
Accessing secondary amine containing fine chemicals and polymers with an earth-abundant hydroaminoalkylation catalyst†
Manfred
Manßen‡
 ,
Sabrina S.
Scott‡
,
Danfeng
Deng
,
Cameron H. M.
Zheng
and
Laurel L.
Schafer
*
,
Sabrina S.
Scott‡
,
Danfeng
Deng
,
Cameron H. M.
Zheng
and
Laurel L.
Schafer
*
Department of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada. E-mail: schafer@chem.ubc.ca
First published on 15th March 2023
Abstract
Titanium-catalyzed hydroaminoalkylation has emerged as an atom-economical, earth-abundant synthesis of N-containing products. Secondary amines are added to diverse alkenes with branched regioselectivity by a catalytic system that is assembled in one step using Ti(NMe2)4 and urea ligand. Both 1,1- and 1,2-disubstituted alkenes are aminated with this titanium catalyst. Sterically enhanced aryl and alkyl amines are installed onto silylether functionalized alkenes to synthesize precursors for biologically active substrates. Regioselective formation of branched amine functionalized materials are prepared from vinyl terminated polypropylene to access aminated materials. Such materials are potentially useful adhesives, compatibilizers, and reactive macromolecules to fabricate functional materials. A life cycle analysis of competing tantalum and titanium catalysts is provided to quantify the environmental impacts and demonstrate how titanium-catalyzed hydroaminoalkylation is a “greener” solution to amine terminated polypropylene synthesis.
Introduction
Transition metal catalysts are under scrutiny as they often are reliant on depleting resources1 and have the potential to release toxic metals2 into the environment. Therefore, the chemistry community has focused efforts to develop catalytic strategies that use earth-abundant, non-toxic transition metals.3 Generally, the first row of transition metals on the periodic table are abundant in the earth's crust and several are considered non-toxic. Of these, industry widely uses titanium, the second most abundant transition metal,4,5 for a variety of important synthetic transformations. The system Cp2TiCl2/MOA is used in Ziegler–Natta polymerisation of ethylene and propylene, producing 10 million tonnes collectively of the respective polymers.6–15 Single electron titanocene chemistry has been used to synthesize a range of N-heterocycles in a single-step.16–19 Sharpless epoxidations rely on Ti(OiPr)4 for its conversion of allylic alcohols to epoxyalcohols and are used in the total synthesis of natural product derivatives and antibiotics.20,21 Titanium is also featured in well-established catalytic transformations such as the Kulinkovich22 and Pauson–Khand reactions,23,24 and more recently in the catalytic synthesis of pyrroles.25–29 Titanium is a broadly useful earth-abundant transition metal catalyst making it a promising candidate to develop its use for other metal-catalyzed transformations.Titanium hydroaminoalkylation catalysts have been developed to synthesize amines from unactivated alkene and secondary amine feedstocks.28–38 Early transition metal catalyzed hydroaminoalkylation is a unique amination method because amines do not need protecting groups nor prefunctionalized coupling partners.32 Hydroaminoalkylation is an atom economical reaction operating by activating the Csp3–H bond α to the amine nitrogen to install across an alkene (Scheme 1a). This transformation is broadly useful for amine synthesis as it is compatible with a breadth of amine and alkene substrates.39 Generally, unprotected amines are challenging to use in catalysis because their polarity, basicity, and nucleophilicity make them incompatible with many catalysts and/or mechanisms that result in catalyst death or undesired byproducts.40 However, amines are prevalent motifs in fine chemicals, agricultural products, pharmaceuticals, and materials.
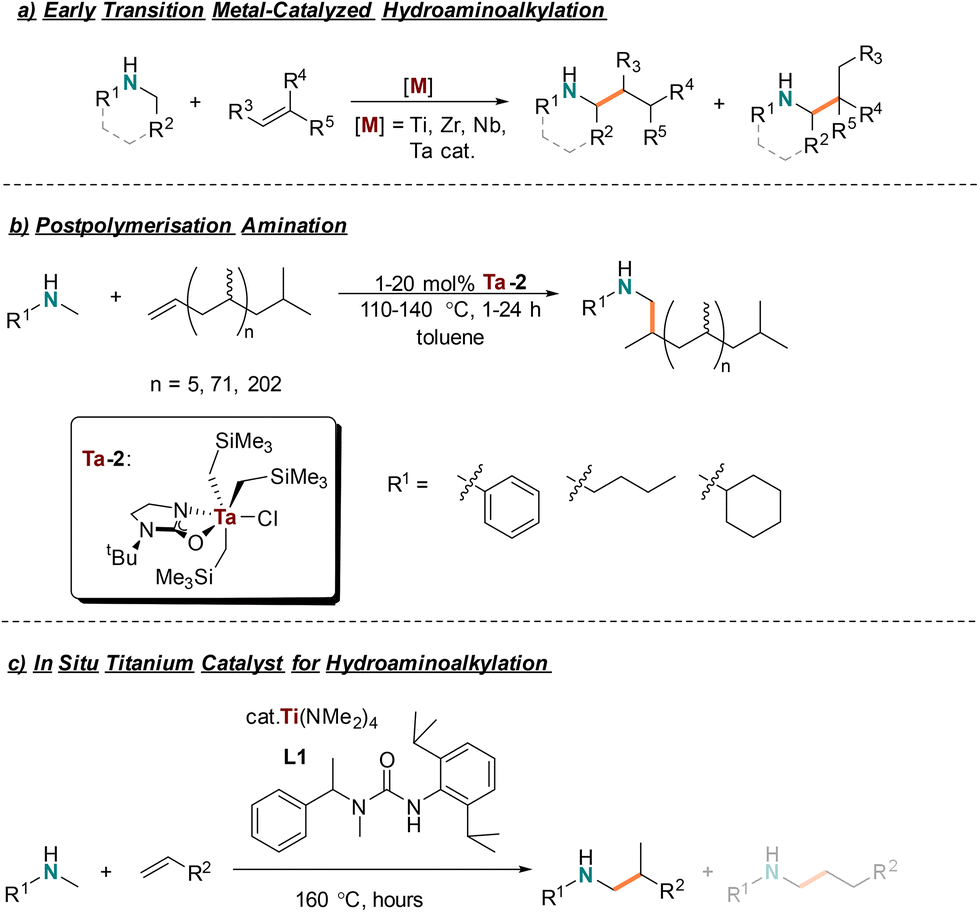 | ||
| Scheme 1 (a) General concept of early transition metal-catalyzed hydroaminoalkylation; (b) postpolymerisation amination using tantalum-catalyzed hydroaminoalkylation; (c) our in situ titanium catalyst system for hydroaminoalkylation. | ||
Industry relies on established methodologies such as hydroformylation followed by reductive amination, otherwise known as hydroaminomethylation, to access these products.40–42 These transformations often rely on rhodium hydroformylation catalysts to access linear products preferentially and can generate stoichiometric waste. Branched selectivity in hydroformylation has been well known for styrene and vinyl acetate for several decades,43 with more recent works using these methods in branched amine synthesis.44 Substrates such as ortho-(diphenylphosphanyl)benzoyl esters,45N-vinylphthalimides,46 methyacrylates, and 2-vinylnaphthalene47 have also been reported with modest branched selectivity in hydroformylation. To date, there are no reported hydroaminomethylation protocols featuring excellent branched selectivity of unactivated alkenes or vinyl terminated polyolefins. Resolving this limitation with hydroaminoalkylation offers an alternative disconnection to access amines and amine terminated polymers. Therefore, regioselective titanium catalyzed hydroaminoalkylation is a promising green methodology to selectively substitute amines and amine containing materials.
To this day, a collection of highly active titanium catalysts for hydroaminoalkylation have been developed – especially by the Doye group.33,35,38,48–55 Recently, our group developed a readily assembled titanium catalyst using commercially available Ti(NMe2)4 and an acyclic urea ligand (L1) (Scheme 1c).31 It has comparable reactivity to previous titanium catalysts and best-in-class tantalum hydroaminoalkylation catalysts,56,57 yet provides a “green” advance because of its simplified ligand synthesis and earth abundant metal precursor choice. As shown previously, this titanium catalyst was useful with aryl, alkyl, silyl, and silylether functionalized terminal alkenes.31 This same catalyst could be used to make monoaminated monomers, known to be useful for the synthesis of amine containing polymers produced by ring opening metathesis polymerisation.58–60 This paper discloses an extended substrate scope, including reactions of both aryl and sterically diverse alkyl amine substrates for use in both terminal and internal alkene hydroaminoalkylation. This work also reports the first example of using a titanium catalyst for challenging postpolymerisation hydroaminoalkylation of vinyl terminated polypropylene (VTPP) to produce branched amine-terminated functionalized oligomers (Scheme 1b).61,62 Unlike small molecule hydroaminomethylation to access branched products, there are no examples of post-polymerization hydroaminomethylation. Such materials are potentially industrially relevant, as they can potentially be used as surface modifiers63–67 and responsive materials.64,68,69 Further, a life cycle analysis (LCA) illustrates how using titanium catalysts and simple ligand syntheses can reduce the environmental impact of amine terminated polypropylene synthesis.
Results and discussion
As previously reported, a titanium catalyst can be prepared in situ from the commercially available and inexpensive titanium amido precursor, Ti(NMe2)4, and a sterically demanding unsymmetric urea ligand L1 (Scheme 1c).31 The electrophilic character at the metal center is enhanced by the electron withdrawing ligand and steric bulk is optimized for improved catalyst reactivity. Ligand L1 is prepared quickly, using appropriate safety measures for working with triphosgene, to access up to 22 g of L1 in a single batch. This catalyst system is known to be effective with various activated and unactivated terminal alkenes.31 Here, an expanded substrate scope with respect to both alkene and amine is presented, including regioselective hydroaminoalkylation of macromolecular VTPP to give uniquely the branched product.Scope of alkenes
The utility of our in situ titanium hydroaminoalkylation catalyst had previously been reported for terminal and strained cyclic alkenes. Here our catalyst was tested with various internal and sterically congested disubstituted alkenes, which are known to be particularly challenging substrates for hydroaminoalkylation. These substrates expand on the alkene scope for the Ti(NMe2)4/L1 system reported in 2021.31 A consideration of the reaction mechanism shows how alkene steric bulk can impact reactivity in the insertion step of the reaction (Scheme 2, A–B).28,29,32,70,71 NMR tube scale reactions (0.5 mmol) were completed with N-methylaniline as the consistent amine substrate.28,30,35,37,38,57,72 The yields of these reactions were determined by 1H NMR spectroscopy by integrating the characteristic signals of the aniline starting material and product (e.g. the resonances of the ortho protons on the aromatic amines at ca. δ = 6.3 ppm) and comparing with the internal standard, 1,3,5-trimethoxybenzene (TMB) resonance at δ = 6.2 or 3.3 ppm. The reaction times and temperatures were optimized by selection of catalyst loading to afford yields ranging from 30% to 99% (Chart 1). | ||
| Scheme 2 General mechanism for the titanium-catalyzed intermolecular hydroaminoalkylation of internal alkenes. | ||
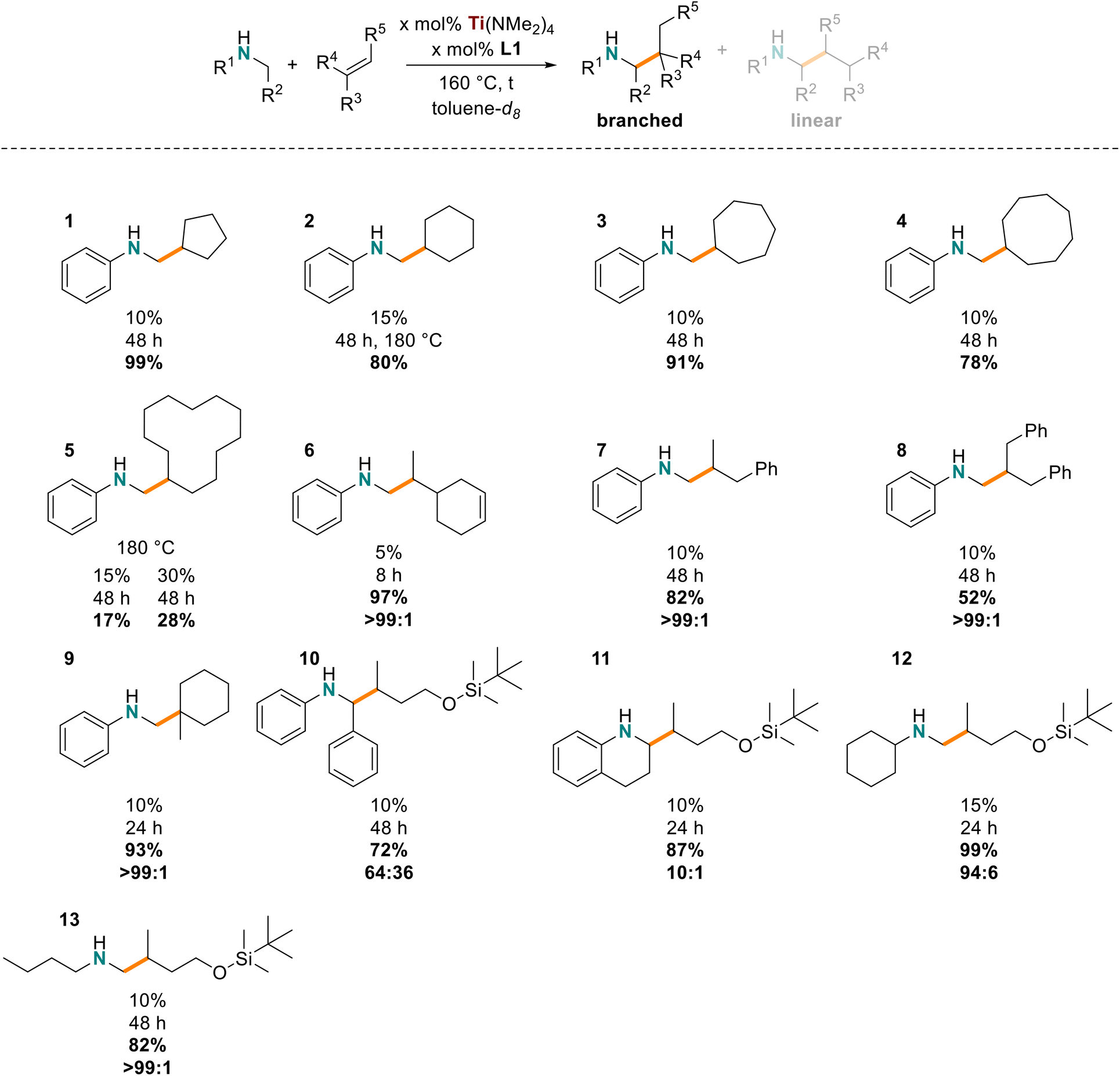 | ||
| Chart 1 Alkene and amine substrate scope (catalyst loadings, reaction times, yields, regioselectivities, major product shown). Reaction conditions: amine (0.5 mmol), alkene (0.5 mmol), toluene-d8 (0.7 mL), 1,3,5-trimethoxybenzene (0.17 mmol), catalyst loadings as shown. Yields determined by 1H NMR spectroscopy and GC analysis. The regioselectivities for each product were determined by GC analysis. All data presented have been collected in duplicate or triplicate to ensure consistency of results. | ||
First, the cyclic internal alkenes were tested (Chart 1, 1–5), and reactions with cyclopentene (1), cyclohexene (2), cycloheptene (3), and cyclooctene (4) had optimal yields (78–99%) with 10–15 mol% catalyst, 160–180 °C, and 48 h.28,37 To form 2, the catalyst loading and temperature had to be slightly increased to 15 mol% and 180 °C to realize 80% yield likely because of the low ring strain in cyclohexene. Notably, the cyclododecene product (5) had much lower yields (15 mol% catalyst: 17% yield; 30 mol% catalyst: 28% yield), which suggests stoichiometric reactivity and not catalytic turnover with this challenging substrate. The reduced reactivity is presumably due to the negligible ring-strain of 10-membered rings and conformers with inaccessible alkenes, pointing to an upper limit of ring size that can be accommodated. Overall, our titanium catalyst is effective for hydroaminoalkylation of 1,2-disubstituted cyclic alkenes, demonstrating an expanded substrate scope and better reactivity with internal alkenes than other Ti(NMe2)4-based catalyst systems.37
As a direct comparison between the internal and terminal alkenes the diene substrate 4-vinylcyclohex-1-ene was used to make one product selectively (6). Catalyst loadings and reaction times were significantly reduced (97%, b/l = 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5 mol% catalyst, 160 °C, 8 h),31 only the hydroaminoalkylation of the terminal C
:1, 5 mol% catalyst, 160 °C, 8 h),31 only the hydroaminoalkylation of the terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond was observed, and gave exclusively the branched product in excellent yields. This reaction confirms that the 1,2-disubstituted cyclic alkenes are less favored than monosubstituted alkenes with this catalyst.
C double bond was observed, and gave exclusively the branched product in excellent yields. This reaction confirms that the 1,2-disubstituted cyclic alkenes are less favored than monosubstituted alkenes with this catalyst.
We then tested the titanium catalyst with unactivated acyclic 1,2-disubstituted alkenes, (Z)-β-methylstyrene (7) and a mixture of (Z)- and (E)-β-benzylstyrene (8). A slight favorability (<10%, by integrating the residual styrene) for hydroaminoalkylation of the (Z)-isomer in producing 8 was observed. The corresponding hydroaminoalkylation products of these reactants were obtained in moderate to good yields with excellent regioselectivities (>99:1). No isomerization to the corresponding allylbenzene was observed by GC and NMR analysis. Other titanium catalysts derived from Ti(NMe2)4, had lower reactivity for the β-methylstyrene substrate (64%, 99:1, 10 mol% catalyst, 180 °C, 96 h) and no reactivity reported for β-benzylstyrene.37 These results suggest that 1,1-disubsituted acyclic alkenes are slightly less reactive than cyclic variants likely due to a lack of ring strain. To complement this series of sterically challenging alkene substrates the sterically bulky 1,1-disubstituted alkene, methylenecyclohexane was tested and shown to give 9. This substrate affords exclusively a β-quaternary center when reacted with N-methylaniline and excellent yields were obtained (93%, b/l > 99:1, 10 mol% catalyst, 160 °C, 24 h). This result is comparable to other reports for titanium37 and offers similar yields but longer reaction times when compared to tantalum catalysts.57
For the titanium catalyst to be widely useful synthetically it needs to be functional group tolerant and react with precursors that minimize the steps required to synthesize biologically active substances. Generally, early transition metals are electrophilic and oxophilic, making them prone to inactivity in the presence of nitriles, carbonyls, and alcohols,28,29,32,35,70 but silyl protected alcohols can be used to prevent decomposition.31,73,74 Thus, (but-3-en-1-yloxy)(tbutyl)dimethylsilane was reacted with N-benzylaniline, 1,2,3,4-tetrahydroquinoline, N-methylcyclohexylamine, and N-methylbutylamine to give their respective products 10–13. In all cases the branched products were favored and good to excellent yields for the hydroaminoalkylation products were obtained (72–99%). As the steric bulk increased at the Csp3 centre α to nitrogen, the regioselectivity dropped significantly (1,2,3,4-tetrahydroquinoline: b/l = 10:1; N-benzylaniline: b/l = 64:36). Whereas excellent regioselectivities were obtained with less bulky N-methylcyclohexylamine (b/l = 94:6) and N-methylbutylamine (b/l > 99:1). These are new pairings of sterically demanding and unactivated amines with (but-3-en-1-yloxy)(tbutyl)dimethylsilane, demonstrating that b/l selectivity can be varied by the steric influence of the amine.31
Fine chemical synthetic application
The silylether of 11 was used in a cyclization reaction to generate the corresponding benzoindolizidine, which falls into an interesting class of bicyclic alkaloids prominent in biologically active compounds75–78 such as antitumor agents,79 antimicrobial compounds,80,81 and sodium channel blockers82 that can have rich structural diversity. β-Methylated amine/N-heterocycle products are often targeted because of their “magic methyl” effects in medicinal chemistry,83 notably our approach delivers β-methylated benzoindolizidine.Compound 11 was prepared on a gram scale using neat reaction conditions in a high-pressure vial (Scheme 3). Hydroaminoalkylation products 11 were obtained as a mixture of regioisomers and diastereomers (1.97 g, 82%) and were used without further purification. Benzoindolizidine 14a was prepared in good yield (80%) by cyclizing the precursor silyl protected amino alcohol products using p-toluenesulfonyl fluoride. The desired 14a product was easily separated by column chromatography from the side product benzoquinolizidine 14b (5%). Interestingly, benzoindolizidine 14a was isolated as a diastereomeric mixture with good selectivity for the anti product (1:4.8 syn/anti, confirmed by NOESY NMR spectroscopy). This diastereoselectivity can be rationalized by the defined stereochemistry of the hydroaminoalkylation product, which can be attributed to the diastereoselective alkene insertion step in the formation of the metallacyclic intermediate of hydroaminoalkylation (Scheme 2, B).32,56,70,73 In summary, our method shows that this titanium catalyst provides a two-step synthesis to biologically relevant benzoindolizidine featuring diastereoselective hydroaminoalkylation and complements other enantioselective synthetic pathways.84–86
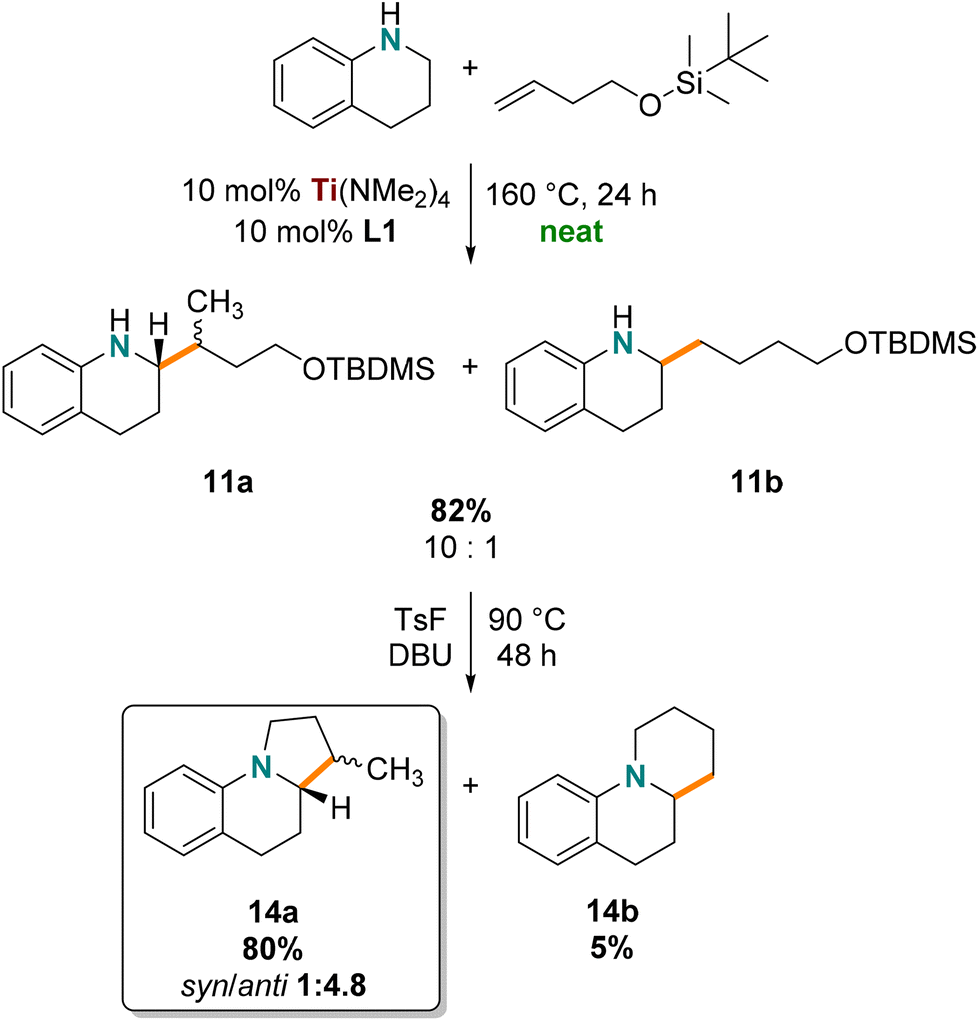 | ||
| Scheme 3 Synthesis of β-methylated benzoindolizidine (branched to linear selectivity given for 11 and diastereoselectivity given for 14). | ||
Functionalized polymers
Postpolymerisation modification strategies for fully saturated hydrocarbon polyolefins suffer from a combination of poor atom-economy, use of toxic transition metals, and lack control over polymer microstructure.87,88 Alkene containing polymers offer a handle for selective functional group incorporation by catalytic hydrofunctionalization reactions, although postpolymerization C–N bond formation, such as in hydroamination, remain elusive.87,88 Alternatively, C–C bond forming reactions like hydroaminomethylation (sequential hydroformylation/reductive amination) can be used in postpolymerization modification to favourably produce the linear product.89,90 To date, there are no examples of postpolymerization modifications featuring exclusively branched selective hydroformylation. As reported, the highest branched selectivity using unactivated alkenes was achieved by Brezny and Landis with a 3:1 b/l ratio from 1-hexene by a rhodium catalyst supported by a phospholane-phosphite ligand.91 Our work on tantalum-catalyzed hydroaminoalkylation of vinyl terminated polypropylene (VTPP) demonstrated that hydroaminoalkylation is a viable postpolymerisation amination method and regioselective formation of the branched products was observed.61 Such postpolymerization modifications can be challenging to catalyze due to the limited solubility and viscosity of the polymer, but the reactivity of the Ti(NMe2)4/L1 system with challenging and diverse alkene substrates31 (vide supra) suggests that this system could also be used with challenging macromolecular substrates for postpolymerisation (Table 1) and would offer a “green” advance.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | VT | R | Mass (g) | Mol% catalyst | Temp (°C) | Yield (g,%) | T g (°C) | T d,50% (°C) |
| Yields determined by in situ monitoring (entries 1–3) or collecting aliquots of the reaction (entries 4–12) and monitoring by 1H NMR spectroscopy. Yields are isolated yields of the materials. Td,50% represents 50% thermal decomposition data. | ||||||||
| 1 | 230 | Ph | 0.10 | 5 | 160 | 0.12, 82% | −63.8 | 234.1 |
| 2 | 230 | n Bu | 0.10 | 10 | 160 | 0.10, 73% | −88.7 | 236.8 |
| 3 | 230 | Cy | 0.10 | 10 | 160 | 0.11, 74% | −77.5 | 233.7 |
| 4 | 350 | Ph | 1.00 | 10 | 160 | 0.88, 68% | −51.1 | 313.3 |
| 5 | 350 | n Bu | 1.00 | 15 | 160 | 0.41, 32% | −69.8 | 271.4 |
| 6 | 350 | Cy | 1.00 | 10 | 160 | 1.04, 77% | −68.3 | 302.4 |
| 7 | 840 | Ph | 5.00 | 10 | 160 | 4.70, 83% | −43.6 | 423.1 |
| 8 | 840 | n Bu | 5.00 | 15 | 160 | 3.77, 68% | −39.2 | 423.2 |
| 9 | 840 | Cy | 5.00 | 20 | 160 | 1.68, 30% | −35.1 | 432.4 |
| 10 | 7200 | Ph | 1.20 | 20 | 160 | 0.92, 76% | −11.7 | 449.5 |
| 11 | 7200 | n Bu | 1.50 | 30 | 180 | 1.15, 76% | −12.5 | 454.3 |
| 12 | 7200 | Cy | 1.25 | 25 | 160 | 0.74, 59% | −9.6 | 446.2 |

VTPPs were chosen as the polymeric alkene because they provide a singular, well-defined alkene to easily monitor in situ by 1H NMR spectroscopy and reactivity can be directly compared with previous tantalum-catalyzed work. From an industrial perspective, this transformation could benefit from a green advance as end-functionalized polymers can serve functions as blend compatibilizers,92,93 surface modifiers63,65–67 and adhesives,64,94 or as reactive95 or responsive macromolecules.68 Four sizes of VTPPs were chosen (Mn = 230, 350, 840, 7200 g mol−1) to investigate if the molecular weight, and physical properties, would impact catalyst activity. The amines chosen were N-methylaniline (HNMePh), N-methylbutylamine (HNMeBu), and N-methylcyclohexylamine (HNMeCy), to vary both electronic and steric factors at the chain termini of the synthesized materials. The resulting amine terminated polypropylene (ATPP) materials are denoted as ATX-R where AT = amine terminated, X = molecular weight, and R = abbreviation for amine substrate.
The reaction conditions with the macromolecules were initially benchmarked using 0.1 g of VT230 and VT350 with one equivalent of the amine substrate. Stoichiometry for all reactions were determined by using Mn as the molecular weight, assuming 100% vinyl termination of each chain. A minimum of 10 h was needed for full conversion to the aminated product at this scale. Additionally, the reactions were found to be sensitive to headspace in the reaction vessel, where no more than 50% of the vessel could be filled with reaction substrates and solvent. This initial optimization showed that 5 mol% Ti(NMe2)4/L1 was suitable for reactions with the activated N-methylaniline and 10 mol% Ti(NMe2)4/L1 loading was required for N-methylbutylamine and N-methylcyclohexylamine (Table 1, entries 1–3).
Amine terminated material synthesis was then scaled to one-gram with VT350, which required a modest increase in catalyst loading to 10 mol% with N-methylaniline and 15 mol% with the more challenging N-methylbutylamine (entries 4 and 5). VT840 was aminated on a five-gram scale with an increase in catalyst loading to 20 mol% for both N-methylbutylamine and N-methylcyclohexylamine (entries 8 and 9). The last sample, VT7200, again needed an increase in catalyst loading to 20 mol%, 30 mol%, and 25 mol% for N-methylaniline, N-methylbutylamine (180 °C), and N-methylcyclohexylamine, respectively (entries 10–12). Although high catalyst loadings were provided for high Mn materials, the wt% fraction of catalyst out of total reactants was low, 2.3 wt% (entry 11); compared to entry 2 which required 15 wt% catalyst. Larger VTPP materials required more solvent to fully solubilize the material and improve reactivity, whereas VT230 and VT350 could be functionalized in solvent-free conditions. Larger materials are more challenging, potentially due to either poor solvation, low concentrations of reactive substrates in solution, or mass transfer issues due to the higher viscosity of the reaction mixture. This trend is also observed with the tantalum catalyzed hydroaminoalkylation which required 20 mol% tantalum catalyst for VTPPs with Mn = 8500 g mol−1.61
Up to 83% (entry 7) isolated yields were obtained for this series of materials. Materials derived from VT230 and VT350 could be purified by filtering through a short silica pad and removing solvents in vacuo. However, AT350-HNMeBu was low yielding because of contaminating L1, which was highly soluble in all butylamine terminated polymers, and could only be partially removed by column chromatography to give a material with a purity of 95%. The thermal degradation event of the ligand that could be observed in the material analyzed by TGA (see ESI†). Materials based on the larger VT840 and VT7200 could be purified by either filtering through a silica pad or precipitating in cold methanol three times and drying in vacuo while heating to 50 °C.
All isolated ATPP materials were characterized by 1H and 13C{1H} NMR, and FTIR spectroscopy, DSC, and TGA. AT230, AT350, and AT840 were also characterized by low-resolution field desorption spectrometry to confirm no chain degradation occurred during the catalytic transformation. In the 1H NMR spectrum of the materials the vinyl resonances at ca. δ = 5.5 and 5.0 ppm were absent in the isolated aminated materials. A new resonance for the methylene α to the amine nitrogen around ca. δ = 3.0 to 2.3 ppm was observed in all the ATPP materials, except AT7200-HNMeBu and AT7200-HNMeCy. The amine end-group was challenging to spectroscopically characterize in these alkyl derivatives due to their low intensities and overlapping signals. The diagnostic doublet of multiplets arises from the geminal protons of the α methylene splitting, if the linear product were formed a triplet would be expected in this region. Derivatives with the N-methylaniline substrate had characteristic 13C{1H} NMR resonances at ca. δ = 51 ppm whereas N-methylbutylamine and N-methylcyclohexylamine substrates had resonances between δ = 54 and 57 ppm for the new methylene carbon α to nitrogen, respectively. Both 1H and 13C{1H} diagnostic resonances are comparable to chemical shifts previously reported where the branched isomer was exclusively observed by DEPT-embedded HSQC NMR spectroscopy.61 In the FTIR spectra of the ATPP materials the CC stretch at 1640 cm−1 and 991 cm−1 is no longer present. The 2° amine N–H stretch of AT230-HNMePh, AT350-HNMePh, and AT840-HNMePh is observed at 3423 cm−1. The alkyl amine materials did not have observable diagnostic N–H stretches in the FTIR spectra.
The thermal characteristics of the materials were characterized by DSC and TGA analysis. There was an overall increase in the glass transition (Tg) of the materials after amination. The most profound increase was in the smaller macromolecular substrates VT230 and VT350, likely due to the largest mass% gained after amination of these materials. The increase in Tg may also be influenced by associative hydrogen bonding interactions through the amine functionality. There is less of an influence on Tg with VT840 and VT7200 due to the lower mass% gained after amination. All materials increased in thermal stability after amination, as characterized by TGA (See ESI†).
Life cycle analysis
Single-step approaches to produce amine functionalized polymers from a hydrocarbon polyolefin are rare. Typical amination strategies are limited because amination methods are multi-step, feature protecting groups, and often produce undesired byproducts.40–42 Previous efforts to produce amine terminated polypropylene (ATPP) using hydroformylation followed by reductive amination gave a mixture of regioisomeric products and efforts featuring tantalum hydroaminoalkylation catalysts (Ta(NMe2)5, Ta(NEt2)2Cl3) required long reaction times and could only be used with a limited amine scope.62 However, hydroaminoalkylation is one of few catalytic amination strategies that can directly install commercial amines onto polymeric alkenes and is the only method that offers good control for the branched product in an atom-economical single step, which we have now achieved with both tantalum and titanium catalysts. Overall, titanium catalyzed hydroaminoalkylation is an improvement as it aligns with several Green Chemistry mandates, where catalyst development focused on earth-abundant transition metals and simple ligand syntheses are key areas for minimizing this reaction's environmental impact. The potential application of this reaction for the commercial synthesis of functionalized oligomers and materials merits a life cycle analysis (LCA).As such, the catalyst based on Ti(NMe2)4/L1 was developed and a life cycle analysis (LCA) was undertaken to demonstrate how it compares with a previously reported optimized tantalum-based hydroaminoalkylation catalyst for the synthesis of branched ATPP (Scheme 4).72 The tantalum and titanium catalyzed hydroaminoalkylation methods were the only ones assessed because these produce the same materials, and LCA requires assessment of the same product to be meaningful. The use of an LCA multivariate assessment tool quantifies the environmental impact of a synthesis, to do this here we are following procedures reported by Mercer et al.96 This includes acidification potential (AP), ozone depletion potential (ODP), smog formation potential (SFP), global warming potential (GWP), human toxicity by ingestion potential (INGTP), human toxicity by inhalation potential (INHTP), persistence (PER), bioaccumulation (ACCU), and abiotic depletion potential (ADP).96 These potentials are scaled based on the mass of a given reagent or solvent that is estimated to be emitted into the environment (or mass of element used for ADP) to produce indices for each metric (Table 2). The indices for each reagent and solvent used in a pathway are then compared to understand problematic aspects of the synthesis, where the summed indices of all reagents and solvents for a single synthetic pathway are used make broad comparisons between alternative routes. An LCA provides insight on hazardous reagents and solvents and where raw material utilization can be improved. This offers a better understanding on the environmental impact of the chemistry than common univariant metrics, like E factor (EF) and atom economy (AE) can afford.
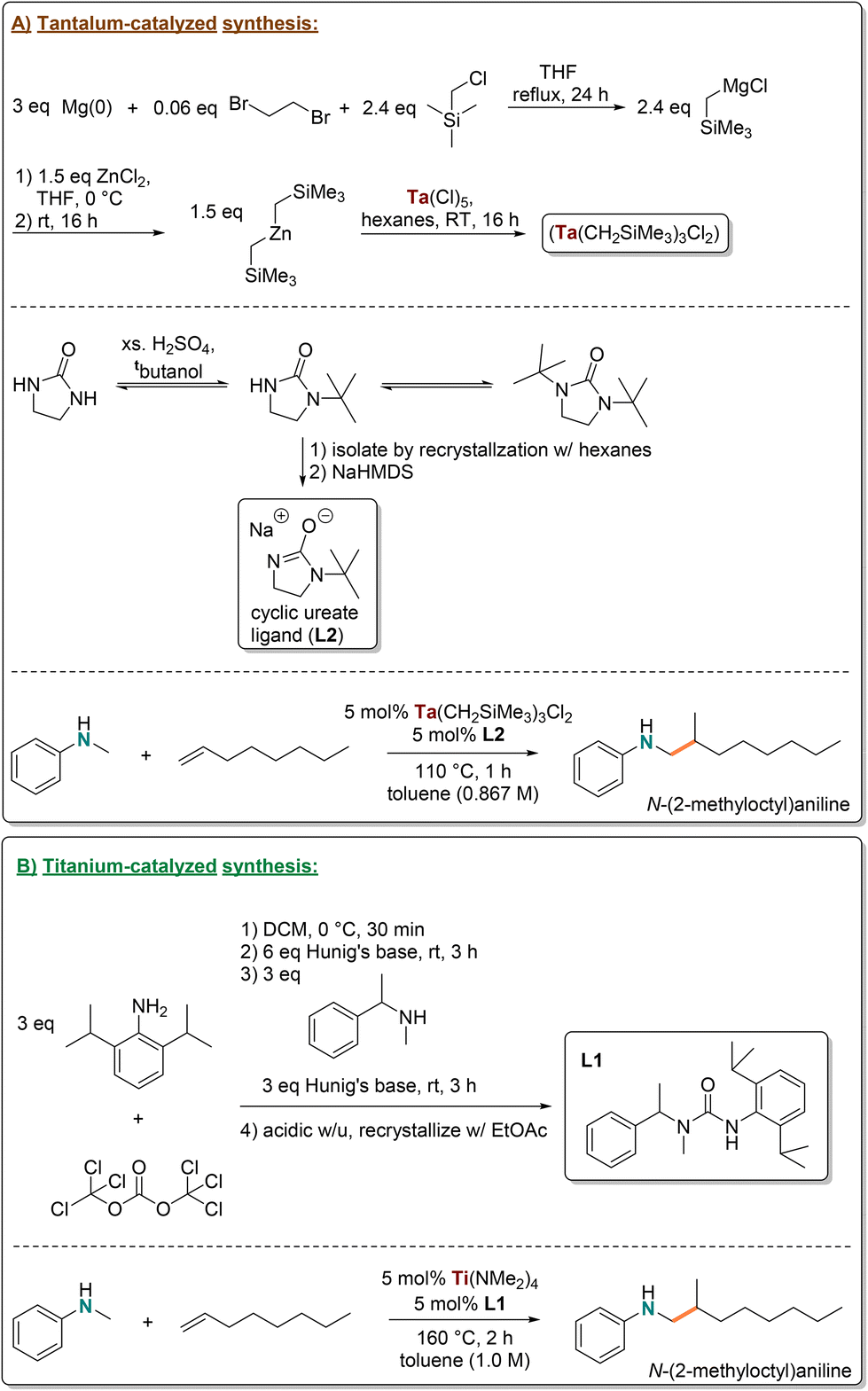 | ||
| Scheme 4 Generalized synthetic procedure to produce 1 kg of N-(2-methyloctyl)aniline from commercially available starting materials for the tantalum-catalyzed route (A) and the titanium-catalyzed route (B). | ||
| Routea | IAP | IOD | ISF | IGW | IINGT | IINHT | PER | ACCU logKow |
IAD |
|---|---|---|---|---|---|---|---|---|---|
|
a IAP = acidification index, IOD = ozone depletion index, ISF = smog formation index, IGW = global warming index, IINGT = human toxicity by ingestion index, IINHT = human toxicity by inhalation index, PER = persistence, ACC logKow = bioaccumulation, IAD = abiotic depletion index.
b Value when sulfuric acid is removed from calculation.
|
|||||||||
| Ta | 80 | 0 | 60 | 70000 |
−600000 (200000b) |
3000 | Months | 4 | 2 |
| Ti | 30 | 0 | 5 | 100 | 5000000000 |
30 | Very long-lived | 4 | 0.005 |
For our assessment, N-methylaniline and 1-octene were the chosen hydroaminoalkylation reactants because both are commonly used hydroaminoalkylation substrates and 1-octene can act as an alkene analogue for the VTPP substrates. The final product was assumed to form in 100% yield (based on reported yields of this reaction with respective catalysts)31,72 to generate 1 kg of N-(2-methyloctyl)aniline product with 5 mol% catalyst (Scheme 4). Reagents and solvents used for in-house synthesis of the metal precursor and ligands, the CO2 generated from energy use, and the alkene and amine starting materials were assessed, making this a gate-to-gate LCA. Further LCA specifications can be found in the ESI.†
To assess which route has an overall lower environmental impact the summed indices of each pathway must be compared (Table 2). LCAs are known to have significant error associated with the calculation,97,98 which is why values are reported to only one significant figure. Large differences between routes need to be present to have confidence in making decisions based on the assessment. An adjustment was made for the INGTP of the tantalum route by removing sulfuric acid from the summation of indices. The sulfuric acid has a negative logKow (n-octanol water partition coefficient), which is used for the assessment of this category. This negative value means sulfuric acid is more soluble in water than n-octanol and this reagent will appear more in water run-off than in soil. Since ca. 117 g of sulfuric acid is estimated to be used in the synthesis of the ligand this value outweighs the positive portion of the summation, where the positive portion represents the amount of the reagents that may remain in the soil if emitted. Therefore, removing the sulfuric acid gives a better understanding of the toxicity of the other reagents adsorbed by the soil and the potential ingestion toxicity of the tantalum route.
With that, the titanium route is less impactful in the five out of nine categories: IAP, ISF, IGW, IINHT and IAD than tantalum; and the two are negligibly different in their IOD and ACCU. Tantalum is worse performing for IAP because of the sulfuric acid emitted in the ligand synthesis; ISF because of the ethylene emitted in producing the Grignard reagent; IGW because of the bis [(trimethylsilyl)methyl]zinc; IINHT because of the bis (trimethylsilyl)amine emitted and greater amounts of solvents used; and IAD because of the (bis(trimethylsilyl)methyl)tantalum(V) chloride precursor synthesis (see ESI†). The titanium is only worse performing in the IINGT and PER because of the amount of hydrochloric gas emitted during its ligand synthesis. Clearly, it would be challenging to improve the environmental impact of the tantalum route because nearly every step in the catalyst synthesis would need to be adjusted. This contrasts with the titanium catalyst, where the only major concern is the hydrochloric gas byproduct.
The LCA is effective with evaluating the impact of organic syntheses but struggles with inorganic compounds due to a lack of resources on the matter. Assumptions can be made to best model the impact of these reagents, but the most accurate parameter for inorganic compounds in the LCA is the IAD. This represents the risk of depletion of an element relative to antimony and this index is based on the total mass of an element used. Ideally for a catalytic system, the IAD of the catalyst should not outweigh the reagents. However, the synthesis of the precursor, (bis(trimethylsilyl)methyl)tantalum(V) chloride, results in a method that is 1000 times more depleting that the titanium-catalyzed synthesis. This is due to the number of metals and halogens used to produce the tantalum catalyst. Even directly comparing the commercial sources of each metal, TaCl5 and Ti(NMe2)4, the tantalum starting material is 2804 times more depleting than the titanium, highlighting the of advantage earth-abundant transition metals.
To complement the LCA, the mass of CO2 produced for distilling, evaporating, and refluxing solvents for the respective syntheses are provided, along with the EF and the reaction mass efficiency (RME) (Table 3). The tantalum-catalyzed route produces 1.4 times as much CO2 as an energy byproduct than the titanium-catalyzed route mostly due to the amount of toluene that must be distilled and refluxed for the optimal hydroaminoalkylation reaction concentrations reported. Further, this calculation only considered the CO2 produced during the energy consumption for operations that require heating. This does not account for the energy required to cool or condense gases, nor the energy required to run vacuum pumps for Schlenk technique. If these were factored, then the tantalum-catalyzed route would produce significantly more CO2 because the synthesis of the metal precursor requires a multi-step air free environment and more reliant on coolants and condensed gases. The EFs for the tantalum route is nearly double that of titanium, but both are in the range of bulk chemical synthesis.99 The AE for hydroaminoalkylation is 100% since the sum of the molecular weight of the reagents are equal to the molecular weight of the product and catalysts and solvents do not contribute to the calculation. This allows for over-generalization of reaction efficiencies, making AE not an effective metric for assessing green advances in catalyst syntheses. A more effective metric for our assessment here is the reaction mass efficiency (RME), where the mass of reactants (including catalysts) can be compared relative to the mass of product obtained. For the tantalum-catalyzed reaction the RME drops to 40% due to the multi-step catalyst synthesis. Whereas the titanium-catalyzed approach maintains a high RME of 80% because there are few steps for mass loss in generating the catalyst, resulting in a more efficient synthesis of amine products.
| Route | CO2 (kg) | EF | RME |
|---|---|---|---|
| Ta | 70 | 7 | 40% |
| Ti | 50 | 5 | 80% |
Conclusions
We demonstrated that the in situ catalyst system based on a commercially available precursor, Ti(NMe2)4, and urea ligand, L1, is an effective catalyst for postpolymerization hydroaminoalkylation to give the branched regioisomer selectively. The LCA highlights titanium's sustainable advantage over tantalum for ATPP synthesis. Sterically enhanced disubstituted alkene substrates and amines were also accessible, enabling a two-step synthesis to prepare a N-heterocycle benzoindolizidine in neat conditions. The diversity of substrates, excellent regioselectivity, simple catalyst design, and quantifiable improvement on standard environmental assessment metrics demonstrate a significant advancement in sustainability of catalytic amine synthesis by using Ti(NMe2)4 with L1.Conflicts of interest
The authors declare a potential financial conflict of interest due to intellectual property that has been filed on the catalysts used in this report.Acknowledgements
The authors thank Dr. Patrick Brant for helpful discussion and Exxon Mobil for donation of PP oligomers. MM thanks the von Humboldt Foundation for a Feodor Lynen Postdoctoral Fellowship. SSS and CHM thank NSERC for doctoral scholarships. DD thanks the National Natural Science Foundation of China for support. LLS thanks the Canada Research Chair program for partial support of this work.References
- H. Bruijn, R. Duin, M. A. J. Huijbregts, J. B. Guinee, M. Gorree, R. Heijungs, G. Huppes, R. Kleijn, A. Koning, L. Oers, A. Wegener Sleeswijk, S. Suh and H. A. U. de Haes, Handbook on Life Cycle Assessment, Springer, Dordrecht, 1 edn, 2002 Search PubMed.
