
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct CO2 to methanol reduction on Zr6-MOF based composite catalysts: a critical review†
Elif
Tezel‡
,
Dag Kristian
Sannes‡
,
Stian
Svelle
 ,
Petra Ágota
Szilágyi
* and
Unni
Olsbye
*
,
Petra Ágota
Szilágyi
* and
Unni
Olsbye
*
Department of Chemistry, Centre for Materials Science and Nanotechnology, University of Oslo, Norway. E-mail: unni.olsbye@kjemi.uio.no; p.a.szilagyi@kjemi.uio.no
First published on 13th October 2023
Abstract
The pressing problem of climate change on account of anthropogenic greenhouse-gas emissions underlines the necessity for carbon capture and utilisation technologies. Several heterogeneous catalyst systems allowing the conversion of waste CO2 into desirable products, such as methanol, have emerged as promising and potentially viable solutions to this perennial problem. In particular, composite catalysts based on hexanuclear zirconium metal–organic framework matrices have shown much promise in the direct conversion of CO2 into value-added and useful products. Herein, we critically review the literature in this area and relate differences in composition, defect chemistry, and structural characteristics, their reaction conditions with their performance and stability in the thermocatalytic hydrogenation of CO2 to methanol, on the basis of both experimental and theoretical studies. We also highlight the obstacles in directly comparing the performance of these systems for CO2 hydrogenation and suggest potential solutions and opportunities for further advancement.
 Elif Tezel | Elif Tezel is a PhD Research Fellow at the University of Oslo, working on investigating the design criteria for active and stable MOF-based catalyst for hydrogenation of CO2 to high-value chemicals. She is a chemical engineer and chemist with a background in thermal and electro-catalyst design, solid oxide cell systems, and hydrogen production processes. She obtained an BSc degree at the Department of Chemical Engineering at Bogazici University, Turkey, and MSc degrees from Georgia State University, USA, and Yildiz Technical University, Turkey. She also worked on electrocatalyst design and solid oxide cell systems at Wayne State University, USA. |
 Dag Kristian Sannes | Dag Kristian Sannes is a PhD student at the University of Oslo (Norway) and a member of the Catalysis Section. He received his master's in chemistry in 2020 from the University of Oslo. His research interest is focused on the synthesis of metal-functionalised metal–organic frameworks for the application of CO2 hydrogenation. |
 Stian Svelle | Dr Stian Svelle has been Professor of chemistry at the University of Oslo, Norway, since 2013. He received his PhD from the same institution in 2004. He does fundamental studies of porous materials and their use in catalysis, especially the conversion of methanol to hydrocarbons, and more recently the direct conversion of methane to methanol. Of further interest is the influence of zeolite catalyst crystal morphology and nanostructure, in both cases aiming for improved performance. He combines kinetic, quantum chemical, and operando studies to investigate individual reaction steps and deactivation phenomena occurring within the catalyst crystals or bodies. |
 Petra Ágota Szilágyi | Dr Petra Ágota Szilágyi is an Associate Professor at the University of Oslo (Norway) and a member of the Catalysis Section. She received PhD degrees in 2008 in chemistry from the University Eötvös Loránd (Hungary) and in Physics/Nanophysics from the University Paul Sabatier (France). Her research interest is focussed on the green synthesis of metal–organic frameworks, and their design and application for processes of energy and environmental importance. |
 Unni Olsbye | Unni Olsbye obtained an MSc in Industrial Chemistry at NTNU, Norway, in 1987, and a PhD in Organic Chemistry at the University of Oslo (UiO), Norway, in 1991. She worked at SINTEF and Nordox Industrier before returning to UiO as associate professor in 2001. She was promoted to full professor in 2002. Her research group focuses on catalysis for sustainable valorization of light molecules (C1–C3), mainly using zeolite- and metal organic framework-based catalysts. Experimental studies of kinetic and mechanistic consequences of single material parameter variation are used as a guiding tool for catalyst design. |
Introduction
Climate change is arguably the most pressing emergency humanity faces. There is a scientific and increasingly societal consensus that it is caused by the emission of greenhouse gases, chiefly but not exclusively carbon-dioxide, which is by large a consequence of anthropogenic activities such as transport, construction, heating, etc.1 While it is clear that there is an urgent need to decarbonise industries and shift the energy paradigm to renewables, in order to avoid a full-scale climate catastrophe, it is becoming evident that solutions must be found to also remove greenhouse gases from the atmosphere.Two distinct methodologies tackle the reduction of atmospheric carbon-dioxide levels: carbon capture and sequestration (CCS) and carbon capture and utilisation (CCU).2–9 Both of the methodologies rely on removing CO2 from the emitter initially, e.g. nascent carbon dioxide from flue gas, or, in a more challenging approach, directly from the atmosphere by various CO2 capture materials, mainly aqueous solutions of amino alcohols such as monoethanolamine (MEA) and diethanolamine (DEA). However, they differ in the way they handle carbon dioxide post-capture; CCS is aimed at storing it in the long term either in cavities or in rocks, while CCU aims at transforming CO2 into more reduced forms of carbon, ideally into value-added substances including methanol and formic acid, through chemical reactions. While it is evident that all technologies able to definitively remove CO2 from the atmosphere must be applied in the short-to-medium term to combat the adverse effects of climate change, CCU offers a more economically viable solution to the paradigm as it ultimately aims at yielding commercialisable products with potential application as fuels or platform chemicals, i.e., the approach offers a way to tackle climate change simultaneously, and the energy crisis or a path to sustainable feedstock sourcing.
The conversion of CO2 takes place thanks to catalysis, be it conventional thermal-, electro-, or photo(electro)-catalysis.10–13 There is a multitude of reduction products that can be achieved, highlighting the highly diverse carbon chemistry involved (Fig. 1). Major products include C1 as well as Cx (x > 1) products; the production of the latter occurs via C–C coupling reactions, which are more frequent in the case of electrocatalysis.
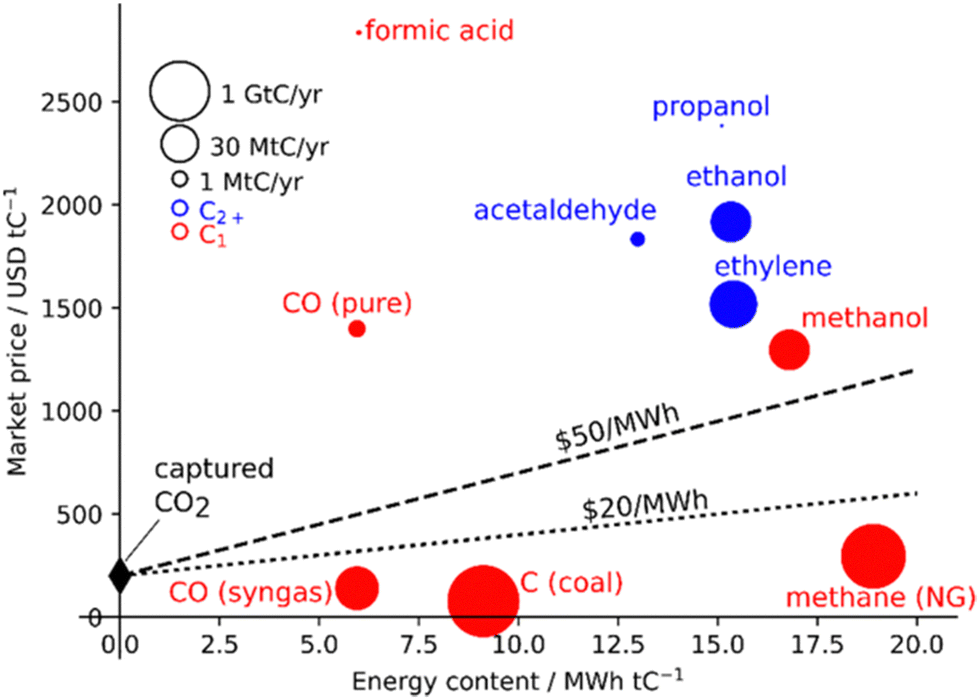 | ||
| Fig. 1 Market price of CO2 conversion products per energy content. Lines represent minimum energy and CO2 costs. Capital costs are not considered. Reprinted with permission from ref. 14. Copyright 2019 American Chemical Society (https://pubs.acs.org/doi/10.1021/acs.chemrev.8b00705). | ||
One of the most exciting outcomes of CO2 upcycling is the prospect of fuel synthesis using a sustainable feedstock whose removal from the atmosphere would also come with several environmental benefits, “two birds with one stone”. This concept is also desirable as it allows for a CO2-neutral fuel technology without massive infrastructural changes. For catalyst selection, it is paramount to evaluate the possible products, in terms of their cost and, particularly for synthetic fuel generation, their energy content (Fig. 1).14
Indeed, the most energetic products per tonne C are CH4 and CH3OH, with the latter having a greater market price, leading to the idea of the methanol economy.15 For this reason, even though products with longer carbon chains may afford higher market value, this review will focus on CO2-to-methanol technologies.
The different catalytic approaches offer distinct advantages, including high selectivity (photocatalysis),16 great adaptability for remote communities (electrocatalysis),17 and the existence of mature technologies for industrial uptake (thermal catalysis).18 While all of these advantages have their merits and all of the approaches should be used in order to meet our climate goals, the last one, i.e., conventional thermal catalysis, offers the greatest impact in the short and short-to-medium terms. In addition, heterogeneous catalysts have the advantage over their homogeneous counterparts of not requiring costly and energy-intensive separation steps for post-catalytic processes, and is therefore the focus of the present critical review.19
The activity for the direct hydrogenation of carbon dioxide to methanol is well documented for several catalysts; chief among these are promoted Cu, Pt, Cu/Zn, etc.20–24 As an example, on the copper catalyst, the CO2 conversion takes place in a site-selective manner, meaning that the catalyst performance will, to a large extent, be determined by the size, shape, and surface chemistry, i.e., the nature and extent of promotion, of the catalyst particles.25 Electronic promotion via strong metal–support interactions (SMSI) has been shown to improve the performance of Cu nanoparticles; as an example, zirconia, a partially reducible substrate, interacts more strongly with the Cu nanoparticles than alumina, which is not reducible, thereby improving catalyst dispersion and, consequently, inhibiting deactivation by sintering.26,27 Porous substrates, e.g. zeolites have also been shown to thwart particle agglomeration.
Another type of porous material, metal–organic frameworks (MOFs) has sparked interest as a way of controlling and limiting the geometry of catalytically active species in their pores.28,29 MOFs are highly desirable catalyst substrates as they display unrivalled topological and chemical diversity, in addition to permanent and highly regular porosity, and crystallinity. They are made up of inorganic nodes interconnected by organic linkers, whose geometry and composition determine the framework topology and chemistry.30 As the pores are part of the crystal structure, they are also highly regular in terms of geometry and composition. Consequently, when employed as moulds for particle growth, they may yield a multitude of supported catalyst particles with identical size and shape, a great advantage in heterogeneous catalysis, where performance often depends on the particle geometry, especially in the case of site-selective reactions. In addition, possible interaction with the MOF material may finely alter the guest particles’ surface chemistry, akin to SMSI, while the open-channel structure also minimises mass-transport issues. For these reasons, we devote this review to the in-depth analysis of MOF-based catalysts for the direct hydrogenation of CO2 to methanol.
As mentioned above, a theoretically infinite number of MOF topologies and chemistries are achievable, although their numbers are limited experimentally. Still, there are over 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures reported, with very different physical and chemical properties.31 To ensure that our analysis is as consistent as possible, we have selected MOFs based on a hexanuclear zirconium node (hence Zr6), which have been the focus of great scientific interest on account of their thermal and chemical stability, which are critical for their successful application as catalyst substrate. While there is a string of Zr6 frameworks, there are three representatives that are particularly explored, namely UiO-66 (with terephthalic acid or 1,4-benzendicarboxylic acid/BDC linker), UiO-67 (with 4,4′-biphenyl 1,1′-dicarboxylic acid/BPDC linker), and MOF-808 (with trimeric acid, or 1,3,5-benzenetricarboxylic acid/BTC linker).32,33 It should be noted however that MOF-808 does not display equally high stability as it is very unstable in aqueous conditions. In addition to their stability, Zr6 MOFs may bear various chemical functional groups,34 can resist structural collapse on defect formation,35,36 and display a rich node chemistry,37 potentially lending itself to promoting reactions, which will be further discussed in this paper as a means of their application. Most importantly, Zr6 MOFs have been shown to be excellent templates and supports for catalytically active species, including those for CO2 reduction to methanol.37–46 In this review, we analyse the status of thermocatalytic CO2 hydrogenation by composite Zr6 MOFs-based catalysts. Firstly, we briefly provide information regarding the fundamentals of the process including, thermodynamics and possible reaction mechanisms. Then, we evaluate trends between the structural and compositional variations in the catalyst support and active sites and their catalytic performance, followed by the impacts of reaction parameters on the performance based on experimental and theoretical studies. Finally, we identify challenges in direct comparison of studies from the literature and suggest possible solutions, and we finally highlight some prospective future research pathways.
000 structures reported, with very different physical and chemical properties.31 To ensure that our analysis is as consistent as possible, we have selected MOFs based on a hexanuclear zirconium node (hence Zr6), which have been the focus of great scientific interest on account of their thermal and chemical stability, which are critical for their successful application as catalyst substrate. While there is a string of Zr6 frameworks, there are three representatives that are particularly explored, namely UiO-66 (with terephthalic acid or 1,4-benzendicarboxylic acid/BDC linker), UiO-67 (with 4,4′-biphenyl 1,1′-dicarboxylic acid/BPDC linker), and MOF-808 (with trimeric acid, or 1,3,5-benzenetricarboxylic acid/BTC linker).32,33 It should be noted however that MOF-808 does not display equally high stability as it is very unstable in aqueous conditions. In addition to their stability, Zr6 MOFs may bear various chemical functional groups,34 can resist structural collapse on defect formation,35,36 and display a rich node chemistry,37 potentially lending itself to promoting reactions, which will be further discussed in this paper as a means of their application. Most importantly, Zr6 MOFs have been shown to be excellent templates and supports for catalytically active species, including those for CO2 reduction to methanol.37–46 In this review, we analyse the status of thermocatalytic CO2 hydrogenation by composite Zr6 MOFs-based catalysts. Firstly, we briefly provide information regarding the fundamentals of the process including, thermodynamics and possible reaction mechanisms. Then, we evaluate trends between the structural and compositional variations in the catalyst support and active sites and their catalytic performance, followed by the impacts of reaction parameters on the performance based on experimental and theoretical studies. Finally, we identify challenges in direct comparison of studies from the literature and suggest possible solutions, and we finally highlight some prospective future research pathways.
Fundamentals of CO2 hydrogenation to methanol
Thermodynamics of CO2 hydrogenation
CO2 hydrogenation is challenging from a thermodynamic standpoint due to its high stability with an enthalpy of formation of ΔHf = −393.5 kJ mol−1. To circumvent these constraints, CO2 utilisation is often combined with a high-energy co-reactant to promote the conversion of CO2.47 H2 or CH4 are common co-reactants, with ΔHf = 0 kJ mol−1 and ΔHf = −76.4 kJ mol−1, respectively. Although the co-reactant facilitates the reaction by contributing its intrinsic chemical energy, it renders the utilisation of CO2 a high energy-demanding process.48 Using H2 as a co-reactant is especially desirable due to the potential of H2 production from renewable energy sources, such as water electrolysis and the importance of methanol as a solvent or reactant for other products.49–51| CO2 + H2 ⇌ CO + H2O ΔH298K = 41.2 kJ mol−1 | (1) |
| CO2 + 3H2 ⇌ CH3OH + H2O ΔH298K = −49.5 kJ mol−1 | (2) |
| CO2 + 4H2 ⇌ CH4 + 2H2O ΔH298K = −165.0 kJ mol−1 | (3) |
We used the Factsage software55 to calculate the equilibrium conversion of CO2 in the 170–300 °C temperature range and a pressure range of 1–50 bars with H2/CO2 ratio of 3:1, which are the typical conditions used for catalytic testing in literature and are similar to conditions used in industry.56 The conversion of CO2 and product selectivity were calculated according to eqn (4) and (5). Methanol formation is an exothermic reaction with an ΔH298K = −49.5 kJ mol−1 (eqn (2)); hence, according to Le Chatelier's principle, the forward reaction is favoured at low temperatures (Fig. 2). Low temperatures provide the additional benefit of suppressing the competing RWGS, which reduces the partial pressure of CO2 and affects the equilibrium by producing water.
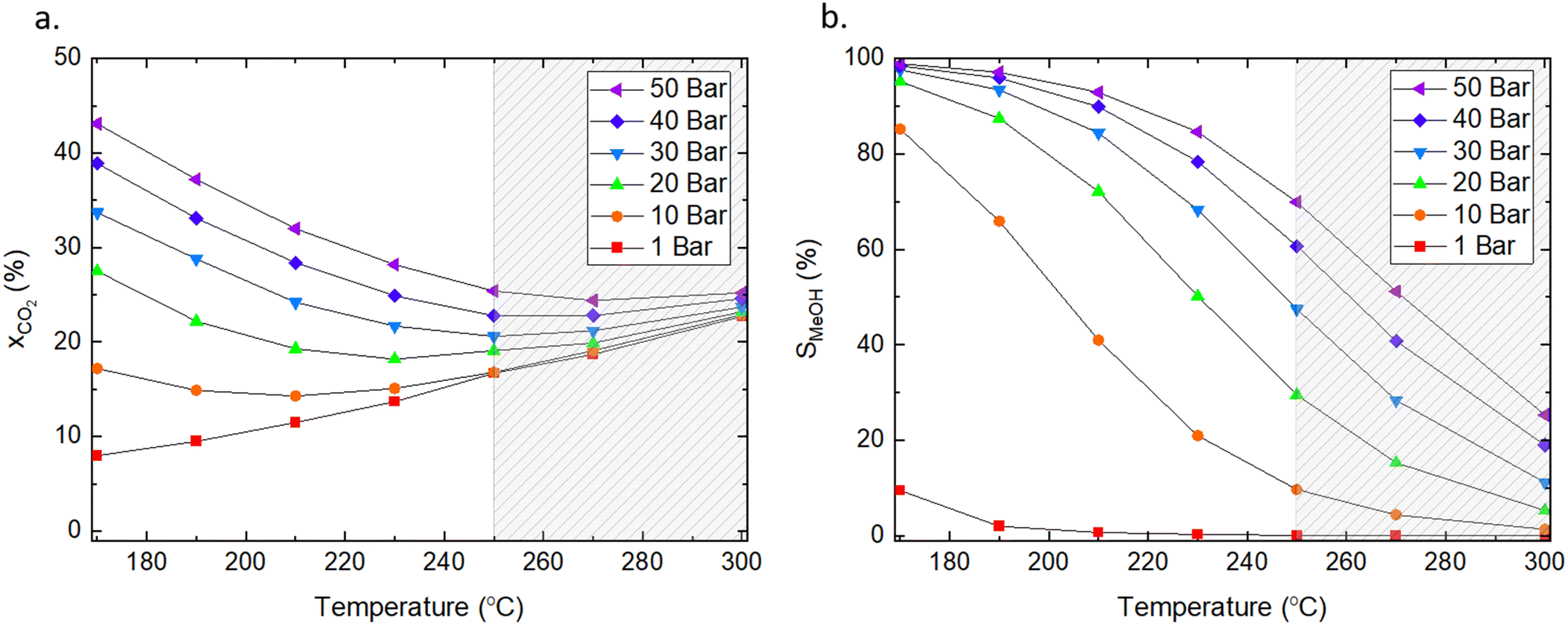 | ||
| Fig. 2 Influence of pressure and temperature on (a) equilibrium conversion of CO2 and (b) selectivity toward methanol at equilibrium. The dashed area indicates the temperature range typically applied for CO2 hydrogenation to methanol in industry. The ratio of H2/CO2 was 3:1, and methane was excluded from the calculations. Factsage was used for the analyses.55 | ||
Conversion of CO2:
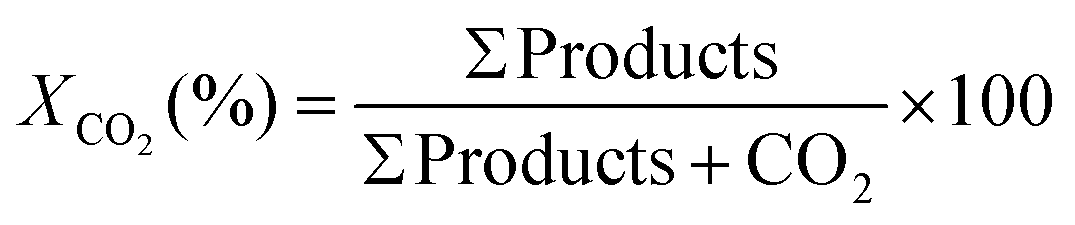 | (4) |
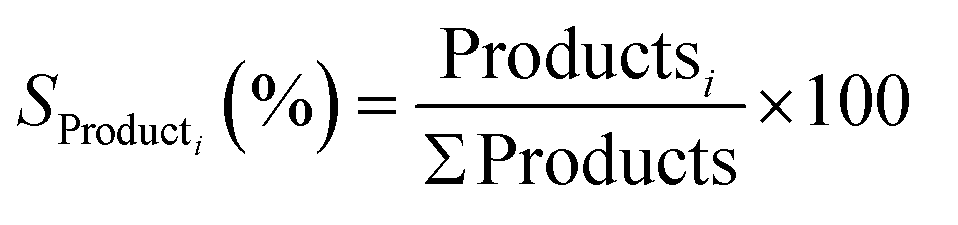 | (5) |
CO2 to methanol reduction mechanism
In order to create highly active and selective catalysts enabling CO2 utilisation, it is crucial to comprehend the mechanism underlying the conversion of CO2 into methanol.57 Two primary routes have been proposed for CO2 hydrogenation to methanol (Fig. 3). The first route proceeds via the hydrogenation of a surface CO species formed by direct C–O bond cleavage of CO2 or from a carboxyl intermediate. The second pathway proceeds through surface formate species formed from hydrogenation of adsorbed CO2 and is often referred to as the formate pathway. The preferred route depends on the catalyst and which intermediates are stabilised on the specific surface. The formate pathway is the prominent mechanism on the Cu/ZnO catalyst; in contrast, on Cu/CeO2, Cu/TiO2 and Cu/ZrO2, the primary mechanism for methanol formation is through hydrogenation of a surface CO intermediate. This is because surface formate species are too strongly adsorbed on these catalysts, effectively poisoning the catalyst surface and suppressing the subsequent hydrogenation. To improve both the activity and selectivity of the catalysts, promoters are often employed, which essentially fine-tune the intermediates’ adsorption energies to reduce kinetic barriers and facilitate effective catalytic pathways.58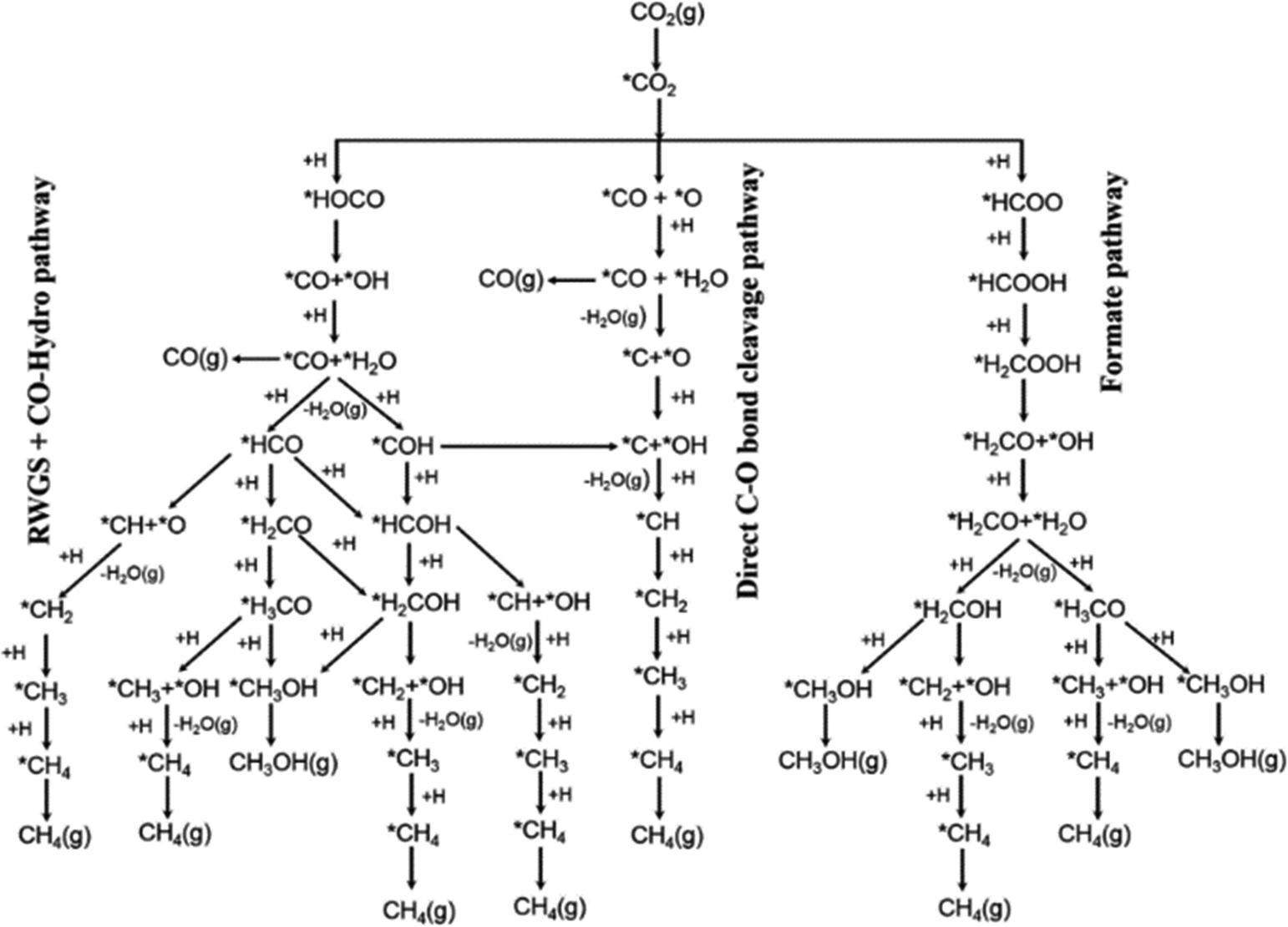 | ||
| Fig. 3 Possible reaction pathways of CO2 hydrogenation to CO, CH3OH, and CH4. *(X) indicates adsorbed species. Reprinted with permission from ref. 57. Copyright 2017 American Chemical Society (https://pubs.acs.org/doi/10.1021/jacs.7b05362). | ||
Among the most studied systems for direct CO2 reduction to methanol is copper supported on Al2O3 and ZrO2 combined with a vast range of promoters such as Zn, Zr, Ce, Al, and Si.59–61 Cu supported on Al2O3 with ZnO as a promoter is often denoted as Cu/ZnO/Al2O3 and has been commercialised on the industrial scale since the 1960s and is therefore often used as a benchmark catalyst for CO2 hydrogenation.56
Experimentally it was found that Cu0 is part of the active sites in Cu/ZnO/Al2O3 for methanol formation, and the performance of the catalyst is linearly dependent on the surface area of the copper nanoparticles (NPs).62–64 The active site for methanol formation has been investigated using density functional theory, and it was found that open Cu surfaces (e.g. Cu(110) and (Cu (100)) partially covered by oxygen are active for CO2 hydrogenation to methanol.65 It has been shown that the methanol synthesis rate over Cu (100) was roughly 30 times faster than on the Cu (110) surface, suggesting that the facets of Cu affect the catalyst's performance. Other theoretical studies have also shown that other facets of Cu may be active for methanol formation. For instance, the stepped Cu (533) surface enhanced the activity compared to the flat Cu (111) surface. Similarly, the step sites on the Cu (997) are more reactive than the terrace sits on Cu (111) for Cu/Zn catalysts.66,67
The size of the Cu NPs is also vital for the activity of the materials. It has been found for Cu NPs smaller than 8 nm, that the turnover frequency (TOF) decreases proportionally with decreasing size. The exact nature of this structure sensitivity for small Cu NPs is unknown but it could be explained by either the stabilisation of a larger fraction of step sites or the removal of unsaturated Cu sites for NPs of Cu above 8 nm.25 Another study showed that the methanol formation rates, normalised by surface copper species, are constant for Cu/ZnO catalysts with Cu NPs varying from 8.5 to 37.3 nm. It was found that the formation of CO is favoured as the size of the Cu NPs is decreased, illustrating that CO2 conversion is site-selective and the selectivity of the materials may be tuned in favour of methanol by increasing the size of the Cu NPs.68
The role of ZnO is still heavily debated, and not all the mechanistic steps are well understood.64 As with many promoters, ZnO acts as a spacer for the Cu NPs, which improves the dispersion and stability of the material.69 Control experiments on pure Cu and Zn catalysts compared to Cu/Zn catalysts show that there is a synergetic interaction between the two.66 DFT calculations performed on a stepped Cu (211) surface partly substituted with Zn atoms showed that Zn increased the adsorption of key intermediates of methanol production and decreased the kinetic barriers, leading to an increase in the rate of methanol formation. Some experimental studies suggest that Zn may migrate to the Cu NPs during reduction, creating active Cu/Zn sites. Cu/Zn active sites were postulated after observing an increase in active sites by increasing the reduction temperature. SMSI have been observed for the Cu/ZnO system using vibrational spectroscopy, thermal desorption of probe molecules and observing the wetting behaviour of Cu/ZnO model systems. Ambient pressure X-ray photoemission spectroscopy (XPS) and high-resolution transmission electron microscopy (HRTEM) show the presence of a 1 nm thick layer of ZnOx on a few of the Cu NPs in the Cu/ZnO/Al2O3 catalyst.64,70–72 The graphite-like ZnOx layer might facilitate the substitution of Cu atoms on the surface of the Cu NPs. Other studies have reported that the ZnOx layer is active for methanol formation. A core–shell Cu/Zn alloy with a ZnOx outer shell was prepared by elaborate synthesis and post-synthesis procedures. The active site was found at the ZnOx outer shell, and 100% methanol selectivity was observed.73 Other studies have suggested that the role of ZnO is to act as a hydrogen reservoir for spillover hydrogen, which may help to keep the surface species of the copper NPs hydrogenated.74
Al2O3 is the commercially used support for the Cu/ZnO system resulting in a catalyst displaying high activity, high product selectivity and low cost. The support material act as a stabilising oxide that increases catalyst stability and maintains the dispersion of Cu and ZnO NPs.75 The acidic sites on the Al2O3 surface are strong enough to adsorb both methoxy and formate species which can react with methanol, by dehydrating it and form either dimethyl ether (DME) or methyl formate, the latter may decompose to methanol and CO, providing an additional source of CO (Fig. 4).76,77 Cu/ZnO/ZrO2 has gained increasing interest in the research community for its higher catalytic activity than Cu/ZnO/Al2O3 and is one of the state-of-the-art catalysts from a research viewpoint.78 The increased activity arising from the application of ZrO2 compared to other support materials may be attributed to its capability to enhance the Cu and ZnO dispersion, i.e. to increase the surface area of the NPs and modify the surface of the copper.79 In addition, ZrO2 is less hydrophilic than Al2O3, which facilitates the desorption of water formed in the course of the reaction, thereby enhancing both the conversion of CO2 and selectivity to methanol by thermodynamically promoting methanol formation.80,81 Studies of the binary systems of Cu/ZnO and Cu/ZrO2 have been investigated in great depth to gain improved insight into the ternary system. It is generally accepted that the binary Cu/ZnO catalyst has higher activity for CO2 hydrogenation, while Cu/ZrO2 shows a higher selectivity to methanol. The ternary system offers a higher yield of methanol than either of the binary systems, although at the expense of a decreased selectivity (Fig. 5). Additional promoters, e.g. Ga2O3 and Y2O3, may increase the activity of the ternary catalyst Cu/ZnO/ZrO2 by increasing Cu dispersion.63
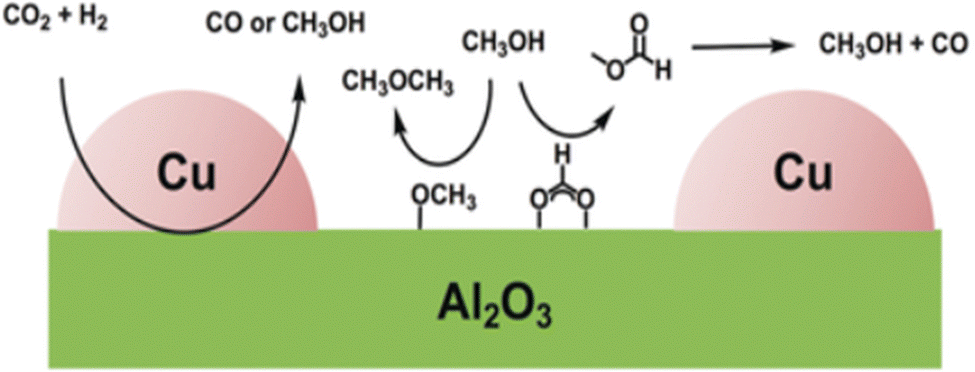 | ||
| Fig. 4 CO2 hydrogenation on Cu/Al2O3. Used with permission of Angewandte Chemie International Edition, from CO2 Hydrogenation on Cu/Al2O3: role of the metal/support interface in driving activity and selectivity of a Bifunctional Catalyst, Lam et al., 2019, 58, 39 permission conveyed through Copyright Clearance Center, Inc. | ||
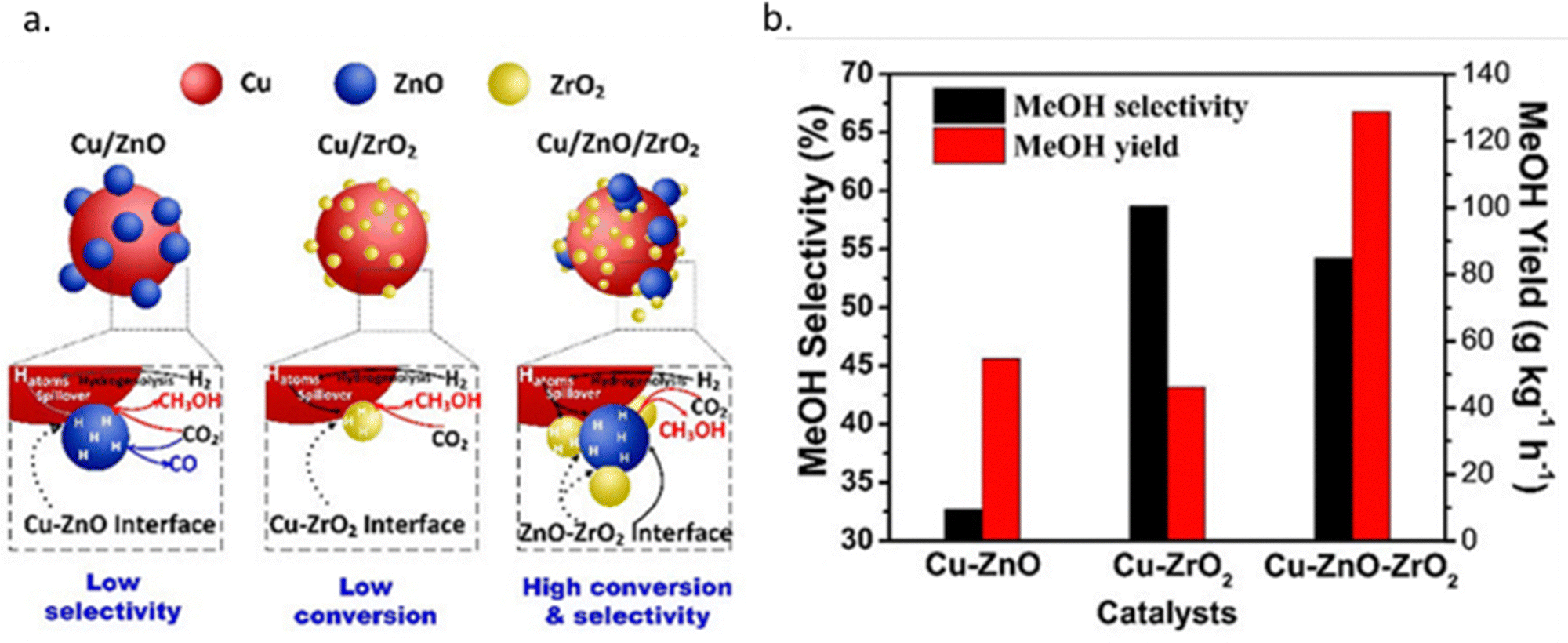 | ||
| Fig. 5 (a) Schematic diagram of the CO2 hydrogenation process over Cu–ZnO, Cu–ZrO2 and Cu–ZnO–ZrO2 catalysts. (b) Methanol yields and methanol selectivity of Cu–ZnO, Cu–ZrO2 and Cu–ZnO–ZrO2 catalysts. Reprinted with permission from ref. 78. Copyright 2019 American Chemical Society (https://pubs.acs.org/doi/full/10.1021/acscatal.9b01943). | ||
The average lifetime of the Cu/ZnO/Al2O3 catalyst under standard operating conditions in industry is typically 2–4 years and a decrease in catalyst activity is observed during this time.56 The sintering of either the Cu or ZnO NPs and the oxidation of Cu cause the catalyst deactivation predominantly.82 Studies on the deactivation suggest that sintering of the Cu NPs does not change the nature of the active sites, as seen by a constant activation energy for methanol formation during deactivation, suggesting the loss of activity is caused by the loss of active sites.83 Experimental studies have shown that after prolonged exposure to the reaction conditions (700 hours), the space-time yield of methanol is reduced by 34.5% over the Cu/Zn/Al2O3 catalyst.84 The study reported an increase in the size of ZnO NPs, but no significant change in the Cu NPs, likely due to the low reaction temperature of 200 °C. Two separate studies found that the deactivation of the catalyst is promoted by the presence of water formed by the RWGS reaction. The size of the NPs in one of these studies, after catalytic testing for 48 hours at 240 °C and 50 bar with varying quantities of water, is shown in Fig. 6, clearly showing an increase in NP size with an increasing quantity of water.83,85
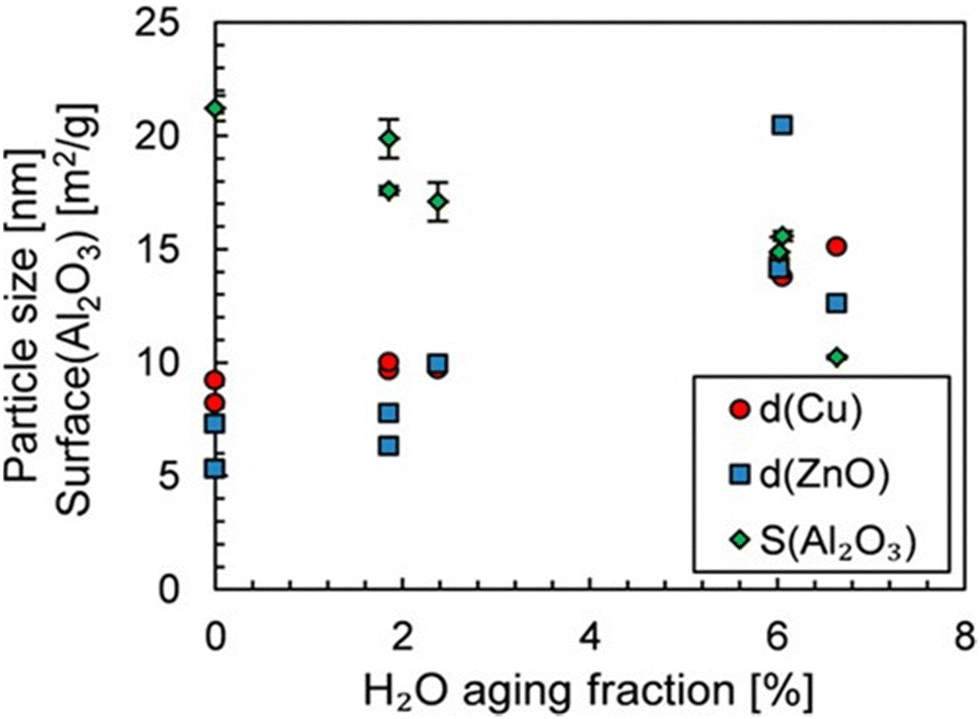 | ||
| Fig. 6 Influence of H2O volume fraction on the Cu and ZnO particle size and Al2O3 surface. The aging period was 48 hours. Reprinted with permission from ref. 85. Copyright 2019 American Chemical Society (https://pubs.acs.org/doi/full/10.1021/acs.iecr.9b01898). | ||
XPS results have also shown that the loss of activity of the material may be due to Cu oxidising to Cu2+ during the reaction. The oxidative nature of CO2 and in situ formed water is likely the reason for the oxidation of Cu.84 These results imply that strong interactions with the support for all constituents are essential for high stability of the catalyst.
MOFs as support materials may provide a satisfactory solution to the problems highlighted above. MOFs are highly crystalline materials, allowing for excellent control of the active sites. Even though MOFs are a relatively new group of hybrid materials, numerous structures have been reported with a large variety of inorganic cations, clusters, and organic linker molecules. The effect of various support materials with an extensive range of chemical properties may be investigated, and the catalyst's active sites may be fine-tuned. The agglomeration of the active species on nonporous supports is the primary deactivation mechanism and MOFs may hinder agglomeration by trapping the NPs inside the framework. The size of NPs may be carefully tuned by changing the size of the pores of the material by changing the length and size of the linker. If the active specie is also grafted inside the framework, it is possible to fine-tune the interaction between the active specie and the support, which could both further hinder agglomeration and hinder oxidation or reduction of the active specie by changing the nature of the framework. However, the pore sizes of MOFs are substantially smaller than the optimum size of the Cu NPs of 8 nm, which may limit the activity of the catalysts. Defects inherent to the MOFs framework, which are described later, may provide effective tools for accommodating the formation of larger NPs.
Finally, MOFs have been shown to be highly tuneable in terms of their hydrophilicity, which may be of relevance in affecting both catalyst stability and selectivity, as has been previously suggested in a bid of rationalising the different behaviours of various oxide supports. For the above-mentioned reasons, it is apparent that MOF-based supports have highly desirable and unique properties when it comes to providing a highly active and selective catalyst for direct CO2-to-methanol reduction.
Experimental catalytic performance trends and mechanistic insights for Zr6 MOF-based catalyst
Zr6-based MOF structures and incorporated metal particles have been successfully studied as composite catalysts for CO2 hydrogenation to methanol in the literature.37–46,86–88 Each study has numerous variables in terms of structural, compositional, and reaction parameters aimed to effectively improve the dispersion of catalytic sites and the adsorption and activation of CO2 for efficient catalysis of CO2 hydrogenation at lower temperatures. Furthermore, the inorganic nodes of MOFs may provide Lewis and Brønsted acid sites, which can contribute to activating CO2 or stabilising certain intermediates. Besides, different catalytic sites can be incorporated into MOFs, and the coordination environment can be adjusted, such as to control the CO2 hydrogenation to value-added products. Even though there is a strong interplay between these parameters and the catalyst performance, determining the optimum catalyst remains a challenge due to the inconsistency of test conditions. Below, we discuss in detail the effects of these parameters on the performance of composite Zr6-based MOF catalysts. Firstly, we review the impact of the structural and compositional variations in the catalyst support and active sites on the catalytic performance. Then we analyse the effects of reaction conditions, including temperature, pressure, and space velocity.Structural and compositional variables-performance relations
The aim of this section is to provide insight into the effects of different structural components of multicomponent systems on the performance of catalysts for CO2 hydrogenation to methanol. The structural properties of MOFs, the nature of the metal active sites, and metal–support interactions are evaluated. Zr-based MOFs, specifically UiO-66, UiO-67, and MOF-808 have differences, including synthesis procedure, linker composition, and presence of non-structural sites. The principle of isoreticularity can be used to design isotopological frameworks with varying pore sizes by using linkers with the same connectivity but different lengths, such as seen for UiO-66 and UiO-67. UiO-66 (Fig. 7a) and UiO-67 (Fig. 7b) consist of 12-connected Zr6(μ3-O)4(μ3-OH)4 clusters which are interconnected by BDC and BPDC linkers, respectively. Even though MOF-808 (Fig. 7c) shares the same Zr6 inorganic node, as UiO-66/67, its crystal structure differs since only six tritopic linkers (BTC) are coordinated to each node, or secondary building unit (SBU). In this topology, charge compensation is provided by an additional six monotopic ligands, which are easily exchangeable.89 Also, various synthesis methods have been reported in the literature, and studies show significant effects of the type of modulator on stability, porosity, thermal behaviour, and defect quantity of MOFs.89,90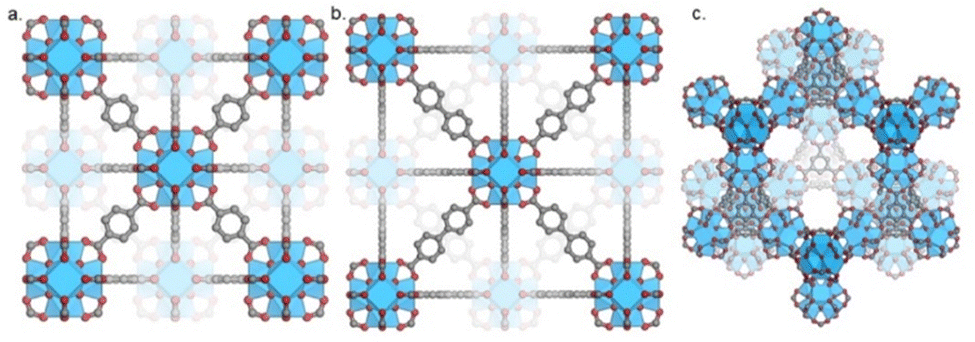 | ||
| Fig. 7 Topology of (a) UiO-66, (b) UiO-67, and (c) MOF-808 frameworks. | ||
Defects in MOFs could be defined as sites that locally break the periodic arrangement of atoms or ions of the static crystalline parent framework due to missing or dislocated atoms or ions.91 Even though various types of defects have been reported, two types of defects exist most commonly: missing linker (Fig. 8a) and missing cluster defects (Fig. 8b). Missing linker defects are formed when linkers are replaced by monodentate capping agents. Typically, these capping agents are monocarboxylates called modulators, which are added during the synthesis to assist the MOF self-assembly into crystallites.92 Similarly, missing cluster defects are formed when a cluster is removed from the framework; this kind of defect results in a relatively large cavity in the structure. While the presence of the cluster defects has been reported in UiO-66 MOF, the linker defects have been investigated in both UiO-66 and UiO-67.90,93–96 The linker defects typically stem from the incomplete ligand exchange reaction between the linker and modulator. The non-defective UiO66/67 MOFs contain a 1:1 ratio between Zr and the linker; however, the ratio was observed to be less than one for materials with missing linker defects. The defect quantity is increased in the UiO-66/67 because of the strong interaction between Zr4+ and carboxylate, which slows down the linker exchange reaction. Missing cluster defects and missing linker defects could affect the properties of the material, including surface area, pore volume, mechanical stability, and thermal stability. Besides, the cluster defects could have an influence on the performance of the catalysts due to diminished Lewis acid sites. The synthesis parameters to influence the concentration of missing linker- and cluster-defects in MOFs are discussed in detail below. Shearer et al. have previously reported an extensive study on synthesis conditions leading to missing linker defects in UiO-66. They found that the missing-linker defect concentration could be reduced by either increasing the synthesis temperature or increasing the linker-to-zirconium ratio, and, additionally, it has been found that increasing the synthesis time leads to a less defective material.90,97 The reduction of missing linker defects when alternating these synthesis variables is attributed to increasing the rate of the exchange reaction between the linker and the modulator, thereby increasing the probability of linkers coordinating with the zirconium nodes and providing more time for self-assembly, respectively. Kaur et al. published an extensive study on the synthesis of UiO-67, including the effect of changing the solvent dimethylformamide (DMF) to Zr ratio. The study showed that missing linker defects were far more prominent for samples synthesised in dilute (DMF:Zr ratio of 300:1) rather than in concentrated reaction mixtures (DMF to Zr ratio of 50:1).98 in addition, due to the tridentate nature of BTC in MOF-808, it is possible to further increase the missing linker defects by changing the tridentate linker with a bidentate linker such as isophthalic acid.99 The effect of synthesis parameters on the formation of missing cluster defects is less readily available; however, it has been found that the synthesis of UiO-66 using a large amount of modulator, especially with a low pKa value, induces missing cluster defects. Clearly, the synthesis conditions play an essential role in tuning missing linker and missing cluster defects.90
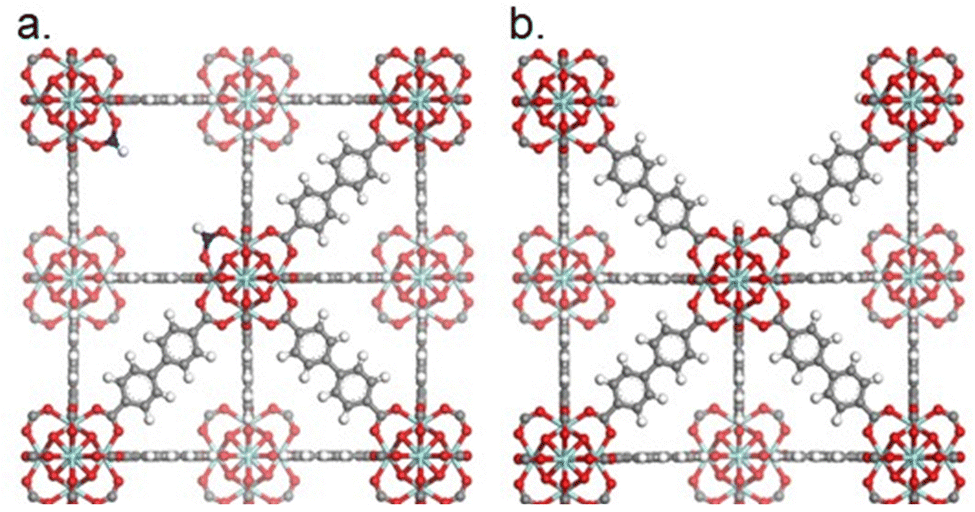 | ||
| Fig. 8 Illustration of the UiO-67 framework. (a) UiO-67 with missing linker defects (two formate groups as capping agents). (b) UiO-67 with a missing cluster defect. Zirconium, oxygen, hydrogen, and carbon are shown in teal, red, white, and grey, respectively. Carbons in the monocarboxylates are shown in blue. Hydrogen atoms on to the nodes are omitted for clarity. Reprinted with permission from ref. 93. Copyright 2023 American Chemical Society (https://pubs.acs.org/doi/10.1021/acs.chemmater.2c03744). | ||
The specific surface areas of ideal Zr6-MOFs with UiO-66 and UiO-67 topologies are around 1187 and 3000, respectively, according to Brunauer–Emmett–Teller (BET) theory.32 Missing clusters tend to yield higher BET; in contrast missing linker defects result in lower specific surface areas,90 while functionalised linkers89 may also impact the BET values. Reported literature values are in the following range: UiO-66 (1100–1600 m2 g−1) and UiO-67 (2100–2700 m2 g−1). For MOF-808, specific surface areas in the range of 1200–2300 m2 g−1 have been reported, depending on the capping agent on the uncoordinated Zr sites.85 Among materials reported as CO2 hydrogenation catalysts in the literature, we note that metal incorporation alters the BET area (Fig. 9). The effect was particularly strong for the incorporation of Cu and Zn in UiO-67, while the incorporation of Pt on UiO-67 had only a small impact on specific surface area (Fig. 9). A significant decrease in the BET surface area indicates that the structural integrity of the MOF has not been preserved, which may affect the accessibility of active sites. No direct correlation is observable between the specific surface area of the MOF support and the catalysts’ CO2 conversion performance (Fig. 9). The reason may be several convoluted factors influencing the results, e.g.; the location of the metal particles in relation to the MOF matrix may be widely different, as both particle size and SMSI can be potentially affected. It stands to reason that when the surface area is low but tangible catalyst activity has been observed, the metal particles would likely be deposited onto the surface of the (partially) collapsed or otherwise altered MOF particles, which then effectively act as a conventional support surface.
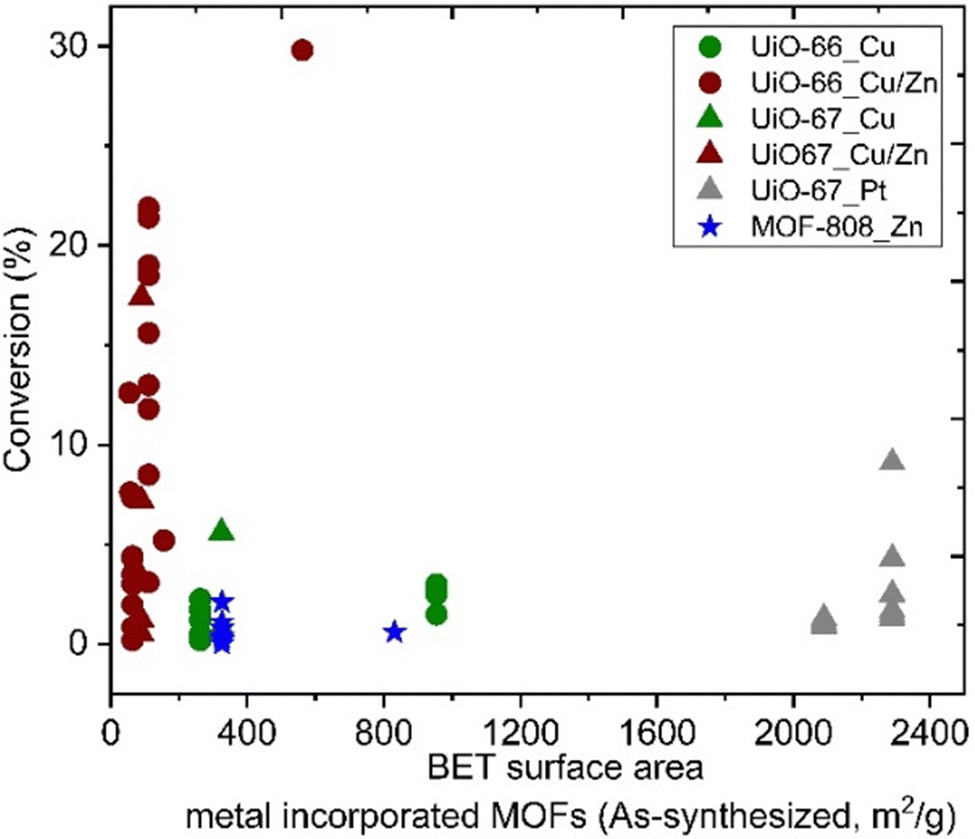 | ||
| Fig. 9 Catalytic conversion versus BET surface area of metal incorporated MOFs. | ||
Fig. 10 shows the correlation between the performance of catalysts for CO2 hydrogenation to methanol and catalyst composition, i.e.; MOF topology and active metal guest. While UiO-66 structures with Cu or Cu/Zn active sites have been most commonly studied, UiO-67 structures have also been reported with Pt, Cu/Zn, and Zn active sites. Besides, the MOF-808 structure was only investigated with Zn active sites. Higher conversions (>10%) have been overwhelmingly achieved with the UiO-66 topology and uniquely with Cu/Zn active components. Although general trends are hard to pinpoint, we note the lack of well-established studies with numerous variations on reaction conditions, inherently making the comparison challenging.
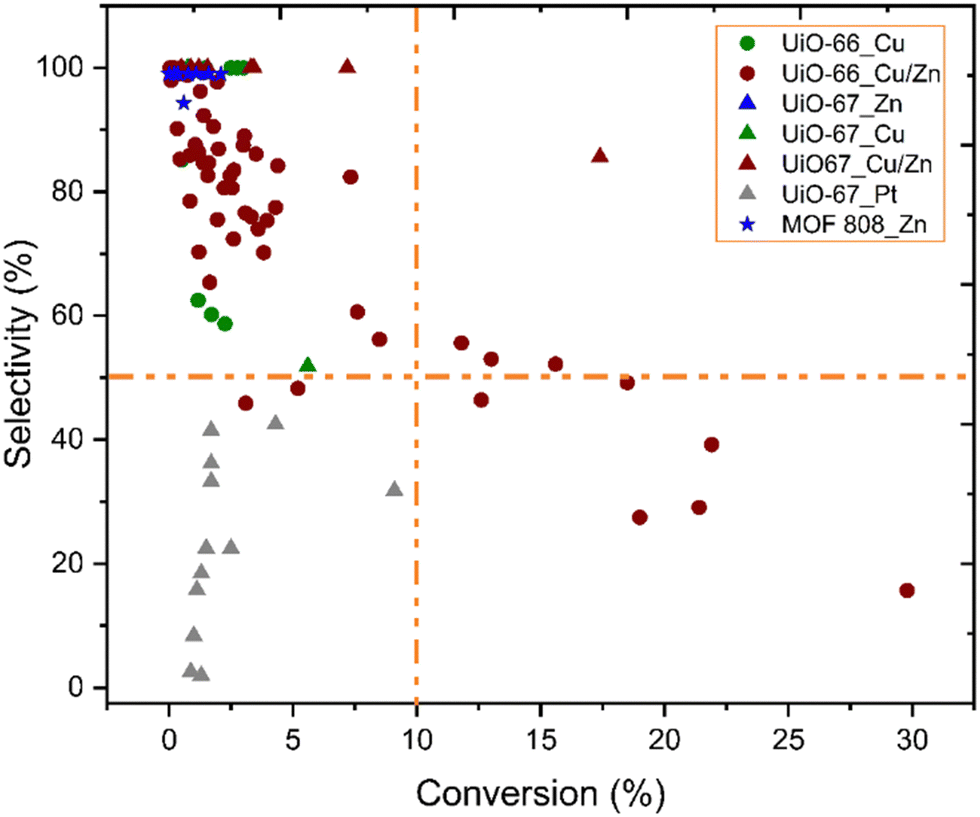 | ||
| Fig. 10 Selectivity toward methanol versus conversion of CO2. While different symbols represent various metal–organic frameworks, different colours represent various incorporated metal active sites.37–46 Orange dashed lines represent 10% conversion and 50% selectivity and have been included to guide the eyes. | ||
In the following, we will first discuss contributions where the MOF structure of Cu containing MOFs was preserved,42,43 and then proceed to studies where the MOF structure collapsed. Finally, we will discuss the performance of Zn-MOF and Pt-MOFs.37,45,46 The catalytic properties for CO2-to-methanol are strongly correlated with the characteristics of the metal oxide-metal interfaces.42 MOF-based composite catalysts incorporated with Cu have been commonly investigated because of the presumed strong interaction between Cu and metal nodes of MOFs. Rungtaweevoranit et al. investigated the support effects on Cu-based catalysts for CO2 hydrogenation to methanol over different support materials, including UiO-66, and reported that Cu particles encapsulated into UiO-66 (Cu⊂UiO-66) show 100% selectivity towards methanol in the 175 °C to 250 °C temperature range and at 10 bar, while the MOF structure was preserved.42 In this study, the MOF was grown around Cu NPs. Interestingly, XPS data indicated a lower oxidation state of Zr after incorporation of Cu, suggesting SMSI between Cu and Zr6 SBUs. The authors concluded that the high yield and selectivity toward methanol stem from Cu–ZrO2 interfaces in the Cu⊂UiO-66 since the oxidised form of Cu may stabilise the formate intermediates while the metallic Cu activates the hydrogenation through hydrogen dissociation.
To shed light on how Cu–ZrO2 interfaces influence methanol production, Zhu et al. modified UiO-66 by creating missing linker defects (Fig. 8b), which could increase the abundance of Cu–ZrO2 interfaces. A direct correlation between the activity and selectivity of the catalysts and the quantity of Cu–ZrO2 interfaces was demonstrated.43 The nature of the Cu–ZrO2 interaction was investigated qualitatively and quantitatively, using two samples denoted Cu/UiO-66-a and Cu/UiO-66-b. The location and nature of the Cu particles in Cu/UiO-66-a and Cu/UiO-66-b were characterised using Cu K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). The results indicated that while 30% of the Cu atoms in Cu/UiO-66-a (1.4 wt% Cu) were bonded to ZrO2 nodes, thereby creating Cu–ZrO2 interfaces, the presence of Cu–ZrO2 interfaces in Cu/UiO-66-b (1.8 wt% Cu) was suggested to be too low to contribute to the X-ray absorption spectra (XAS). On the other hand, the particle size of metallic Cu was almost identical for both samples. The significantly better performance of Cu/UiO-66-a compared with Cu/UiO-66-b (Fig. 11a) was suggested to highlight the importance of Cu–ZrO2 interface sites for methanol production through CO2 hydrogenation.
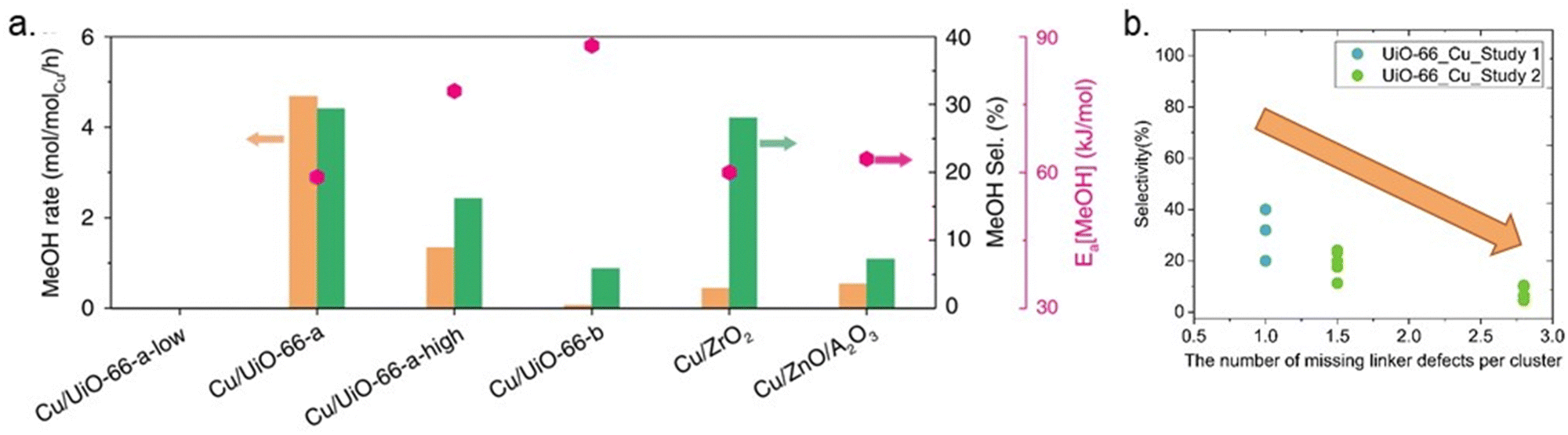 | ||
| Fig. 11 (a) Rates of methanol (MeOH) production (orange bars), methanol selectivity (green bars) on selected catalysts at 250 °C and 32 bar, and activation energy (Ea, pink points). Reproduced from ref. 43 (https://www.nature.com/articles/s41467-020-19438-w) with the permission of unrestricted use (https://creativecommons.org/licenses/by/4.0/). (b) Selectivity toward methanol versus the number of missing linker defects per cluster.40,43 | ||
Even though the previous study highlighted the importance of missing linker defects of UiO-66 on the activity of catalysts, the optimum amount of defect concentration has yet to be investigated. In another example, Ye et al. theoretically examined the quantitative influences of missing-linker defects on the activation of H2 and CO2 for hydrogenation of CO2 to methanol in Cu@UiO-66 at the Cu–ZrO2 interface and reported that the presence and number of linker defects could affect the kinetics of the reaction, catalytic efficiency, and side-product formation.87 They highlighted the necessity of an optimum number of missing linkers, which is approximately 5–7 per unit cell or 1.25–1.75 per cluster. Notably, too many missing linker defects would result in the too strong adsorption of CO2, thereby inhibiting efficient CO2 hydrogenation. Some support for these theoretical results is found in the literature, although the studies are scarce and several parameters may vary between them. A few datasets collected from the literature are shown in Fig. 11b. The results indicate that an increase in defect concentrations beyond one missing linker per cluster leads to decreased methanol selectivity.
According to data represented in Fig. 10, the performance of catalysts with Cu/Zn active sites, regardless of the MOF structure, was the most promising catalysts, and a few of these catalysts exhibited over 50% selectivity and 10% conversion, while those containing only Cu did not, even though reaction conditions were similar. The synergetic effect between Cu and Zn has been studied for CO2 hydrogenation to methanol since Cu/ZnO/Al2O3 is the commonly used catalyst for methanol production in the industry, as mentioned previously. An et al. investigated Cu/ZnOx nanoparticles anchored into UiO-67 for hydrogenation of CO2. Despite losing the porous structure of UiO-67 (Fig. 9), as evidenced by the loss of surface area, they observed enhanced catalytic activity and selectivity towards methanol (100%), which they suggested was due to SMSI between the MOF nodes and the Cu/ZnOx nanoparticles, as well as to confinement effects within the small pores of the MOF.38 The confinement is related to the structural and functional properties of the MOF cavity, which affect the environment and accessibility of catalytically active sites, and the shape of the cavity can be tuned by modifications, including linker functionalisation.100 In addition to the effect of the Zr nodes, the synergy of Cu and ZnO could also play an important role in CO2 hydrogenation to methanol. The importance of the Cu–ZrO2 interface for methanol formation is already discussed above. Also, the catalytic stability for 100 hours was reported. While the selectivity toward methanol was still 100%, the activity slightly decreased during 100 hours of reaction.
Density functional theory (DFT) calculations were performed to understand the adsorption properties of CO2 on the different sites on Cu decorated UiO-66 (Fig. 12).43 Based on the calculated CO2 adsorption energies, the Cu–Zr4+ (ZrO2) interfacial sites showed ΔEads of −80.8 kJ mol−1, stronger than that of Cu, Zr–O and Zr–O–Zr. This indicates that the adsorption of CO2 at the Cu–ZrO2 interfacial sites is significantly stronger than on the Cu NPs. The identification of the active interfacial sites allowed for examining the possible reaction mechanisms. CO2 is activated by charge transfer from the copper particles to its carbon atom, while the partially negative oxygen interacts with the high valent cation, Zr4+. An increase in the Cu–ZrO2 interfacial sites enhances the activity of the catalyst; however, when only Cu–ZrO2 interfaces (without additional Cu particles) were present, only the CO2-to-CO reduction was catalysed and not the methanol production, since Cu is needed for the H2 dissociation. On the other hand, in the presence of ZnOx, CO2 was strongly adsorbed and activated on ZnOx in addition to unsaturated Zr sites in the SBU, while hydrogen was activated on Cu to provide hydrogen atom by dissociation, leading to hydrogen spillover to react with the activated CO2 which is already shown in the previous study.38
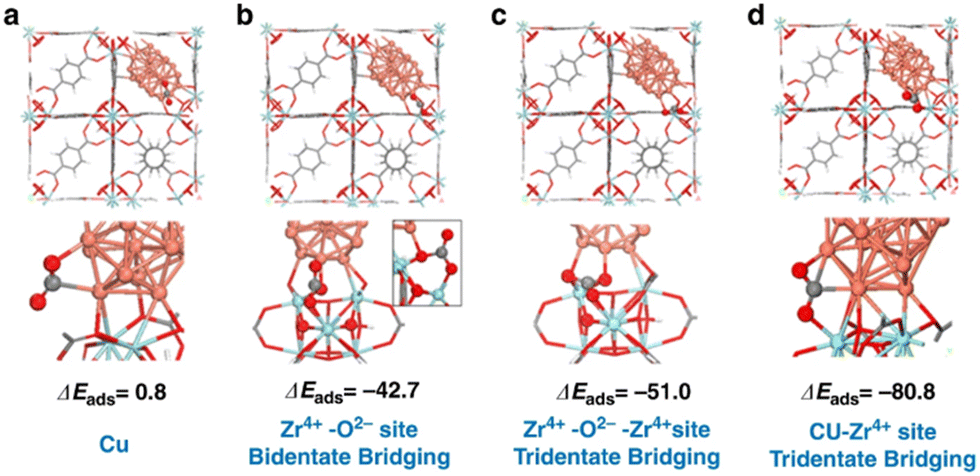 | ||
| Fig. 12 Adsorption energies (ΔEads in kJ mol−1) of CO2 at the Cu/UiO-66 interface (a) Cu only sites, (b) Zr4+–O2 sites in a bidentate bridging mode, (c) Zr4+–O2–Zr4+ sites in a tridentate bridging mode, and (d) Cu–Zr4+ (ZrO2) interfacial sites. Reproduced from ref. 43 (https://www.nature.com/articles/s41467-020-19438-w) with the permission of unrestricted use (https://creativecommons.org/licenses/by/4.0/). | ||
Also, some studies investigated the effects of the particle size of metal sites in MOF-based composite catalysts on CO2 hydrogenation to methanol, which could have significant effects on the performance of the catalyst.38,40,42–44 For example, Yang et al. synthesised ultra-small Cu/ZnOx nanoparticles within 1.2–2 nm in UiO-66 via the double solvent method for CO2-to- methanol hydrogenation to overcome the agglomeration of metal particles and phase separation between Cu and ZnOx at the high reaction temperatures, and consequently resulting in improved stability during the reaction.44 The reported Cu particle size for MOF-based composite catalysts containing Cu or Cu/Zn at different reaction temperatures is displayed in Fig. 13. Although a clear trend in the effect of the varying Cu size on CO2 conversion is not apparent in Fig. 13a, we would like to point out that the size of Cu particles for most catalysts that provided conversion over 10% was 4 nm. Furthermore, 2 nm is the most common particle size for Cu in MOF-based composite catalysts that exhibited selectivity toward methanol over 50% (Fig. 13b). It should be noted that the latter size corresponds well with the pore size of the UiO-67 framework, indicating that the catalyst particles were embedded in the pores of the supporting framework, at least prior to the catalyst testing. We already noted that the size of the Cu NPs on Al2O3 was crucial for the catalyst activity for methanol production, and a decrease in the size of Cu NPs smaller than 8 nm decreased the turnover frequency (TOF),25 thereby supporting our findings. Even though the confinement effects increased the activity of the catalysts, decreasing the size of metal NPs could cause adverse effects on the activity.38,41
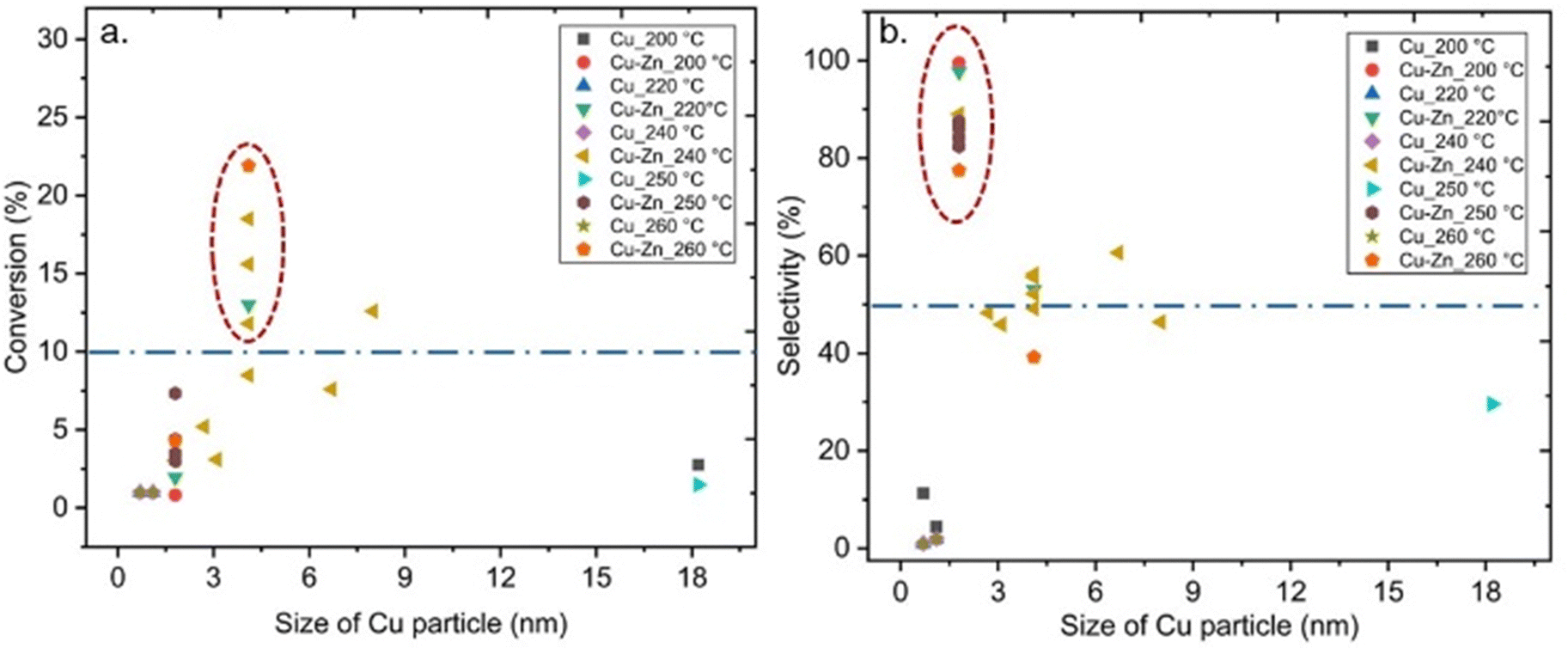 | ||
| Fig. 13 (a) Conversion of CO2versus size of Cu particle. (b) Selectivity towards methanol versus size of Cu particle. Blue dashed lines represent 10% conversion and 50% selectivity. | ||
The particle size of the Cu particles could be varied by different parameters, including Cu loading. XANES and EXAFS analyses revealed that low loading (0.04 wt% Cu) of Cu in Cu/UiO-66 only yielded atomically dispersed Cu on the Zr nodes in Cu/UiO-66, whereas higher Cu loading (7.6 wt% Cu) led to the formation of metallic Cu nanoparticles mainly with a small proportion of Cu–ZrO2 interfaces only.43 In addition, when the size of the Cu NPs was increased by further increasing the Cu loading, a decrease in methanol yield was observed. The influence of Cu loading on the particle size of Cu is reported in Fig. 14a. No direct correlation is observed between the size of Cu metal and Cu loading for the catalysts containing only Cu active sites. On the other hand, an increase in Cu loading increases the size of Cu particles for the catalysts containing Cu and Zn (Fig. 14a-inset). Regardless of the effect on the metal particle size, the extent of guest loading itself could affect the performance since an increase in loading could increase the number of active sites, however, there are no obvious trends between Cu metal loading and conversion or selectivity (Fig. 14b and c, respectively).
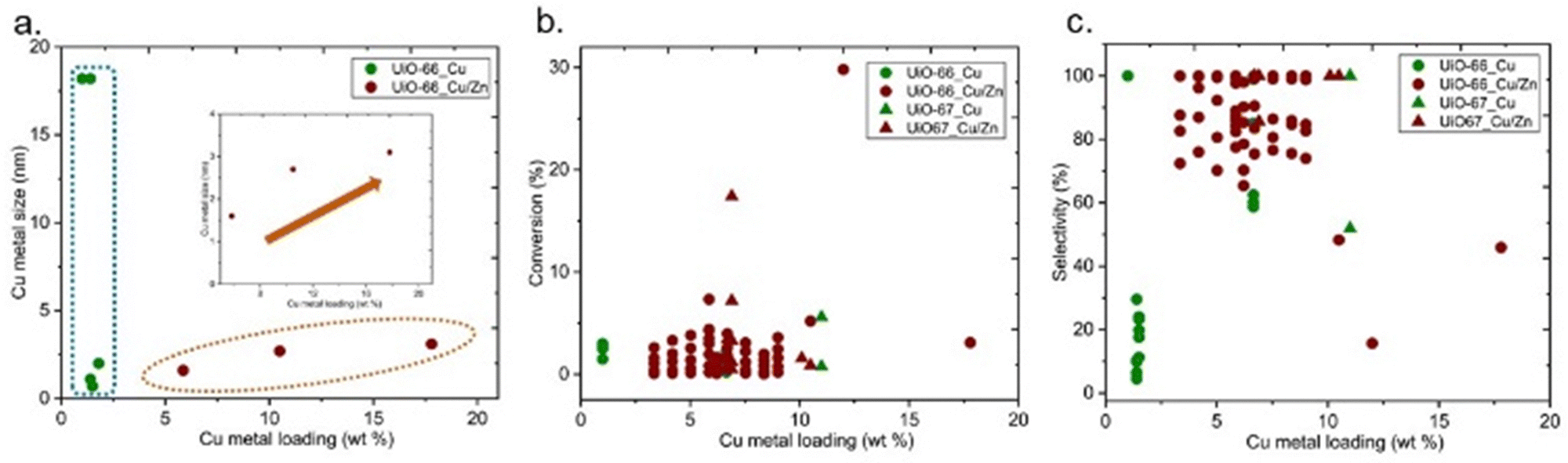 | ||
| Fig. 14 (a) Cu metal size versus Cu metal loading (b) conversion of CO2versus Cu metal loading (b) selectivity towards methanol versus Cu metal loading. | ||
In summary, our critical literature analysis shows that catalyst particles may be active when embedded in the pores of MOFs or otherwise encapsulated therein, even though this size regime is well below what is considered optimal – for Cu the least.25 In fact, small, 2 nm-sized nanoparticles have demonstrated the highest methanol selectivity, which suggests some degree of contribution from the MOF host. To further evaluate this, we continue our discussion with the possibility for MOF-metal SMSI and its impact on the direct hydrogenation of CO2 to methanol.
Zhang et al. investigated the construction of Zn2+–O–Zr4+ motifs in MOF-808 to gain better insights into the structural requirement for CO2 hydrogenation to methanol.37 The Zn2+–O–Zr4+ active sites on the Zr6 nodes were formed in situ after exchanging the μ3-OH proton with ZnEt2. The MOF-808-Zn catalyst exhibited over 99% selectivity towards methanol at 250 °C, the yield was stable for 100 hours, and the activity of the catalyst did not decrease during the reuse of MOF-808-Zn for a second time. No structural change and aggregation of Zn was observed according to powder diffraction (PXRD) patterns and transmission electron microscopy (TEM) images. In addition, XAS results supported the presence of Zn2+–O–Zr4+ centres, and hydrogen isotope scrambling showed H2 scission, presumably by the Zn2+ centres. Furthermore, they suggested that open Zr4+ sites were critical for methanol production, as this was not observed on Zn2+ centres supported on Zr nodes of other MOFs without open Zr4+ sites, which was reported in previous studies. In situ diffuse reflectance infrared spectra and DFT calculations indicated that the activated CO2 reacted with atomic hydrogen, forming a formate intermediate on the Zn2+ sites. This process is followed by hydrogenation to dioxo methylene, formaldehyde, methoxy, and eventually methanol. High catalytic efficiency and stability were attributed to the strong synergy between Zn2+ and unsaturated Zr4+ on the Zn–O–Zr interface.
Lastly, Gutterød et al. studied Pt NPs embedded inside UiO-67 with mixed linkers of BDC and BPDC. They reported 13% and 40% selectivity toward methanol at the mild reaction conditions of 170 °C and 8 or 30 bar, respectively.45,46 The Pt NPs are believed to be decorated by Zr clusters close to the Pt, which are partially or fully detached from the framework during the growth of the Pt NP. The synergy between the Pt NPs and the unsaturated Zr nodes is key to producing methanol, as demonstrated by combining coupling Steady-State Isotope Transient Kinetic Analysis (SSITKA) and operando IR analysis with DFT calculations. The proposed mechanism for CO2 hydrogenation to methanol occurs through the formate pathway at the interface of a zirconia node with a missing linker defect concentration of 0.4 per cluster, providing the necessary unsaturated sites, and the platinum NPs. Firstly, CO2 is adsorbed on the zirconium cluster near the Pt NPs, which adsorb and dissociate hydrogen molecules. Then, the hydride transfers to the CO2 molecule, forming a formate specie, which coordinates in a bidentate manner after removing the hydroxyl group (Fig. 15). The rate-determining step of the mechanism is one of the subsequent hydrogenation steps of the formate species. The importance of the interface between the Pt NPs and the Zr node with a missing linker defect was further established when Gutterød et al. investigated the effect of reducing the number of missing linker defects in another study on Pt NP-containing UiO-67. The formation rate of methanol was diminished as the amount of missing linker defects was reduced, which strongly supports the hypothesis that the unsaturated Zr4+ sites are actively engaged in the catalytic cycle.
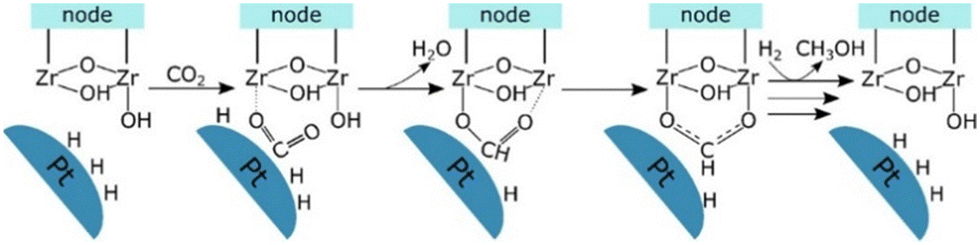 | ||
| Fig. 15 Schematic presentation of the postulated reaction mechanism of CO2 hydrogenation to the formate intermediate in CH3OH formation at the Pt–Zr node interface. Reprinted with permission from ref. 45. Copyright 2020 American Chemical Society (https://pubs.acs.org/doi/10.1021/jacs.9b10873). | ||
In summary, methanol formation has occurred on Cu, Cu/Zn, Zn, and Pt metal containing Zr6-MOFs. The role of the metal is to dissociate H2, while the subsequent H transfer to CO2 and consequent HxCO2 intermediate formation takes place on the open Zr sites. Considering that the integrity of the MOF structure does not appear to be the limiting factor, as a significant decrease in surface area41 and linker functionalisation40 did not drastically affect the activity of the catalysts, it is hypothesised that it is the metal guest–ZrO2 interface, which is the key site for the reaction to occur.
Reaction parameters-performance relationships
In addition to the nature of the catalysts discussed above, the reaction conditions, specifically the reaction temperature and pressure, play crucial roles in the methanol production from CO2 hydrogenation. According to the literature data on Zr6 MOF-based composite catalysts, the trends between reaction conditions (temperature and pressure) on CO2 conversion and selectivity toward methanol are reported in Fig. 16. Overall, there is a significant divergence in the reaction conditions reported, including but not restricted to the applied reaction temperatures and pressures varying from 170 °C to 300 °C and from 1 to 40 bar, respectively. In general, an increase in reaction temperature increases the reaction rate and thus conversion. On the other hand, CO2 hydrogenation to methanol is an exothermic reaction and thermodynamically favourable at lower temperatures. However, lower reaction temperatures would not be sufficient to overcome the activation barrier for the reaction. The experimental results show that over 10% conversion of CO2 could be achieved above 240 °C (Fig. 16a). Even though the 10% conversion of CO2 at 240 °C is below the thermodynamic limitations, which is approximately 20% at 30 bar, it is well above the average of the reported conversions using Zr6-MOF-based catalysts. On the other hand, no common trend is observed for selectivity to methanol (Fig. 16b) as a function of temperature, even though it is expected that higher reaction temperatures could have adverse effects on the selectivity from thermodynamics considerations. Although theoretical thermodynamic calculations could provide information regarding the optimum reaction temperature, the fact that the optimum reaction parameters also depend on the type of catalytic system used needs to be considered. The methanol production from the hydrogenation of CO2 is favourable at high pressures, in accordance with Le Chatelier's principle (eqn (2)). According to previous experimental data, the conversion of CO2 is above 10% at pressures over 30 bar (Fig. 16c), and the selectivity towards methanol is below 50% under 30 bar, except for one data point, which showed 100% selectivity at 10 bar by using Cu NPs encapsulated into UiO-66 as catalyst (Fig. 16d). These trends indicated that pressure plays a vital role for methanol production.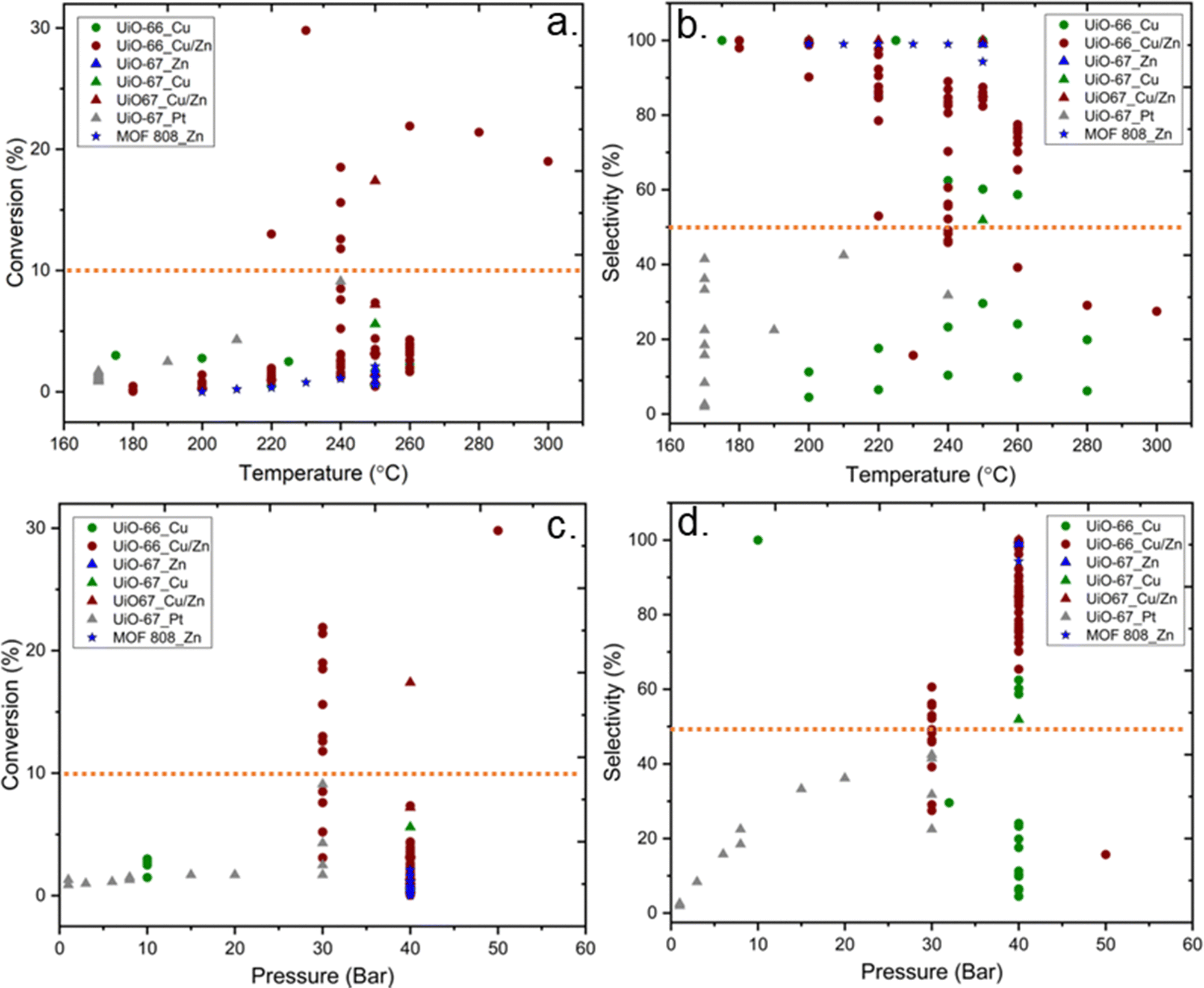 | ||
| Fig. 16 The effects of temperature and pressure on the performance (a) conversion versus temperature (b) selectivity toward methanol versus temperature (c) conversion versus pressure (d) selectivity toward methanol versus pressure. Orange dashed lines represent 50% selectivity toward methanol and 10% conversion. | ||
It has been reported that in addition to the reaction temperature and pressure, space velocity of reactant feed also has an impact on the conversion and product selectivity; however, the lack of information, including reactor dimensions, catalyst mass and flow rate, does not allow proper performance comparison among the reported studies. A table containing the type of MOF structure, active metals, and reaction conditions is provided in the ESI.†
Summary and outlook
In this review, we summarise the effects of structural properties and reaction parameters on CO2 hydrogenation to methanol using Zr6-based MOF structures with incorporated metal particles as composite catalysts by the critical analysis of the reported experimental and theoretical data. Although a direct overarching correlation between the structural properties and catalyst performance cannot be established for UiO-66, UiO-67 and MOF 808, such matrices have been found to be valuable substrates overall. In addition, the data underlines a crucial impact of the nature of the active sites on methanol production. The results suggest that the incorporation of the active metals into the MOF structures often results in structural deformation, laid bare by a drastic decrease in the BET surface area. A direct quantity of Cu–ZrO2 interfaces in MOFs had a significant impact on the activity and selectivity of catalysts for CO2 hydrogenation to methanol since the oxidised form of Cu may stabilise the formate intermediates while the metallic Cu activates the hydrogenation through dissociation of hydrogen. The MOFs with incorporated Cu/Zn active sites show the best performance. Not only the Cu–ZrO2 interfaces but also the synergy of Cu and ZnOx could play an important role in the reaction. In addition, the effect of the NPs size on performance is observable. While the literature suggested 8 nm as the optimum size of active metals on conventional substrates, the optimum size for the metal particles for Zr6-MOF-based catalysts was found to be around 2–4 nm. In addition to structural characteristics, the influence of reaction parameters on performance has also been hereby analysed. While an increase in temperature and pressure increased the conversion of CO2, only the pressure increase obviously enhanced the selectivity toward methanol.To conclude, recent studies provided a better understanding of Zr6-MOF-based composite catalysts for the hydrogenation of CO2 to methanol, highlighting the MOF-based catalysts as promising for the reaction because of their unique features. The strong interaction between metal NPs and metal oxide clusters stabilised the NPs and prevented their agglomeration, thus enhancing catalyst stability for up to 100 hours. The well-defined local structure around the SBU in MOFs provided a unique opportunity to reveal the structural requirement for synergistic catalysis. The MOFs allowed tuning the catalytic properties of the material, including the size of active metal sites and quantity of metal–Zr6 SBUs interface, by creating missing linker defects. On the other hand, the pore size of MOF structure could be a limitation to obtaining optimum size active metal, and encapsulating metal NPs into MOFs could allow tuning and increasing the size. In addition, MOFs effectively improved the dispersion of catalytic sites because of their rich porous structure and high surface area.
One thing that has been laid bare through our critical review of previous literature is the lack of standardised methodologies enabling the comparison of data reported by various groups. This not only makes writing a review difficult, but it also hinders developing an in-depth understanding of the performance of catalysts, and, thus ultimately, to optimise catalysts for the direct CO2-to-methanol conversion. For this reason, we suggest some practices for data acquisition and reporting, both on the materials and testing conditions, post-synthesis, during operation, and post-mortem, to improve the comparability of test results.
Firstly, it should be recalled that the defect chemistry of Zr6-MOFs affects the materials’ properties, stability, and catalytic performance. For this reason, it is of the utmost importance that the nature and concentration of defects be analysed in order to evaluate the catalyst performance. Although Zr6-MOFs practically do not exist in a perfect crystalline, defect-free form, their defect chemistry is fairly well understood and explored, consequently, with a careful analysis of their composition including a combined nuclear magnetic resonance (NMR) spectroscopy and thermogravimetric analysis (TGA) profiling, one can accurately establish the nature and concentration of defects, which is fundamental to rationalising catalyst performance in systems containing these materials. It is, therefore, crucial that efforts are being made to this end and that the very least, post-digestion NMR and TGA data is reported on the synthesised MOF-containing catalyst.
It should be noted that there needs to be more in situ and operando data reported in the literature. This is problematic as without the understanding of the catalyst behaviour in its active form, it is impossible to rationally improve, let alone optimise and upscale, the materials. We realise that such experiments can and should be carried out at large-scale facilities and they may be challenging in pinpointing active species, while the reconciliation of the space-time velocities achievable in experimental conditions at the laboratory test rigs and the synchrotron beamlines may also be a limiting factor for developing mechanistic insights. Nevertheless, an increased volume of data obtained in well-designed and well characterised conditions, and supported through theoretical modelling is necessary to uncover the underlying reaction mechanism. Regarding the catalyst performance, we would like to emphasise that both conversion and selectivity, at distinct conditions (for instance at 200 °C and 30 bar) should be reported in order to allow for an intelligible comparison of the various catalysts. It would also be helpful if a set of standard experimental conditions were to be established.
Finally, when it comes to the stability of the catalyst, which is another crucial key performance indicator, the scientific reports are very few and far between. In particular, there are precious few publications analysing the catalyst for an extended period of time. Crucially, the post-catalysis and post-mortem characterisation of the catalysts should be reported, including crystal integrity, as inferred from PXRD and BET surface area analysis, and compared with the as-synthesised sample, thermal and/or elemental analysis, and spectroscopic evaluation including but not restricted to NMR. Furthermore, comparing the TEM micrographs of the MOF-supported catalyst particles before and after catalysis is a reliable way of pinpointing significant sintering of the metal particles. Evidently, other techniques, such as EXAFS and atomic pair distribution function, can also be successfully applied to this end.
In summary, we believe that following the above basic principles in terms of catalyst characterisation and catalyst testing would contribute significantly to developing insights into the structure–composition–performance relationships, which is arguably the best way to enable the rational design of more efficient catalysts. In addition, our analysis of the available literature also highlights that Zr6-based frameworks are desirable support materials of catalysts for the direct CO2-to-methanol conversion.
Author contributions
E. T. and D. S. contributed equally to writing the original draft. P. A. S., S. S. and U. O. all contributed to reviewing & editing, and conceptualising the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the primary financial support from the Faculty of Mathematics and Naturals Sciences through their Sustainable Development Initiative. Additionally, D. K. S. and U. O. acknowledge the Research Council of Norway for funding through grant no. 288331 (CO2LO). The authors thank Erlend Aunan for providing illustrations in Fig. 7.References
- S. Bilgen, Renewable Sustainable Energy Rev., 2014, 38, 890–902 CrossRef CAS.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- H.-J. Ho, A. Iizuka and E. Shibata, Ind. Eng. Chem. Res., 2019, 58, 8941–8954 CrossRef CAS.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS PubMed.
- B. L. Salvi and S. Jindal, SN Appl. Sci., 2019, 1, 885 CrossRef CAS.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- A.-H. Liu, R. Ma, C. Song, Z.-Z. Yang, A. Yu, Y. Cai, L.-N. He, Y.-N. Zhao, B. Yu and Q.-W. Song, Angew. Chem., Int. Ed., 2012, 51, 11306–11310 CrossRef CAS PubMed.
- Z.-Z. Yang, L.-N. He, Y.-N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971–3975 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- B. Hu, C. Guild and S. L. Suib, J. CO2 Util., 2013, 1, 18–27 CrossRef CAS.
- A. Galadima and O. Muraza, Renewable Sustainable Energy Rev., 2019, 115, 109333 CrossRef CAS.
- W. Zhang, Z. Jin and Z. Chen, Adv. Sci., 2022, 9, 2105204 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, 2009, pp. 233–278 DOI:10.1002/9783527627806.ch12.
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168–175 CrossRef CAS.
- S. Perathoner and G. Centi, ChemSusChem, 2014, 7, 1274–1282 CrossRef CAS PubMed.
- G. Leonzio, J. CO2 Util., 2018, 27, 326–354 CrossRef CAS.
- F. Zaera, Coord. Chem. Rev., 2021, 448, 214179 CrossRef CAS.
- S. Zhang, Z. Wu, X. Liu, K. Hua, Z. Shao, B. Wei, C. Huang, H. Wang and Y. Sun, Top. Catal., 2021, 64, 371–394 CrossRef CAS.
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS.
- R. van den Berg, G. Prieto, G. Korpershoek, L. I. van der Wal, A. J. van Bunningen, S. Lægsgaard-Jørgensen, P. E. de Jongh and K. P. de Jong, Nat. Commun., 2016, 7, 13057 CrossRef CAS PubMed.
- X. Yang, X. Ma, X. Yu and M. Ge, Appl. Catal., B, 2020, 263, 118355 CrossRef CAS.
- F. Wang, J. Jiang and B. Wang, Catalysts, 2019, 9, 477 CrossRef CAS.
- X.-J. Kong and J.-R. Li, Engineering, 2021, 7, 1115–1139 CrossRef CAS.
- L. Feng, K.-Y. Wang, X.-L. Lv, T.-H. Yan and H.-C. Zhou, Natl. Sci. Rev., 2019, 7, 1743–1758 CrossRef PubMed.
- M. Li, D. Li, M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS PubMed.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Nat. Commun., 2020, 11, 4068 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- R. J. Marshall, Y. Kalinovskyy, S. L. Griffin, C. Wilson, B. A. Blight and R. S. Forgan, J. Am. Chem. Soc., 2017, 139, 6253–6260 CrossRef CAS PubMed.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef CAS PubMed.
- J. Zhang, B. An, Z. Li, Y. Cao, Y. Dai, W. Wang, L. Zeng, W. Lin and C. Wang, J. Am. Chem. Soc., 2021, 143, 8829–8837 CrossRef CAS PubMed.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS PubMed.
- Z. G. Duma, X. Dyosiba, J. Moma, H. W. Langmi, B. Louis, K. Parkhomenko and N. M. Musyoka, Catalysts, 2022, 12, 401 CrossRef CAS.
- C. E. Pompe and P. Á. Szilágyi, Faraday Discuss., 2021, 231, 371–383 RSC.
- J. Yu, G. Chen, Q. Guo, X. Guo, P. Da Costa and D. Mao, Fuel, 2022, 324, 124694 CrossRef CAS.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS PubMed.
- Y. Zhu, J. Zheng, J. Ye, Y. Cui, K. Koh, L. Kovarik, D. M. Camaioni, J. L. Fulton, D. G. Truhlar, M. Neurock, C. J. Cramer, O. Y. Gutiérrez and J. A. Lercher, Nat. Commun., 2020, 11, 5849 CrossRef CAS PubMed.
- Y. Yang, Y. Xu, H. Ding, D. Yang, E. Cheng, Y. Hao, H. Wang, Y. Hong, Y. Su, Y. Wang, L. Peng and J. Li, Catal. Sci. Technol., 2021, 11, 4367–4375 RSC.
- E. S. Gutterød, A. Lazzarini, T. Fjermestad, G. Kaur, M. Manzoli, S. Bordiga, S. Svelle, K. P. Lillerud, E. Skúlason, S. Øien-Ødegaard, A. Nova and U. Olsbye, J. Am. Chem. Soc., 2020, 142, 999–1009 CrossRef PubMed.
- E. S. Gutterød, S. H. Pulumati, G. Kaur, A. Lazzarini, B. G. Solemsli, A. E. Gunnæs, C. Ahoba-Sam, M. E. Kalyva, J. A. Sannes, S. Svelle, E. Skúlason, A. Nova and U. Olsbye, J. Am. Chem. Soc., 2020, 142, 17105–17118 CrossRef PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221–231 CrossRef CAS.
- A. M. El-Zeftawy, J. King Saud Univ., Eng. Sci., 1995, 7, 209–254 CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- A. J. Shih, M. C. O. Monteiro, F. Dattila, D. Pavesi, M. Philips, A. H. M. da Silva, R. E. Vos, K. Ojha, S. Park, O. van der Heijden, G. Marcandalli, A. Goyal, M. Villalba, X. Chen, G. T. K. K. Gunasooriya, I. McCrum, R. Mom, N. López and M. T. M. Koper, Nat. Rev. Methods Primers, 2022, 2, 84 CrossRef CAS.
- K. Stangeland, H. Li and Z. Yu, Ind. Eng. Chem. Res., 2018, 57, 4081–4094 CrossRef CAS.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
- J. Wambach, A. Baiker and A. Wokaun, Phys. Chem. Chem. Phys., 1999, 1, 5071–5080 RSC.
- C. W. Bale, E. Bélisle, P. Chartrand, S. A. Decterov, G. Eriksson, A. E. Gheribi, K. Hack, I. H. Jung, Y. B. Kang, J. Melançon, A. D. Pelton, S. Petersen, C. Robelin, J. Sangster, P. Spencer and M.-A. Van Ende, FactSage Thermochemical Software and Databases, 2010–2016, Calphad, 2016, vol. 54, pp. 35–53, https://www.factsage.com.
- J. Sehested, J. Catal., 2019, 371, 368–375 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- N. A. Sholeha, H. Holilah, H. Bahruji, A. Ayub, N. Widiastuti, R. Ediati, A. A. Jalil, M. Ulfa, N. Masruchin, R. E. Nugraha and D. Prasetyoko, S. Afr. J. Chem. Eng., 2023, 44, 14–30 Search PubMed.
- W. J. Lee, A. Bordoloi, J. Patel and T. Bhatelia, Catal. Today, 2020, 343, 183–190 CrossRef CAS.
- P. Gao, F. Li, N. Zhao, F. Xiao, W. Wei, L. Zhong and Y. Sun, Appl. Catal., A, 2013, 468, 442–452 CrossRef CAS.
- L. Zhang, Y. Zhang and S. Chen, Appl. Catal., A, 2012, 415–416, 118–123 CrossRef CAS.
- M. Behrens and R. Schlögl, Z. Anorg. Allg. Chem., 2013, 639, 2683–2695 CrossRef CAS.
- S. Natesakhawat, J. W. Lekse, J. P. Baltrus, P. R. Ohodnicki, Jr., B. H. Howard, X. Deng and C. Matranga, ACS Catal., 2012, 2, 1667–1676 CrossRef CAS.
- V. D. B. C. Dasireddy and B. Likozar, Renewable Energy, 2019, 140, 452–460 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- T. Koitaya, S. Yamamoto, Y. Shiozawa, Y. Yoshikura, M. Hasegawa, J. Tang, K. Takeuchi, K. Mukai, S. Yoshimoto, I. Matsuda and J. Yoshinobu, ACS Catal., 2019, 9, 4539–4550 CrossRef CAS.
- D. Kopač, B. Likozar and M. Huš, Appl. Surf. Sci., 2019, 497, 143783 CrossRef.
- A. Karelovic and P. Ruiz, Catal. Sci. Technol., 2015, 5, 869–881 RSC.
- F. Zhang, X. Xu, Z. Qiu, B. Feng, Y. Liu, A. Xing and M. Fan, Green Energy Environ., 2022, 7, 772–781 CrossRef CAS.
- R. Naumann d’Alnoncourt, X. Xia, J. Strunk, E. Löffler, O. Hinrichsen and M. Muhler, Phys. Chem. Chem. Phys., 2006, 8, 1525–1538 RSC.
- J. D. Grunwaldt, A. M. Molenbroek, N. Y. Topsøe, H. Topsøe and B. S. Clausen, J. Catal., 2000, 194, 452–460 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- Y. Choi, K. Futagami, T. Fujitani and J. Nakamura, Appl. Catal., A, 2001, 208, 163–167 CrossRef CAS.
- H. Y. Chen, S. P. Lau, L. Chen, J. Lin, C. H. A. Huan, K. L. Tan and J. S. Pan, Appl. Surf. Sci., 1999, 152, 193–199 CrossRef CAS.
- K. Xiao, Q. Wang, X. Qi and L. Zhong, Catal. Lett., 2017, 147, 1581–1591 CrossRef CAS.
- R. S. Schiffino and R. P. Merrill, J. Phys. Chem., 1993, 97, 6425–6435 CrossRef CAS.
- E. Lam, J. J. Corral-Pérez, K. Larmier, G. Noh, P. Wolf, A. Comas-Vives, A. Urakawa and C. Copéret, Angew. Chem., Int. Ed., 2019, 58, 13989–13996 CrossRef CAS PubMed.
- K. Li and J. G. Chen, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS.
- H. Li, L. Wang, X. Gao and F.-S. Xiao, Ind. Eng. Chem. Res., 2022, 61, 10446–10454 CrossRef CAS.
- I. U. Din, M. S. Shaharun, M. A. Alotaibi, A. I. Alharthi and A. Naeem, J. CO2 Util., 2019, 34, 20–33 CrossRef CAS.
- F. Arena, K. Barbera, G. Italiano, G. Bonura, L. Spadaro and F. Frusteri, J. Catal., 2007, 249, 185–194 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- M. B. Fichtl, D. Schlereth, N. Jacobsen, I. Kasatkin, J. Schumann, M. Behrens, R. Schlögl and O. Hinrichsen, Appl. Catal., A, 2015, 502, 262–270 CrossRef CAS.
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Ind. Eng. Chem. Res., 2019, 58, 9030–9037 CrossRef CAS.
- A. Prašnikar, A. Pavlišič, F. Ruiz-Zepeda, J. Kovač and B. Likozar, Ind. Eng. Chem. Res., 2019, 58, 13021–13029 CrossRef.
- G. Chen, J. Yu, G. Li, X. Zheng, H. Mao and D. Mao, Int. J. Hydrogen Energy, 2023, 48, 2605–2616 CrossRef CAS.
- J. Ye, M. Neurock and D. G. Truhlar, J. Phys. Chem. C, 2022, 126, 13157–13167 CrossRef CAS.
- E. S. Gutterød, S. Øien-Ødegaard, K. Bossers, A.-E. Nieuwelink, M. Manzoli, L. Braglia, A. Lazzarini, E. Borfecchia, S. Ahmadigoltapeh, B. Bouchevreau, B. T. Lønstad-Bleken, R. Henry, C. Lamberti, S. Bordiga, B. M. Weckhuysen, K. P. Lillerud and U. Olsbye, Ind. Eng. Chem. Res., 2017, 56, 13206–13218 CrossRef.
- E. Aunan, C. W. Affolter, U. Olsbye and K. P. Lillerud, Chem. Mater., 2021, 33, 1471–1476 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- O. Halbherr and R. A. Fischer, Defects and Disorders in MOF, The Chemistry of Metal–Organic Frameworks, 2016, pp. 795–822 DOI:10.1002/9783527693078.ch26.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- D. K. Sannes, S. Øien-Ødegaard, E. Aunan, A. Nova and U. Olsbye, Chem. Mater., 2023, 35, 3793–3800 CrossRef CAS.
- X. Feng, J. Hajek, H. S. Jena, G. Wang, S. K. P. Veerapandian, R. Morent, N. De Geyter, K. Leyssens, A. E. J. Hoffman, V. Meynen, C. Marquez, D. E. De Vos, V. Van Speybroeck, K. Leus and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 3174–3183 CrossRef CAS PubMed.
- P. Chammingkwan, G. Y. Shangkum, L. T. T. Mai, P. Mohan, A. Thakur, T. Wada and T. Taniike, RSC Adv., 2020, 10, 28180–28185 RSC.
- Y. Feng, Q. Chen, M. Jiang and J. Yao, Ind. Eng. Chem. Res., 2019, 58, 17646–17659 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- G. Kaur, S. Øien-Ødegaard, A. Lazzarini, S. M. Chavan, S. Bordiga, K. P. Lillerud and U. Olsbye, Cryst. Growth Des., 2019, 19, 4246–4251 CrossRef CAS.
- H. H. Mautschke, F. Drache, I. Senkovska, S. Kaskel and F. X. Llabrés i Xamena, Catal. Sci. Technol., 2018, 8, 3610–3616 RSC.
- K. Hemmer, M. Cokoja and R. A. Fischer, ChemCatChem, 2021, 13, 1683–1691 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00345k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |