peak at around 402.5 eV. The edges and defect sites afford C atoms that are much more chemically reactive than that in the plane of perfect graphene for the formation of nitrogen-containing groups.34
To further confirm that defect sites are responsible for the enhanced performance, the XPS results for PG and DG-800 were compared. As presented in Fig. S16,† only a small amount of N was doped in PG, mostly in the form of pyrrolic N. Combined with the nearly perfect basal plane of PG, it is credible that N was predominantly incorporated into the edges of PG, an unavoidable defective domain in graphene. Considering the NRR, the basal plane was found to be inert and N2 molecules cannot be effectively absorbed.35 This comparison highlights the necessity of creating a favorable environment, such as defect sites, for molecular chemisorption. In addition, DFT calculations were employed to describe the N2 adsorption and activation on PG and defect sites of DG, in which both side-on and end-on configurations were considered. N2 adsorption on DG is stronger than that on PG due to the more negative energy value of DG,36 which manifests that the NRR activity benefits from the defects in DG (Fig. 4d and e). The N2 molecules on the edge of DG are the most active, where the N–N bond is elongated to 1.116 Å, resulting in more efficient charge transfer.
Next, the evolution of nitrogen species during the reaction was monitored to study the reaction mechanism (Fig. 4c). As illustrated in Fig. S17,† we speculated that the possible configurations to form pyrrolic N and graphitic N are armchair edges and point defects, respectively. Obviously, the possible defect sites for N-dopants are much more than the two mentioned above, which are still challenging to exactly identify.37 Thus, we focused on the edge sites and point defects to study the process concomitant with the NRR. On one hand, the peak of graphitic N appeared with nearly a stable area, which is associated with a decrease in the amount of point defects available for the incorporation of N atoms as the reaction proceeds (Fig. 4c). This implies that point defects are active in the primary stage. Then, once the point defects are “saturated” with N atoms, these sites are not active anymore. A previous study also reported that graphitic nitrogen is NRR-inactive since it is difficult for N2 to adsorb on graphitic N-doped carbon.38 On the other hand, pyrrolic N shows constant improvement with time (Fig. 4c and Table S4†). Mostly originating from rich edges, it can be predicted that more and more pyrrolic nitrogen will be detected as electrolysis continues.
Lastly, we supposed that the yielded NH3, instead of N2, serves as the nitrogen source for N doping. Then, a parallel experiment was designed to answer the above conjecture, which was carried out by replacing the N2 flow and H2SO4 electrolyte with an Ar flow and (NH4)2SO4 electrolyte, respectively, while keeping the reaction time (6 h) and catalyst (DG-800) constant. As expected, pyrrolic and graphitic N emerged in the parallel experiment (Fig. S18†), where NH4+ was the only nitrogen source for the generation of pyrrolic and graphitic N. Thus, the formation of nitrogen-containing groups can be understood as follows, N2 is firstly electrochemically reduced to NH3 and then the product NH4+ is incorporated into the graphene lattices in the form of pyrrolic N and graphitic N.39 Specifically, the existence of pyrrolic N and graphitic N is additional proof of the N2 reduction reaction, which represent the defect sites of graphene.
Conclusions
In summary, we demonstrated that N2 can be efficiently reduced to NH3 by metal-free defect graphene under ambient conditions. The defect engineering based on a salt melt endowed DG-800 with an excellent faradaic efficiency of 8.51% at −0.4 V vs. RHE for the NRR, where the well-controlled active centers of defects and edges play a crucial role. The observation of N doping in graphene after NRR provided additional proof of the successful N2 reduction and bridges the NRR activity with the defect sites of graphene. This study highlights the significant contribution of the inherent activity of carbon in the NRR and provides different insight to understand N2 fixation. Moving forward, precise studies such as investigation of the different roles of defect structures in NRR will be challenging but also exciting. Further, we believe that the combination of defect engineering and other promising strategies will greatly favor the development of high efficiency NRR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their appreciations for the financial support from the National Natural Science Foundation of China (11575084, 51602153), the Natural Science Foundation of Jiangsu Province (BK20160795), the Fundamental Research Funds for the Central Universities (No. NE2018104), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed
.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS
- G. Chen, X. Cao, S. Wu, X. Zeng, L. Ding, M. Zhu and H. Wang, J. Am. Chem. Soc., 2017, 139, 9771–9774 CrossRef CAS
- Y. Yao, H. Wang, X. Yuan, H. Li and M. Shao, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS
- B. H. R. Surrnto, D. Wang, L. M. Azofra, M. Harb, L. Cavallo, R. Jalili, D. R. G. Mitchell, M. Chatti and D. R. MacFarlane, ACS Energy Lett., 2019, 4, 430–435 CrossRef
- M. Legare, G. Belanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–899 CrossRef CAS
- W. Xia, J. Li, T. Wang, L. Song, H. Guo, H. Gong, C. Jiang, B. Gao and J. He, Chem. Commun., 2018, 54, 1623–1626 RSC
- L. Song, J. Tang, T. Wang, C. Wu, Y. Ide, J. He and Y. Yamauchi, Chem.–Eur. J., 2019, 25, 6807–6813 CrossRef CAS PubMed
- H. Wang, X. Li, L. Gao, H. Wu, J. Yang, L. Cai, T. Ma, C. Tung, L. Wu and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 192–197 CrossRef CAS
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu, O. Zhuo, X. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 CrossRef CAS
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2014, 588–593 Search PubMed
- L. Tao, M. Qiao, R. Jin, Y. Li, Z. Xiao, Y. Wang, N. Zhang, C. Xie, Q. He, D. Jiang, G. Yu, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 1019–1024 CrossRef CAS PubMed
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS
- W. Lei, D. Portehault, R. Dimova and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 7121–7127 CrossRef CAS PubMed
- X. Li, X. Hao, M. Zhao, Y. Wu, J. Yang, Y. Tian and G. Qian, Adv. Mater., 2013, 25, 2200–2204 CrossRef CAS
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237–8265 RSC
- W. Xia, J. Tang, J. Li, S. Zhang, K. C. W. Wu, J. He and Y. Yamauchi, Angew. Chem., Int. Ed., 2019, 58, 13354–13359 CrossRef CAS
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed
- M. S. Dresselhaus, A. Jorio, A. G. Souza Filho and R. Saito, Philos. Trans. R. Soc., A, 2010, 368, 5355–5377 CrossRef CAS PubMed
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S. Z. Qiao, Adv. Mater., 2019, 1902709 CrossRef PubMed
- Y. Wang, L. Tao, Z. Xiao, R. Chen, Z. Jiang and S. Wang, Adv. Funct. Mater., 2018, 28, 1705356 CrossRef
- C. Young, R. R. Salunkhe, J. Tang, C. Hu, M. Shahabuddin, E. Yanmaz, M. S. A. Hossain, J. H. Kim and Y. Yamauchi, Phys. Chem. Chem. Phys., 2016, 18, 29308–29315 RSC
- S. H. Lee, J. Kim, D. Y. Chung, J. M. Yoo, H. S. Lee, M. J. Kim, B. S. Mun, S. G. Kwon, Y. Sung and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 2035–2045 CrossRef CAS
- H. Cheng, L. Ding, G. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef
- J. Zheng, Y. Lyu, M. Qiao, R. Wang, Y. Zhou, H. Li, C. Chen, Y. Li, H. Zhou, S. P. Jiang and S. Wang, Chem, 2019, 5, 617–633 CAS
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed
- W. Grunert, R. Feldhaus, K. Anders, E. S. Shpiro, G. V. Antoshin and K. M. Minachev, J. Electron Spectrosc. Relat. Phenom., 1986, 40, 187–192 CrossRef
- L. Sygellou, Appl. Surf. Sci., 2019, 476, 1079–1085 CrossRef CAS
- J. L. Smith, R. G. Herman, C. R. Terenna, M. R. Galler and K. Klier, J. Phys. Chem. A, 2004, 108, 39–46 CrossRef CAS
- H. Tan, Y. Li, X. Jiang, J. Tang, Z. Wang, H. Qian, P. Mei, V. Malgras, Y. Bando and Y. Yamauchi, Nano Energy, 2017, 36, 286–294 CrossRef CAS
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed
- W. Zhang, X. Jiang, X. Wang, Y. V. Kaneti, Y. Chen, J. Liu, J. Jiang, Y. Yamauchi and M. Hu, Angew. Chem., Int. Ed., 2017, 56, 8435–8440 CrossRef CAS
- X. Wang, X. Li, L. Zhang, Y. Yoon, P. K. Weber, H. Wang, J. Guo and H. Dai, Science, 2009, 324, 768–771 CrossRef CAS
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S. Qiao, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS
- Z. Hou, X. Wang, T. Ikeda, K. Terakura, M. Oshima, M. Kakimoto and S. Miyata, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165439 CrossRef
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS
- F. Lou, M. E. M. Buan, N. Muthuswamy, J. C. Walmsley, M. Rønning and D. Chen, J. Mater. Chem. A, 2016, 4, 1233–1243 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta10071g |
| This journal is © The Royal Society of Chemistry 2020 |