- G. Nordberg, B. Fowler and M. Nordberg, Handbook on the Toxicology of Metals, 4 edn, Elsevier, 2015 Search PubMed.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- A. J. Hunt, T. J. Farmer and J. H. Clark, Element Recovery and Sustainability, The Royal Society of Chemistry, 2013, pp. 1–28 Search PubMed.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics, CRC Press, 2017 Search PubMed.
- R. A. Collins, A. F. Russell and P. Mountford, Applied Petrochemical Research, Springer, 2015, pp. 153–171 Search PubMed.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 541–547 CrossRef CAS.
- G. Wilke, Angew. Chem., Int. Ed., 2003, 42, 5000–5008 CrossRef CAS PubMed.
- G. Natta, Angew. Chem., 1956, 68, 393–403 CrossRef CAS.
- D. S. Breslow and N. R. Newburg, J. Am. Chem. Soc., 1957, 79, 5072–5073 CrossRef CAS.
- G. Natta, P. Pino, G. Mazzanti and U. Giannini, J. Am. Chem. Soc., 1957, 79, 2975–2976 CrossRef CAS.
- K. H. Reichert and K. R. Meyer, Makromol. Chem., 1973, 169, 163–176 CrossRef CAS.
- W. P. Long and D. S. Breslow, Liebigs Ann. Chem., 1975, 1975, 463–469 CrossRef.
- A. Andresen, H.-G. Cordes, J. Herwig, W. Kaminsky, A. Merck, R. Mottweiler, J. Pein, H. Sinn and H.-J. Vollmer, Angew. Chem., Int. Ed. Engl., 1976, 15, 630–632 CrossRef.
- H. Sinn, W. Kaminsky, H.-J. Vollmer and R. Woldt, Angew. Chem., Int. Ed. Engl., 1980, 19, 390–392 CrossRef.
- A. Gansäuer, S. Hildebrandt, A. Michelmann, T. Dahmen, D. vonLaufenberg, C. Kube, G. D. Fianu and R. A. Flowers II, Angew. Chem., Int. Ed., 2015, 54, 7003–7006 CrossRef PubMed.
- S. Hildebrandt and A. Gansäuer, Angew. Chem., Int. Ed., 2016, 55, 9719–9722 CrossRef CAS PubMed.
- R. B. Richrath, T. Olyschläger, S. Hildebrandt, D. G. Enny, G. D. Fianu, R. A. Flowers II and A. Gansäuer, Chem. – Eur. J., 2018, 24, 6371–6379 CrossRef CAS PubMed.
- F. Mühlhaus, H. Weißbarth, T. Dahmen, G. Schnakenburg and A. Gansäuer, Angew. Chem., Int. Ed., 2019, 58, 14208–14212 CrossRef PubMed.
- J. J. Li, Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications Fifth Edition, ed. J. J. Li, Springer International Publishing, Cham, 2014, pp. 552–554 Search PubMed.
- A. Pfenninger, Synthesis, 1986, 89–116 CrossRef CAS.
- S. Okamoto, Chem. Rec., 2016, 16, 857–872 CrossRef CAS PubMed.
- O. Geis and H.-G. Schmalz, Angew. Chem., Int. Ed., 1998, 37, 911–914 CrossRef CAS PubMed.
- J. Blanco-Urgoiti, L. Añorbe, L. Pérez-Serrano, G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC.
- Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS PubMed.
- H.-C. Chiu and I. A. Tonks, Angew. Chem., Int. Ed., 2018, 57, 6090–6094 CrossRef CAS PubMed.
- H.-C. Chiu, X. Y. See and I. A. Tonks, ACS Catal., 2019, 9, 216–223 CrossRef CAS PubMed.
- M. Manßen and L. L. Schafer, Chem. Soc. Rev., 2020, 49, 6947–6994 RSC.
- L. L. Schafer, M. Manßen, P. M. Edwards, E. K. J. Lui, S. E. Griffin and C. R. Dunbar, Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2020, vol. 74, pp. 405–468 Search PubMed.
- M. Manßen, N. Lauterbach, J. Dörfler, M. Schmidtmann, W. Saak, S. Doye and R. Beckhaus, Angew. Chem., Int. Ed., 2015, 54, 4383–4387 CrossRef PubMed.
- M. Manßen, D. Deng, C. H. M. Zheng, R. C. DiPucchio, D. Chen and L. L. Schafer, ACS Catal., 2021, 11, 4550–4560 CrossRef.
- M. Manßen and L. L. Schafer, Trends Chem., 2021, 3, 428–429 CrossRef.
- D. Geik, M. Rosien, J. Bielefeld, M. Schmidtmann and S. Doye, Angew. Chem., Int. Ed., 2021, 60, 9936–9940 CrossRef CAS PubMed.
- M. Rosien, I. Toben, M. Schmidtmann, R. Beckhaus and S. Doye, Chemistry, 2020, 26, 2138–2142 CrossRef CAS PubMed.
- J. Bielefeld and S. Doye, Angew Chem., Int. Ed., 2020, 59, 6138–6143 CrossRef CAS PubMed.
- J. Bielefeld and S. Doye, Angew. Chem., Int. Ed., 2017, 56, 15155–15158 CrossRef CAS PubMed.
- J. Dörfler, T. Preuß, C. Brahms, D. Scheuer and S. Doye, Dalton Trans., 2015, 44, 12149–12168 RSC.
- J. Dörfler, T. Preuß, A. Schischko, M. Schmidtmann and S. Doye, Angew. Chem., Int. Ed., 2014, 53, 7918–7922 CrossRef PubMed.
- R. C. DiPucchio, S.-C. Rosca and L. L. Schafer, J. Am. Chem. Soc., 2022, 144, 11459–11481 CrossRef CAS PubMed.
- A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- P. Kalck and M. Urrutigoïty, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- F. Agbossou, J.-F. Carpentier and A. Mortreux, Chem. Rev., 1995, 95, 2485–2506 CrossRef CAS.
- L. Routaboul, C. Buch, H. Klein, R. Jackstell and M. Beller, Tetrahedron Lett., 2005, 46, 7401–7405 CrossRef CAS.
- B. Breit, C. U. Grünanger and O. Abillard, Eur. J. Org. Chem., 2007, 2497–2503 CrossRef CAS.
- G. Parrinello, R. Deschenaux and J. K. Stille, J. Org. Chem., 1986, 51, 4189–4195 CrossRef CAS.
- C. G. Arena, F. Nicolò, D. Drommi, G. Bruno and F. Faraone, J. Chem. Soc., Chem. Commun., 1994, 19, 2251–2252 RSC.
- J. Bielefeld, E. Kurochkina, M. Schmidtmann and S. Doye, Eur. J. Inorg. Chem., 2019, 2019, 3713–3718 CrossRef CAS.
- S. H. Rohjans, J. H. Ross, L. H. Lühning, L. Sklorz, M. Schmidtmann and S. Doye, Organometallics, 2018, 37, 4350–4357 CrossRef CAS.
- L. H. Lühning, M. Rosien and S. Doye, Synlett, 2017, 2489–2494 Search PubMed.
- L. H. Lühning, C. Brahms, J. P. Nimoth, M. Schmidtmann and S. Doye, Z. Anorg. Allg. Chem., 2015, 641, 2071–2082 CrossRef.
- J. Dörfler, B. Bytyqi, S. Hüller, N. M. Mann, C. Brahms, M. Schmidtmann and S. Doye, Adv. Synth. Catal., 2015, 357, 2265–2276 CrossRef.
- J. Dörfler and S. Doye, Angew. Chem., Int. Ed., 2013, 52, 1806–1809 CrossRef PubMed.
- T. Preuß, W. Saak and S. Doye, Chem. – Eur. J., 2013, 19, 3833–3837 CrossRef PubMed.
- R. Kubiak, I. Prochnow and S. Doye, Angew. Chem., Int. Ed., 2010, 49, 2626–2629 CrossRef CAS PubMed.
- R. C. DiPucchio, K. E. Lenzen, P. Daneshmand, M. B. Ezhova and L. L. Schafer, J. Am. Chem. Soc., 2021, 143, 11243–11250 CrossRef CAS PubMed.
- R. C. DiPucchio, S.-C. Roşca and L. L. Schafer, Angew. Chem., Int. Ed., 2018, 57, 3469–3472 CrossRef CAS PubMed.
- M. R. Perry, T. Ebrahimi, E. Morgan, P. M. Edwards, S. G. Hatzikiriakos and L. L. Schafer, Macromolecules, 2016, 49, 4423–4430 CrossRef CAS.
- N. Kuanr, T. Tomkovic, D. J. Gilmour, M. R. Perry, S.-J. Hsiang, E. van Ruymbeke, S. G. Hatzikiriakos and L. L. Schafer, Macromolecules, 2020, 53, 2649–2661 CrossRef CAS.
- N. Kuanr, D. J. Gilmour, H. Gildenast, M. R. Perry and L. L. Schafer, Macromolecules, 2022, 55, 3840–3849 CrossRef CAS.
- S. S. Scott, S.-C. Roşca, D. J. Gilmour, P. Brant and L. L. Schafer, ACS Macro Lett., 2021, 10, 1266–1272 CrossRef CAS PubMed.
- J. R. Hagadorn, D. J. Crowther, R. N. Ganesh and P. Brant, US8669326B2, 2014.
- C. Jalbert, J. T. Koberstein, A. Hariharan and S. K. Kumar, Macromolecules, 1997, 30, 4481–4490 CrossRef CAS.
- D. J. Gilmour, T. Tomkovic, N. Kuanr, M. R. Perry, H. Gildenast, S. G. Hatzikiriakos and L. L. Schafer, ACS Appl. Polym. Mater., 2021, 3, 2330–2335 CrossRef CAS.
- S. H. Ryu and A. M. Shanmugharaj, J. Chem. Eng., 2014, 244, 552–560 CrossRef CAS.
- S. Stankovich, D. A. Dikin, O. C. Compton, G. H. B. Dommett, R. S. Ruoff and S. T. Nguyen, Chem. Mater., 2010, 22, 4153–4157 CrossRef CAS.
- D. L. Allara, S. V. Atre, C. A. Elliger and R. G. Snyder, J. Am. Chem. Soc., 1991, 113, 1852–1854 CrossRef CAS.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- P. Sabourian, M. Tavakolian, H. Yazdani, M. Frounchi, T. G. M. van de Ven, D. Maysinger and A. Kakkar, J. Controlled Release, 2020, 317, 216–231 CrossRef CAS PubMed.
- P. M. Edwards and L. L. Schafer, Chem. Commun., 2018, 54, 12543–12560 RSC.
- D. J. Gilmour, J. M. P. Lauzon, E. Clot and L. L. Schafer, Organometallics, 2018, 37, 4387–4394 CrossRef CAS.
- P. Daneshmand, S.-C. Roşca, R. Dalhoff, K. Yin, R. C. DiPucchio, R. A. Ivanovich, D. E. Polat, A. M. Beauchemin and L. L. Schafer, J. Am. Chem. Soc., 2020, 142, 15740–15750 CrossRef CAS PubMed.
- P. R. Payne, P. Garcia, P. Eisenberger, J. C. H. Yim and L. L. Schafer, Org. Lett., 2013, 15, 2182–2185 CrossRef CAS PubMed.
- P. M. Edwards and L. L. Schafer, Org. Lett., 2017, 19, 5720–5723 CrossRef CAS PubMed.
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 139–165 RSC.
- J. P. Michael, Nat. Prod. Rep., 2001, 18, 520–542 RSC.
- J. P. Michael, The Alkaloids: Chemistry and Biology, ed. H.-J. knölker, Academic Press, 2016, vol. 75, pp. 1–498 Search PubMed.
- D. S. Seigler, Plant Secondary Metabolism, ed. D. S. Seigler, Springer US, Boston, MA, 1998, pp. 546–567 Search PubMed.
- J. Chen, H. Lv, J. Hu, M. Ji, N. Xue, C. Li, S. Ma, Q. Zhou, B. Lin, Y. Li, S. Yu and X. Chen, Cancer Lett., 2016, 381, 391–403 CrossRef CAS PubMed.
- T. P. T. Cushnie, B. Cushnie and A. J. Lamb, Int. J. Antimicrob. Agents, 2014, 44, 377–386 CrossRef CAS PubMed.
- V. Samoylenko, M. K. Ashfaq, M. R. Jacob, B. L. Tekwani, S. I. Khan, S. P. Manly, V. C. Joshi, L. A. Walker and I. Muhammad, J. Nat. Prod., 2009, 72, 92–98 CrossRef CAS PubMed.
- J. W. Daly, E. McNeal, F. Gusovsky, F. Ito and L. E. Overman, J. Med. Chem., 1988, 31, 477–480 CrossRef CAS PubMed.
- E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
- J. Guo and S. Yu, Org. Biomol. Chem., 2015, 13, 1179–1186 RSC.
- A. Zidan, M. Cordier, A. M. El-Naggar, N. E. A. Abd El-Sattar, M. A. Hassan, A. K. Ali and L. El Kaïm, Org. Lett., 2018, 20, 2568–2571 CrossRef CAS PubMed.
- S. Zhao, M. J. Totleben, J. P. Freeman, C. L. Bacon, G. B. Fox, E. O'Driscoll, A. G. Foley, J. Kelly, U. Farrell, C. Regan, S. A. Mizsak and J. Szmuszkovicz, Bioorg. Med. Chem., 1999, 7, 1637–1646 CrossRef CAS PubMed.
- C. M. Plummer, L. Li and Y. Chen, Polym. Chem., 2020, 11, 6862–6872 RSC.
- N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267–275 RSC.
- M. P. McGrath, E. D. Sall and S. J. Tremont, Chem. Rev., 1995, 95, 381–398 CrossRef CAS.
- G. Menendez Rodriguez, M. M. Díaz-Requejo and P. J. Pérez, Macromolecules, 2021, 54, 4971–4985 CrossRef CAS.
- A. C. Brezny and C. R. Landis, Acc. Chem. Res., 2018, 51, 2344–2354 CrossRef CAS PubMed.
- C. R. López-Barrón, P. Brant, M. Shivokhin, J. Lu, S. Kang, J. A. Throckmorton, T. Mouton, T. Pham and R. C. Savage, Macromolecules, 2018, 51, 5720–5731 CrossRef.
- C. A. Orr, J. J. Cernohous, P. Guegan, A. Hirao, H. K. Jeon and C. W. Macosko, Polymer, 2001, 42, 8171–8178 CrossRef CAS.
- S. Kobayashi, J. Song, H. C. Silvis, C. W. Macosko and M. A. Hillmyer, Ind. Eng. Chem. Res., 2011, 50, 3274–3279 CrossRef CAS.
- B. Lin, D. Lawler, G. P. McGovern, C. A. Bradley and C. E. Hobbs, Tetrahedron Lett., 2013, 54, 970–974 CrossRef CAS.
- S. M. Mercer, J. Andraos and P. G. Jessop, J. Chem. Educ., 2012, 89, 215–220 CrossRef CAS.
- G. Koller, U. Fischer and K. Hungerbühler, Ind. Eng. Chem. Res., 2000, 39, 960–972 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00011g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |