ConMedNP: a natural product library from Central African medicinal plants for drug discovery†
Fidele
Ntie-Kang
*abc,
Pascal Amoa
Onguéné
d,
Michael
Scharfe
c,
Luc C.
Owono Owono
e,
Eugene
Megnassan
f,
Luc Meva'a
Mbaze
d,
Wolfgang
Sippl
c and
Simon M. N.
Efange
b
aCEPAMOQ, Faculty of Science, University of Douala, P. O. Box 8580, Douala, Cameroon. E-mail: ntiekfidele@gmail.com; Tel: +237 77915473
bChemical and Bioactivity Information Centre, Department of Chemistry, University of Buea, P. O. Box 63, Buea, Cameroon
cDepartment of Pharmaceutical Sciences, Martin-Luther University of Halle-Wittenberg, Wolfgang-Langenbeck Str. 4, 06120, Halle(Saale), Germany
dDepartment of Chemistry, Faculty of Science, University of Douala, P. O. Box 24157, Douala, Cameroon
eLaboratory for Simulations and Biomolecular Physics, Advanced Teachers Training College, University of Yaoundé I, P.O. Box 47, Yaoundé, Cameroon
fLaboratory of Fundamental and Applied Physics, University of Abobo-Adjamé, Abidjan 02 BP 801, Cote d'Ivoire
First published on 28th October 2013
Abstract
We assess the medicinal value and “drug-likeness” of ∼3200 compounds of natural origin, along with some of their derivatives which were obtained through hemisynthesis. In the present study, 376 distinct medicinal plant species belonging to 79 plant families from the Central African flora have been considered, based on data retrieved from literature sources. For each compound, the optimised 3D structure has been used to calculate physicochemical properties which determine oral availability on the basis of Lipinski's “Rule of Five”. A comparative analysis has been carried out with the “drug-like”, “lead-like”, and “fragment-like” subsets, containing respectively 1726, 738 and 155 compounds, as well as with our smaller previously published CamMedNP library and the Dictionary of Natural products. A diversity analysis has been carried out in comparison with the DIVERSet™ Database (containing 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 651 compounds) from ChemBridge. Our results prove that drug discovery, beginning with natural products from the Central African flora, could be promising. The 3D structures are available and could be useful for virtual screening and natural product lead generation programs.
651 compounds) from ChemBridge. Our results prove that drug discovery, beginning with natural products from the Central African flora, could be promising. The 3D structures are available and could be useful for virtual screening and natural product lead generation programs.
1 Introduction
Phytomedicine is a part of health care systems around the world,1 and its importance is underscored by the fact that natural products (NPs) play an increasingly important role in the drug discovery process.2–4 This is particularly the case when the knowledge of the ethnobotanical uses of medicinal plants is coupled with the bioassay-guided isolation and characterization of active principles contained in plant materials.5 Even though NPs of plant origin often fail the famous Lipinski “drug-like” test,6 they are often rich in stereogenic centres and cover segments of chemical space which are typically not occupied by a majority of synthetic molecules and drugs.7,8 In general, pure natural products contain more oxygen atoms and chiral centres, and have less aromatic atoms on average, when compared with “drug-like” molecules.9 Additionally, the structural and hence physico-chemical properties of the isolated phytochemicals are often fine-tuned by chemical synthesis, leading to “drug-like” molecules with desirable ADME/T (absorption, distribution, metabolism, excretion, and toxicity) properties. These are often referred to as natural product inspired drugs.10–12 Thus, modern drug discovery programs often resort to natural sources to guide the careful design of “drug-like” leads from suitable scaffolds, often by synthetic modifications of the latter.11–13 Additionally, the use of computer-aided drug design (CADD) methods has become a very important part of the drug discovery process. This involves virtual screening (VS) by the docking of huge compound databases against validated drug targets and evaluating the interactions between the potential binders and the receptor site by the use of mathematical scoring methods, followed by the careful selection of virtual hit compounds to be screened by biological assays.14–16 The effectiveness of such methods is often evaluated by the ability of the scoring method to clearly discriminate the active compounds from inactive ones.11,15–19 When this objective is achieved, the number of compounds to undergo biological screening is considerably narrowed down and hence the cost of discovery of a drug drastically reduced.20 Some success stories of natural product drug discovery involving virtual screening have been recorded in the literature.21–23 The adoption of this drug discovery strategy has however necessitated the development of databases of virtual compounds.24–32 In a recent paper, we presented the CamMedNP compound library, containing 3D structures of 1859 NPs and their derivatives,33 which was motivated by the richness of the Cameroonian flora and its great potential as a source of drugs.34,35 In the pursuit of our effort to develop a 3D structural library for NPs from African medicinal plants for virtual screening, this study has now been extended to include NPs derived from medicinal plants from other countries located in the Congo Basin (Burundi, Central African Republic, Chad, Congo, Equatorial Guinea, Gabon, the Democratic Republic of Congo, Rwanda and the Republic of São Tomé é Príncipe). The corresponding 3D structural library has been named ConMedNP and is available in the ESI† (additional files 1–4), and could be virtually screened in drug discovery programs. This compound library could be used by research groups involved in CADD to carry out protein-ligand docking, pharmacophore mining, and VS against validated drug targets. Since these plants have been used traditionally in the treatment of several medical disorders, the aim of VS would be to identify suitable compound scaffolds which could be subjected to further investigation in the search for lead compounds for the treatment of these and other diseases. To the best of our knowledge, a 3D structural library of NPs from the Central African flora has not been previously reported.2 Results and discussion
2.1 Origin and description of plant metabolites
The plant sources from which the 3177 plant metabolites have been isolated or derived are 376 species belonging to 79 families, from the Congo Basin rainforest in Central Africa, 31.60% of the compounds being isolated or derived from plants in Central Africa for the very first time. Most of the compounds were isolated from plants of the Leguminosae (14.38%), Moraceae (9.44%), and Guttiferae (9.40%) families.36 A detailed analysis of the composition of each plant family and general classification of the collected compounds has been published separately.36 It was estimated that the 72.63% of the compounds had not been previously tested by biological assays. Meanwhile the rest of the secondary metabolites have previously shown a wide range of in vitro biological activities, some of which have been described elsewhere.33–372.2 Discussion of property distribution and Lipinski's criteria
It is worth mentioning that the identification of lead compounds often involves the development of compound libraries with a high level of molecular diversity within the limits of significant “drug-like” properties. In addition, Lipinski's “rule” was extracted from chemical libraries from the World Dug Index (WDI) as a criterion to evaluate likely oral bioavailability.38,39 This “rule” omitted the highly valuable class of natural products, since Lipinski had initially postulated that the “Rule of Five” was not respected by NPs. However, NP libraries have been previously analysed comparatively using this “rule” in order to have a rough idea of the extent of “drug-likeness” of a compound library to be used in virtual screening.6 It is on these grounds that Lipinski's criteria38 are often used for the evaluation of “drug-likeness” of compounds within the designed libraries. Thus, Lipinski's “Rule of Five” (ro5) is often considered as a useful filter for the elimination of compounds not likely to be orally available in the early stages of drug discovery protocols.39 In summary, Lipinski's ro5 defines a “drug-like” molecule as one with high likelihood to be orally available, for which the molar weight (MW) ≤ 500 Daltons (Da), the logarithm of the octan-1-ol–water partition coefficient (logPo/w or logP) ≤ 5, the number of hydrogen bond acceptors (HBA) ≤ 10 and the number of hydrogen bond donors (HBD) ≤ 5. An additional rule for the number of rotatable bonds (NRB) is often added to this ro5, such that NRB ≤ 5. An evaluation of “lead-likeness” is often carried out using more stringent criteria, following the “Rule of 3.5” (150 ≤ MW ≤ 350; logPo/w ≤ 4; HBD ≤ 3; HBA ≤ 6),40–43 and “fragment-likeness” using even more stringent criteria, following the “Rule of 2.5” (MW ≤ 250; −2 ≤ logPo/w ≤ 3; HBD < 3; HBA < 6; NRB < 3).44Fig. 1 shows the distribution of the number of violations of the ro5 within the ConMedNP library. Plots of the distribution of these physical properties for the total ConMedNP library, along with the standard subsets are shown in Fig. 2, while a summary of the average values of the physical indicators of the ro5 for the various subsets as well as the number of generated tautomers are given in Table 1. The number of rotatable bonds (NRB) within the ConMedNP library was used as an additional criterion to test for the favourable drug metabolism and pharmacokinetics (DMPK) outcomes, based on the observation that naturally occurring compounds often exhibit a wide range of flexibility, from rigid conformationally constrained molecules to very flexible compounds. It was noted that 50.02% of the compounds within ConMedNP were Lipinski compliant and 79.61% showed < 2 violations (Fig. 1), while the maximum in the distribution of the NRB was between 1 and 2 for the total ConMedNP library, as well as for the “drug-like” and “lead-like” subsets (Fig. 2A). It was also noted that the distribution of MW showed a maximum value between 401 and 500 Da (Fig. 2B), showing a curve similar to those previously reported for other “drug-like” NP libraries in the literature,6,33,45 with about 21% of MW > 500 Da. Meanwhile, the “drug-like”, “lead-like” and “fragment-like” subsets showed maxima respectively in the regions 301–400 Da, 201–300 Da and 101–200 Da. The distribution of the calculated logPo/w values showed a roughly Gaussian shaped curve with a maximum value centred at 3.5 logPo/w units, while the maxima of the “drug-like”, “lead-like” and “fragment-like” subsets were at 2.5, 1.5 and 0.5 units respectively (Fig. 2C). Some exceptionally large logPo/w values (up to > 26 units) were observed and could be attributed to the fact that the training database/algorithm used to calculate logPo/w in MOE may not suit the types and combinations of functional groups found in natural products.6 It is worth mentioning that the logPo/w calculator used in MOE is based on a linear atom type model which takes implicit hydrogen atoms into consideration. This model has been previously validated using 1827 molecules in the training set (r2 = 0.93, RMSE = 0.39), but the reliability of the predictions could be dependent on the chemical space of the initial dataset used in validating the linear model.46,47 It should however be noted that, inspite of this limitation, 67.61% of the compounds currently in the ConMedNP library had logPo/w values < 5 units. The peaks of the HBA and HBD were respectively at 5 acceptors and 1 donor and both curves fell off rapidly to maximum numbers of 70 and 37 respectively (Fig. 1E and F). It was also noted that only 10.13% of the compounds in ConMedNP had HBA > 10 and only about 9.79% had HBD > 5. Further calculated descriptors include the number of heavy atoms (HA, for which the atomic number Z > 1), the logarithm of water solubility (logSwat),48 the molar refractivity (MR),49 the total polar surface area (TPSA),50 and the total hydrophobic surface area (THSA). These distributions are also shown in Fig. 2 for the ConMedNP dataset, as well as for the standard subsets. These results clearly point out the fact that compounds within the “drug-like” subset have interesting polarities and hydrophobicities, which fall within the acceptable limits for favourable bioavailabilities. Additionally, the pairwise comparison displaying the mutual relationship between the MW versus the calculated logPo/w, HBA, HBD and the NRB are specified in Fig. 3. These plots show that the areas with the highest population densities fall within the “Lipinski region of interest” (MW < 500, −2 < logPo/w < 5, HBA < 10 and HBD < 5), and for which NRB < 5.
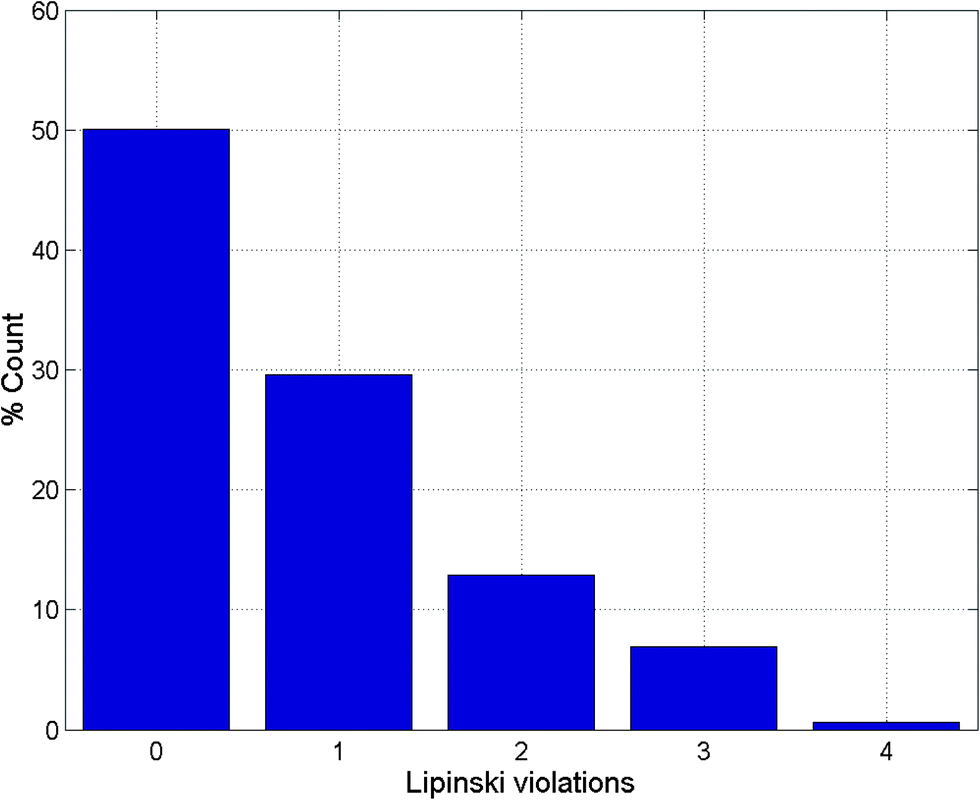 | ||
| Fig. 1 Histogram of Lipinski violations as a percentage of the ConMedNP dataset. | ||
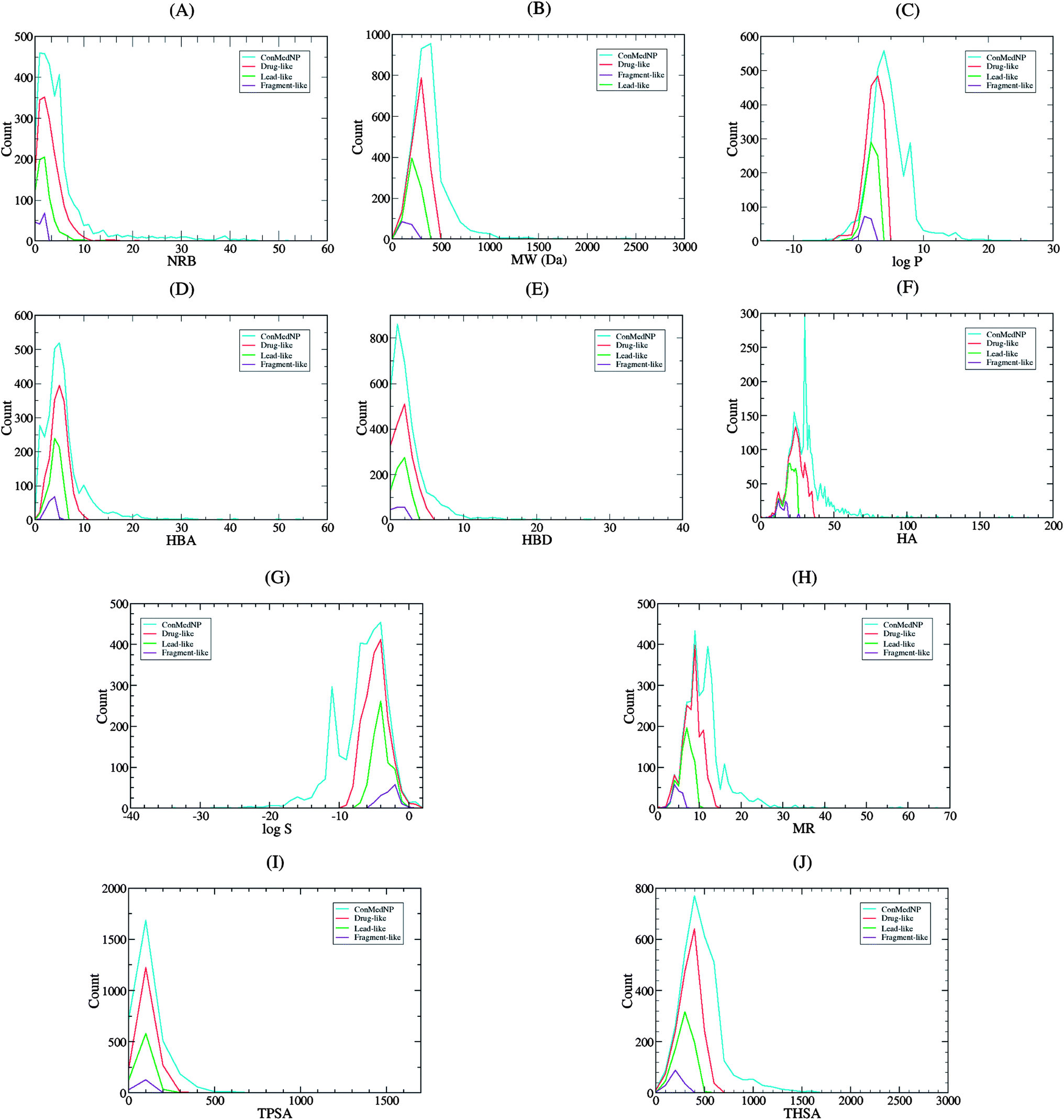 | ||
| Fig. 2 Graph distribution of features that determine “drug-likeness” for the total, “drug-like”, “lead-like” and “fragment-like” subsets derived from ConMedNP library (A, B, C, D, E, F, G, H, I and J). Comparative distribution curves of the NRB, MW, logP, HBA, HBD, HA, logS, MR, TPSA and THSA, respectively for the 3177 compounds currently in ConMedNP (cyan), alongside the “drug-like” (red), “lead-like” (green) and “fragment-like” (violet) subsets. For subfigures B, C, G, H, I and J, the x-axis label is the lower limit of binned data, e.g. 0 is equivalent to 0 to 100. | ||
| Library name | Library size | Totaumers | MW (Da) | log P |
HBA | HBD | NRB |
|---|---|---|---|---|---|---|---|
|
a MW, mean of molar weight; logP, mean of logarithm of the calculated octan-1-ol–water partition coefficient; HBA, mean number of hydrogen bond acceptors; HBD, mean number of hydrogen bond donors; NRB, mean number of rotatable bonds.
|
|||||||
| ConMedNP | 3177 | 7838 | 426.70 | 4.18 | 5.85 | 2.39 | 5.31 |
| Drug-like | 1726 | 3900 | 326.16 | 2.87 | 4.97 | 1.79 | 2.96 |
| Lead-like | 738 | 1610 | 269.58 | 2.48 | 4.17 | 1.49 | 2.01 |
| Fragment-like | 155 | 355 | 192.12 | 1.74 | 3.31 | 1.08 | 1.14 |
| CamMedNP | 1859 | 5286 | 421.63 | 4.07 | 6.00 | 2.40 | 5.51 |
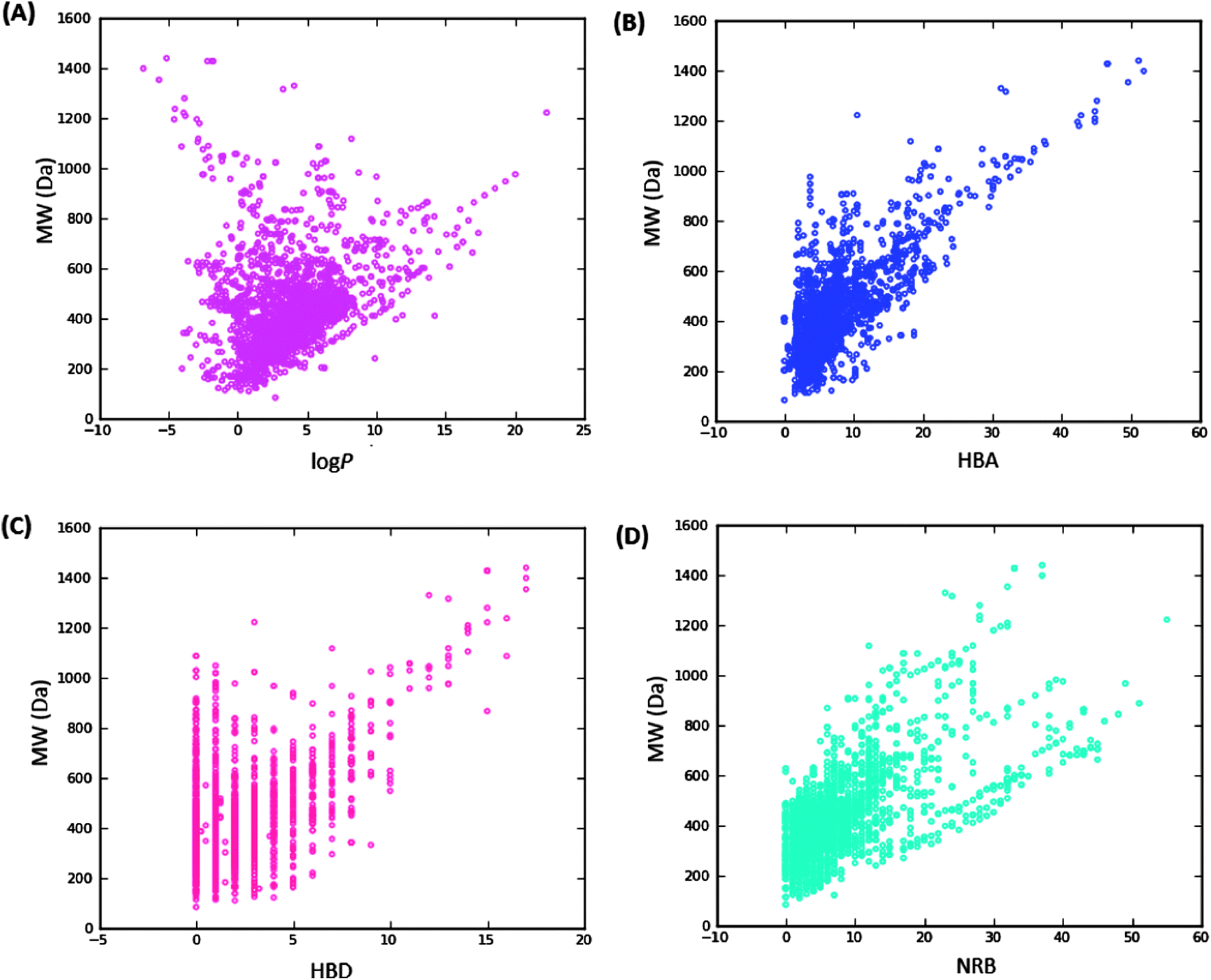 | ||
| Fig. 3 Pairwise comparison of mutual relationships between molecular descriptors: A = the distribution of the calculated logPo/wversus MW, B = HBA versus MW, C = HBD versus MW and D = NRB versus MW. | ||
2.3 Comparison with the Dictionary of Natural Products and the CamMedNP library
For our ConMedNP dataset, the distributions of the compound MWs, lipophilicity (logPo/w), HBA, and HBD have been calculated and used to compare the “drug-likeness” of the ConMedNP library with 126140 compounds from the Dictionary of Natural Products (DNP),51 which have been previously analysed, and retrieved from the literature.6 This comparison has been carried out, side by side, with the same properties for our previously published CamMedNP library33 and shown in Fig. 4. In these histograms, we show only data that falls within the “Lipinski region of interest” (MW < 500, −2 < logPo/w < 5, HBA < 10, and HBD < 5), and the values are expressed as a percentage count of their respective datasets. In all cases the distributions of ConMedNP were enhanced for the Lipinski properties (peaks of the distributions moved to more “drug-like” properties) when compared to the DNP. This is particularly noticeable for 301 ≤ MW ≤ 500, 3 ≤ logPo/w ≤ 5, 4 ≤ HBA ≤ 7 and 1 ≤ HBD ≤ 3. The MW distribution of ConMedNP peaks at 401–500 Da, while those of the other two datasets peak at 301–400 Da (Fig. 4A). Below the range 301 ≤ MW ≤ 500, the percentages were reduced for the ConMedNP when compared to the DNP. This same observation is true for logPo/w < 3, HBA < 4 and HBD < 1. This improved profile for MW is exactly what is desirable for a more “drug-like library”, according to Lipinski's criteria. The proportions of CamMedNP and ConMedNP which satisfy Lipinski's MW property (<500 Da) were respectively 77.94% and 78.91%, compared to 73.04% for the DNP. The distribution maxima for calculated logPo/w (Fig. 4B) were similar for our two datasets, appearing between logPo/w values of 3–4 (for both CamMedNP and ConMedNP), while DNP gives a value between 2 and 3. A similar trend was observed for the MW distribution. This showed an enhancement of 13.89% for MW values between 301 and 500 Da of ConMedNP over the DNP, comparable with the enhancement of 11.19% of CamMedNP over the DNP. The enhancements of our two datasets over the DNP for logPo/w values in the range 2 ≤ logPo/w ≤ 5 were calculated to be 13.15% and 13.11 respectively for CamMedNP and ConMedNP. For HBA (Fig. 4C), ConMedNP showed an improvement of 11.86% over the DNP within the range 4 ≤ HBA ≤ 7, which was significantly lower than the corresponding enhancement of 18.65%, which the CamMedNP library had shown over the DNP.28 For the HBD, it was noticed that our two datasets (CamMedNP and ConMedNP) both showed enhancements over the DNP within the range 1 ≤ HBD ≤ 3, with respective percentages of 10.39% and 11.50%. The peak of the distribution for the HBA for both CamMedNP and ConMedNP is at 5 acceptors (respectively 18.45% and 16.30%), while that of the DNP is at 4 acceptors (14.15%). This gives us a significant increase in 6 or 7 acceptors for the CamMedNP library and a corresponding 4 or 5 acceptors for the ConMedNP library, when compared to the DNP (Fig. 4C). Similarly, the peak of the distribution for the HBD for the ConMedNP is at 1 acceptor (27.07%) with a significant increase in 5 or 6 donors as compared to the DNP (Fig. 4D). The overall summary of the four Lipinski parameters for the three datasets thus reveals that both CamMedNP and ConMedNP libraries are more “drug-like” than the DNP. This is an indication that the chances of finding “lead-like” molecules with improved DMPK properties within these libraries are quite significant. Data is currently available on the various plant sources from which the compounds in ConMedNP were derived, their ethnobotanical uses, geographical regions of collection of plant material, dates of collection, literature sources, known experimentally measured biological activities of compounds and sample availability. The ConMedNP library is constantly being updated; meanwhile a MySQL platform to facilitate the searching of the ConMedNP database and ordering of samples is being set up by our group. However, 3D structures of the compounds, as well as their physico-chemical properties that were used to evaluate “drug-likeness”, for the total library, as well as the “drug-like”, “lead-like” and “fragment-like” subsets can be freely downloaded as additional files accompanying this publication (respectively additional files 1–4).
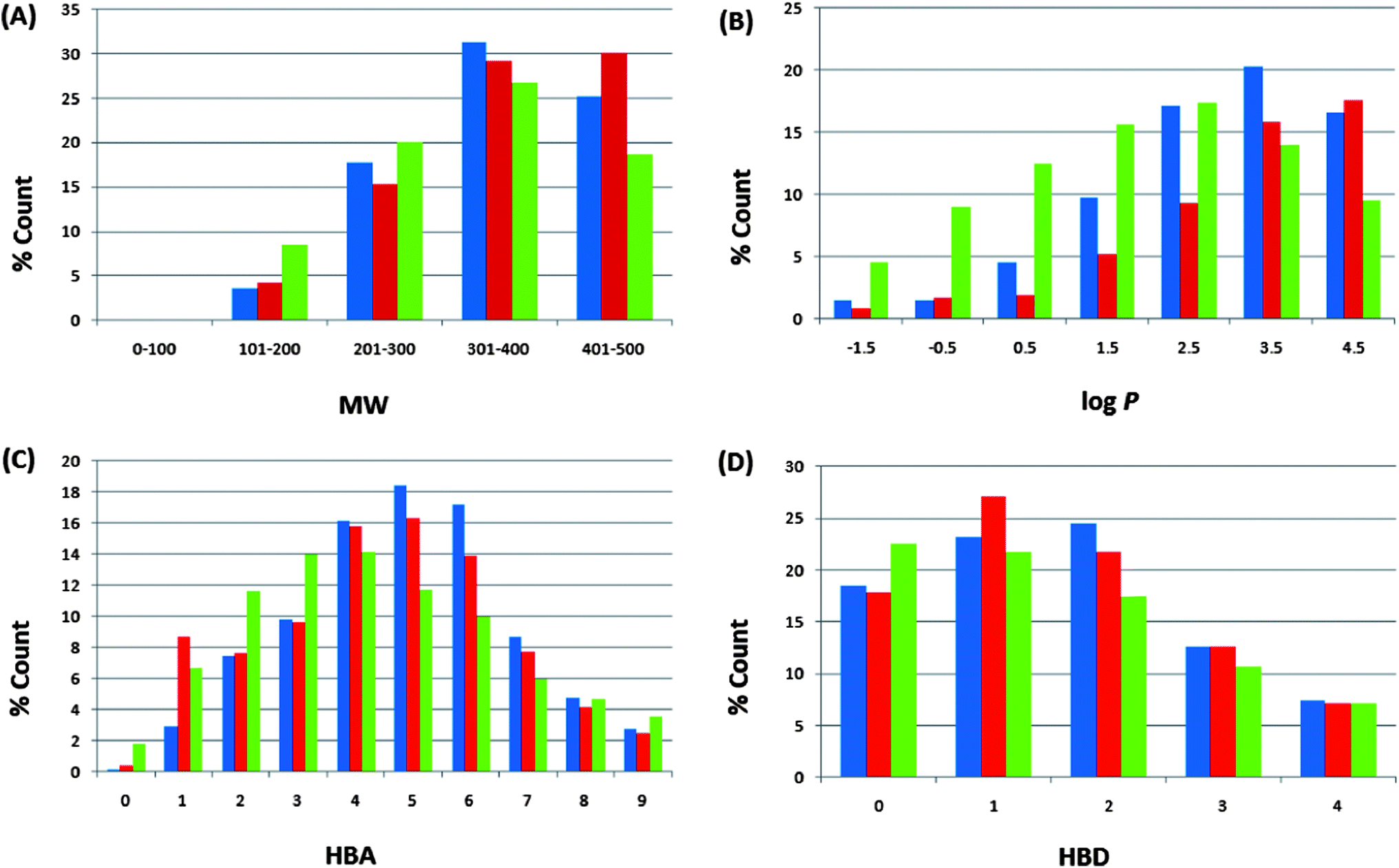 | ||
| Fig. 4 Comparison of property distribution for the three datasets (CamMedNP, ConMedNP and the DNP): A = molar weight, B = logarithm of octan-1-ol–water partition coefficient, C = number of hydrogen bond acceptors, and D = number of hydrogen bond donors. DNP in green, ConMedNP in red and CamMedNP in blue. For subfigure B, the x-axis label is the lower limit of binned data, e.g. −2 is equivalent to −2 to −1. | ||
2.4 Diversity analysis
In order to reduce redundancy and enhance the coverage of biological activity and chemical space, a dataset for virtual screening must have the requirement of diversity. In this case, we carried out a simple molecular descriptor comparison with a relatively larger known diverse library (the DIVERSet™ database, containing 48651 compounds) from the ChemBridge Corporation.52 Histograms showing the calculated descriptors (MW, HBA, HBD, logPo/w, NR, NRB, NN, NO, NRB and TPSA) are shown in Fig. 5 for ConMedNP (in light green) and the ChemBridge dataset (in red). The regions shown in dark green represent regions of intersection. The MW of the ConMedNP dataset stretches well beyond 1000 Da, while that of the ChemBridge dataset is restricted to the range 200 ≤ MW ≤ 500 Da. This observation could be explained by the complexity and large sizes of some of the compounds within the natural product library. The large proportion of very large and complex NPs in ConMedNP, could also explain the mean molar weight (=427 Da), when compared to those of the standard “drug-like”, “lead-like” libraries and typical drugs (=310 Da).53 This same explanation holds for the trend which is observed in the distributions of logPo/w, HBD, NCC, NO, NRB, NR, TPSA and HBA for ConMedNP, when compared with the ChemBridge dataset. It was generally observed that the ConMedNP dataset covers another physicochemical space than the ChemBridge Diversity dataset. Principal component analysis (PCA) was as well used as a means of comparing the extent of diversity of the two datasets. This consists in reducing the dimensionality of the calculated descriptors by linearly transforming the data, by calculating a new and smaller set of descriptors, which are uncorrelated and normalised (mean = 0, variance = 1). The PCA scatter plot of the previously calculated physicochemical properties of the ConMedNP (light green) and ChemBridge Diverset database (red), shown in Fig. 6, is a visual representation of the molecules in the respective datasets, as described by the 3 selected principal components (PC1, PC2 and PC3). Each point shown corresponds to a molecule, the spread of the points representing the diversity of the respective datasets. The first three principal components (PCs) explain 83% (ConMedNP) and 64% (ChemBridge) of the variance of the individual datasets. The larger number of outliers in the case of the ConMedNP dataset (away for the centre and towards the sides of the cube) indicates a wider sampling of the chemical space compared to the ChemBridge Diverset collection.
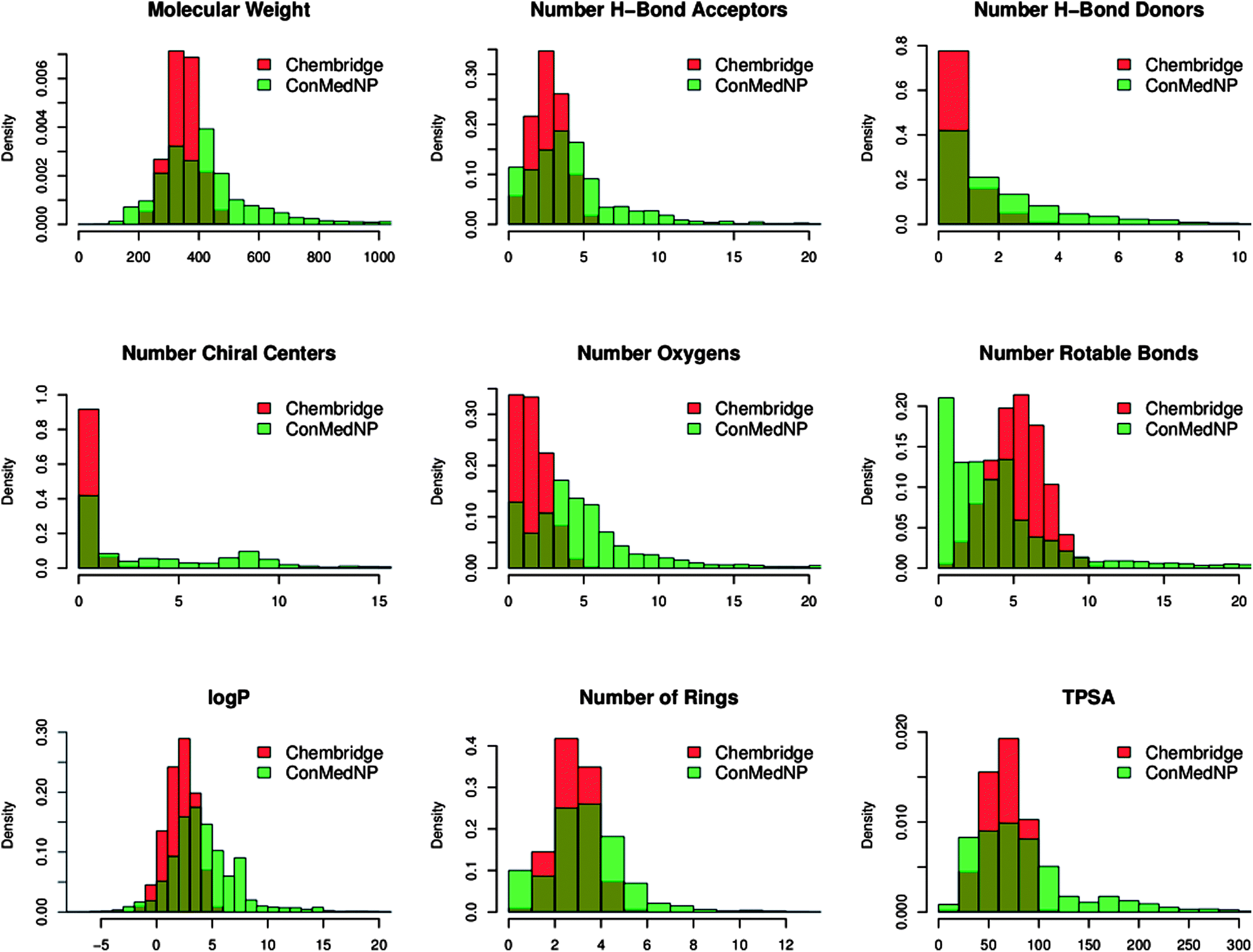 | ||
| Fig. 5 A simple descriptor-based comparison of the ConMedNP database and the ChemBridge Diversity database. Comparison of typical physicochemical property distributions (MW, HBA, HBD, NCC, NO, NRB, logP, NR and TPSA) in the ConMedNP (green) and ChemBridge Diverset (red) database. All histograms and scatterplots were generated with the R software.72 | ||
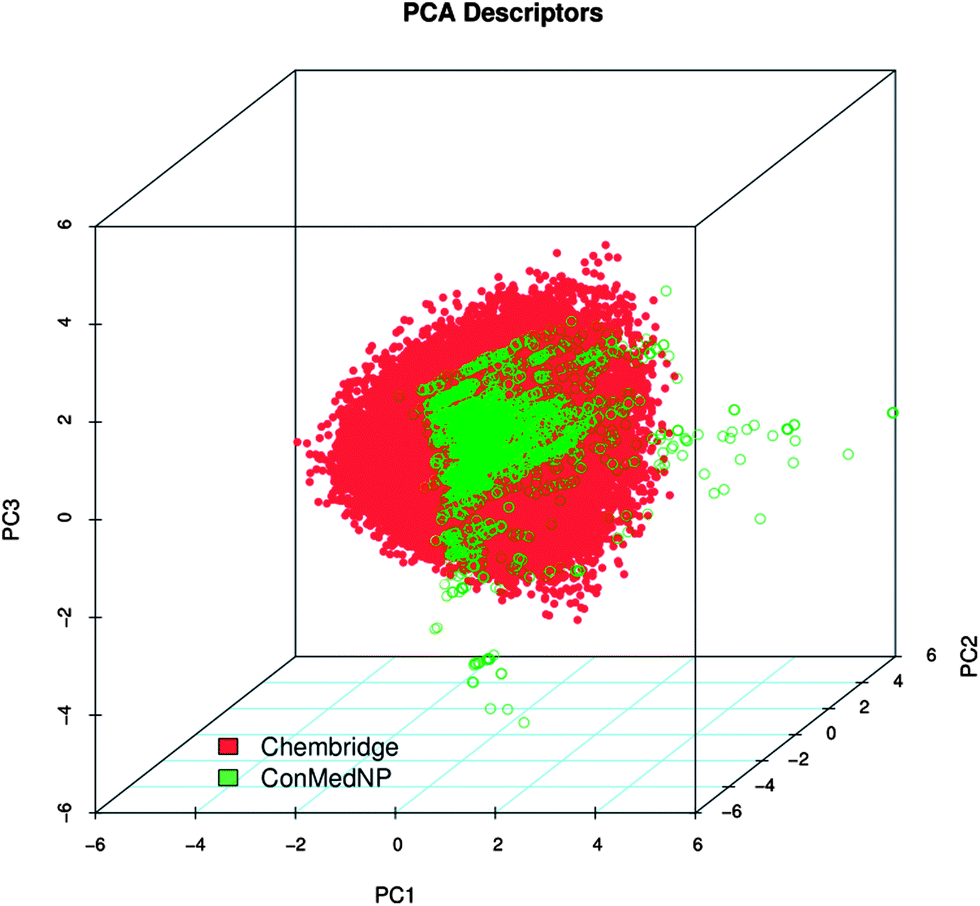 | ||
| Fig. 6 A principal component analysis (PCA) plot, showing the comparison of the chemical space defined by the NPs in ConMedNP (green) and the chemical space represented by NPs in the ChemBridge diversity (red) databases. | ||
2.5 Pharmacokinetic profiling
Drug metabolism and pharmacokinetic property prediction was carried out using molecular descriptors computed with the QikProp software.54 QikProp computes physically relevant descriptors, and uses them to perform ADMET predictions. An overall ADME-compliance score – drug-likeness parameter (indicated by #stars), was used to assess the pharmacokinetic profiles of the compounds within the CamMedNP library. The #stars parameter indicates the number of property descriptors computed by QikProp that fall outside the optimum range of values for 95% of known drugs. The methods implemented were developed by Jorgensen and Duffy.55–57 Among the 24 computed molecular descriptors related to the absorption, distribution, metabolism, excretion and toxicity of drugs are; the logarithm of blood–brain partition coefficient,58–60 aqueous solubility,55,57 logarithm of predicted IC50 for blockage of HERG K+ channels,61,62 predicted binding to human serum albumin,63 predicted permeability across Caco-2 cells in Boehringer–Ingelheim scale (in nm s−1),64–66 the predicted apparent Madin–Darby canine kidney (MDCK) cell permeability (in nm s−1),65 the predicted index of cohesion interaction in solids,57 the predicted skin permeability,67,68 and the number of likely metabolic steps the compounds can undergo. This survey demonstrated that about 45% of the compounds within the ConMedNP database are compliant, having properties which fall within the range of ADME properties of 95% of currently known drugs, while > 69% of the compounds have ≤ 2 violations. Moreover, about 73% of the compounds within the corresponding “drug-like” subset showed compliance. A summary of the percentage compliances for 14 selected ADMET-related molecular descriptors is shown in Table 2, while a detailed analysis of these results has been published separately.69| Library name | logB/Ba |
BIPcaco-2b (nm s−1) | S mol c (Å2) | S mol,hfob d (Å2) | V mol e (Å3) | logSwatf (S in mol L−1) |
logKHSAg |
|---|---|---|---|---|---|---|---|
| Total library | 88.53 | 37.28 | 90.36 | 91.32 | 91.03 | 72.46 | 81.01 |
| Drug-like | 99.35 | 43.99 | 99.35 | 99.82 | 98.99 | 90.39 | 99.35 |
| Lead-like | 99.72 | 53.70 | 99.72 | 100.00 | 99.72 | 99.31 | 99.59 |
| Fragment-like | 100.00 | 33.11 | 95.45 | 100.00 | 92.21 | 97.40 | 98.05 |
| Library name | MDCKh | Indcohi | Globj | ro3k | logHERGl |
logKpm |
# metabn |
|---|---|---|---|---|---|---|---|
| a Logarithm of predicted blood/brain barrier partition coefficient (range for 95% of drugs: −3.0 to 1.0). b Predicted apparent Caco-2 cell membrane permeability in Boehringer–Ingelheim scale, in nm s−1 (range for 95% of drugs: < 5 low, > 500 high). c Total solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 300–1000 Å2). d Hydrophobic portion of the solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 0–750 Å2). e Total volume of molecule enclosed by solvent-accessible molecular surface, in Å3 (probe radius 1.4 Å) (range for 95% of drugs: 500–2000 Å3). f Logarithm of aqueous solubility (range for 95% of drugs: −6.0 to 0.5). g Logarithm of predicted binding constant to human serum albumin (range for 95% of drugs: −1.5 to 1.5). h Predicted apparent MDCK cell permeability in nm s−1 (< 25 poor, > 500 great). i Index of cohesion interaction in solids (0.0 to 0.05 for 95% of drugs). j Globularity descriptor (0.75 to 0.95 for 95% of drugs). k Percentage compliance to Jorgensen’s Rule of Three. l Predicted IC50 value for blockage of HERG K+ channels (concern < −5). m Predicted skin permeability (−8.0 to −1.0 for 95% of drugs). n Number of likely metabolic reactions (range for 95% of drugs: 1–8). | |||||||
| Total library | 47.14 | 94.92 | 89.88 | 43.57 | 58.35 | 92.09 | 81.52 |
| Drug-like | 59.61 | 99.14 | 97.70 | 73.52 | 63.62 | 95.93 | 91.89 |
| Lead-like | 59.86 | 100.00 | 97.81 | 93.56 | 76.03 | 97.53 | 96.44 |
| Fragment-like | 63.33 | 100.00 | 92.21 | 100.00 | 100.00 | 98.05 | 92.21 |
2.6 Searching for most common substructures
The most common substructure selection (MCSS) panel for compound selection (Fig. 7) is based on substructures that can be synthetically combined and are common in “drug-like” molecules, allowing a direct selection and identification of compounds containing such substructures. The panel highlights the large diversity of the rings present in the NPs of ConMedNP.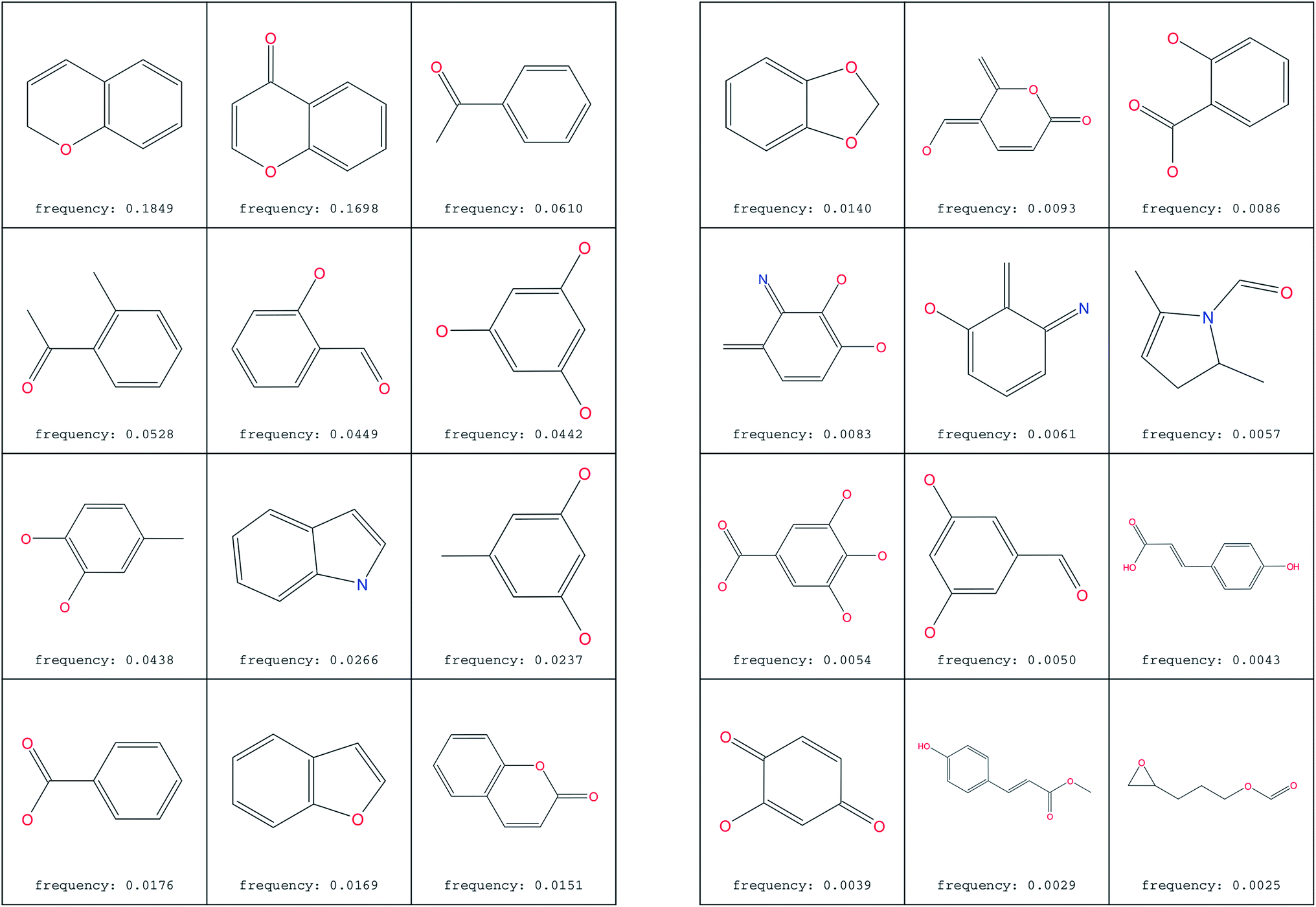 | ||
| Fig. 7 MCSS panel in ConMedNP, featuring the most common cyclic structures included in the database. | ||
2.7 MACCS key clustering
A clustering with a threshold of 70% Tanimoto similarity within a cluster leads to 1374 clusters (of 2954 compounds) An identical clustering of 2954 randomly selected compounds from Chembridge lead to 2859 clusters. But the high number of clusters are not surprising, because we used the “DiverseSet” of the Chembridge database.2.8 Utility and compound availability
The 3D structures of the compounds, as well as their computed physico-chemical properties that were used to predict “drug-likeness” and pharmacokinetic profiles, can be freely downloaded as a ESI† accompanying this publication (additional files 1–4). In addition, information about compound sample availability can be obtained on request from the authors of this paper or from the pan-African Natural Products Library (p-ANAPL) project, which has a mandate to make a physical collection of NPs from plants growing in Africa and make them available for biological screening.70,713 Experimental methods
3.1 Data sources
The plant sources, geographical collection sites, chemical structures of pure compounds, were retrieved from literature sources comprising of 31 PhD theses and journal articles, with references ranging from 1971 to 2013. This constitutes a total of 657 journal references, 2 textbook chapters, as well as 10 unpublished conference presentations (from personal communication with the authors). A full list of journals consulted is given in the ESI† (additional file 5). In each journal webpage, an author name search was carried out, as well as institution names and country name searches. The articles hit were sorted out with respect to geographical location of the plant material harvested. Articles reporting compounds derived from plants harvested from the 10 countries under this study were selected, data retrieved from them and compiled on an excel sheet for further analysis.3.2 Generation of 3D models, optimization and calculation of molecular descriptors
Based on the reported chemical structures of the NPs, all 3D molecular structures were generated using the builder module and graphical user interface of the MOE software73 running on a Linux workstation with a 3.5 GHz Intel Core2 Duo processor. Energy minimization was subsequently carried out using the MMFF94 force field74 until a gradient of 0.01 kcal mol−1 was reached. The derived 3D structures of the compounds were then saved as.mol2 files subsequently included into a MOE database (.mdb) file and converted to other file formats (.sdf,.mol,.mol2 and .ldb), which are suitable for use in several virtual screening workflow protocols. The MW, NRB, logPo/w, logS, HBA, HBD, THSA, TPSA, NO, NCC, NR and number of Lipinski violations were calculated using the molecular descriptor calculator included in the QuSAR module of the MOE package,73 while molecular descriptors related to drug metabolism and pharmacokinetic profiles were computed using the QikProp software54 running in normal mode. The ChemBridge Diverset dataset (48651 compounds) was downloaded from the official ChemBridge webpage.52 The LibMCS program of JKlustor75 was used for maximum common substructure clustering of the ConMedNP database. In the MCSS search, only structures with MW ≤ 600 were included, since MCSS clustering is only feasible on small molecules. This means, only 2785 of the compounds of the ConMedNP were analyzed for MCSS. The compounds were fragmented using the RECAP algorithm.76 It is noteworthy that the provided 3D structures are those published in the literature, based on NMR and other spectroscopic data. In order to facilitate the virtual screening procedure for inexperienced modelers, the preliminary treatment of input ligand structures by assignment of biologically relevant protonation states at physiological pH (5 ≤ pH ≤ 9.5) and tautomer generation was also carried out. Group I metals in simple salts were disconnected, strong acids were deprotonated, strong bases protonated, while topological duplicates and explicit hydrogens were added. For extremely large NPs, only the largest molecular fragments were retained and a cutoff of up to 100 tautomers per molecular structure was taken.
4 Conclusions
Virtual screening workflows often involve docking a compound library toward the binding site of a target receptor and using scoring functions and binding free energy calculations to identify putative binders. The availability of 3D structures of the compounds to be used for docking is therefore of utmost importance. To the best of our knowledge, ConMedNP represents the largest “drug-like”, “lead-like”, “fragment-like” and diverse collection of 3D structures of NPs from the Central African forest, readily available for download. This dataset has the advantages that it is relatively small, “drug-like”, diverse and easily assessable for virtual screening purposes. Thus the availability of such structures within ConMedNP, as well as their calculated physico-chemical properties and indicators of “drug-likeness” will facilitate the drug discovery process from leads that have been identified from Central African medicinal plants. A typical example for a drug discovery effort for a wide range of diseases beginning from a Chinese natural products chemical library has been recently described.77 Additionally, fragment-based ligand and drug discovery predominantly employs small sp2-rich compounds covering well-explored regions of chemical space.78 Natural-product-derived fragments with high structural diversity, which resemble natural scaffolds and are rich in sp3-configured centres are often employed in the design of new chemical entities to be employed as drugs.78–80 Thus, the small “fragment-like” subset of 155 compounds, derived from ConMedNP, could serve as a suitable base line for fragment-based drug design projects.Acknowledgements
Financial support is acknowledged from the German Academic Exchange Service (DAAD) to FNK for his stay in Halle, Germany for part of his PhD and from the ICTP through the ICTP/OEA-AC71 program. Computational facilities were jointly provided by the Molecular Simulations Lab, Department of Chemistry, University of Buea and the Department of Pharmaceutical Sciences, Martin-Luther University of Halle-Wittenberg, Halle (Saale), Germany. The assistance of Dr Philip N. Judson (Chemical and Bioactivity Information Centre, Leeds, UK) is acknowledged for proofreading the draft manuscript.Notes and references
- O. Akerele, Herbalgram, 1993, 28, 13 Search PubMed.
- O. Potterat and M. Hamburger, Drug discovery and development with plant-derived compounds, in Progress in drug research: natural compounds as drugs, ed. F. Petersen and R. Amstutz, Basel: Birhäusser, Verlag AG, 2008, pp. 45–118 Search PubMed.
- Y. W. Chin, M. J. Balunas, H. B. Chai and A. D. Kinghorn, AAPS J., 2006, 8(2), E239 CAS.
- J. W. H. Li and J. C. Vederas, Science, 2009, 325, 161 CrossRef PubMed.
- B. R. Holmstedt and J. G. Bruhn, Ethnopharmacology – a challenge, in Ethnobotany, Evolution of a Discipline, ed. R. E. Schultes and S. von Reis, Portland, Oregon, Dioscorides Press, 1995, pp. 338–342 Search PubMed.
- R. J. Quinn, A. R. Carroll, M. B. Pham, P. Baron, M. E. Palframan, L. Suraweera, G. K. Pierens and S. Muresan, J. Nat. Prod., 2008, 71, 464 CrossRef CAS PubMed.
- S. Wetzel, A. Schuffenhauer, S. Roggo, P. Ertl and H. Waldmann, Chimia Int. J. Chem., 2007, 61, 355 CrossRef CAS.
- K. Grabowski, K.-H. Baringhaus and G. Schneider, Nat. Prod. Rep., 2008, 25, 892 RSC.
- K. Grabowski and G. Schneider, Curr. Chem. Biol., 2007, 1, 115 CAS.
- S. M. N. Efange, Natural products: a continuing source of inspiration for the medicinal chemist, in Advances in Phytomedicine, Vol. 1, Ethnomedicine and Drug Discovery, ed. M. M. Iwu and J. C. Wootton, Amsterdam, The Netherlands, Elsevier Science, 2002, pp. 61–69 Search PubMed.
- A. L. Harvey, Drug Discovery Today, 2008, 13, 894 CrossRef CAS PubMed.
- D. J. Newman, J. Med. Chem., 2008, 51, 2589 CrossRef CAS PubMed.
- L. O. Haustedt, C. Mang, K. Siems and H. Schiewe, Curr. Opin. Drug Discovery Dev., 2006, 9, 445 CAS.
- T. I. Oprea and H. Matter, Curr. Opin. Chem. Biol., 2004, 8, 349 CrossRef CAS PubMed.
- C. Sangma, D. Chuakheaw, N. Jongkon, K. Saenbandit, P. Nunrium, P. Uthayopas and S. Hannongbua, Comb. Chem. High Throughput Screening, 2005, 8(5), 417 CrossRef CAS.
- F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206 CrossRef CAS PubMed.
- G. Klebe, Drug Discovery Today, 2006, 11, 580 CrossRef CAS PubMed.
- H. Kubinyi, Curr. Opin. Drug Discovery Dev., 1998, 1, 4–15 CAS.
- C. Bissantz, G. Folkers and D. Rognan, J. Med. Chem., 2000, 43, 4759 CrossRef CAS PubMed.
- J. A. DiMasi, R. W. Hansen and H. G. Grabowsk, J. Health Econ., 2003, 22, 151 CrossRef.
- J. M. Rollinger, H. Stuppner and T. Langer, Prog. Drug Res., 2008, 65, 213 Search PubMed.
- J. M. Rollinger, S. Haupt, H. Stuppner and T. Langer, J. Chem. Inf. Comput. Sci., 2004, 44(2), 480 CrossRef CAS PubMed.
- J. Shen, X. Xu, F. Cheng, H. Liu, X. Luo, J. Shen, K. Chen, W. Zhao, X. Shen and H. Jiang, Curr. Med. Chem., 2003, 10(21), 2327 CrossRef CAS.
- M. Fullbeck, E. Michalsky, M. Dunkel and R. Preissner, Nat. Prod. Rep., 2006, 23, 347 RSC.
- M. Dunkel, M. Fullbeck, S. Neumann and R. Preissner, Nucleic Acids Res., 2006, 34, D678 CrossRef CAS PubMed.
- X. Qiao, T. Hou, W. Zhang, S. Guo and X. Xu, J. Chem. Inf. Comput. Sci., 2002, 42, 481 CrossRef CAS PubMed.
- J. Lei and J. Zhou, J. Chem. Inf. Comput. Sci., 2002, 42, 742 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2004, 21, 1 RSC.
- X. Lucas, C. Senger, A. Erxleben, B. A. Grüning, K. Döring, J. Mosch, S. Flemming and S. Günther, Nucleic Acids Res., 2013, 41, D1130 CrossRef CAS PubMed.
- P. Daisy, S. K. Singh, P. Vijayalakshmi, C. Selvaraj, M. Rajalakshmi and S. Suveena, Bioinformation, 2011, 6(4), 167 CrossRef.
- D. Pitchai, R. Manikkam, S. R. Rajendran and G. Pitchai, Bioinformation, 2010, 5(2), 43 CrossRef.
- C. Y. Chen, PLoS One, 2011, 6(1), e15939 CAS.
- F. Ntie-Kang, J. A. Mbah, L. M. Mbaze, L. L. Lifongo, M. Scharfe, J. Ngo Hanna, F. Cho-Ngwa, P. A. Onguéné, L. O. O. Owono, E. Megnassan, W. Sippl and S. M. N. Efange, BMC Complementary Altern. Med., 2013, 13, 88 CrossRef PubMed.
- V. Kuete and T. Efferth, Front. Pharmacol., 2010, 1, 123 CrossRef PubMed.
- V. Kuete, Planta Med., 2010, 76, 1479 CrossRef CAS PubMed.
- D. Zofou, F. Ntie-Kang, W. Sippl and S. M. N. Efange, Nat. Prod. Rep., 2013, 30, 1098 RSC.
- F. Ntie-Kang, L. L. Lifongo, L. M. Mbaze, N. Ekwelle, L. C. O. Owono Owono, E. Megnassan, P. N. Judson, W. Sippl and S. M. N. Efange, BMC Complementary Altern. Med., 2013, 13, 147 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 253 CrossRef.
- S. J. Teague, A. M. Davis, P. D. Leeson and T. I. Opea, Angew. Chem., Int. Ed., 1999, 38, 3743 CrossRef CAS.
- M. M. Hann and T. I. Oprea, Curr. Opin. Chem. Biol., 2004, 8, 255 CrossRef CAS PubMed.
- T. I. Oprea, J. Comput.-Aided Mol. Des., 2002, 16, 325 CrossRef CAS.
- G. Schneider, Curr. Med. Chem., 2002, 9, 2095 CrossRef CAS.
- M. L. Verdonk, J. C. Cole, M. L. Hartshorn, C. W. Murray and R. D. Taylor, Proteins, 2003, 52, 609 CrossRef CAS PubMed.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS PubMed.
- S. A. Wildman and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1999, 39, 868 CrossRef CAS.
-
P. Labute, MOE logP (octanol–water) model. Available as a source code in MOE, 1998, unpublished.
- T. J. Hou, K. Xia, W. Zhang and X. J. Xu, J. Chem. Inf. Comput. Sci., 2004, 44, 266 CrossRef CAS PubMed.
- P. Labute. MOE molar refractivity model. Available as a source code in MOE 1998, unpublished.
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714 CrossRef CAS PubMed.
- Chapman and Hall/CRC Press, Dictionary of Natural Products on CD-Rom, London, 2005 Search PubMed.
- ChemBridge Corporation, http://chembridge.com/.
- V. Khanna and S. Ranganathan, J. Cheminf., 2011, 3, 30 CAS.
- QikProp, version 3.4, Schrödinger, Inc., New York, 2011 Search PubMed.
- W. L. Jorgensen and E. M. Duffy, Adv. Drug Delivery Rev., 2002, 54, 355 CrossRef CAS.
- E. M. Duffy and W. L. Jorgensen, J. Am. Chem. Soc., 2000, 122, 2878 CrossRef CAS.
- W. L. Jorgensen and E. M. Duffy, Bioorg. Med. Chem. Lett., 2000, 10, 1155 CrossRef CAS.
- J. M. Luco, J. Chem. Inf. Comput. Sci., 1999, 39, 396 CrossRef CAS.
- J. Kelder, P. D. Grootenhuis, D. M. Bayada, L. P. Delbresine and J. P. Ploemen, Pharm. Res., 1999, 16, 1514 CrossRef CAS.
- Ajay, G. W. Bermis and M. A. Murkco, J. Med. Chem., 1999, 42, 4942 CrossRef CAS PubMed.
- A. Cavalli, E. Poluzzi, F. De Ponti and M. Recanatini, J. Med. Chem., 2002, 45, 3844 CrossRef CAS PubMed.
- F. De Ponti, E. Poluzzi and N. Montanaro, Eur. J. Clin. Pharmacol., 2001, 57, 185 CrossRef CAS.
- G. Colmenarejo, A. Alvarez-Pedraglio and J.-L. Lavandera, J. Med. Chem., 2001, 44, 4370 CrossRef CAS PubMed.
- M. Yazdanian, S. L. Glynn, J. L. Wright and A. Hawi, Pharm. Res., 1998, 15, 1490 CrossRef CAS.
- J. D. Irvine, L. Takahashi, K. Lockhart, J. Cheong, J. W. Tolan, H. E. Selick and J. R. Grove, J. Pharm. Sci., 1999, 88, 28 CrossRef CAS PubMed.
- P. Stenberg, U. Norinder, K. Luthman and P. Artursson, J. Med. Chem., 2001, 44, 1927 CrossRef CAS PubMed.
- R. O. Potts and R. H. Guy, Pharm. Res., 1992, 9, 663 CrossRef CAS.
- R. O. Potts and R. H. Guy, Pharm. Res., 1995, 12, 1628 CrossRef CAS.
- F. Ntie-Kang, L. L. Lifongo, J. A. Mbah, L. C. O. Owono, E. Megnassan, L. M. Mbaze, P. N. Judson, W. Sippl and S. M. N. Efange, In Silico Pharmacol, 2013, 1, 12 CrossRef.
- K. Chibale, M. Davies-Coleman and C. Masimirembwa, Drug discovery in Africa: impacts of genomics, natural products, traditional medicines, insights into medicinal chemistry, and technology platforms in pursuit of new drugs, Springer, 2012 Search PubMed.
- pan-ANAPL: pan-African Natural Products Library, http://www.linkedin.com/groups/pANPL-4098579/about.
- R. Core Team, R.: A Language and Environment for Statistical Computing. R. Foundation for Statistical Computing, Vienna, 2012, http://www.R-project.org Search PubMed.
- Chemical Computing Group Inc., Molecular Operating Environment Software, Montreal, 2010, http://www.chemcomp.com/ Search PubMed.
- A. T. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef.
- ChemAxon, JChem software, version 5.11.3, 2012, https://www.chemaxon.com/jchem/doc/user/LibMCS.html Search PubMed.
- X. Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hann, J. Chem. Inf. Comput. Sci., 1998, 38, 511 CrossRef CAS.
- J. Gu, Y. Gui, L. Chen, G. Yuan, H.-Z. Lu and X. Xu, PLoS One, 2013, 8, e62839 CAS.
- B. Over, S. Wetzel, C. Grütter, Y. Nakai, S. Renner, D. Rauh and H. Waldmann, Nat. Chem., 2013, 5, 21 CrossRef CAS PubMed.
- M. Congreve, G. Chessari, D. Tisi and A. J. Woodhead, J. Med. Chem., 2008, 51, 3661 CrossRef CAS PubMed.
- R. A. E. Carr, M. Congreve, C. W. Murray and R. C. Rees, Drug Discovery Today, 2005, 10, 987 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra43754j |
| This journal is © The Royal Society of Chemistry 2014 |