
Sulfur covalently bonded to porous graphitic carbon as an anode material for lithium-ion capacitors with high energy storage performance†
Yue
Sun‡
ad,
Junpeng
Ma‡
b,
Xinyue
Yang
b,
Liping
Wen
 a,
Weidong
Zhou
bc and
Jianxin
Geng
*bc
a,
Weidong
Zhou
bc and
Jianxin
Geng
*bc
aCAS Key Laboratory of Bio-inspired Materials and Interfacial Science, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, 29 Zhongguancun East Road, Haidian District, Beijing 100190, China
bBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, 15 Beisanhuan East Road, Chaoyang District, Beijing 100029, China. E-mail: jianxingeng@mail.buct.edu.cn
cState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, 15 Beisanhuan East Road, Chaoyang District, Beijing 100029, China
dUniversity of Chinese Academy of Sciences, 19A Yuquan Road, Shijingshan District, Beijing 100049, China
First published on 15th November 2019
Abstract
Sulfur covalently bonded to porous graphitic carbon (CB–S@PGC) as an anode for lithium-ion capacitors is shown to offer an ultrawide operating potential window of 4 V. When coupled with activated carbon as a cathode, the CB–S@PGC electrode exhibits a markedly high specific capacitance (412 F g−1 at 0.2 A g−1) and superiorly high capacity retention. And CB–S@PGC//lithium cobalt oxide cells exhibit high power and energy densities (e.g., a power density of 1665 W kg−1 is obtained at an energy density of 411 W h kg−1).
With the overwhelming use of portable electronic equipment, electric vehicles, and large-scale smart grids, energy storage devices with high energy density, high power density and long cycle life are urgently needed.1,2 While supercapacitors (SCs) exhibit high power density and long cycle life due to their rapid and reversible accommodation of charges at the interfaces of the electrolyte and electrode,3,4 lithium-ion batteries (LIBs) show high energy density by utilizing faradaic processes at the cathode (Li+ insertion, intercalation or reaction).5–7 However, each of these two types of devices also has drawbacks due to their energy storage mechanisms. To complementarily utilize the advantages of SCs and LIBs, it is highly significant to combine their energy storage mechanisms to develop a new type of energy storage device that exhibits high energy density and high power density: lithium-ion capacitors (LICs) are such devices,8–10 in which an SC-type electrode (charge accommodation) and an LIB-type electrode (Li+ insertion/intercalation) are assembled into one unit with an electrolyte containing Li salt. Progress has recently been achieved mainly using transition metal compounds such as TiO2, Li4Ti5O12, Nb2O5, Li3VO4, Ni–Co oxide and MXene,11–17 and heteroatom-hybridized carbon and Si/C composites18–20 as anode materials for LICs. The use of transition metal compounds may be limited due to their complicated syntheses, low earth abundance, and/or environmental unfriendliness.
Sulfur, having a superiorly high theoretical specific capacity (1675 mA h g−1) and energy density (2567 W h kg−1) for storing lithium,21–24 is environmentally benign and highly earth abundant. In lithium–sulfur (Li–S) batteries, sulfur stores lithium on the basis of multi-electron-transfer electrochemistry (S8 + 16Li+ + 16e− → 8Li2S), i.e., one sulfur atom (with a relative atomic mass of 32.06) reacts with two lithium atoms. By contrast, one molecule of the aforementioned transition metal compounds, for example Li3VO4 (with a relative formula mass of 135.76) also reacts with at most two lithium atoms.15 Therefore, the gravimetric lithium storage density of sulfur is roughly four times higher than those of transition metal compounds. However, a number of obstacles, including the electrical insulation of sulfur, the volume changes of sulfur in charge/discharge processes, and the “shuttle effect” due to the dissolution of lithium polysulfides in the electrolyte, severely limit the practical use of Li–S batteries. To overcome these issues, a variety of strategies have been proposed, including encapsulation of sulfur with various carbon materials,25–29 doping of a cathode framework with heteroatoms or transition metal oxides to improve its affinity for lithium polysulfides,30,31 and creation of covalent C–S bonds32–38 and sulfur-containing complexes.39 Although achievements have been made in designing sulfur-containing materials for Li–S batteries, the structural features of these materials are not satisfactorily compatible with the LIC requirements. Therefore, to design sulfur-containing materials that fulfill the structural requirements of LIC anodes is a great challenge.
Based on the mechanism of LICs, an ideal anode material should satisfy the following requirements: (1) a high specific capacity for storing lithium, (2) fast lithiation/delithiation reactions, and (3) a stable structure during lithiation/delithiation processes. While requirement (1) is basically decided by the type of the lithiation/delithiation reactions of anode materials, requirements (2) and (3) are related to the structural features of the anode materials investigated. Therefore, we speculate that rationally designing the structures of sulfur/carbon composites might satisfy the aforementioned requirements, thus serving as good anode materials for LICs. Herein, we report the use of sulfur covalently bonded to hierarchically porous graphitic carbon (CB–S@PGC) as an anode to create an LIC system. The CB–S@PGC composite was found to be capable of enduring an ultrawide potential window of up to 4 V; in the operating potential window, the lithiation/delithiation reactions resulted in a significant contribution to pseudocapacitance. As a result, the CB–S@PGC electrode exhibited a markedly high specific capacitance (412 F g−1 at 0.2 A g−1) when coupled with AC and high energy and power densities when coupled with lithium cobalt oxide (LCO) (in particular, a high power density of 1665 W kg−1 was achieved at an energy density of 411 W h kg−1). The nanoscale distribution of the sulfur particles offered fast lithiation/delithiation processes, leading to less dependence of the redox reaction-capacitance on the scan rate. More importantly, the CB–S@PGC electrode exhibited a superiorly high capacity retention rate (no drop detected after 5000 cycles). The development of sulfur-containing materials as anode materials for LICs circumvents the issues faced by transition metal compounds, such as complicated syntheses, low earth abundance, and environmental unfriendliness. Therefore, this research introduces a new approach for developing new anode materials for LICs.
CB–S@PGC was synthesized by the in situ synthesis of sulfur via oxidation of Na2S by Fe(NO3)3 in a PGC that was prepared using cheap starch as a carbon source and NaCl as a hard template for pores (see Materials and methods and Fig. S1 for details†). Fig. 1a schematically explains the formation of C–S bonds between sulfur and the PGC framework in the synthesis process of sulfur nanoparticles. The oxidation of Na2S by Fe(NO3)3 results in the formation of a free radical HS˙, which can be transformed into a radical anion S˙− and a disulfide ion S22−; subsequent oxidation leads to the formation of a group of radical anions (S3˙−, S4˙−, S6˙−, etc.) and negatively charged polysulfides (S32−, S42−, S62−, etc.).40 These reactive species may attack the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds or the oxygen-containing functional groups of PGC and lead to covalent attachment of the sulfur nanoparticles to the PGC framework. Finally, unbonded sulfur was removed by washing with CS2 and the product was denoted as CB–S@PGC. As a control, the unwashed sample containing free sulfur was denoted as S@PGC.
C bonds or the oxygen-containing functional groups of PGC and lead to covalent attachment of the sulfur nanoparticles to the PGC framework. Finally, unbonded sulfur was removed by washing with CS2 and the product was denoted as CB–S@PGC. As a control, the unwashed sample containing free sulfur was denoted as S@PGC.
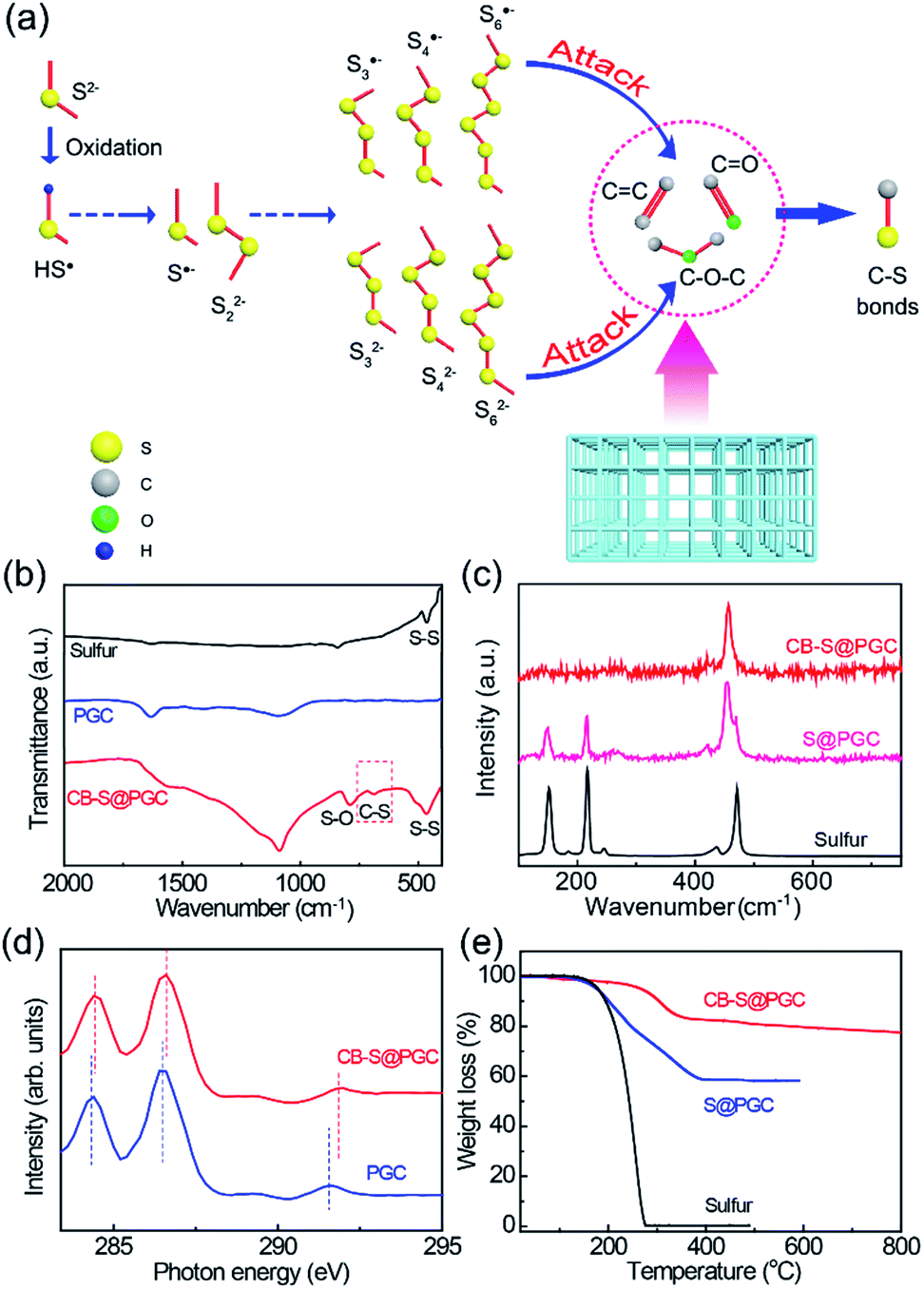 | ||
| Fig. 1 Synthesis and molecular structure characterization. (a) Schematic illustration of the formation of covalent C–S bonds between sulfur nanoparticles and the PGC framework. (b) FT-IR spectra of sulfur, PGC, and CB–S@PGC. (c) Raman spectra of sulfur, S@PGC, and CB–S@PGC. (d) Carbon K-edge XANES spectra of PGC and CB–S@PGC. (e) TGA curves of sulfur, S@PGC, and CB–S@PGC. | ||
The structures of CB–S@PGC were studied using FT-IR, Raman, X-ray absorption near-edge structure (XANES) spectroscopy. The FT-IR data revealed the presence of C–S bonds in CB–S@PGC, as the characteristic signal corresponding to C–S vibrations was found at 678 cm−1 (Fig. 1b). Fig. 1c shows the Raman spectra recorded for pure sulfur, S@PGC, and CB–S@PGC. Pure sulfur showed characteristic signals at 151, 216, and 471 cm−1 corresponding to the e2, a1, and e1 vibrational modes of S8 rings, respectively.41 By contrast, a new peak was detected at 456 cm−1 in the spectrum of the S@PGC composite, consistent with sulfur chains tethered to the PGC framework. Moreover, in the Raman spectrum recorded for CB–S@PGC, only the characteristic signal at 456 cm−1 was detected, indicating that all of the sulfur in CB–S@PGC was covalently bonded to the PGC framework. Furthermore, the detection of the characteristic D and G bands at 1356 and 1607 cm−1 in the Raman spectrum confirmed the conversion of starch to graphitic carbon (Fig. S2†).
To confirm the presence of C–S bonds, CB–S@PGC was further investigated using X-ray absorption near-edge structure (XANES) spectroscopy. Fig. 1d shows the carbon K-edge XANES spectra recorded for PGC and CB–S@PGC. Compared with the characteristic signals of PGC, the signals associated with 1s → π* of CC species, 1s → π* of CO/C–O species, and 1s → σ* of CC/C–O species were shifted to higher energy regions in the spectrum of CB–S@PGC (284.3, 286.5, and 291.6 eV for the former vs. 284.4, 286.6, and 291.9 eV for the latter, respectively).42 Furthermore, the S 2p X-ray photoelectron spectroscopy data reconfirmed the presence of C–S bonds in CB–S@PGC (Fig. S3†). Collectively, the aforementioned data proved that the sulfur nanoparticles were covalently bonded to the PGC framework in the CB–S@PGC composite. As demonstrated later, the covalent C–S bonds between sulfur and the PGC framework would be significantly important to offer a high capacity retention rate of the CB–S@PGC electrode. The sulfur content of CB–S@PGC was investigated using thermogravimetric analysis (TGA) (Fig. 1e). Although the sulfur content of CB–S@PGC was determined to be ca. 21.4 wt%, sulfur's high gravimetric lithium storage density as well as other features may still maintain their superiorities. The sulfur removal temperatures recorded for CB–S@PGC were significantly higher than those recorded for pure sulfur because of the existence of covalent C–S bonds between the sulfur nanoparticles and the PGC framework.36
The porous structures of the CB–S@PGC composite were studied by N2 adsorption–desorption analysis. While PGC consisted of hierarchical porous structures including micro-, meso-, and macropores, the micro- and mesopores were filled with sulfur in S@PGC (Fig. 2a). The N2 adsorption–desorption isotherm of CB–S@PGC is located between that of PGC and S@PGC (Fig. 2a), indicative of the removal of free sulfur and the preservation of the covalently bonded sulfur in the PGC framework. The pore size distributions of these samples (Fig. 2b) were consistent with the N2 adsorption–desorption isotherms. The specific surface area of CB–S@PGC was calculated to be 650.4 m2 g−1, which was close to that of PGC (819.0 m2 g−1) but significantly larger than that of S@PGC (53.29 m2 g−1). Therefore, the sulfur nanoparticles covalently bonded to the PGC framework contribute to pseudocapacitance via the lithiation/delithiation reactions, and the large specific surface area of the composite offers structural superiority for electrochemical double-layer capacitance. The crystallinity of sulfur in CB–S@PGC was investigated using X-ray diffraction (XRD) (Fig. 2c). Comparing the XRD patterns of sulfur and PGC, it was found that the sulfur in CB–S@PGC exhibited amorphous characteristics. This finding was ascribed to the restrained molecular packing of the sulfur chains for crystallization in CB–S@PGC because they were covalently tethered to the PGC framework.
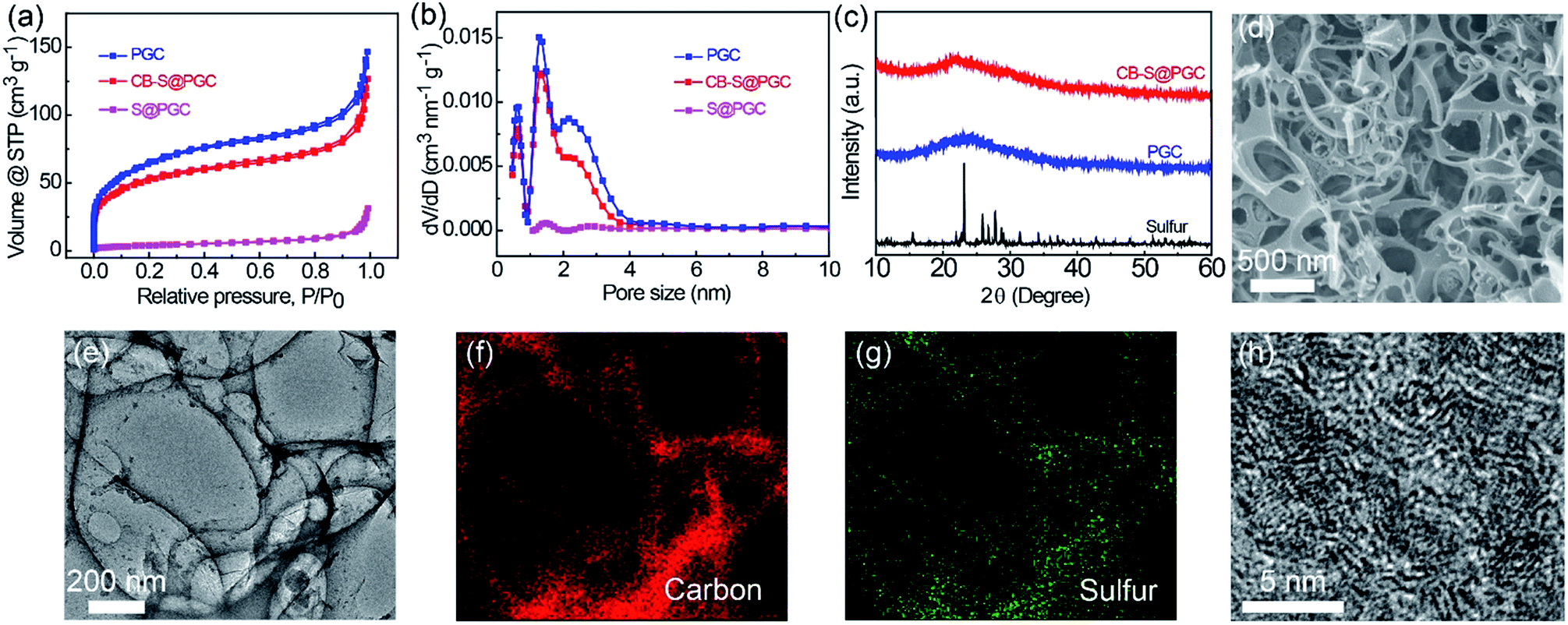 | ||
| Fig. 2 Porous structure and morphology characterization. (a) N2 adsorption–desorption isotherms and (b) pore size distributions of PGC, S@PGC and the CB–S@PGC composites. (c) XRD patterns of pure sulfur, PGC, and the CB–S@PGC composite. (d) SEM image of the CB–S@PGC composite. (e) TEM image of the CB–S@PGC composite and (f and g) the corresponding EDS elemental maps of (f) carbon and (g) sulfur collected from the entire area shown in panel (e). (h) HRTEM image of the PGC. | ||
The porous structures of the CB–S@PGC composite were further investigated by electron microscopy. Scanning electron microscopy (SEM) observations indicated that CB–S@GPC contained interconnected macropores (Fig. 2d); furthermore, transmission electron microscopy (TEM) images showed that nanoscale sulfur particles were uniformly attached to the surfaces of the PGC framework (Fig. 2e). These data demonstrated that the size of the sulfur particles was well controlled on the nanoscale, which would facilitate the diffusion of Li+ and promote the usage rate of sulfur. Energy-dispersive X-ray spectra (EDS) collected via TEM confirmed the homogeneous distribution of sulfur in the composite, as the distributions of carbon and sulfur coincided with the morphology of CB–S@PGC (Fig. 2f and g). HRTEM observation confirmed the presence of micropores in the PGC and the lattice fringes of PGC (with a spacing of 0.22 nm) (Fig. 2h).
To date, the compositional and structural features of CB–S@PGC have been clearly elaborated. From an electrode dynamics viewpoint, while the macropores are beneficial for Li+ transportation, the covalent bonding between the sulfur nanoparticles and the PGC framework inhibits the migration of polysulfide species in the electrolyte,36,43,44 potentially leading to an enhancement in LIC cycling stability. The electrochemical kinetics of the CB–S@PGC electrode was first evaluated by assembling Li–S half cells using CB–S@PGC as the cathode and metallic lithium as the anode. As shown in the cyclic voltammetry (CV) curve obtained at a scan rate of 0.2 mV s−1 (Fig. 3a), the two cathodic current peaks (C1 and C2) observed at ca. 2.38 and 2.03 V were consistent with the two-step discharging process that corresponds to the reduction of sulfur to polysulfides (Li2Sn, 4 ≤ n ≤ 8) and the final reduction of polysulfides to the solid products, Li2S2 and Li2S, respectively.45–48 The anodic peak (A) observed at 2.46 V represented the oxidation process of Li2S2/Li2S to S.45,46 Notably, although the sweep rate was increased from 0.2 to 2.4 mV s−1, the shapes of the three redox peaks remained virtually the same, and the polarization was not serious, indicating the fast electrochemical kinetics of the lithiation/delithiation processes.
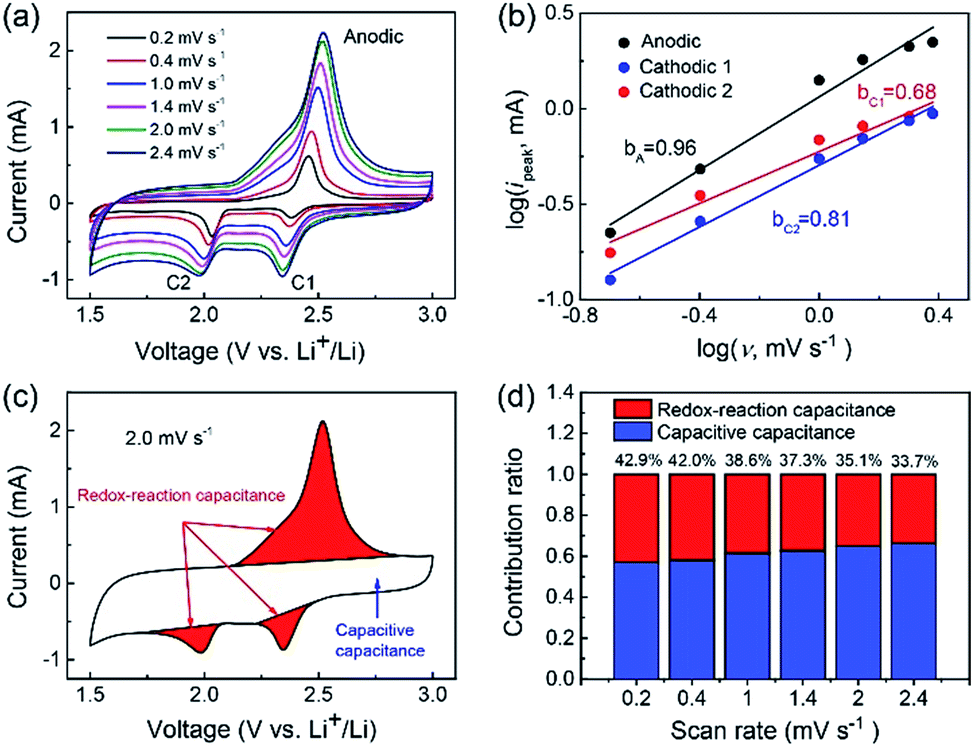 | ||
| Fig. 3 Electrochemical kinetic analysis of the CB–S@PGC electrode using Li–S half cells. (a) CV curves recorded at various scan rates ranging from 0.2 to 2.4 mV s−1. (b) log(Ipeak) vs. log(ν) plots for determining the values of parameter b for the cathodic and anodic processes. (c) A CV curve illustrating the different contributions to the CB–S@PGC electrode. (d) Contribution ratio of the redox-reaction and capacitive capacitances at various scan rates. | ||
To obtain a deeper understanding of the lithium storage process, the storage mechanism of the CB–S@PGC electrode was analyzed using eqn (1) and (2),49,50 which describe the relationship between the recorded current (i) and the scan rate (ν):
| i = avb | (1) |
log(i) = b × log(v) + log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a | (2) |
The contributions of the capacitive capacitance and redox-reaction capacitance were quantified using the CV curves recorded at various scan rates. The CV curve is composed of a roughly rectangular area and redox peaks (Fig. 3c): these two parts are attributed to capacitive capacitance and redox-reaction capacitance, respectively. Following the reported procedure,12 the contributions of these two mechanisms to the whole capacitance were calculated by integrating the areas of the respective regions (Fig. 3d). The relatively larger contribution of capacitive capacitance was due to the conversion of nonpolar surfaces to polar surfaces of the carbon due to the formation of C–S bonds. In addition, although the contribution of the redox-reaction capacitance gradually decreased from 42.9% to 33.7% as the scan rate increased from 0.2 to 2.4 mV s−1, the decrease was relatively small, implying the fast lithiation/delithiation processes of the sulfur nanoparticles due to the structural features of the CB–S@PGC composite (as shown in Fig. 2).
LIC cells were prepared using CB–S@PGC as the anode, AC as the cathode, and a LiTFSI solution in DOL/DME (1:1 v/v, 1 M) as the electrolyte (Fig. 4a). Unlike the traditional symmetric AC//AC supercapacitors showing rectangle-shaped CV curves, the CB–S@PGC//AC LIC cell exhibited marked redox peaks due to the faradaic reactions on the sulfur-based anode, which provided a significant contribution to pseudocapacitance. To obtain the optimum potential window for the CB–S@PGC electrode, the cells were measured with different voltage ranges. Fig. 4b shows a series of CV curves recorded from 0 V gradually to an increased upper potential (i.e., from 1.5 to 1.9 V). Polarization was observed when the potential was elevated to 1.9 V. With the upper potential fixed at 1.8 V, the lower potential was gradually decreased from −1.6 to −2.4 V (Fig. 4c). Polarization was observed at −2.4 V. Therefore, the operating potential window of the CB–S@PGC electrode with respect to AC was optimized to be from −2.2 to 1.8 V, which is wider than those of other anode materials.3,12–14,51 LIC cells were also prepared using PGC as the anode, AC as the cathode, and a LiTFSI solution in DOL/DME as the electrolyte (Fig. S4†). It is observed that the PGC electrode did not show a peak at ca. −0.8 V in the cathodic process and the peak at ca. 0.8 V in the anodic process as the CB–S@PGC electrode did, implying that these peaks were due to the lithiation/delithiation of sulfur in the CB–S@PGC electrode and the other redox peaks were ascribed to the lithiation/delithiation of graphitic carbon.
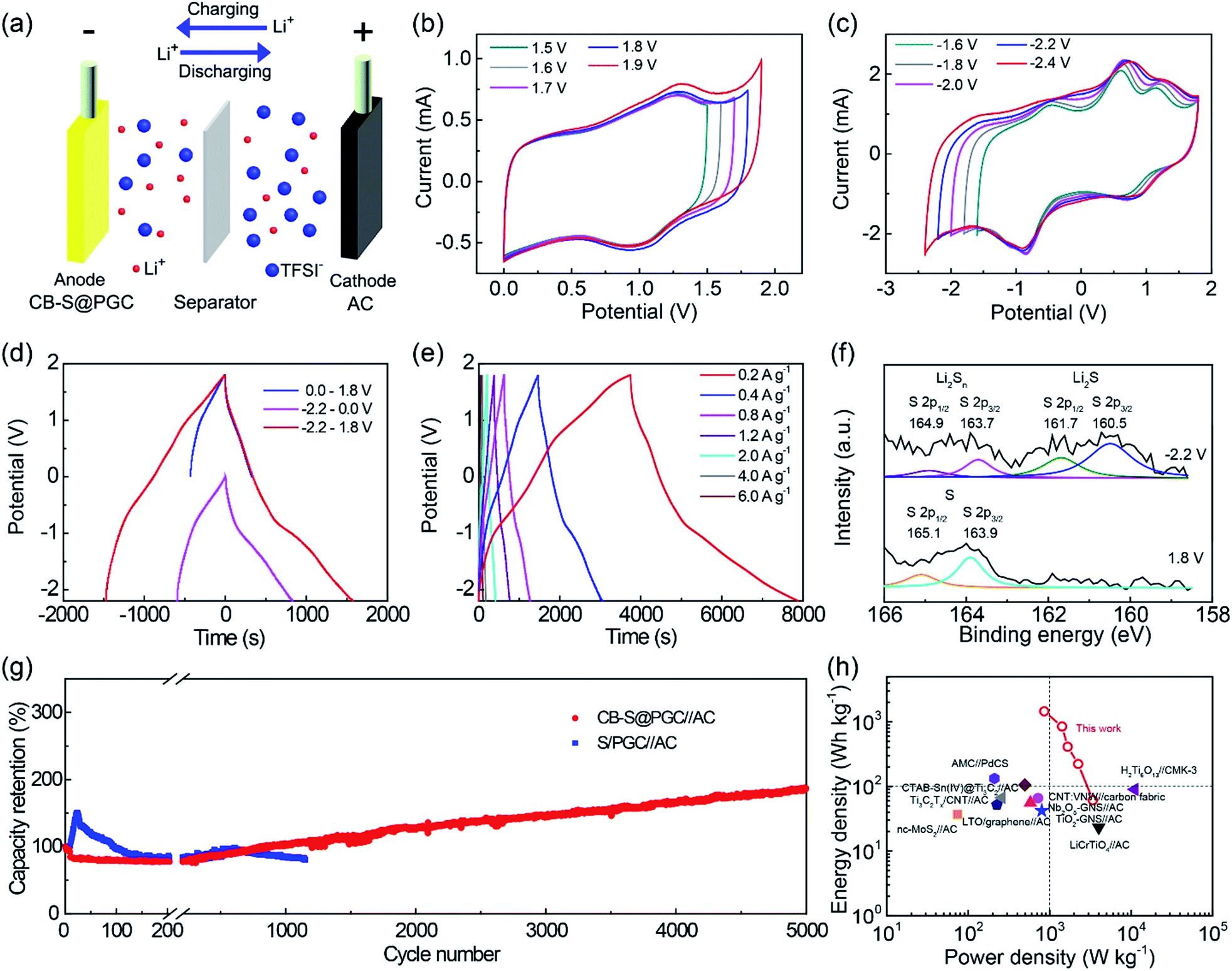 | ||
| Fig. 4 Electrochemical characterization of the CB–S@PGC electrode. (a) Schematic illustration of a CB–S@PGC//AC LIC cell. (b and c) CV curves of the CB–S@PGC electrode recorded at a scan rate of 10 mV s−1 in various potential windows. (d) Galvanostatic charge/discharge curves recorded at a current density of 0.4 A g−1 in various potential windows. (e) Galvanostatic charge/discharge curves recorded from −2.2 to 1.8 V at different current densities. (f) S 2p XPS spectra of the anode material when it was charged at −2.2 and 1.8 V with respect to AC. (g) Cycling stabilities of the CB–S@PGC and S/PGC electrodes measured at 4.0 A g−1. (h) Ragone plot comparing the performance of the CB–S@PGC//LCO cell and other materials reported in the literature.10,12,14,17,51,55–58,60 | ||
It is noteworthy that the potential window of the CB–S@PGC electrode is a relative value with respect to the AC electrode.17,52–54 Operating voltage windows spanning from a negative potential to a positive potential were previously reported with other materials.3,17 It can be tuned to fully locate either in the positive range or in the negative range by selecting another counter (also as a reference) electrode. For example, when LCO that has a higher potential than CB–S@PGC was used as a cathode, the potential window of the CB–S@PGC electrode was found to appear from −4 to 0 V (Fig. S5†).
Fig. 4d shows the galvanostatic charge/discharge curves recorded for the CB–S@PGC electrode with respect to the AC electrode in various potential windows at a current density of 0.4 A g−1. Consistent with the CV data, the charge/discharge time increased as the operating potential window expanded. Fig. 4e shows the charge/discharge curves recorded at current densities from 0.2 to 6.0 A g−1. Based on the mass of CB–S@PGC, the specific capacitances of the CB–S@PGC electrode were calculated as 412, 316, 260, 234, 209, 174 and 156 F g−1 at current densities of 0.2, 0.4, 0.8, 1.2, 2.0, 4.0, and 6.0 A g−1, respectively. The specific capacitances are several times higher than those reported in ref. 12, 14, 51 and 55–58. In contrast, for the PGC electrode, the specific capacitances were measured to be 187, 144, 133, 129, 114, 90, and 72 F g−1 at current densities of 0.2, 0.4, 0.8, 1.2, 2.0, 4.0, and 6.0 A g−1, respectively. Furthermore, symmetric supercapacitors were fabricated using the PGC or CB–S@PGC composite as an electrode material (Fig. S6 and S7†), and their specific capacitances (39.0 and 42.5 F g−1 measured at 0.2 A g−1 for PGC and CB–S@PGC, respectively) were found to be much smaller than those recorded for the CB–S@PGC electrode, showing the superiority of the hybrid energy storage mechanism of the CB–S@PGC electrode. The reaction mechanism of sulfur in the CB–S@PGC electrode was confirmed by monitoring the species formed in the electrochemical processes. As shown in Fig. 4f, Li2S was detected when the cell was scanned to −2.2 V, followed by the disappearance of the species when the cell was scanned to 1.8 V. Such findings unambiguously supported the proposed mechanism for sulfur being used as an anode material in the CB–S@PGC electrode.
The long-term charge/discharge cycling test indicated that the CB–S@PGC electrode was very stable, as the capacity was found to plateau during the initial 200 cycles, followed by a steady increase (no drop detected) in the following 5000 cycles (Fig. 4g). By contrast, the S/PGC electrode, which utilized an S/PGC composite prepared by thermally impregnating sulfur into PGC, exhibited a sharp increase in capacity during the initial 20 cycles, followed by a fast decay and fluctuation. Lithiation of sulfur takes place at the interfaces of the sulfur particles and electrolyte and the reaction speed is slower than that of insertion-type materials. Therefore, the sulfur inside particles might not be fully utilized initially (evidence for such an assumption is that the sulfur in the CB–S@PGC electrode was not entirely converted to Li2S when the electrode was charged to −2.2 V in the first cycle, as shown in Fig. 4f). Due to the diffusion of the electrolyte in the electrode materials the sulfur inside particles could be utilized during the following charge/discharge cycles, leading to gradually increased capacitance. The capacity increase was reproducible and displayed by all of our CB–S@PGC electrodes measured. A similar phenomenon was also reported with a metal compound system.59 Such a stability is superiorly competitive to other materials as anodes.11,13–15,51,55–58,60 On the other hand, for the S/PGC electrode, the diffusion of formed lithium polysulfides led to capacity decay after the initial 20 cycles. To visually demonstrate the superiority of the CB–S@PGC electrode over the S/PGC electrode in cycling stability, these two electrodes were analysed in a transparent beaker. In contrast to the marked change in color of the electrolyte containing the S/PGC electrode after one cycle, the electrolyte containing the CB–S@PGC electrode remained colorless (Fig. S8†). This finding demonstrated the significant role of the covalent C–S bonds in maintaining the outstanding cycling stability of the CB–S@PGC electrode.
The galvanostatic charge/discharge curves of CB–S@PGC//LCO cells were collected at different current densities (Fig. S9†), from which the energy and power densities were calculated. As summarized in the Ragone plot (i.e., the relationship of energy density vs. power density) (Fig. 4h), the CB–S@PGC//LCO cells yielded high energy and power densities, with the highest energy density of 1454 W h kg−1 achieved at a power density of 858 W kg−1 and the highest power density of 3385 W kg−1 achieved at an energy density of 61 W h kg−1. Under a balanced condition, an energy density of 411 W h kg−1 was achieved at a power density of 1665 W kg−1. The energy and power densities are significantly higher than those delivered by most of the anode materials reported.10,12,14,17,51,55–58 Such outstanding energy storage performance of the CB–S@PGC composite can be ascribed to its unique structural features.
Conclusions
In summary, we propose that sulfur covalently bonded to hierarchically porous carbon can be used as an anode material for high-performance LICs. When coupled with AC, the CB–S@PGC electrode exhibits a markedly high specific capacitance (412 F g−1 at 0.2 A g−1) due to the lithiation/delithiation processes of sulfur. Importantly, the CB–S@PGC electrode exhibits a superiorly high capacity retention rate. With an ultrawide operating potential window of 4 V, the CB–S@PGC//LCO cell exhibits high energy and power densities; in particular, a high energy density of 411 W h kg−1 is achieved at a power density of 1665 W kg−1. The findings of this research are expected to open a new avenue for developing new anode materials for LICs as well as for devising new structures for advanced energy storage devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51773211 and 21961160700), the “National High Level Talents Special Support Plan” of China, and Beijing Municipal Science and Technology Commission. Authors also thank the BL08U1-A beamline of the Shanghai Synchrotron Radiation Facility (SSRF) for carbon K-edge XANES measurements.References
- X. Xia, D. Chao, Z. Fan, C. Guan, X. Cao, H. Zhang and H. J. Fan, Nano Lett., 2014, 14, 1651–1658 CrossRef CAS PubMed.
- J. Jiang, P. Nie, B. Ding, Y. Zhang, G. Xu, L. Wu, H. Dou and X. Zhang, J. Mater. Chem. A, 2017, 5, 23283–23291 RSC.
- L. Shen, L. Yu, H. B. Wu, X.-Y. Yu, X. Zhang and X. W. Lou, Nat. Commun., 2015, 6, 6694 CrossRef CAS PubMed.
- Z. Yang, H. Guo, X. Li, Z. Wang, J. Wang, Y. Wang, Z. Yan and D. Zhang, J. Mater. Chem. A, 2017, 5, 15302–15309 RSC.
- Y. Zhou, X. Rui, W. Sun, Z. Xu, Y. Zhou, W. J. Ng, Q. Yan and E. Fong, ACS Nano, 2015, 9, 4628–4635 CrossRef CAS PubMed.
- B. Li, Z. Xiao, M. Chen, Z. Huang, X. Tie, J. Zai and X. Qian, J. Mater. Chem. A, 2017, 5, 24502–24507 RSC.
- Y. Sun, J. Tang, F. Qin, J. Yuan, K. Zhang, J. Li, D.-M. Zhu and L.-C. Qin, J. Mater. Chem. A, 2017, 5, 13601–13609 RSC.
- Y. Ma, H. Chang, M. Zhang and Y. Chen, Adv. Mater., 2015, 27, 5296–5308 CrossRef CAS PubMed.
- B. Li, S. Li, Y. Jin, J. Zai, M. Chen, A. Nazakat, P. Zhan, Y. Huang and X. Qian, J. Mater. Chem. A, 2018, 6, 21098–21103 RSC.
- J. B. Cook, H.-S. Kim, T. C. Lin, C.-H. Lai, B. Dunn and S. H. Tolbert, Adv. Energy Mater., 2017, 7, 1601283 CrossRef.
- K. Naoi, S. Ishimoto, Y. Isobe and S. Aoyagi, J. Power Sources, 2010, 195, 6250–6254 CrossRef CAS.
- H. Kim, M.-Y. Cho, M.-H. Kim, K.-Y. Park, H. Gwon, Y. Lee, K. C. Roh and K. Kang, Adv. Energy Mater., 2013, 3, 1500–1506 CrossRef CAS.
- E. Lim, H. Kim, C. Jo, J. Chun, K. Ku, S. Kim, H. I. Lee, I.-S. Nam, S. Yoon, K. Kang and J. Lee, ACS Nano, 2014, 8, 8968–8978 CrossRef CAS PubMed.
- J. Luo, W. Zhang, H. Yuan, C. Jin, L. Zhang, H. Huang, C. Liang, Y. Xia, J. Zhang, Y. Gan and X. Tao, ACS Nano, 2017, 11, 2459–2469 CrossRef CAS PubMed.
- L. Shen, H. Lv, S. Chen, P. Kopold, P. A. van Aken, X. Wu, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1722142 Search PubMed.
- L. Yu, B. Guan, W. Xiao and X. W. Lou, Adv. Energy Mater., 2015, 5, 1500981 CrossRef.
- S. D. Perera, B. Patel, N. Nijem, K. Roodenko, O. Seitz, J. P. Ferraris, Y. J. Chabal and K. J. Balkus Jr, Adv. Energy Mater., 2011, 1, 936–945 CrossRef CAS.
- Q. Xia, H. Yang, M. Wang, M. Yang, Q. Guo, L. Wan, H. Xia and Y. Yu, Adv. Energy Mater., 2017, 7, 1701336 CrossRef.
- B. Li, F. Dai, Q. Xiao, L. Yang, J. Shen, C. Zhang and M. Cai, Adv. Energy Mater., 2016, 6, 1600802 CrossRef.
- R. V. Salvatierra, D. Zakhidov, J. Sha, N. D. Kim, S.-K. Lee, A.-R. O. Raji, N. Zhao and J. M. Tour, ACS Nano, 2017, 11, 2724–2733 CrossRef CAS PubMed.
- Y. Qiu, W. Li, W. Zhao, G. Li, Y. Hou, M. Liu, L. Zhou, F. Ye, H. Li, Z. Wei, S. Yang, W. Duan, Y. Ye, J. Guo and Y. Zhang, Nano Lett., 2014, 14, 4821–4827 CrossRef CAS PubMed.
- Z. Cao, J. Zhang, Y. Ding, Y. Li, M. Shi, H. Yue, Y. Qiao, Y. Yin and S. Yang, J. Mater. Chem. A, 2016, 4, 8636–8644 RSC.
- J. Ye, F. He, J. Nie, Y. Cao, H. Yang and X. Ai, J. Mater. Chem. A, 2015, 3, 7406–7412 RSC.
- J. Sun, Y. Sun, M. Pasta, G. Zhou, Y. Li, W. Liu, F. Xiong and Y. Cui, Adv. Mater., 2016, 28, 9797–9803 CrossRef CAS PubMed.
- Y. Zhao, W. Wu, J. Li, Z. Xu and L. Guan, Adv. Mater., 2014, 26, 5113–5118 CrossRef CAS PubMed.
- X. Ye, J. Ma, Y.-S. Hu, H. Wei and F. Ye, J. Mater. Chem. A, 2016, 4, 775–780 RSC.
- J. Moon, J. Park, C. Jeon, J. Lee, I. Jo, S.-H. Yu, S.-P. Cho, Y.-E. Sung and B. H. Hong, Nanoscale, 2015, 7, 13249–13255 RSC.
- J. Sun, J. Ma, J. Fan, J. Pyun and J. Geng, APL Mater., 2019, 7, 020904 CrossRef.
- G. Zhou, J. Sun, Y. Jin, W. Chen, C. Zu, R. Zhang, Y. Qiu, J. Zhao, D. Zhuo, Y. Liu, X. Tao, W. Liu, K. Yan, H. R. Lee and Y. Cui, Adv. Mater., 2017, 29, 1603366 CrossRef PubMed.
- T. Zhao, Y. Ye, X. Peng, G. Divitini, H. K. Kim, C. Y. Lao, P. R. Coxon, K. Xi, Y. Liu and C. Ducati, Adv. Funct. Mater., 2016, 26, 8418–8426 CrossRef CAS.
- R. Carter, L. Oakes, N. Muralidharan, A. P. Cohn, A. Douglas and C. L. Pint, ACS Appl. Mater. Interfaces, 2017, 9, 7185–7192 CrossRef CAS PubMed.
- J. Fanous, M. Wegner, J. Grimminger, A. Andresen and M. R. Buchmeiser, Chem. Mater., 2011, 23, 5024–5028 CrossRef CAS.
- S. Wei, L. Ma, K. E. Hendrickson, Z. Tu and L. A. Archer, J. Am. Chem. Soc., 2015, 137, 12143–12152 CrossRef CAS PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- A. Chang, Q. Wu, X. Du, S. Chen, J. Shen, Q. Song, J. Xie and W. Wu, Chem. Commun., 2016, 52, 4525–4528 RSC.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
- H.-X. Ji and R. S. Ruoff, Acta Phys.-Chim. Sin., 2016, 32, 797–799 CAS.
- S. Qi, J. Sun, J. Ma, Y. Sun, K. Goossens, H. Li, P. Jia, X. Fan, C. W. Bielawski and J. Geng, Nanotechnology, 2019, 30, 024001 CrossRef PubMed.
- P. Jia, T. Hu, Q. He, X. Cao, J. Ma, J. Fan, Q. Chen, Y. Ding, J. Pyun and J. Geng, ACS Appl. Mater. Interfaces, 2019, 11, 3087–3097 CrossRef CAS PubMed.
- R. Steudel, Ind. Eng. Chem. Res., 1996, 35, 1417–1423 CrossRef CAS.
- D. W. Scott, F. H. Kruse and J. P. McCullough, J. Mol. Spectrosc., 1964, 13, 313–320 CrossRef CAS.
- K. G. Latham, M. I. Simone, W. M. Dose, J. A. Allen and S. W. Donne, Carbon, 2017, 114, 566–578 CrossRef CAS.
- Y. Z. Zhang, S. Liu, G. C. Li, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2014, 2, 4652–4659 RSC.
- A. Konarov, Z. Bakenov, H. Yashiro, Y.-K. Sun and S.-T. Myung, J. Power Sources, 2017, 355, 140–146 CrossRef CAS.
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- L. Sun, W. Kong, Y. Jiang, H. Wu, K. Jiang, J. Wang and S. Fan, J. Mater. Chem. A, 2015, 3, 5305–5312 RSC.
- S. Li, Y. Cen, Q. Xiang, M. K. Aslam, B. Hu, W. Li, Y. Tang, Q. Yu, Y. Liu and C. Chen, J. Mater. Chem. A, 2019, 7, 1658–1668 RSC.
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Adv. Mater., 2017, 29, 1702093 CrossRef PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- P. Yu, G. Cao, S. Yi, X. Zhang, C. Li, X. Sun, K. Wang and Y. Ma, Nanoscale, 2018, 10, 5906–5913 RSC.
- H. Itoi, H. Hasegawa, H. Iwata and Y. Ohzawa, Sustainable Energy Fuels, 2018, 2, 558–565 RSC.
- Y. Xia, T. S. Mathis, M.-Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS PubMed.
- X. Lang, A. Hirata, T. Fujita and M. Chen, Adv. Energy Mater., 2014, 4, 1301809 CrossRef.
- V. Aravindan, W. Chuiling and S. Madhavi, J. Mater. Chem., 2012, 22, 16026–16031 RSC.
- L. Kong, C. Zhang, S. Zhang, J. Wang, R. Cai, C. Lv, W. Qiao, L. Ling and D. Long, J. Mater. Chem. A, 2014, 2, 17962–17970 RSC.
- W. S. V. Lee, X. Huang, T. L. Tan and J. M. Xue, ACS Appl. Mater. Interfaces, 2018, 10, 1690–1700 CrossRef CAS PubMed.
- G. Wang, C. Lu, X. Zhang, B. Wan, H. Liu, M. Xia, H. Gou, G. Xin, J. Lian and Y. Zhang, Nano Energy, 2017, 36, 46–57 CrossRef CAS.
- H. Wang, Y. Zhang, H. Ang, Y. Zhang, H. T. Tan, Y. Zhang, Y. Guo, J. B. Franklin, X. L. Wu, M. Srinivasan, H. J. Fan and Q. Yan, Adv. Funct. Mater., 2016, 26, 3082–3093 CrossRef CAS.
- Y. Wang, Z. Hong, M. Wei and Y. Xia, Adv. Funct. Mater., 2012, 22, 5185–5193 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, the synthetic route for the CB–S@PGC composite, the Raman spectra of sulfur, S@PGC, and CB–S@PGC composites, the S 2p XPS spectra of the CB–S@PGC composite and sulfur, electrochemical characterization of the PGC electrode, CV curves of CB–S@PGC and LCO electrodes and the CB–S@PGC//LCO cell, galvanostatic charge/discharge curves recorded for a symmetric supercapacitor fabricated using PGC and CB–S@PGC as electrode materials, photographs of CB–S@PGC and S/PGC electrodes during charge/discharge cycles, galvanostatic charge/discharge curves recorded for the CB–S@PGC//LCO cell. See DOI: 10.1039/c9ta09347h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |