DOI:
10.1039/C9SC00195F
(Edge Article)
Chem. Sci., 2019,
10, 3756-3764
Controlling a burn: outer-sphere gating of hydroxylamine oxidation by a distal base in cytochrome P460†
Received
13th January 2019
, Accepted 14th February 2019
First published on 6th March 2019
Abstract
Ammonia oxidizing bacteria (AOB) use the cytotoxic, energetic molecule hydroxylamine (NH2OH) as a source of reducing equivalents for cellular respiration. Despite disproportionation or violent decomposition being typical outcomes of reactions of NH2OH with iron, AOB and anammox heme P460 proteins including cytochrome (cyt) P460 and hydroxylamine oxidoreductase (HAO) effect controlled, stepwise oxidation of NH2OH to nitric oxide (NO). Curiously, a recently characterized cyt P460 variant from the AOB Nitrosomonas sp. AL212 is able to form all intermediates of cyt P460 catalysis, but is nevertheless incompetent for NH2OH oxidation. We now show via site-directed mutagenesis, activity assays, spectroscopy, and structural biology that this lack of activity is attributable to the absence of a critical basic glutamate residue in the distal pocket above the heme P460 cofactor. This substitution is the only distinguishing characteristic of a protein that is otherwise effectively structurally and spectroscopically identical to an active variant. This highlights and reinforces a fundamental principal of metalloenzymology: metallocofactor inner-sphere geometric and electronic structures are in many cases insufficient for imbuing reactivity; a precisely defined outer coordination sphere contributed by the polypeptide matrix can be the key differentiator between a metalloenzyme and an unreactive metalloprotein.
Introduction
Nature has mastered selective redox chemistry using earth-abundant first-row transition metals, affording a wealth of inspiration toward human leverage of similar chemistry for a sustainable chemical future.1–3 Nature's redox enzymes, be they metalloenzymes or otherwise, make critical use of well-designed outer coordination spheres in order to tune properties such as reduction potentials.4–7 They also make use of these motifs to impart substrate chemo- and regioselectivity as exemplified by the cytochromes P450.8,9 This tuning is crucial for successful biological energy transduction, where metabolic enzymes such as those in the Krebs cycle must effect controlled release of energy from chemical fuel via product selectivity. Nitrification—a nitrogen-based energy transduction pathway in which ammonia (NH3) serves as the input fuel—affords a fresh arena in which to study this phenomenon. A key step of nitrification is the 3-electron oxidation of cytotoxic and energetic hydroxylamine (NH2OH) to form nitric oxide (NO).10,11 Nature has evolved Fe-based enzymes bearing heme P460 cofactors that effect the controlled release of electrons from NH2OH.12,13 This is remarkable given that more commonly reported reaction outcomes between NH2OH and Fe comprise disproportionation or uncontrolled production of ill-defined mixtures bearing a variety of gaseous oxynitrogen species.14–17 Consequently, P460 cofactors represent a valuable system from which to glean insights into controlled redox chemistry.
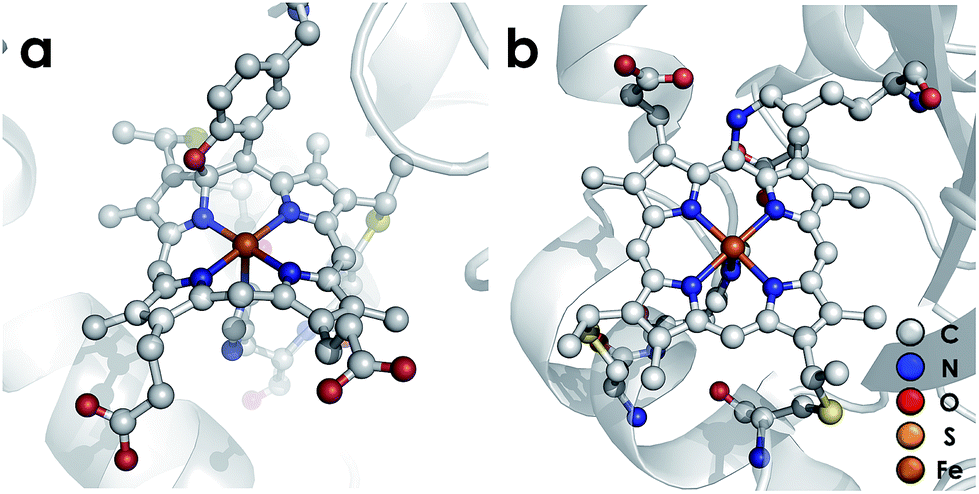
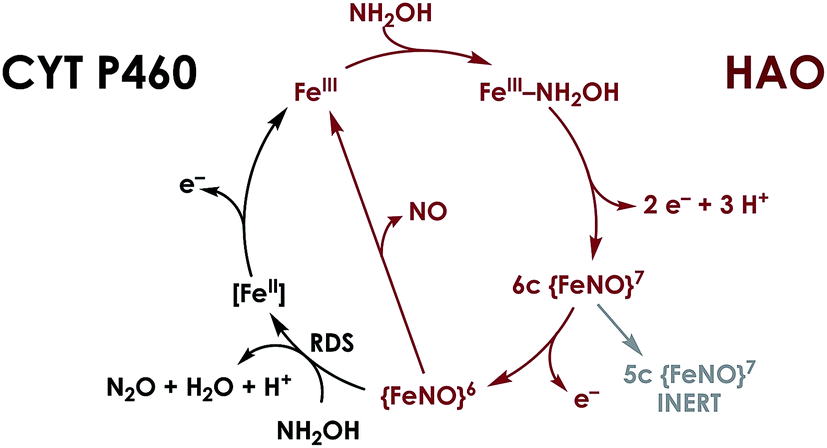
P460 cofactors are modified c-type hemes found in enzymes from aerobic ammonia oxidizing (AOB), anaerobic ammonia oxidizing (anammox), methanotrophic, and—possibly—denitrifying bacteria.18,19 P460 hemes are distinguished from canonical c-hemes by additional covalent porphyrin-amino acid cross-links. In octaheme enzymes, such as hydroxylamine oxidoreductase (HAO), two cross-links are contributed by a tyrosine, which binds through its phenoxy O and its ortho-disposed C3 to the porphyrin meso- and α-pyrrolic carbons, respectively (Fig. 1a). In monoheme cytochrome (cyt) P460 enzymes, a lysine sidechain forms a single N–C bond to a macrocyclic meso carbon (Fig. 1b). The cofactor macrocycle remains dibasic in either case,12,20 although the cross-link and macrocycle ruffling together afford cofactors with significantly red-shifted UV-vis absorption profiles whose absorptivities are diminished relative to standard c-hemes.20,21 To date, heme P460 cofactors have been shown to effect selective oxidation of NH2OH to NO through formation of a sequence of Fe-nitrosyl intermediates (Fig. 2). In HAO catalysis, the NO product rapidly dissociates and is ultimately oxidized by AOB to nitrite (NO2−).10 In monoheme cyt P460 enzymes, the NO product remains bound to heme P460 for a sufficient duration to make possible nucleophilic attack by a second equivalent of NH2OH to form N2O.22 The establishment of selective oxidation of NH2OH to NO by hemes P460 overturns decades of convention in which these cofactors were incorrectly implicated as the enzymatic source of NO2− produced by AOB.10
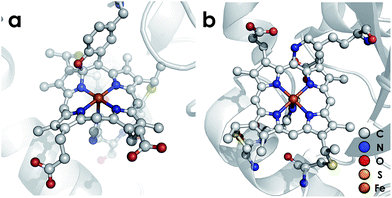 |
| | Fig. 1 Heme P460 cofactors found in N. europaea (a) hydroxylamine oxidoreductase (PDBID: 4FAS) and (b) cytochrome P460 (PDBID: 2JE3). Adapted from ref. 22. | |
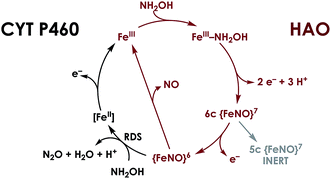 |
| | Fig. 2 Working mechanism for NH2OH oxidation by heme P460 cofactors. The steps indicated in black distinguish cyt P460, in which the {FeNO}6 persists and can undergo nucleophilic attack by NH2OH. In HAO, no {FeNO}6 has been observed, presumably due to facile release of NO, its stoichiometric product of NH2OH oxidation. Adapted from ref. 23. | |
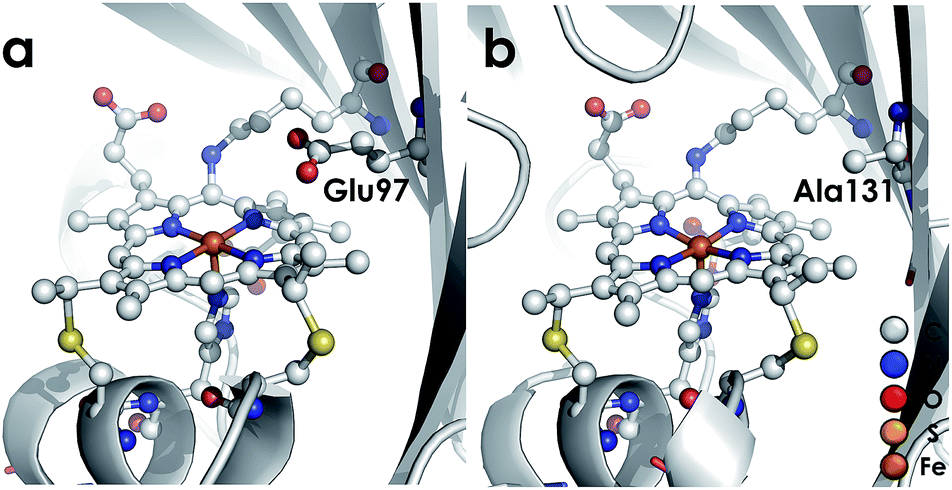
We recently reported the 1.45 Å crystal structure of a cyt P460 variant from Nitrosomonas sp. AL212.20 We noted that, despite its cofactor exhibiting identical inner-sphere structural features as well as spectroscopic features to that of the N. europaea variant used in our prior mechanistic studies, the AL212 cyt P460 was not competent for NH2OH oxidation. Now expanding our perspective to include the outer coordination sphere surrounding the AL212 heme P460 cofactor, we have found that a key difference from the N. europaea variant is the substitution of an alanine residue for a glutamate at position 131 (position 97 in N. europaea, Fig. 3). Carboxylic acid residues have been shown to greatly impact the activity of heme proteins.24,25 Further, we hypothesized that this glutamate could operate as a proton relay during oxidation of FeIII–NH2OH to {FeNO}7—a reaction that necessarily involves proton transfer. Through characterization of an array of new variants of both N. europaea and AL212 cyt P460, we now show that this glutamate residue is absolutely required for oxidation of heme P460 FeIII–NH2OH adducts.
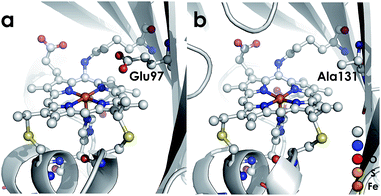 |
| | Fig. 3 Cyt P460 from N. europaea (a, PDB ID: 2JE3) and Nitrosomonas sp. AL212 (b, PDB ID: 6AMG). The presence of a distal carboxylate (Glu97) in the N. europaea variant marks a key difference from the NH2OH oxidation-incompetent AL212 variant, which has an alanine (Ala131) occupying the position. | |
Results and discussion
Comparison of WT N. europaea and AL212 cyt P460s
WT AL212 cyt P460, although incompetent for turnover of NH2OH, supports stable heme P460 FeIII–NH2OH and {FeNO}6 species that were previously identified as pathway intermediates in the N. europaea cyt P460 catalytic cycle for NH2OH oxidation to NO/N2O (Fig. 2). We explored whether any significant differences in NH2OH or NO binding affinities distinguish catalytically inactive cyt P460s. Representative titration experiments are plotted in Fig. S2,† and aggregate binding affinities are compiled in Table 1. The 298 K Kd for NH2OH binding to FeIII cyt P460 at pH 8.0 is 9 ± 1 mM for the WT N. europaea variant. The WT AL212 variant exhibits a modest, 2-fold diminution in Kd to 18 ± 1 mM. Meanwhile, binding affinities for NO at pH 8.0 are effectively identical: for N. europaea this Kd is 10 ± 2 μM, while for AL212 it is 8 ± 1 μM. Similarities between the proteins extend to FeII/III reduction potentials, which we determined using potentiometric titrations. The FeII/III reduction potential for N. europaea cyt P460 is −400 ± 5 mV vs. NHE, while it is −424 ± 7 mV for AL212 (Fig. S2† and Table 1). UV-vis absorption spectra and EPR parameters are, likewise, effectively identical between the two variants (Fig. S3† and Table 1). Overall, these similarities are not surprising given that the overall cofactor structures in the two cyt P460 variants are superimposable (RMSD = 0.074 Å, Fig. S5†).
Table 1 Spectroscopic and thermodynamic properties of cyt P460 variants under discussion
| Variant |
Reduction Potential (mV vs. NHE) |
NH2OH Kd (mM) |
NO Kd (μM) |
Resting FeIII component 1 (S = 5/2) |
Resting FeIII component 2 (S = 5/2) |
FeIII–NH2OH (S = 1/2) |
|
g
eff values |
E/D |
g
eff values |
E/D |
g values |
| WT N. europaea |
−400 ± 5 |
9 ± 1 |
10 ± 2 |
6.57, 5.09, 1.97 |
0.03 |
N/A |
N/A |
2.75, 2.28, 1.54 |
| WT N. sp. AL212 |
−424 ± 7 |
18 ± 1 |
8 ± 1 |
6.39, 5.13, 1.97 |
0.03 |
6.00, 5.52, 1.99 |
0.012 |
2.84, 2.25, 1.44 |
| AL212 Ala131Glu |
−428 ± 2 |
16 ± 5 |
5 ± 1 |
6.40, 5.14, 1.97 |
0.03 |
6.00, 5.51, 1.99 |
0.012 |
2.86, 2.27, 1.46 |
| AL212 Ala131Gln |
−406 ± 2 |
15 ± 3 |
6 ± 1 |
6.51, 5.12, 1.97 |
0.03 |
6.03, 5.53, 1.99 |
0.012 |
2.78, 2.28, 1.49 |
| AL212 Ala131Leu |
−381 ± 10 |
12 ± 3 |
2 ± 1 |
6.40, 5.11, 1.98 |
0.03 |
6.00, 5.48, 1.99 |
0.012 |
2.80, 2.27, 1.46 |
| AL212 Ala131Asp |
−388 ± 7 |
19 ± 7 |
4 ± 1 |
6.40, 5.12, 1.97 |
0.03 |
6.03, 5.50, 1.99 |
0.012 |
2.86, 2.25, 1.44 |
Introduction of an outer-sphere glutamate imbues NH2OH oxidase activity to AL212 cyt P460
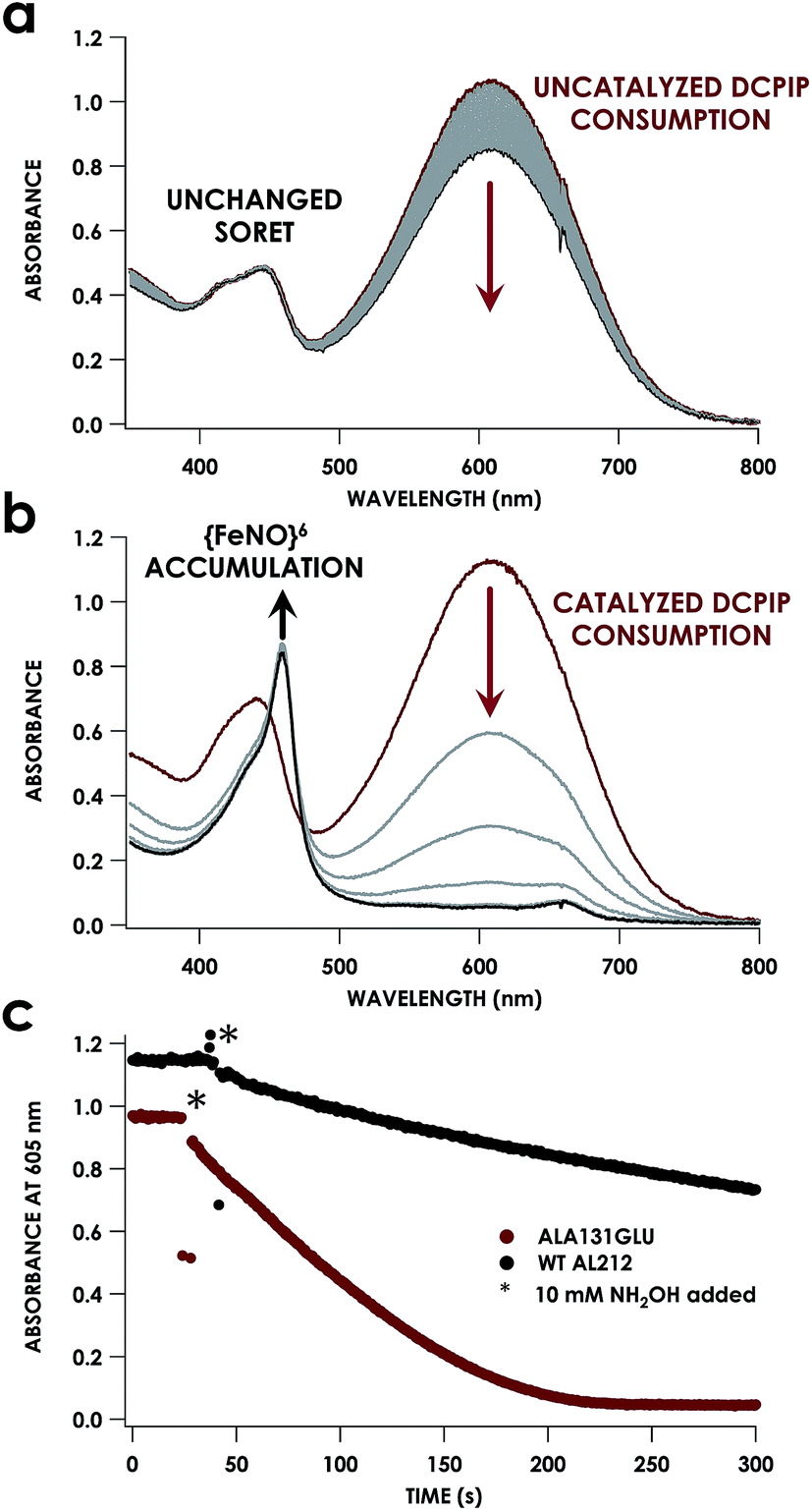
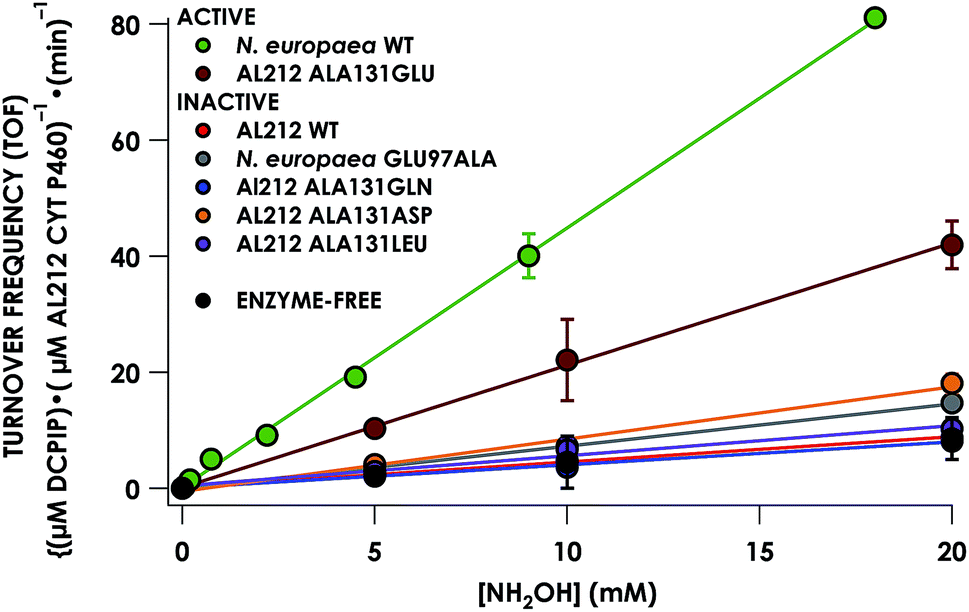
Despite inner-sphere structural, spectroscopic, and thermodynamic identity between the two cyt P460 cofactors and their adducts, the WT AL212 cyt P460 FeIII–NH2OH adduct is redox inactive using either 2,6-dichlorophenolindophenol (DCPIP, +217 mV), phenazine methosulfate (PMS, +80 mV), or hexaammonium ruthenium(III) chloride ([Ru(NH3)6]Cl3, −8.3 mV) as oxidants. When 10 mM NH2OH and 70 μM DCPIP were added to WT AL212 cyt P460, no cyt P460 intermediates (i.e. {FeNO},6 {FeNO}7) were observed (Fig. 4a). Only basal, background consumption of DCPIP—monitored by following its absorbance at 605 nm—occurs, which can also be observed in the absence of any cyt P460: 0.44 ± 0.19 μM DCPIP·mM−1 NH2OH·min−1. By contrast, with 1 μM WT AL212 cyt P460 this process occurs with a specific activity of 0.43 ± 0.02 μM DCPIP·mM−1 NH2OH·min−1 (Fig. 5).
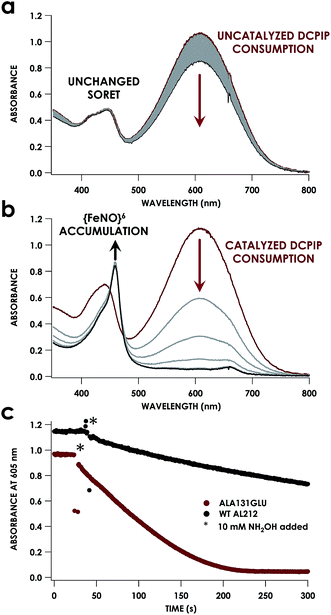 |
| | Fig. 4 UV-vis absorption traces showing reactions of 12 μM WT N. sp. AL212 cyt P460 (a) or AL212 Ala131Glu (b) with 10 mM NH2OH and 70 μM DCPIP. Reactions were carried out at 25 °C in 50 mM sodium phosphate (pH 8.0). Reactions were initiated after collection of red initial traces by addition of NH2OH, with subsequent gray scans collected at 15 s intervals. (c) Represents a kinetic trace of the absorbance of DCPIP at 605 nm for both WT AL212 and Ala131Glu under the conditions in (a) and (b), with scans every 0.5 seconds. | |
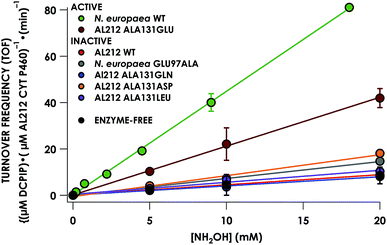 |
| | Fig. 5 Steady-state NH2OH oxidation activity plot for all investigated cyt P460 variants. Assay conditions were 1 μM cyt P460, 6 μM phenazine methosulfate (PMS), and 70 μM DCPIP. NH2OH concentrations ranged from 0–20 mM. Assays were carried out anaerobically in 50 mM sodium phosphate, pH 8.0, at 25 °C. Each data point is the average of at least three trials, with error bars representing one standard deviation. The data series in black represents NH2OH-dependent rates of DCPIP consumption under enzyme-free but otherwise identical conditions. | |
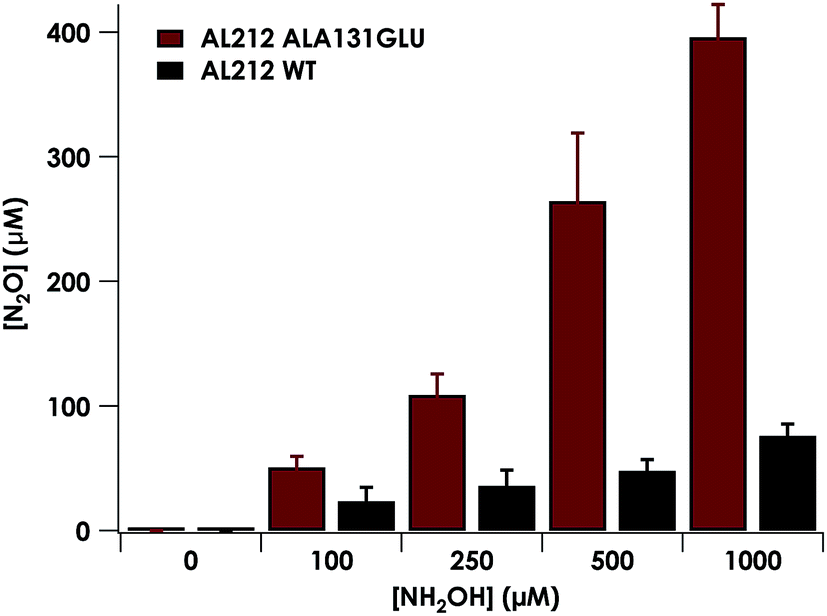
Introduction of glutamate to position 131 in the AL212 cyt P460 imbued catalytic competence for FeIII–NH2OH oxidation to NO and subsequent generation of N2O by the AL212 protein. Again using 10 mM NH2OH and 70 μM DCPIP, the Ala131Glu AL212 variant exhibits rapid formation of the cyt P460 {FeNO}6 species concomitant with rapid oxidant consumption (Fig. 4b). Using DCPIP as oxidant, Ala131Glu AL212 cyt P460 oxidizes NH2OH with a specific activity of 2.1 ± 0.1 μM DCPIP·μM−1 cyt P460·mM−1 NH2OH·min−1. This value is ca. half of that of the wild-type N. europaea variant: 4.5 ± 0.1 μM DCPIP·μM−1 cyt P460·mM−1 NH2OH·min−1 (Fig. 4). This observation was corroborated by GC analysis; under turnover conditions, Ala131Glu AL212 cyt P460 will stoichiometrically convert NH2OH to N2O much like that of cyt P460 from N. europaea. WT AL212 cyt P460 shows a basal level of N2O production attributed to background consumption of NH2OH by DCPIP (Fig. 6).
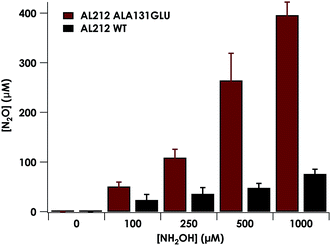 |
| | Fig. 6 Plot of N2O production as a function of NH2OH concentration by AL212 cyt P460 variants. Assay conditions were 5 μM cyt P460, 1 mM DCPIP, and NH2OH concentrations ranging from 0–1 mM. Assays were carried out anaerobically in 200 mM HEPES, pH 8.0. Each data point is the average of at least three trials, with error bars representing one standard deviation. | |
To explore the role of the residue at position 131 in NH2OH oxidation catalysis, we generated several other Ala131X variants of AL212 cyt P460: Ala131Gln, Ala131Asp, and Ala131Leu. None of these variants exhibited NH2OH oxidation activity above basal levels using DCPIP as oxidant (Fig. 5). This emphasizes the importance of a Glu residue in the outer-sphere of the heme P460 cofactor for catalytic competence and selectivity. We hypothesize that the inactivity of the Ala131Asp variant is due to the inability of the shorter side chain to effectively interact with bound NH2OH. In further accord with our hypothesis, Glu97Ala substitution obviates NH2OH oxidation in the N. europaea variant (Fig. 5). We note that these additional variants are all able to support FeIII–NH2OH and {FeNO}6 adducts when treated with either NH2OH or an NO-donor, respectively. All of these additional variants show similar UV-vis absorption, NH2OH/NO binding affinities, and FeII/III reduction potentials to the two WT proteins (Fig. S4† and Table 1).
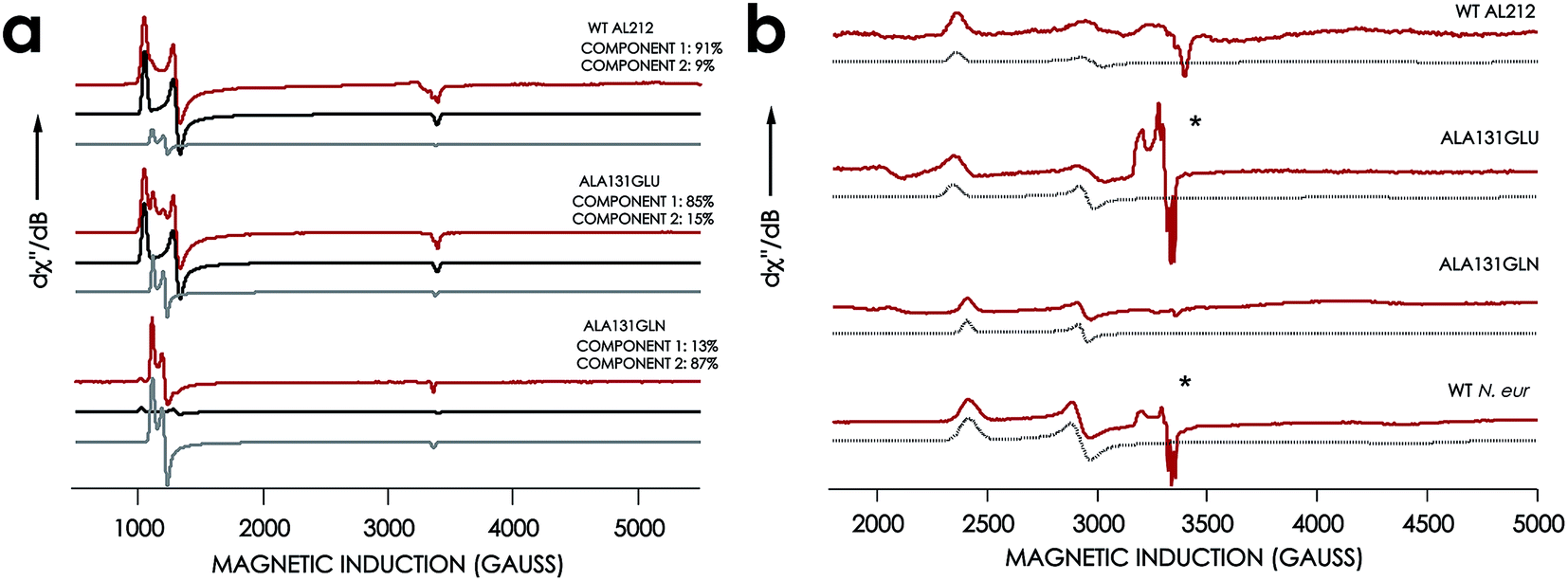
EPR spectra were obtained for the resting FeIII forms of each new cyt P460 variant (Fig. 7, Table 1). All exhibit effectively axial high-spin (S = 5/2) spectra. Interestingly, the spectra of all AL212 variants contain two spin systems, although the abundances of the two components differ in each variant. We hypothesize that these two components arise due to the presence of multiple conformations of a Phe side-chain in the distal NH2OH/NO binding pocket (vide infra).
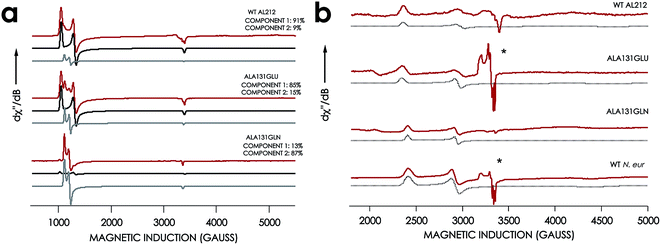 |
| | Fig. 7 EPR spectra of cyt P460 variants. (a) 10 K X-band (9.40 GHz) EPR spectrum (red) and simulated spectra (component 1 in black and component 2 in grey) of FeIII WT N. sp. AL212 cyt P460, FeIII Ala131Glu, FeIII Ala131Gln recorded at 633 μW microwave power. (b) EPR spectrum of NH2OH-bound Ala131Glu, Ala131Gln, and N. europaea cyt P460 under the same conditions. *Indicates the presence of contamination from the {FeNO}7 intermediate in both Ala131Glu and N. europaea cyt P460. | |
The X-band EPR spectra of the NH2OH-bound forms of each variant are characteristic of low-spin FeIII (S = 1/2). Each FeIII–NH2OH variant presents a single spin system, with spin Hamiltonian parameters similar to WT N. europaea cyt P460 FeIII–NH2OH adduct (Fig. 7). The EPR spectrum obtained for the FeIII–NH2OH species for the AL212 Ala131Glu cyt P460 variant also shows trace (ca. 5% abundance) amounts of an {FeNO}7 intermediate. We had observed this species previously when characterizing the N. europaea cyt P460 FeIII–NH2OH adduct, and we attributed its presence to possible O2 contamination during sample freezing, which would react with the FeIII–NH2OH adduct to form the {FeNO}7 intermediate.22,26 Interestingly, we do not observe this {FeNO}7 contaminant in any of the other variants despite maintaining an identical preparative method across the series. Moreover, in contrast to the catalytically active variants, intentionally exposing the FeIII–NH2OH adducts of the catalytically-incompetent variants to O2 does not result in an increase of the {FeNO}7 signal. These results provide further evidence that a properly-positioned, basic residue in the distal pocket of a cyt P460 is required for NH2OH oxidation catalysis.
Structural studies of AL212 cyt P460 Ala131X variants
That glutamine substitution fails to activate AL212 cyt P460 for NH2OH oxidation, despite serving as an essentially isostructural, polar surrogate for glutamate, suggests that hydrogen bonding to NH2OH is insufficient to promote catalysis. Rather, a suitably positioned basic sidechain is required for activity. To ensure that Gln131 can interact with Fe-bound NH2OH but that this interaction is insufficient for activity, we undertook structural studies of the AL212 cyt P460 Ala131Glu and Ala131Gln variants (Table 2). In addition, we were able to obtain structures of the AL212 cyt P460 Ala131Gln FeIII–NH2OH and the Ala131Glu {FeNO}6 adducts. The global structures of all variants including the resting FeIII WT AL212 cyt P460 are superimposable, with the largest pairwise RMSD values being 0.294 Å between WT AL212 and Ala131Gln. As expected, the largest structural deviations are encountered in the distal pocket above the heme P460 cofactor (Fig. S6†).
Table 2 X-ray data collection and crystal structure refinement statistics
|
|
FeIII Ala131Glu |
Ala131Glu–NO |
FeIII Ala131Gln |
Ala131Gln–NH2OH |
| Wavelength (Å) |
0.979 |
0.979 |
0.979 |
0.979 |
| Temperature (K) |
100 K |
100 K |
100 K |
100 K |
| Space group |
P1211 |
P1211 |
P1211 |
P1211 |
|
a (Å) |
48.1 |
48.7 |
48.0 |
48.0 |
|
b (Å) |
80.4 |
80.0 |
80.9 |
80.9 |
|
c (Å) |
120.0 |
119.1 |
120.6 |
120.6 |
|
α (deg) |
90.0 |
90.0 |
90.0 |
90.0 |
|
β (deg) |
95.9 |
92.6 |
96.0 |
96.0 |
|
γ (deg) |
90.0 |
90.0 |
90.0 |
90.0 |
| Reflections |
62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 608 608 |
63916 |
39644 |
41095 |
| Number of reflections in Rwork set |
59530 |
60759 |
38189 |
39122 |
| Number of reflections in Rfree set |
3078 |
3157 |
1455 |
1973 |
| Resolution (Å) |
119.6–1.97 |
48.6–1.97 |
67.1–2.30 |
29.4–2.25 |
|
R
merge (%) |
3.7 (78.8) |
3.0 (84.5) |
4.7 (118) |
5.5 (115) |
|
R
meas (%) |
5.3 (111) |
4.3 (120) |
6.6 (98.8) |
7.3 (165) |
| CC1/2 |
0.99 (0.72) |
0.93 (0.68) |
0.87 (0.66) |
0.99 (0.59) |
| Completeness (%) |
96.1 (84.9) |
98.8 (96.6) |
96.7 (98.3) |
94.8 (57.8) |
| Redundancy |
1.9 (1.9) |
2.0 (1.9) |
2.0 (2.0) |
1.9 (1.8) |
|
I/σ (I) |
32.0 (1.1) |
38.5 (0.9) |
28.0 (2.1) |
13.6 (0.5) |
|
R
work
|
20.6 (43.4) |
20.8 (41.6) |
24.2 (33.2) |
19.5 (34.9) |
|
R
free
|
24.9 (48.4) |
24.0 (45.0) |
29.9 (41.5) |
24.2 (40.0) |
|
|
RMSD from ideality
|
| Bonds (Å) |
0.01 |
0.01 |
0.01 |
0.01 |
| Angles (deg) |
1.04 |
1.01 |
1.07 |
1.02 |
| Average B factors (Å2) |
53.0 |
57.1 |
50.4 |
52.4 |
|
|
Ramachandran plot
|
| Allowed regions (%) |
100 |
100 |
100 |
100 |
| Disallowed regions (%) |
0 |
0 |
0 |
0 |
| PDBID |
6EOX
|
6E17
|
6EOZ
|
6EOY
|
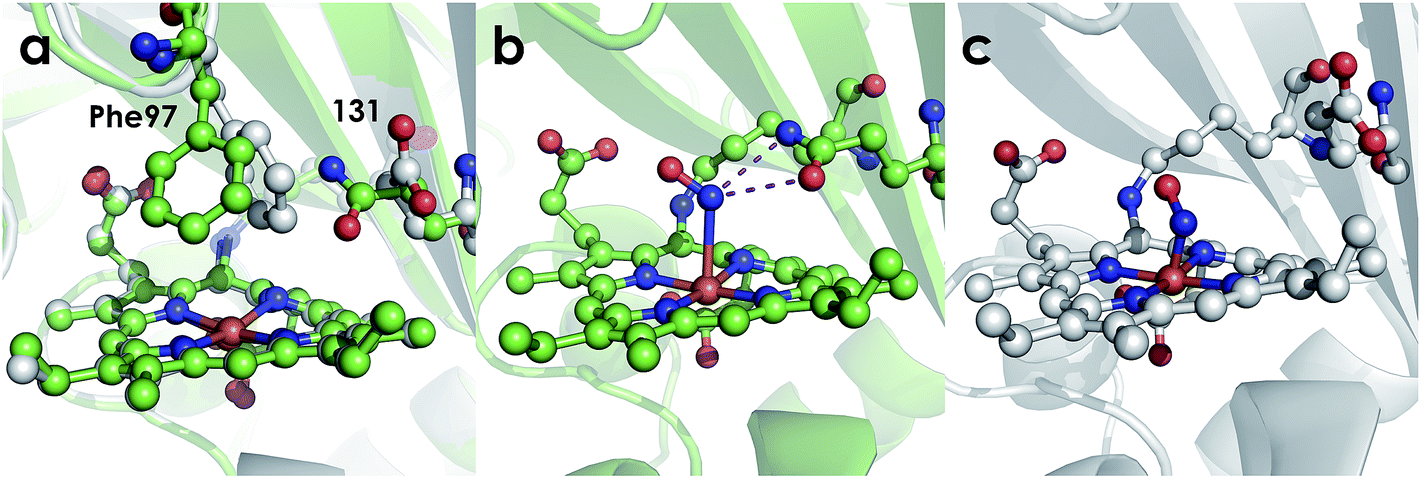
The active site structures of the Ala131Glu and Ala131Gln variants show that both residues at position 131 are positioned to participate in hydrogen bonding interactions with Fe-bound substrate. This point is made clearest by the NH2OH-bound structure of the Ala131Gln variant (Fig. 8), which demonstrates that amino acids with side-chains isostructural to Glu placed at position 131 can interact directly with bound NH2OH. The Fe–N bond distances for the Ala131Gln–NH2OH are longer than anticipated (∼2.5–2.7 Å instead of the anticipated distance closer to 2.0 Å), but we attribute this to the resolution of the structure, at which the e.s.d. in distances is estimated to be 0.4 Å, or the possibility that photoreduction leading to a ferrous center has occurred. Regardless, the composite omit map reveals electron density consistent with a bound NH2OH (Fig. S4†). Though we were unable to obtain quality data for a structure of Ala131Glu soaked with NH2OH, we anticipate a high degree of similarity of the FeIII–NH2OH adducts based on their effectively identical EPR spectra (Fig. 7).
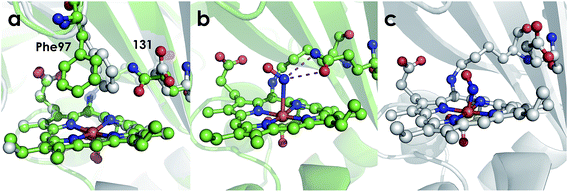 |
| | Fig. 8 Cyt P460 active site views showing orientation of residue 131. (a) Comparison of 1.97 Å structure of Ala131Glu (white, PBDID: 6EOX) with 2.30 Å structure of Ala131Gln (green, PDBID: 6EOZ) active sites with Phe76 and residue 131 highlighted in each. (b) 2.25 Å structure of Ala131Gln with NH2OH bound (PDB ID: 6EOY, chain A). (c) 1.97 Å structure of Ala131Glu with NO bound (PDB ID: 6E17 1017, chain A). | |
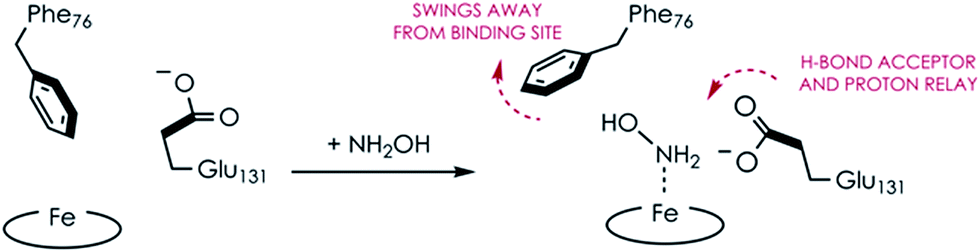
Comparing the active sites of the Ala131Glu/WT AL212 and Ala131Gln with NH2OH bound, it is possible to see molecular motions which may help to accommodate substrate binding and enforce the involvement of residue 131. Specifically, with no substrate bound, Phe76 sits directly above the P460 cofactor. When NH2OH binds, this Phe76 can reorient as is seen in the Ala131Gln structures. The residue in position 131 (Gln, in this case) can also then reposition to interact with the bound substrate (Fig. 8 and Scheme 1). This is reminiscent of a conserved, carboxylate-containing Asp residue in N. europaea HAO (NeHAO) as well as in the multi-heme, P460-containing anammox enzyme kustc1061 from Kuenenia stuttgartiensis.27 In the NH2OH-soaked structures of NeHAO and kustc1061, this aspartate and a nearby histidine residue are shown to interact with the bound substrate. The authors suggested, in fact, that these residues likely participate in shuttling protons during catalysis.27 This further supports the notion that carboxylate-containing residues are important for heme P460 NH2OH oxidation catalysis. Due to the chemical differences in the sidechains of alanine and glutamine, we hypothesize that this residue may serve as a proton relay during proton-coupled electron transfer events in the cyt P460 catalytic cycle. This would also explain why the corresponding Ala131Gln variant is not able to oxidize NH2OH, despite being able to stabilize the FeIII–NH2OH adduct. Though structurally very similar to glutamate, glutamine is far less basic. Thus, despite Gln131 being able to hydrogen-bond with NH2OH, it is unable to accept protons during catalysis, which is required for turnover. This implies that after NH2OH binding, a step involving proton transfer is necessary for the reaction to proceed. Based on the position of the NH2OH in the crystal structure, the first proton to be abstracted is likely from the N atom of NH2OH. This would lead to an NHOH radical species analogous to that proposed during turnover of cytochrome P450 nitric oxide reductase (P450nor), which reduces two molecules of NO to N2O in denitrifying bacteria and fungi.28–30 If true, this provides an interesting link between an enzyme that produces N2O through oxidation (cyt P460) and one that produces N2O through reduction (P450nor). Specific details concerning the nature and sequence of proton- and electron-transfers involving NH2OH are beyond the scope of the present work, however, and will be explored in detail in a subsequent kinetics and computational study.
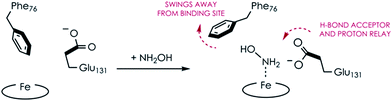 |
| | Scheme 1 Proposed N. sp. AL212 Ala131Glu cyt P460 molecular motions accompanying substrate binding. | |
Interestingly, the resting FeIII form of Ala131Gln also has the Phe76 residue rotated away from the heme, but in this case the Gln131 sidechain is oriented inward towards the heme. While it is unclear why the Gln and not the Glu variant prefers this orientation in the crystal structure, it could explain why a NH2OH-bound Ala131Gln structure was more easily attainable, as these residues were already in an ideal conformation to accept substrate. From the EPR, it is unlikely that H2O is bound in the Ala131Gln cyt P460 as it is also high spin, though the distribution of the two components in all AL212 variants seems to be related to the mutation, and, thus, may be related to the relative orientation preference of Phe76/residue131. That is, where WT AL212 is almost entirely component 1, which would correspond to Phe76 sitting directly over the Fe, Ala131Glu is more of a mixture of the two, and is suggestive of dynamics in which the Glu residue can occasionally push the Phe76 out of the way and reorient towards the Fe center (component 2). The EPR spectrum obtained for Ala131Gln comprises almost entirely component 2, in accord with the crystal structure. The dynamics of this capping Phe residue, as well as its role in modulating substrate/product binding affinities will be addressed in subsequent work.
Despite our inability to obtain a structure of NH2OH-bound Ala131Glu, we were successful in determining this variant's structure with NO bound. Though crystals of Ala131Glu were soaked in an NO-donor (leading to an {FeNO}6), the NO bond distances and angles (Table 3) in some chains agree more closely with an {FeNO}7 configuration.26,31,32 This suggests that perhaps the {FeNO}6 unit was photoreduced upon data collection, as has been proposed for similar heme-nitrosyl crystal structures in the NO-sensing protein nitrophorin.33,34 Given that the bond lengths and angles are also not the same in all chains, it may suggest that more photoreduction occurred at one site than at the other, or that the resolution is not high enough to discriminate between {FeNO}6 and {FeNO}7 intermediates. Additionally, the Fe–N distances are much shorter than anticipated.31 This is also likely a consequence of the modest resolutions of the structures and not a meaningful reflection of the bond distances. Alternative structural techniques such as extended X-ray absorption fine structure (EXAFS) coupled with vibrational studies achieved using either conventional (FTIR or resonance Raman) or nuclear resonance approaches (NRVS) will be required to better probe the structure and electronics of these nitrosyl species. However, the present structures suffice to explore the outer coordination sphere. Case in point, it is clear that Glu131 does not interact with the bound NO, however this may not represent the conformation of the residue when the {FeNO}6 is formed during NH2OH oxidation instead of when exogenous NO is added.
Table 3 Bond distances and angles for substrate-bound crystal structures
|
|
Ala131Glu–NO |
Ala131Gln–NH2OH |
| Fe–N distance in Å (chain A, B, C, D) |
1.50, 1.39, 1.57, 1.78 |
2.70, 2.75, 2.70, 2.70 |
| Fe–N–O angle in deg. (chain A, B, C, D) |
146.36, 152.19, 134.99, 108.40 |
121.22, 133.54, 141.47, 137.43 |
Functional diversity within the cyt P460 family
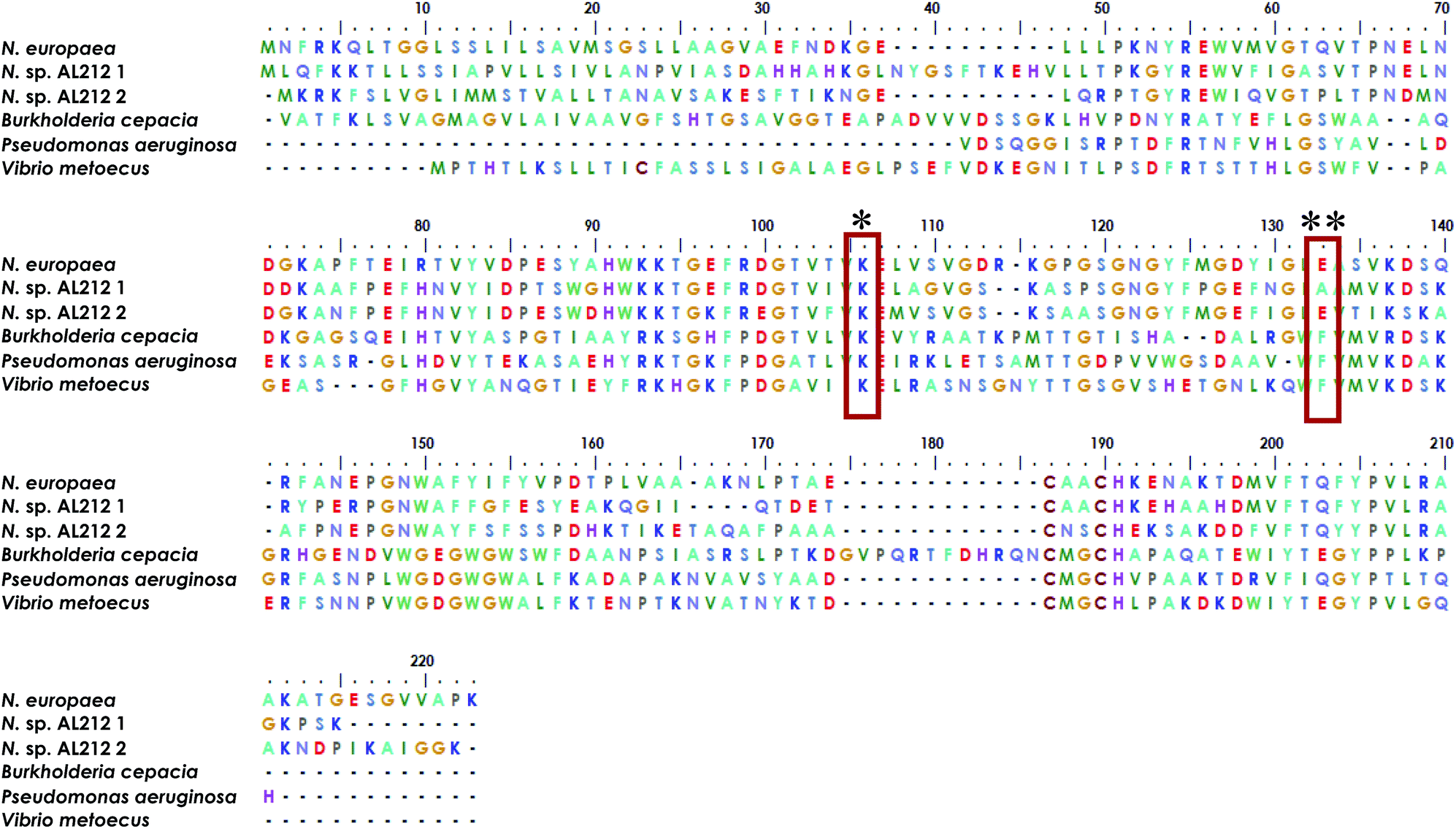
The observation of a wild type cyt P460 that is unable to oxidize NH2OH forces us to reconsider the native function of this enzyme and to consider the possibility of different classes within the cyt P460 family that are tuned toward different substrate specificities. Prior density functional theory analysis showed that π-donation from the Lys N of the hallmark cyt P460 cross-link raises the energy of orbitals associated with the P460 cofactor.20 We postulated that the ability of cyt P460 to directly oxidize Fe-bound NH2OH could be due to the higher energy heme P460 orbitals imposed by the cross-link, which would allow for increased NH2OH character in the singly occupied molecular orbital. This effect, however, does not have to be specific for NH2OH-oxidation. That is, the characteristic heme P460 cross-link defines the oxidative capabilities of the cyt P460 enzyme, and the substrate preference/specificity could be tuned by residues in the outer coordination sphere, such as the residue in position 131, as shown in the present work. This theory accords with the evolutionary trajectory of HAO into oxidative chemistry. HAO belongs to a larger family of multiheme c enzymes, including hydrazine oxidoreductase (HZO), octaheme cytochrome c nitrite reductase (ONR), and pentaheme cytochrome c nitrite reductase (NrfA). It is believed that the presence of additional protein cross-links to the catalytic heme are what distinguish enzymes with oxidative chemistries from those with reductive chemistries.36,37 Comparative homology of currently annotated cyt P460 sequences would suggest distinct classes of cyt P460 which contain different residues (e.g. glutamate/aspartate, alanine, phenylalanine, Fig. 9) in this crucial second sphere position. In fact, the Nitrosomonas sp. AL212 genome contains two cyt P460 sequences (Fig. 9), one that contains Ala131 (the focus of this study) and one that is predicted to have a glutamate in this position (though the latter also contains a CXXCH heme-binding motif different from the CAACH motif observed in the former AL212 cyt P460 and N. europaea cyt P460).38 It is likely that this second cyt P460 behaves in a manner similar to N. europaea cyt P460 and is responsible for some of the N2O produced by the organism, which may be a means by which NH2OH or NO burdens are alleviated.39–41 The function of the presently studied, catalytically-inactive cyt P460 in AL212 is thus mysterious. This may allude to distinct groups of cyt P460 that perform oxidative chemistry but are tuned for different substrates. Of course, we acknowledge that there are many differences among these cyt P460 genes aside from the predicted residue in this specific position, however, this study suggests that cyt P460 enzymes are capable of functions aside from NH2OH oxidation, hence their appearance in the genomes of organisms that are not AOB.18,19 The phylogenetics of the cyt P460 family afford a prolific ground for subsequent studies.
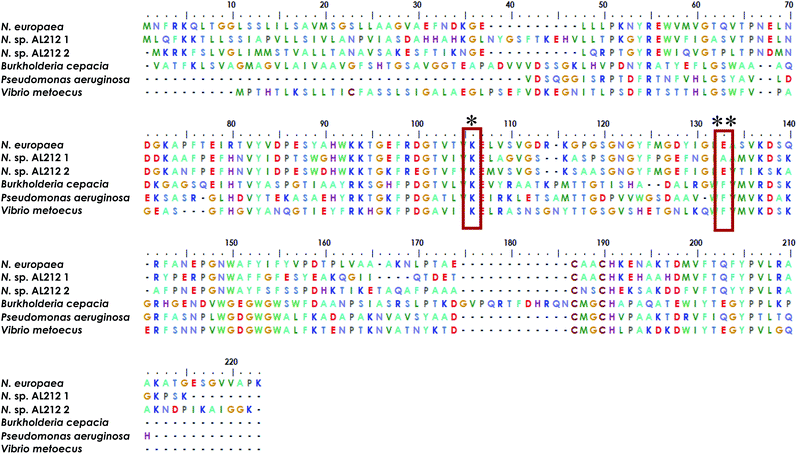 |
| | Fig. 9 Sequence alignment generated using MEGA X software35 showing homology between cyt P460 genes from N. europaea, the two cyt P460 sequences from N. sp. AL212, and genes predicted from mammalian pathogenic bacteria Burkholderia cepacia, Pseudomonas aeruginosa, and Vibrio metoecus. * indicates conserved Lys cross-link, ** indicates the critical second-sphere Ala/Glu/Phe residue. | |
Conclusions
Despite having heme P460 cofactors with identical spectroscopic, electrochemical, and substrate binding properties, N. europaea cyt P460 is competent for NH2OH oxidation and N2O formation, while N. sp. AL212 cyt P460 is inactive. What distinguishes these two proteins is an outer-sphere glutamate in the N. europaea variant, reinforcing the notion that a cofactor alone is insufficient for driving specific reactivity—the protein matrix and, specifically, placement of key residues in the outer sphere of a cofactor, differentiates a protein from an enzyme. The distal cyt P460 glutamate most likely functions as a proton-relay during catalysis, evidenced by the fact that hydrogen-bonding to Fe-bound NH2OH (as seen in the Ala131Gln variant) is insufficient to enable catalysis. Moreover, neither WT N. europaea nor N. sp. AL21 Ala131Glu cyt P460 operate within a pH range at which this distal glutamate is likely to be protonated. Precise details concerning the role of the distal glutamate will be explored in a future, more detailed kinetics and computational study. Our discovery of the existence of cyt P460s without NH2OH oxidation competence suggests the possibility of alternative roles for these enigmatic proteins and may facilitate an explanation for the breadth of organism classes encoding cyt P460s.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Alfred. P. Sloan Foundation (Fellowship to K. M. L.) and the Department of Energy (DOE) Office of Science (Early Career Award DE-SC0013997 to K. M. L.). S. H. M. is supported by the National Science Foundation Graduate Research Program (DGE-1650441). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line was funded by a NIH-ORIP HEI grant (S10 RR029205). The Eiger 16M detector on 24-ID-E beam line was funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This work is also based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation under award DMR-1332208, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award GM-103485 from the National Institute of General Medical Sciences, National Institutes of Health. We thank Mike Fenwick for assistance with X-ray data collection. EPR measurements were performed at the National Institutes of Health (NIH) ACERT with assistance from Boris Dzikhovsky. ACERT is supported by NIH/National Institute of General Medical Sciences (NIGMS) under award number P41 GM103521.
References
- V. Artero, Nat. Energy, 2017, 2, 17131 CrossRef CAS.
- J. W. Jurss, R. S. Khnayzer, J. A. Panetier, K. A. El Roz, E. M. Nichols, M. Head-Gordon, J. R. Long, F. N. Castellano and C. J. Chang, Chem. Sci., 2015, 6, 4954–4972 RSC.
- J. D. Megiatto Jr, D. D. Mendez-Hernandez, M. E. Tejeda-Ferrari, A. L. Teillout, M. J. Llansola-Portoles, G. Kodis, O. G. Poluektov, T. Rajh, V. Mujica, T. L. Groy, D. Gust, T. A. Moore and A. L. Moore, Nat. Chem., 2014, 6, 423–428 CrossRef PubMed.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS PubMed.
- P. Hosseinzadeh, N. M. Marshall, K. N. Chacon, Y. Yu, M. J. Nilges, S. Y. New, S. A. Tashkov, N. J. Blackburn and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 262–267 CrossRef CAS PubMed.
-
K. M. Lancaster, in Molecular Electronic Structures of Transition Metal Complexes I, Springer, 2011, pp. 119–153 Search PubMed.
- K. M. Lancaster, S. Sproules, J. H. Palmer, J. H. Richards and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 14590–14595 CAS.
- S. Takahashi, Y. S. Yeo, Y. Zhao, P. E. O'Maille, B. T. Greenhagen, J. P. Noel, R. M. Coates and J. Chappell, J. Biol. Chem., 2007, 282, 31744–31754 CrossRef CAS PubMed.
- G. Zocher, M. E. Richter, U. Mueller and C. Hertweck, J. Am. Chem. Soc., 2011, 133, 2292–2302 CrossRef CAS PubMed.
- J. D. Caranto and K. M. Lancaster, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8217–8222 CAS.
- A. B. Hooper and K. R. Terry, Biochim. Biophys. Acta, 1979, 571, 12–20 CrossRef CAS.
- P. Cedervall, A. B. Hooper and C. M. Wilmot, Biochemistry, 2013, 52, 6211–6218 CAS.
- A. R. Pearson, B. O. Elmore, C. Yang, J. D. Ferrara, A. B. Hooper and C. M. Wilmot, Biochemistry, 2007, 46, 8340–8349 CrossRef CAS PubMed.
- P. Davis, M. G. Evans and W. C. E. Higginson, J. Chem. Soc., 1951, 0, 2563–2567 RSC.
- I. K. Choi, Y. Liu, Z. Wei and M. D. Ryan, Inorg. Chem., 1997, 36, 3113–3118 CrossRef CAS PubMed.
- S. E. Bari, V. T. Amorebieta, M. M. Gutierrez, J. A. Olabe and F. Doctorovich, J. Inorg. Biochem., 2010, 104, 30–36 CAS.
- A. B. McQuarters, L. E. Goodrich, C. M. Goodrich and N. Lehnert, Z. Anorg. Allg. Chem., 2013, 639, 1520–1526 CrossRef CAS.
- B. O. Elmore, D. J. Bergmann, M. G. Klotz and A. B. Hooper, FEBS Lett., 2007, 581, 911–916 CAS.
- D. J. Bergmann, J. A. Zahn, A. B. Hooper and A. A. DiSpirito, J. Bacteriol., 1998, 180, 6440–6445 CAS.
- M. A. Smith and K. M. Lancaster, Biochemistry, 2018, 57, 334–343 CAS.
- A. C. Vilbert, J. D. Caranto and K. M. Lancaster, Chem. Sci., 2018, 9, 368–379 RSC.
- J. D. Caranto, A. C. Vilbert and K. M. Lancaster, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14704–14709 CAS.
- K. M. Lancaster, J. D. Caranto, S. H. Majer and M. A. Smith, Joule, 2018, 2, 421–441 CrossRef CAS.
- R. E. Berry, M. N. Shokhirev, A. Y. Ho, F. Yang, T. K. Shokhireva, H. Zhang, A. Weichsel, W. R. Montfort and F. A. Walker, J. Am. Chem. Soc., 2009, 131, 2313–2327 CrossRef CAS PubMed.
- E. R. Derbyshire, S. Deng and M. A. Marletta, J. Biol. Chem., 2010, 285, 17471–17478 CrossRef CAS PubMed.
- L. E. Goodrich, F. Paulat, V. K. Praneeth and N. Lehnert, Inorg. Chem., 2010, 49, 6293–6316 CAS.
- W. J. Maalcke, A. Dietl, S. J. Marritt, J. N. Butt, M. S. M. Jetten, J. T. Keltjens, T. R. M. Barends and B. Kartal, J. Biol. Chem., 2014, 289, 1228–1242 CAS.
- A. B. McQuarters, E. J. Blaesi, J. W. Kampf, E. E. Alp, J. Zhao, M. Hu, C. Krebs and N. Lehnert, Inorg. Chem., 2019, 58, 1398–1413 CrossRef CAS PubMed.
- A. Daiber, T. Nauser, N. Takaya, T. Kudo, P. Weber, C. Hultschig, H. Shoun and V. Ullrich, J. Inorg. Biochem., 2002, 88, 343–352 CrossRef CAS PubMed.
- C. Riplinger, E. Bill, A. Daiber, V. Ullrich, H. Shoun and F. Neese, Chemistry, 2014, 20, 1602–1614 CAS.
- N. Lehnert, W. R. Scheidt and M. W. Wolf, Struct. Bonding, 2014, 154, 155–223 CrossRef CAS.
- W. R. Scheidt and M. K. Ellison, Acc. Chem. Res., 1999, 32, 350–359 CAS.
- A. Weichsel, J. F. Andersen, S. A. Roberts and W. R. Montfort, Nat. Struct. Biol., 2000, 7, 551–554 CAS.
- F. A. Walker, J. Inorg. Biochem., 2005, 99, 216–236 CrossRef CAS PubMed.
- S. Kumar, G. Stecher, M. Li, C. Knyaz and K. Tamura, Mol. Biol. Evol., 2018, 35, 1547–1549 CrossRef CAS PubMed.
- M. G. Klotz, M. C. Schmid, M. Strous, H. J. M. O. den Camp, M. S. M. Jetten and A. B. Hooper, Environ. Microbiol., 2008, 10, 3150–3163 CrossRef CAS PubMed.
- D. Haase, B. Hermann, O. Einsle and J. Simon, Mol. Microbiol., 2017, 105, 127–138 CrossRef CAS PubMed.
- Y. Suwa, J. M. Norton, A. Bollmann, M. G. Klotz, L. Y. Stein, H. J. Laanbroek, D. J. Arp, L. A. Goodwin, O. Chertkov, B. Held, D. Bruce, J. C. Detter, J. C. Detter, R. Tapia and C. S. Han, J. Bacteriol., 2011, 193, 5047–5048 CrossRef PubMed.
- L. Y. Stein, Methods Enzymol., 2011, 486, 131–152 CAS.
- O. Perez-Garcia, S. G. Villas-Boas, S. Swift, K. Chandran and N. Singhal, Water Res., 2014, 60, 267–277 CrossRef CAS PubMed.
- L. Y. Stein, Curr. Opin.
Chem. Biol., 2018, 49, 9–15 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, representative trials for Kd and reduction potential determination, UV-vis absorption comparison figure, structural comparison figures. See DOI: 10.1039/c9sc00195f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Sean H.
Majer
,
Sean H.
Majer