
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-click chemistry: a bridge between biocatalysis and click chemistry
Diego F. Rodríguez
a,
Yanina Moglie
 b,
César A. Ramírez-Sarmiento
cd,
Sachin Kumar Singh
e,
Kamal Dua
fg and
Flavia C. Zacconi
*ach
b,
César A. Ramírez-Sarmiento
cd,
Sachin Kumar Singh
e,
Kamal Dua
fg and
Flavia C. Zacconi
*ach
aFacultad de Química y de Farmacia, Pontificia Universidad Católica de Chile, Chile. E-mail: fzacconi@uc.cl
bDepartamento de Química, INQUISUR, Universidad Nacional del Sur (UNS)-CONICET, Argentina
cInstitute for Biological and Medical Engineering, Schools of Engineering, Medicine and Biological Sciences, Pontificia Universidad Católica de Chile, Santiago, Chile
dANID – Millennium Science Initiative Program, Millennium Institute for Integrative Biology (iBio), Santiago, Chile
eSchool of Pharmaceutical Sciences, Lovely Professional University, Phagwara 144411, Punjab, India
fDiscipline of Pharmacy, Graduate School of Health, University of Technology Sydney, NSW 2007, Australia
gFaculty of Health, Australian Research Centre in Complementary and Integrative Medicine, University of Technology Sydney, Ultimo, Australia
hCentro de Investigaciones en Nanotecnología y Materiales Avanzados, CIEN-UC, Pontificia Universidad Católica de Chile, Santiago, Chile
First published on 12th January 2022
Abstract
The fields of click chemistry and biocatalysis have rapidly grown over the last two decades. The development of robust and active biocatalysts and the widespread use of straightforward click reactions led to significant interactions between these two fields. Therefore the name bio-click chemistry seems to be an accurate definition of chemoenzymatic reactions cooperating with click transformations. Bio-click chemistry can be understood as the approach towards molecules of high-value using a green and sustainable approach by exploiting the potential of biocatalytic enzyme activity combined with the reliable nature of click reactions. This review summarizes the principal bio-click chemistry reactions reported over the last two decades, with a special emphasis on small molecules. Contributions to the field of bio-click chemistry are manifold, but the synthesis of chiral molecules with applications in medicinal chemistry and sustainable syntheses will be especially highlighted.
 Diego F. Rodríguez | Diego F. Rodríguez Sanchez studied Chemistry at Universidad del Valle, Colombia. He received a grade of meritorious thesis in 2015 on his research in the synergy effect in enzymatic catalysis under the supervision of Prof. Godoy. In 2017, he started his PhD in Chemistry at Pontificia Universidad Católica de Chile, where he joined Zacconi's research group. His research focuses on the design and development of news safe and environmentally-friendly synthetic methodologies to obtain novel anticoagulants that inhibit the human factor Xa. Moreover, he received the PhD fellowship from the Consejo Nacional de Investigación Científica y Tecnológica, CONICYT, Chile. |
 Yanina F. Moglie | Dr Yanina Moglie obtained her BSc and PhD in Chemistry from the Universidad Nacional del Sur (UNS), Argentina. Dr Moglie received the award for the best PhD thesis in Organic Chemistry from the Sociedad Argentina de Investigación en Química Orgánica (SAIQO). After her postdoctoral stay at the University of Alicante (Spain), in 2014 she became a CONICET Associate Researcher of the Instituto de Química del Sur (INQUISUR) and Full Professor of Organic Chemistry (UNS). Her research focuses on the development of new synthetic methodologies involving metal nanoparticles and their application in organic synthesis mainly in the preparation of bioactive compounds. |
 César A. Ramírez-Sarmiento | Dr César A. Ramírez-Sarmiento obtained his PhD in Sciences at Universidad de Chile, Chile. Currently, Dr Ramírez-Sarmiento in an Assistant Professor at the Institute for Biological and Medical Engineering, Pontificia Universidad Católica de Chile. Dr Ramírez-Sarmineto research focuses on working on protein folding, function, and evolution. The computational and experimental research developed by his group focuses on the discovery and engineering of plastic-degrading enzymes for their utilization as biocatalysts and on the study of structural interconversions in transcription regulators for their repurposing as biosensors. |
 Sachin Kumar Singh | Dr Sachin Kumar Singh, a Professor at School of Pharmaceutical Sciences (Lovely Professional University, India) holds his PhD degree in Pharmaceutical Sciences (Karpagam University, India). Dr Singh has published more than 175 research papers, 11 book chapters, and he has filed 10 patents. He has a keen desire to bring his formulations into market for patients suffering from diabetes and he is set to transfer the technologies developed to pharma and nutraceutical industry. In 2020, he received Dr B.C. Deb memorial award by Indian Science Congress Association and the Bharat Shiksha Ratan Award given by Global Society for Health and Educational Growth, India. |
 Kamal Dua | Dr Kamal Dua, Lecturer, Discipline of Pharmacy, Graduate School of Health, University of Technology Sydney, has a research experience of more than 12 years working in the field of drug delivery targeting inflammatory diseases. Dr Dua is also a Node Leader of Drug Delivery Research in the Centre for Inflammation at Centenary Institute/UTS, where the targets identified from the research projects are pursued developing novel formulations as the first step towards translation into clinics. Dr Dua research in two complementary areas; drug delivery and immunology, specifically addressing how these disciplines can advance one another helping community to live longer and healthier. |
 Flavia C. Zacconi | Dr Flavia C. Zacconi obtained her PhD in Chemistry from the Universidad Nacional del Sur (UNS), Argentina. Currently, her Associate Professor position is share between the Faculty of Chemistry and Pharmacy and the Institute for Biological and Medical Engineering, Pontificia Universidad Católica de Chile. She has a research experience in organic chemistry, medicinal chemistry, and biomaterials. Dr Zacconi research focuses on the intersections of organic chemistry, medicinal chemistry, engineering, and biological chemistry, developing multi- and interdisciplinary projects to understand the complexity of bioactive systems. Additionally, Dr Zacconi acts as a reviewer for international journals and national and international projects; and she has published research papers, book chapters, and books. |
1. Introduction
Chemoenzymatic syntheses are powerful methodologies for the creation of complex molecular structures and functionalities.1 The development of processes which combine biocatalysis and chemical synthesis has rapidly increased over recent years. The two main pillars of innovation in the field are the generation of more active, selective, and stable biocatalysts,2 and the development of new synthetic procedures in water, allowing for highly efficient, and sustainable one-pot procedures.3The term click chemistry was introduced by Sharpless in 2001, comprising highly efficient and reliable reactions enabling the rapid construction of structural and functional diversity through the union of small building blocks. Commonly these reactions have very high inherent driving forces, thus avoiding tedious chromatographic work-up.4 Evidently, the concept of click chemistry is largely inspired by nature's “synthetic toolbox”, and it is not surprising that the merger of click chemistry and biocatalysis exhibits great potential for the synthesis of complex molecular structures.
The increasing demand for sustainable processes is fostering the development and establishment of new synthetic tools. Among these tools, click chemistry and biocatalysis are of utmost importance, since they are characterized by their high selectivity and orthogonality, mild reaction conditions avoiding protecting groups, efficiency, reliability, and user-friendliness.5 The growing interest in click chemistry and biocatalysis in recent years is reflected by the drastic increase in the number of publications on the topic (Fig. 1). It is not surprising that the combination of these two disciplines, which we propose to call bio-click chemistry, has given rise to a myriad of highly interesting developments and has led to a significant impact on the synthesis of structurally diverse molecules, such as chiral amino alcohols, triazoles, amides, cycloadducts, thioethers, and others. In addition, bio-click methodologies show a synergy oftentimes allowing for shorter, stereoselective and efficient synthetic routes.
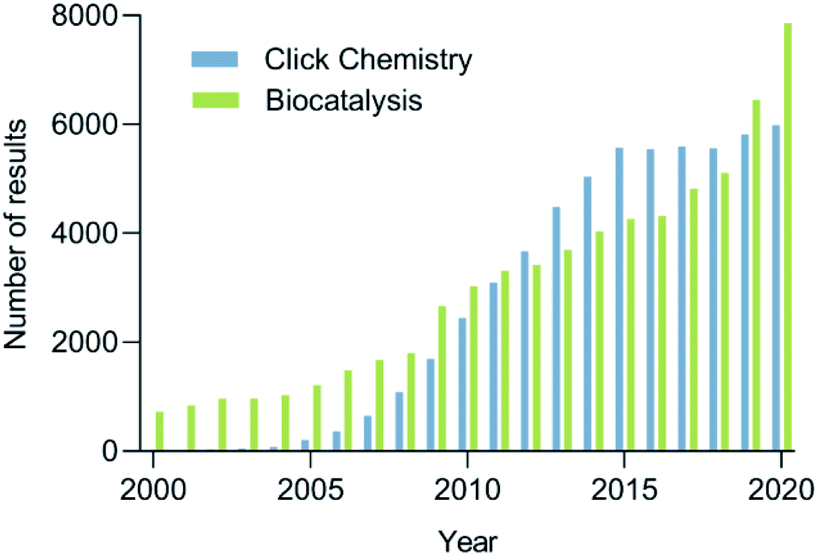 | ||
| Fig. 1 The number of results to the search for click chemistry (blue) and biocatalysis (green) in Scopus per year between 2000–2020. | ||
The present review focuses on syntheses in which biocatalytic processes and click reactions were used in combination. Recent reviews have focused on specific applications of click chemistry and biocatalysis in the in situ generation of inhibitors,6,7 the immobilization and modification of enzymes,8 or the generation of semisynthetic enzymes.9 However, the discrete topic of bio-click chemistry has yet to be approached in depth. We seek to fill this gap and discuss the most important aspects of the development of bio-click methodologies, as well as their main advantages and limitations.
2. Bio-click chemistry
Organic chemistry has a sheer endless set of reactions allowing for the generation of molecular diversity and complexity. Chlorophyll,10 taxol,11,12 and human lysozyme13 are only a few examples of highly important complex molecules obtained through elegant chemical syntheses. Albeit highly sophisticated, organic synthesis is undergoing constant improvements14 especially through the development of more selective, efficient, and sustainable reactions. The interconnection of different disciplines, such as biocatalysis and click chemistry, is essential to achieve these goals (Fig. 2).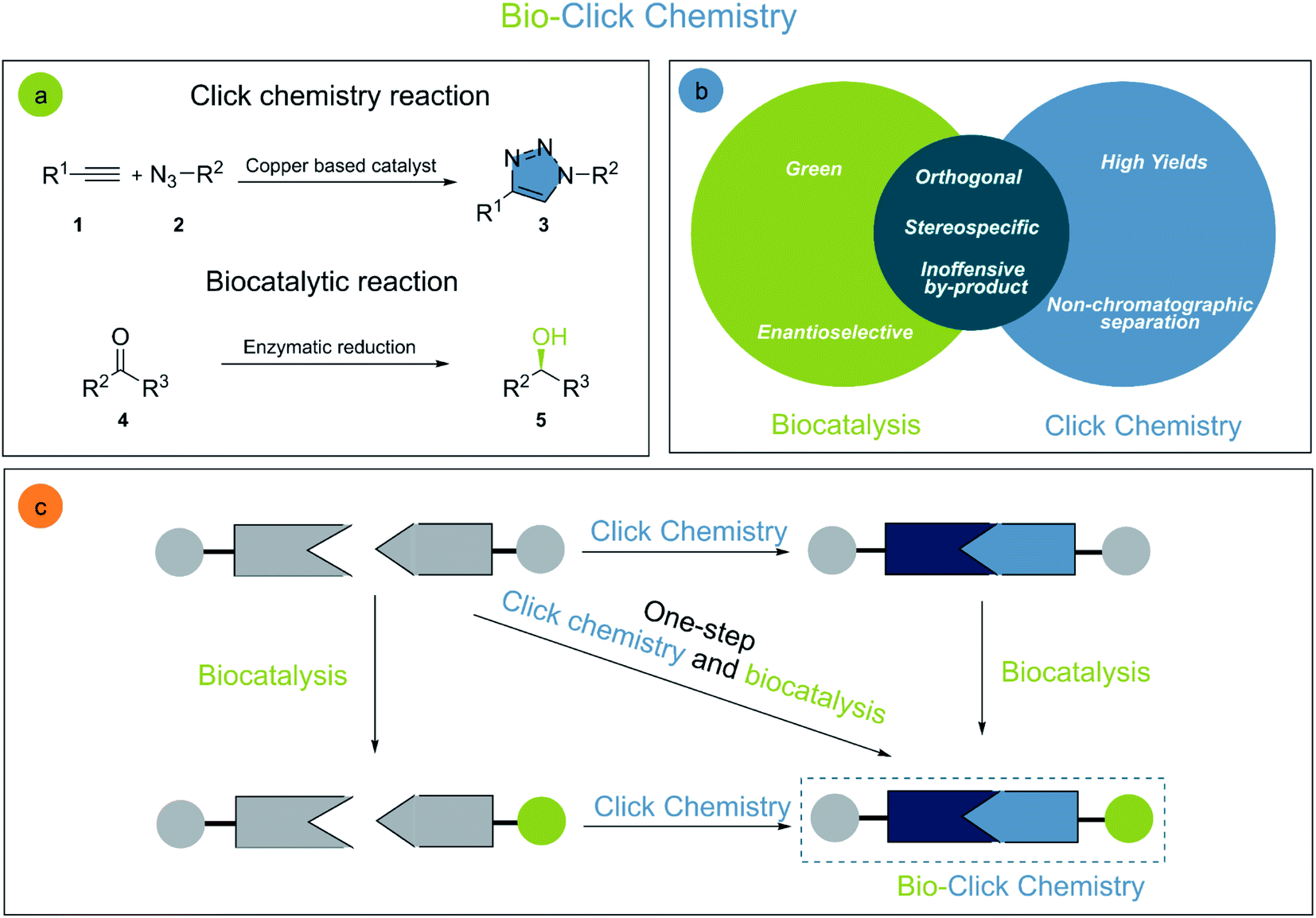 | ||
| Fig. 2 (a) General click chemistry and biocatalysis reactions, (b) bio-click chemistry interface and (c) schematic representation of bio-click chemistry cooperation for the synthesis of small molecules. | ||
Click chemistry is generally outlined by a group of reactions with desirable characteristics for biocompatible reactions or bioconjugations: reliability, effectiveness, protecting group-free, high yielding, and easy to purify (often without any chromatography). The fact that click chemistry has become an integral part of the synthetic toolbox is demonstrated by its success in chemical synthesis, drug discovery, medicinal chemistry,15 molecular labelling,16 polymer synthesis,17 and others.
The cutting-edge example for click chemistry is (i) the copper-catalyzed 1,3-dipolar cycloaddition of terminal alkynes and organic azides (CuAAC). Nevertheless, many other transformations are classified as click reactions as well (ii) the Diels–Alder reaction, (iii) additions to alkenes, (iv) nucleophilic opening of strained rings, (v) non-aldol carbonyl transformations, and (vi) addition reactions to alkynes.18 It has to be clarified, that although click reactions are regio- and stereospecific, they are not necessarily enantioselective.4 This shortcoming can be overridden by the merger of click chemistry with enantioselective biocatalysts such as enzymes.
Biocatalysis comprises the use of purified enzymes, cell-free extracts or whole microorganisms as catalysts in a vast array of transformations, rendering them as excellent and sustainable methods for the construction of complex molecular structures, while fulfilling green chemistry principles.19 However, enzymes have inherent limitations often preventing their widespread use in synthesis: rather low operational stability, inhibition by substrates or products, limited tolerance of non-natural substrates and limited applicability in organic solvents. Innovative technologies, such as protein engineering and enzyme immobilization, have emerged to overcome these drawbacks and limitations.2 Many enzymes obtained by protein engineering are more robust, stable and capable to transform non-natural substrates. Immobilized enzymes generally present outstanding operational stability, an easy removal from the reaction medium and the capability of repetitive recycling.
Enzymatic processes allowing for the synthesis of complex molecules under highly sustainable and green conditions are increasingly popular. Various drug molecules commonly synthesized through traditional catalytic processes are eventually produced by chemoenzymatic methods, thus improving process efficiencies and substantially reducing waste generation.20,21
A state-of-the-art example is the biocatalytic synthesis of the HIV treatment candidate islatravir, as reported by Huffman and co-workers.22 In this process, five enzymes obtained by directed evolution (two immobilized enzymes) and four auxiliary enzymes were applied towards the generation of islatravir from simple achiral building blocks in a three-step cascade reaction (Fig. 3). The target molecule was obtained in 51% overall yield, with high atom economy, using water as solvent, avoiding intermediate purification and in less than half of the number of steps compared to the conventional chemical synthesis.22
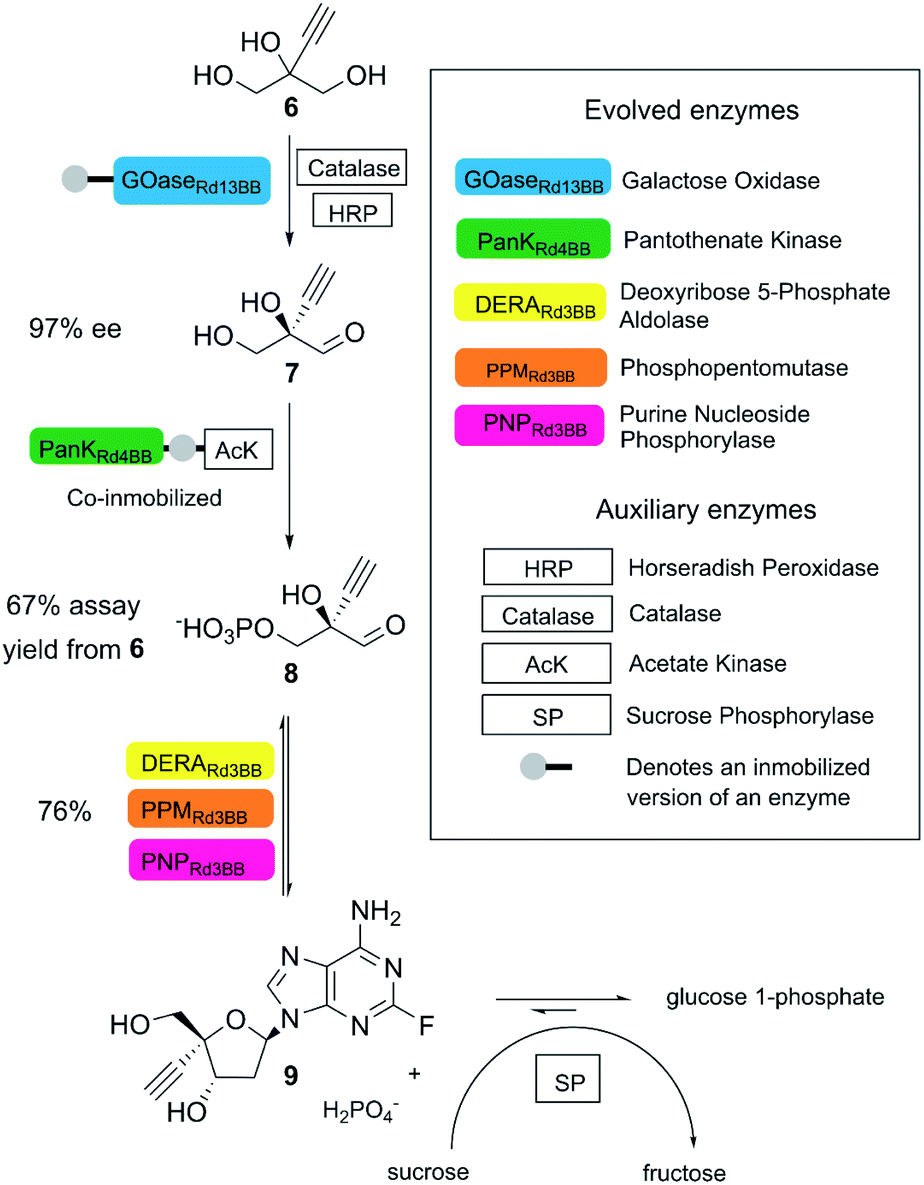 | ||
| Fig. 3 Biocatalytic synthesis of islatravir. (This figure has been adapted from ref. 22. With permission from Science Journal, copyright 2020). | ||
Although, Caruso et al. used the term bio-click chemistry in a study towards the functionalization of polymer drug vehicles,23 a more exhaustive review of the interface between click chemistry and biocatalysis is necessary. Considering the potential for the development of highly efficient, sustainable, and selective processes, a profound impact of bio-click methods on the field can be envisaged.
It should be noted that the term “bio-click chemistry” differs markedly from click chemistry, as it interconnects the latter with biocatalytic processes. In many cases, this allows for the development of greener processes. However, it must be clarified that the examples covered in this review are based not only on sustainability or higher yields, but rather cover a broad spectrum of cases allowing us to show the current advantages and limitations of bio-click chemistry.
3. Bio-click reactions
Click chemistry is a dynamic and growing field, continually leading to new transformations. Many of these developments are predominantly focused on bioconjugation and applications in materials science.24 Examples for new transformations characterised as click reactions are the sulfur(VI) fluoride exchange (SuFex)25 and the synthesis of azides from primary amines26 respectively. Nevertheless, interactions of the latter with biocatalysis for the creation of new molecules are still missing. Significant cooperation between click chemistry and biocatalysis has been reported for CuAAC-, Diels–Alder-, epoxide-opening- and thiol-Michael reactions, and these studies will be discussed in further detail.3.1 CuAAC and biocatalysis
CuAAC is probably the most representative click reaction. It allows for the synthesis of 1,2,3-triazoles from organic azides and alkynes in the presence of a copper-(I)-catalyst (Scheme 1). The 1,2,3-triazole ring has shown great value as a pharmacophore, and several reviews focused on the potential of this invaluable motif in medicinal chemistry.15,27 Regarding the bio-click approach, the main focus in literature is the combination of CuAAC with oxidoreductases and hydrolases respectively.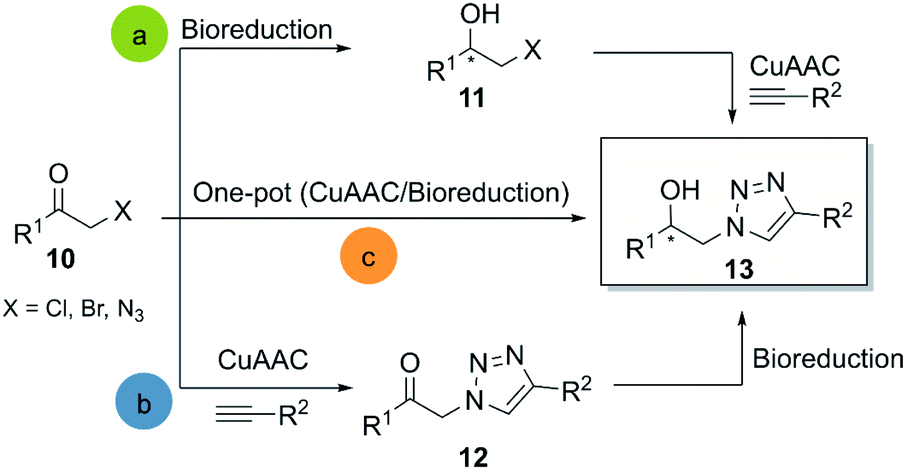 | ||
| Scheme 1 General methodologies for the chemo-biocatalytic synthesis of β-hydroxytriazoles. | ||
In 2008, Hua and co-workers reported the use of a recombinant carbonyl reductase from Candida magnoliae (CMCR) and an alcohol dehydrogenase from Saccharomyces cerevisiae (Ymr226c), with glucose dehydrogenase as cofactor-regeneration enzyme, for the enzymatic reduction of α-azidoacetophenone.29 Both enzymes were able to catalyze the formation of the corresponding chiral alcohol in excellent optical purities, good yields and with wide substrate scope. After the successful bioreduction of 2-azidoacetophenones 14, the desired 1,2,3-triazoles 17 were obtained in high yields, using CuSO4 and sodium ascorbate as catalytic system (Scheme 2a).
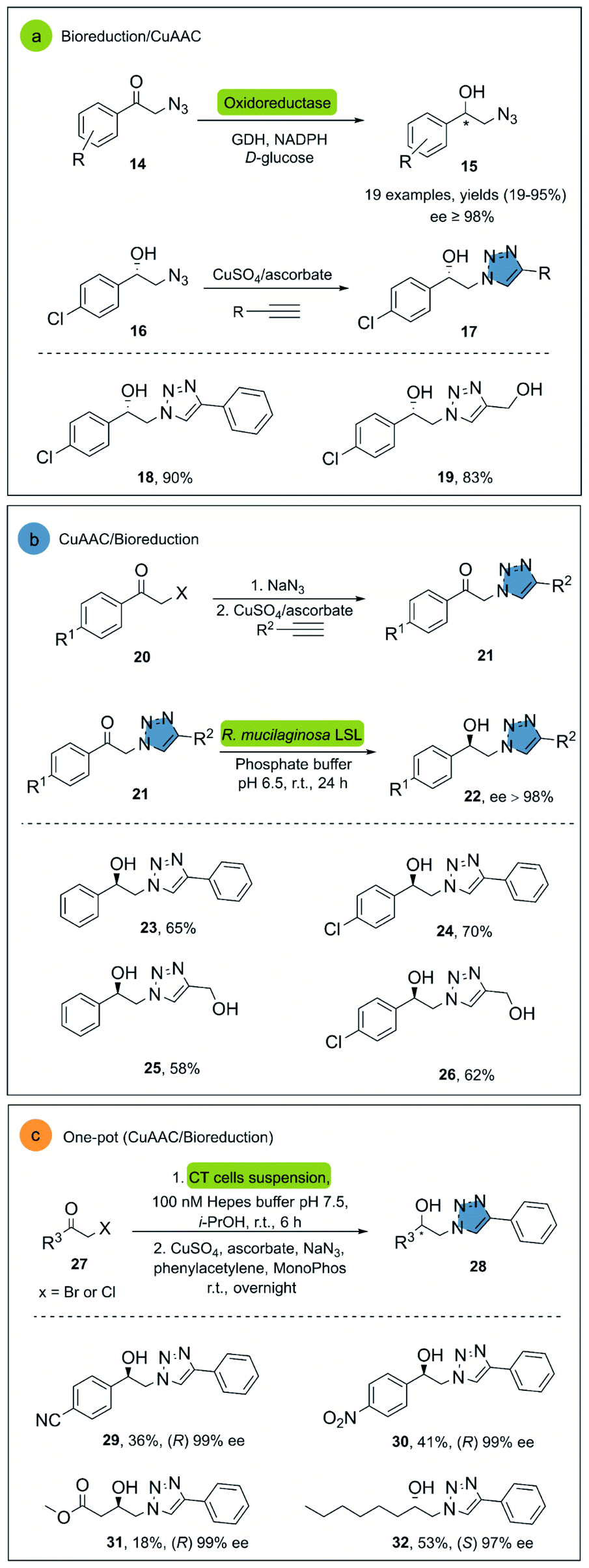 | ||
| Scheme 2 (a) Synthesis of β-blocker analogues containing 1,2,3-triazoles moieties, (b) stereoselective reduction of β-ketotriazoles with R. mucilaginosa LSL, (c) one-pot whole-cell catalysis and CuAAC for the conversion of α-haloketones to β-hydroxytriazoles. | ||
Another interesting example is the direct synthesis of enantioenriched β-hydroxytriazoles from the corresponding haloketones and β-ketotriazoles.30 In this approach, a whole-cell procedure of wild type Rhodotorula mucilaginosa LSL successfully catalyzed the one-pot enantioselective bioreduction of the substrate in water at room temperature, although only four examples were reported under these conditions (Scheme 2b). Notably, the reactants necessary for the ketotriazole formation, such as sodium azide and copper sulfate, did not affect the R. mucilaginosa LSL system in its catalytic activity. This represents an important advantage, avoiding purification steps and the manipulation of organic azides, often exhibiting explosive properties.31
Janssen and co-workers described an alternative system for the synthesis of β-hydroxytriazoles using a one-pot methodology.32 The developed system consists of the E. coli strain MC1061, which overexpresses enzymes such as alcohol dehydrogenases (ADH) and halohydrin dehalogenases (HHDH) with different stereospecificities. The construct that overexpressed (R)-stereospecific (ADH) and (HHDH) was called CT-cells and produce (R)-enantiomers, while the BT-cells form the (S)-enantiomers in high enantiomeric purity, albeit only moderate yields. The catalyst for the click reaction was CuSO4 with sodium ascorbate in the presence of MonoPhos as ligand, increasing the CuAAC reaction rate33 (Scheme 2c).
Chiral diols are a group of compounds widely used as organocatalysts and intermediates for the preparation of chiral heterocycles.34,35 Gotor and co-workers reported the synthesis of dihydroxytriazoles 37 through a highly efficient chemoenzymatic procedure under mild reaction conditions.36 In this two-stage one-pot procedure, the recombinant enzyme ADH-A from Rhodococcus rubber overexpressed in E. coli (Prelog-enzyme) and a commercially available LBADH from Lactobacillus brevis (anti-Prelog-enzyme) were used for the bioreduction of several alkynones 33 and α-azido ketones 34 in a phosphate buffer with 2-propanol. A copper wire was used as a reusable catalyst for the click reaction, supplemented by a catalytic amount of copper sulfate (Scheme 3).
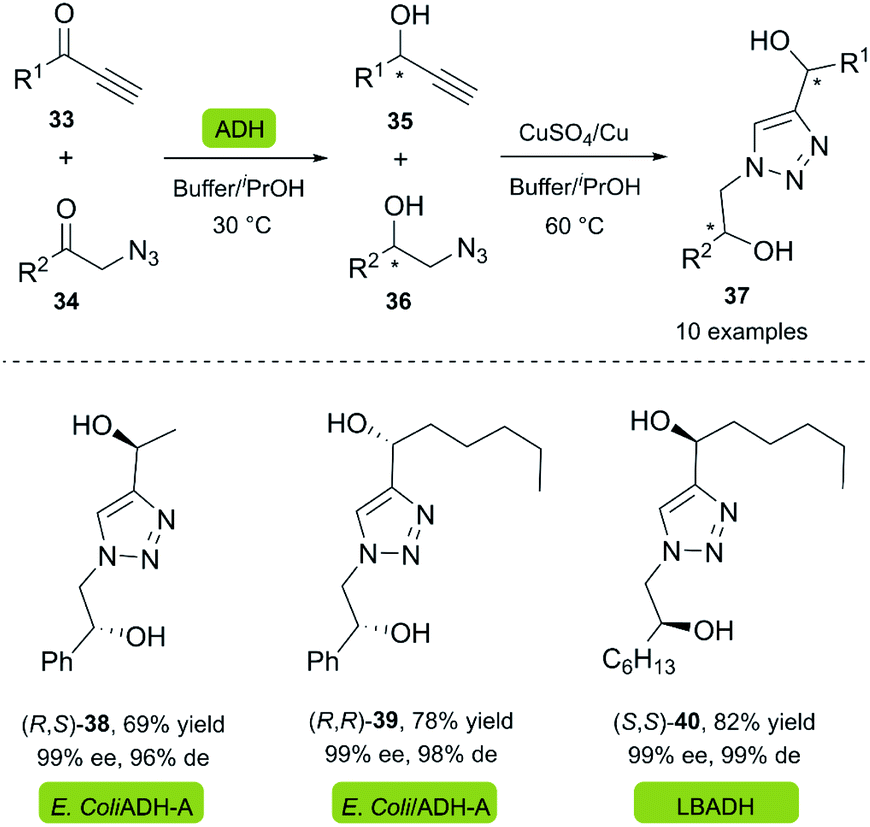 | ||
| Scheme 3 Synthesis of dihydroxytriazoles from chemoenzymatic approach. | ||
A powerful methodology for asymmetric ketone reduction was reported by Omori and co-workers in 2013, using whole cells from plant tissue from carrots (Daucus carota). The main advantage of this approach is its simplicity, economy of the process and the easy availability. The reaction features the one-pot bioreduction of azidoacetophenones (p,m-substituted) 41 with the subsequent CuAAC reaction using the Sharpless–Fokin catalyst (Scheme 4).37 The products were obtained in moderate yields but excellent enantiomeric excess (>99%). However, a disadvantage of this process is the rate of the bioreduction, requiring 5–7 days for completion. This impedes the implementation of a single-stage one-pot procedure, since the click reaction occurs very fast, and non-reduced triazoles are insoluble in water.
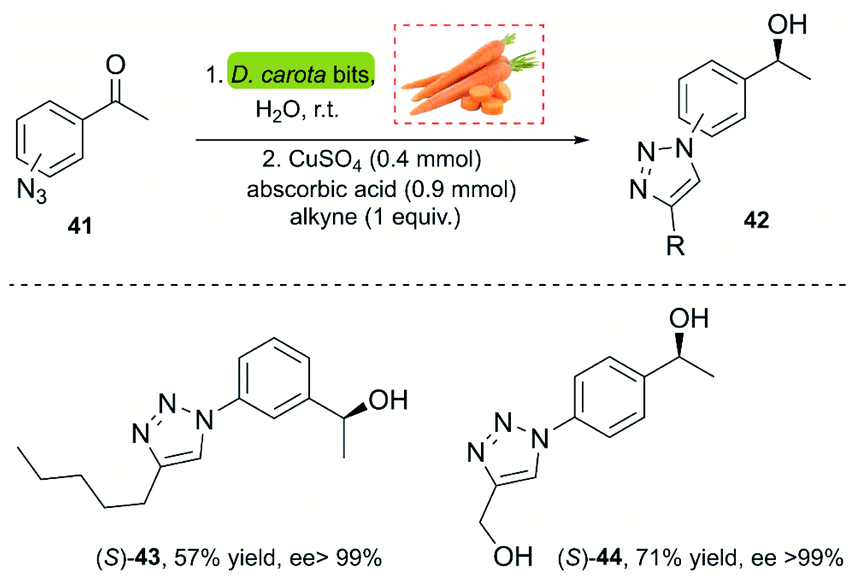 | ||
| Scheme 4 A chemoenzymatic methodology for the synthesis of chiral hydroxytriazole. | ||
Dioxygenases are versatile biocatalysts that have been used in the chemoenzymatic synthesis of chiral hydroxyazides. In 2011, the Stefani group reported the enzymatic dihydroxylation of arenes 45 catalyzed by whole-cell P. putida F39/D en route to chiral azides 47. The subsequent click reaction between 47 and several alkynes was achieved with the Sharpless–Fokin catalyst in t-BuOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O or toluene:H2O,38 and the corresponding triazolylconduritols 48 were obtained in good to excellent yields (Scheme 5).
:H2O or toluene:H2O,38 and the corresponding triazolylconduritols 48 were obtained in good to excellent yields (Scheme 5).
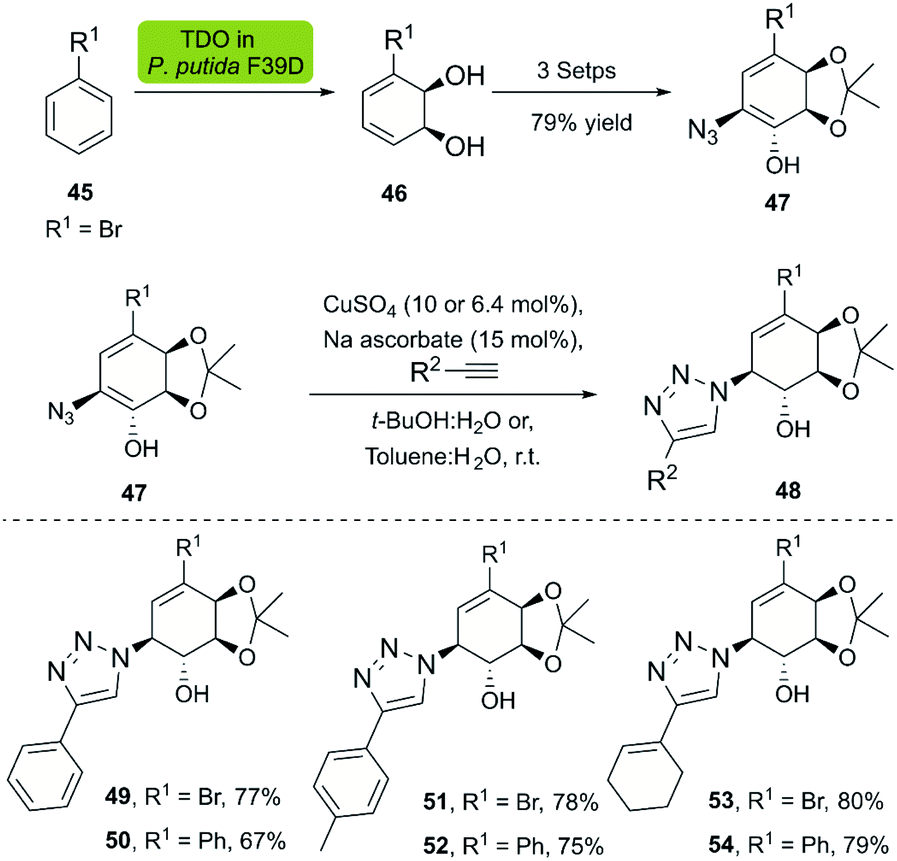 | ||
| Scheme 5 Chemo-enzymatic synthesis of the bromoazidoconduritol derivative, and synthesis of triazolylconduritols. | ||
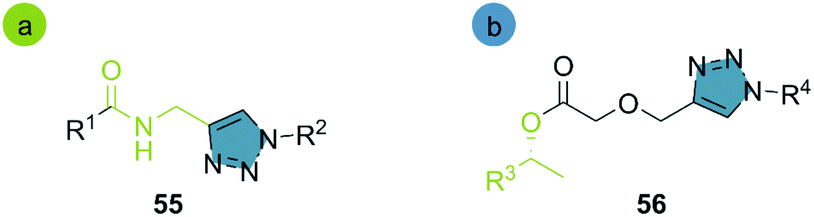 | ||
| Scheme 6 Representative structures obtained by chemoenzymatic hydrolases/CuAAC reactions in (a) peptidomimetic synthesis (b) kinetic resolution of racemates. | ||
Enzymatic aminolysis is an excellent environmentally-friendly alternative to coupling reactions generally used in amide synthesis, and its importance is expected to increase over the coming years.39 However, the enzymatic formation of the peptide bond usually requires a high amount of biocatalyst, which is a major drawback. The sustainability of these processes was greatly improved with the implementation of immobilized enzymes, which are usually very stable and easily removed from the reaction medium, playing a crucial role in green amide syntheses.
Due to their structural and electronic similarity, 1,2,3-triazoles can act as peptidomimetic amides. Specifically, the 1,4-disubstituted 1,2,3-triazoles are bioisosteres of trans-amide compounds.40 Due to the importance of peptides as pharmacologically active compounds, the triazol-ring represents an excellent alternative for the synthesis of peptides with increased metabolic stability, as well as biologically-active natural product analogs difficult to obtain through conventional synthesis.
In 2013, the Müller group reported the first enzymatic aminolysis catalyzed with lipases and a subsequent CuAAC reaction.41 For the aminolysis reaction, different commercially available immobilized lipases were studied. However, only Novozym® 435 and Immobead® 150 successfully catalyzed the reaction of methyl esters 57 with propargyl amine 58. Novozym® 435 (Candida antarctica lipase B [CALB] immobilized in Lewatit VP OC 1600) proved to be superior and achieved 68% conversion in 24 h. The optimized reaction conditions can be applied to a broad range of substrates and generally achieve good yields. The biocatalytic synthesis of propargylamides was then coupled to the CuAAC reaction using CuO2/benzoic acid as catalyst, and H2O:MeOH (1:1) as solvent system. The amide-triazole products 66–67 were obtained with yields between 51–85% (Scheme 7).
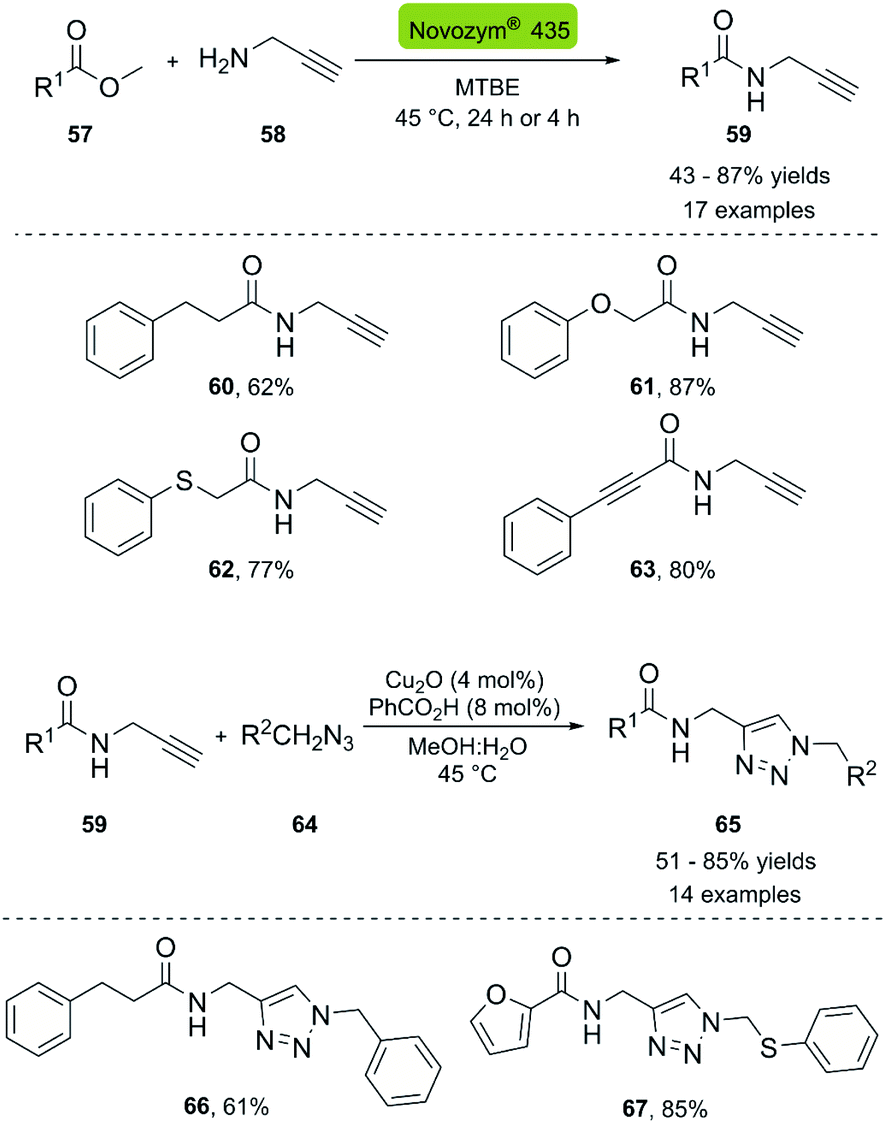 | ||
| Scheme 7 Synthesis of propargyl amides with CAL-B (Novozym® 435) and consecutive three-component synthesis of the amido methylsubstituted 1,2,3-triazoles. | ||
Based on the aforementioned studies, the Müller group reported the consecutive seven-component synthesis of triamides with a triazole moiety in 2019.42 This approach incorporated the biocatalytic aminolysis-CuAAC reaction in a new 5-stage multicomponent one-pot synthesis. The reactions featured (i) the synthesis of a diamide with a methyl ester group, from a 4-component Ugi-reaction, (ii) the propargylamide synthesis catalyzed by Novozym® 435, (iii) a CuAAC reaction and (iv) a Suzuki cross-coupling to the final product. This sequential one-pot procedure allowed to synthesize compound 70 in 36% overall yield. The described method allows for the generation of compound libraries, in an easy way without the need for intermediate purification (Scheme 8).
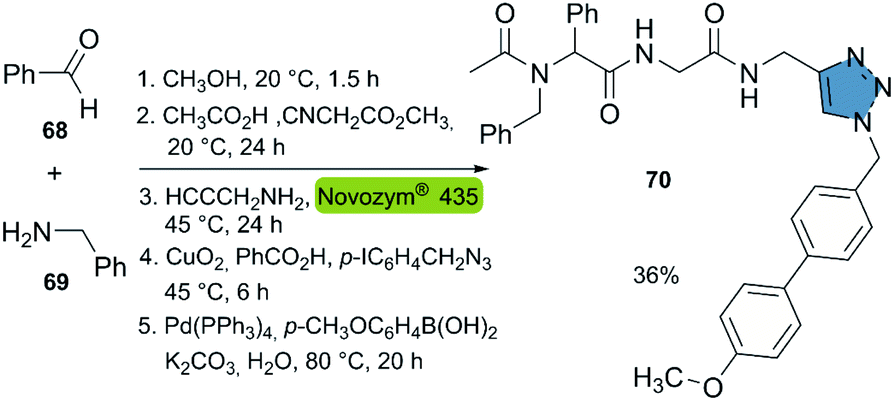 | ||
| Scheme 8 Consecutive seven-component sequence: Ugi reaction, CALB-catalyzed aminolysis, CuAAC, Suzuki coupling for the synthesis of product 70. | ||
A second application of the hydrolases combined with CuAAC are the kinetic resolutions of racemic mixtures. In this context, Büyükadali et al. described the one-pot synthesis of chiral benzothiophenyltriazoles and benzofuranyltriazoles in 2015.43 This bio-click approach produces the enantiomerically enriched homopropargylic alcohols (+) 72 using the commercially available TLIM (immobilized Thermomyces lanuginosus lipase). TLIM proved superior to other lipases, such as Novozym® 435 concerning both, enantioselectivity and reaction time. Furthermore, the reaction occurs in vinyl acetate, which acts as an acyl donor and solvent (Scheme 9). Unfortunately, the amount of biocatalyst necessary was 1:1% w/w with respect to the substrate, and recycling experiments of the catalyst were not reported. The concatenation of the TLIM-mediated enzymatic resolution and a click reaction was achieved through a subsequent one-pot multicomponent CuAAC reaction using CuSO4·5H2O/Na ascorbate, NaN3, L-proline, and Na2CO3, furnishing the corresponding triazole derivatives (+) 79 with good yields (Scheme 9). The in situ generation of the organic azide eliminates the need of an intermediate purification process.
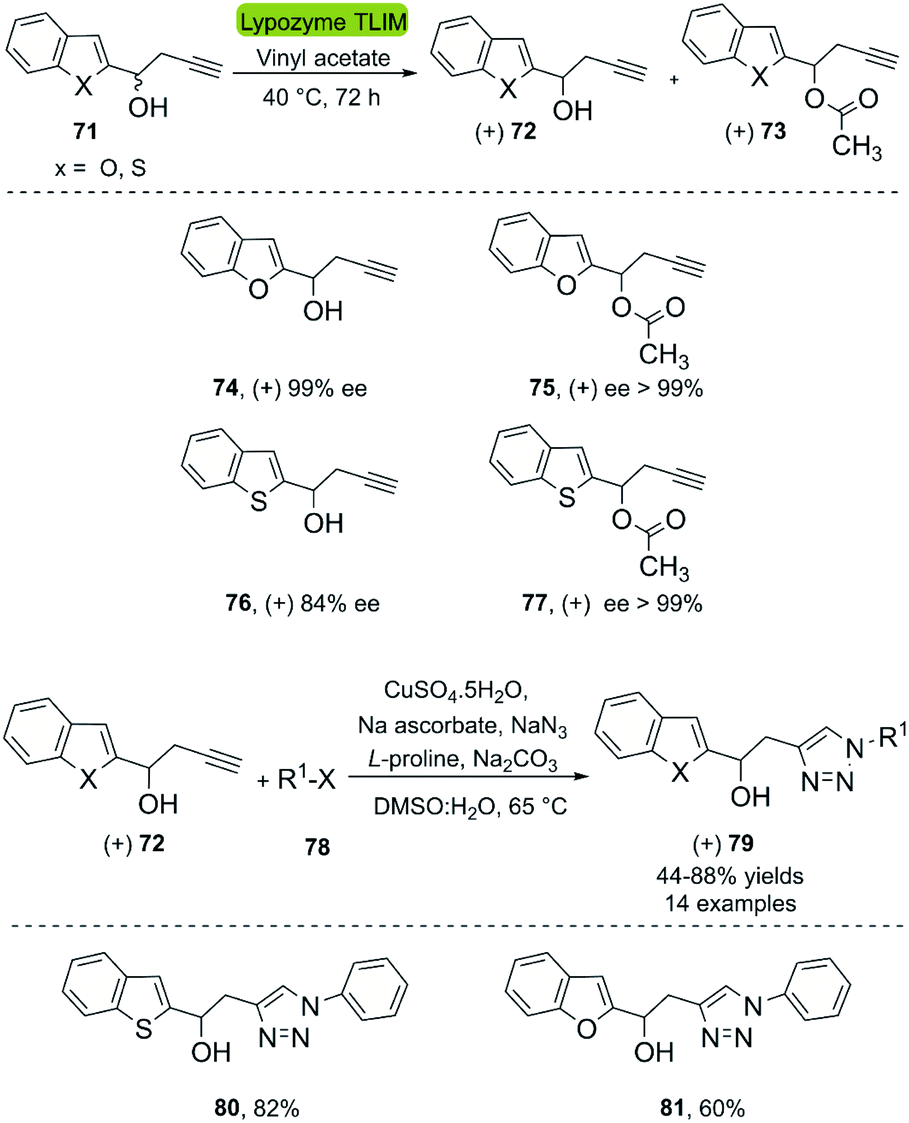 | ||
| Scheme 9 Enzymatic resolution of racemic homopropargylic alcohols with TLIM and multicomponent synthesis of 1,4-disubstituted 1,2,3-triazole derivatives. | ||
In 2018, Moisă et al. reported a new bio-click approach for the separation of racemic secondary alcohols 82 by simple extraction.44 This methodology uses the enantioselectivity of the lipases of Novozym® 435 and Pseudomonas fluorescens (L-AK) for the catalysis of an O-acylation of heteroarylethanols with different esters 83a–d as acylating agents. The corresponding (R)-product 84 is then subjected to a CuAAC reaction with azido-functionalized tertiary amine 85 in the presence of CuI, furnishing the triazole quantitatively. The ionizable triazole ester was efficiently extracted with an aqueous acetic acid solution, while the (S)-82 alcohol was recovered from the organic phase. The alcoholysis of the triazole derivative in the presence of ethanol was catalyzed by CALBSWCNT (CALB covalently immobilized on single-walled carbon nanotubes) and allowed for the obtention of the (R)-82 alcohol (Scheme 10). Up-scaling of the process to a multi-gram scale revealed, that there was neither a change in the selectivity of the enzymes nor in the efficiency, as reflected in virtually unchanged isolated yields of the products (>99%).
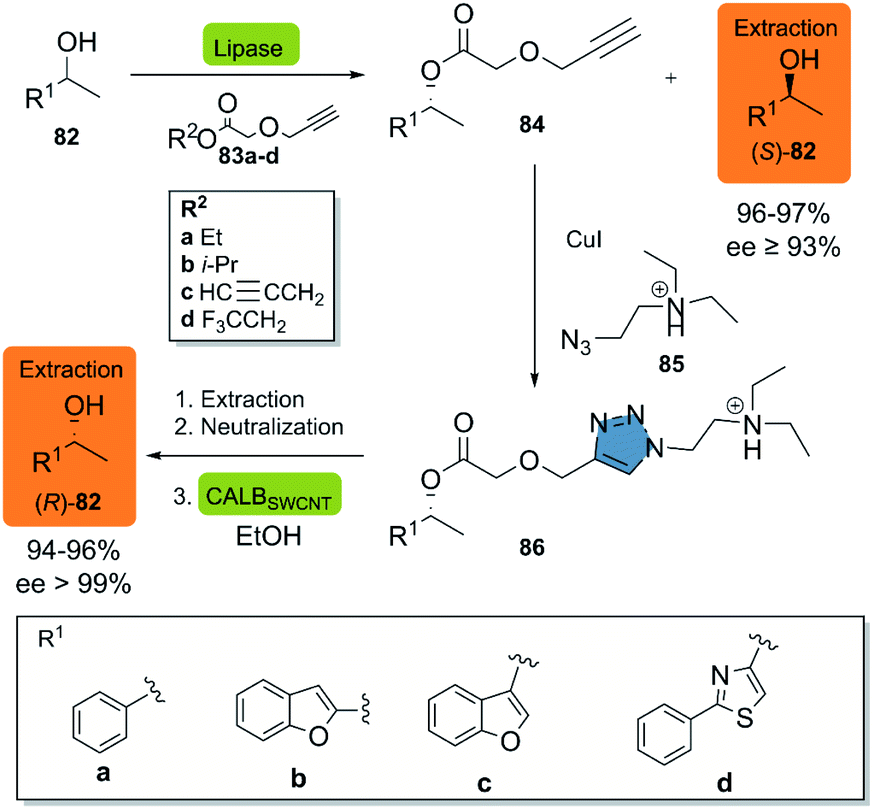 | ||
| Scheme 10 Production of the two enantiomeric forms of (hetero)arylethanols by an enzymatic KR/click reaction-based separation process. This figure has been Adapted from ref. 44 with permission from the Royal Society of Chemistry. | ||
3.2 Diels–Alder reactions and biocatalysis
The Diels–Alder (DA) reaction is an outstanding tool for the synthesis of cyclohexenes and other ring structures. Involving the cycloaddition of a diene and a dienophile, it has been extensively used for the synthesis of complex molecules and is one of the most powerful transformations in organic chemistry (Scheme 11). Moreover, the DA reaction is stereospecific, shows a high atom economy and reliability, and can be carried out in environmentally-friendly solvents.45,46 A hetero-Diels–Alder-reaction (HAD) is equally well studied, and is a reliable alternative for the synthesis of ring structures including heteroatoms.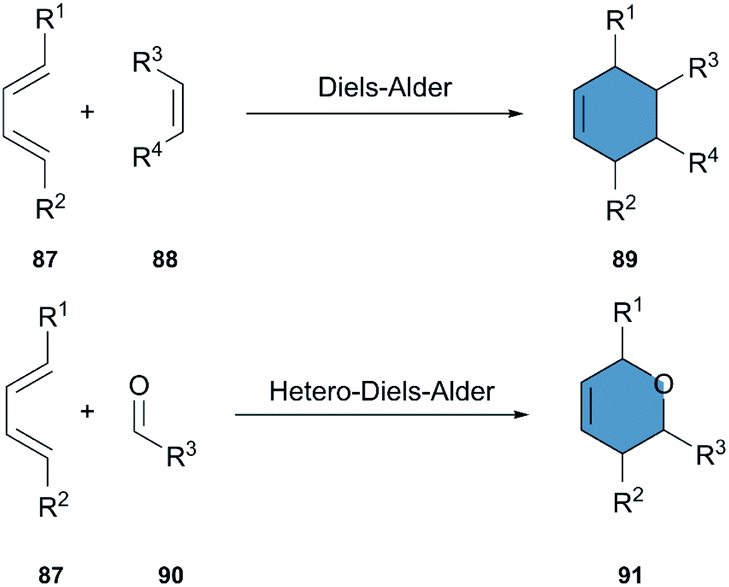 | ||
| Scheme 11 Diels–Alder reaction and hetero-Diels–Alder reaction. | ||
In contrast to the CuAAC reaction, the DA reaction can be carried out without a catalyst. However, the use of Lewis acids allows to decrease the activation energy of the DA reaction, increasing its rate and lowering the necessary reaction temperature.47–49 A further increase of the reaction rate can be achieved by the incorporation of EWG-groups in the dienophile and EDG-groups in the diene, favoring the electronic distribution necessary for the reaction to occur.
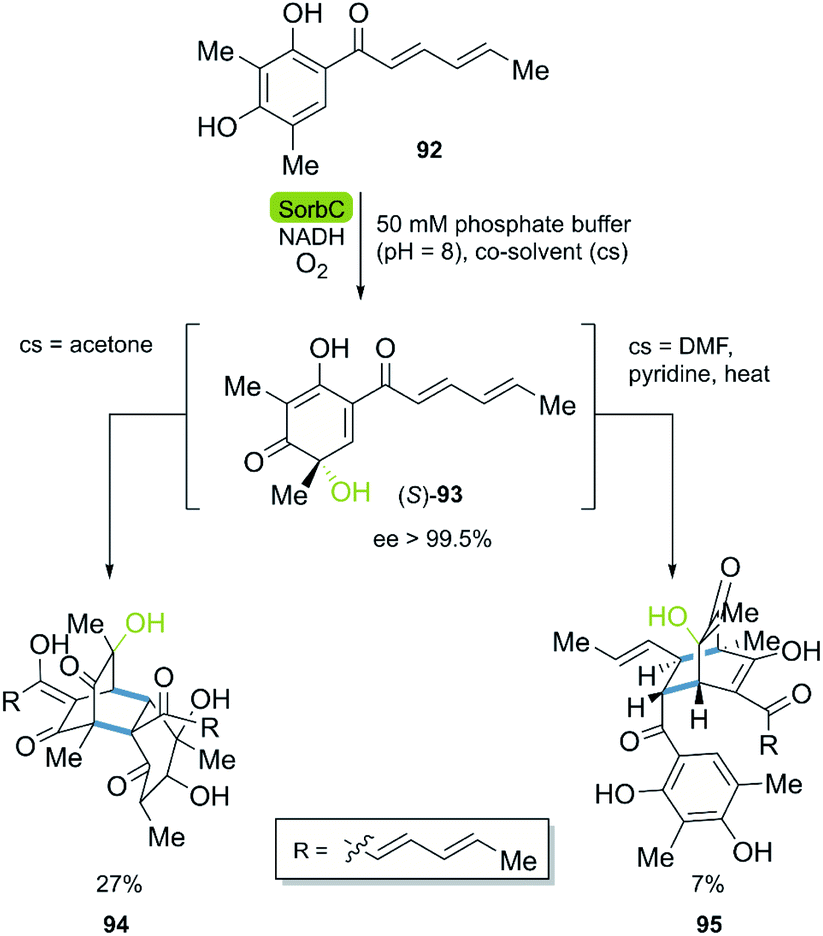 | ||
| Scheme 12 Key step of the stereoselective enzymatic total synthesis of bisorbicillinol 94 and sorbiquinol 95. | ||
In 2018, the groups of Gulder and Narayan independently reported a chemoenzymatic approach towards the synthesis of new sorbicillin derivates.53,54 In these investigations, the use of the SorbC enzyme once again furnished sorbicillinol 93 by oxidative dearomatization as a key step. The use of CH2Cl2 enabled the extraction of 93; and its subsequent reaction with different dienophiles allowed the production of sorbicillin derivates in a simple chemoenzymatic process. The scope of products was very versatile, giving access to urea sorbicillinoid 96, sorbicatechol A 97, and rezishanone C 98 (Scheme 13).
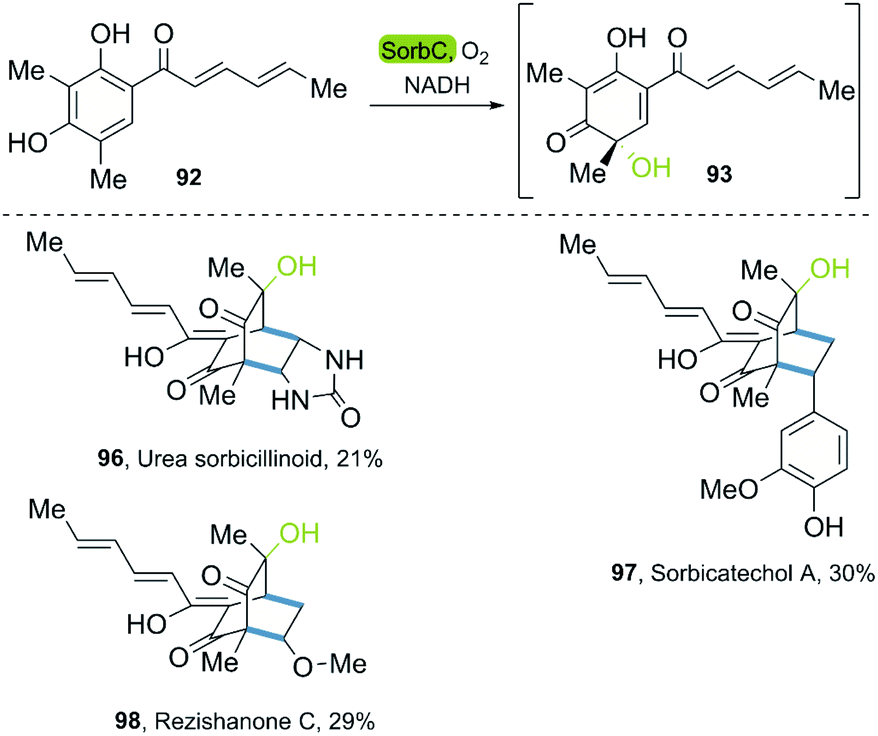 | ||
| Scheme 13 Chemoenzymatic synthesis of the sorbicillinoids via enantioselective oxidative dearomatization of sorbicillin with SorbC and DA reaction. | ||
The Narayan-group synthesized the urea sorbicillinoid 96 via a DA reaction between sorbicillin and bisacylated urea and following by the addition of LiOH. Furthermore, they described different monooxygenases, such as Trop B and AzaH, for the catalysis of the oxidative dearomatization of resorcinols, allowing for the synthesis of valuable ortho-quinol products,54 a process that was adapted to gram scale.
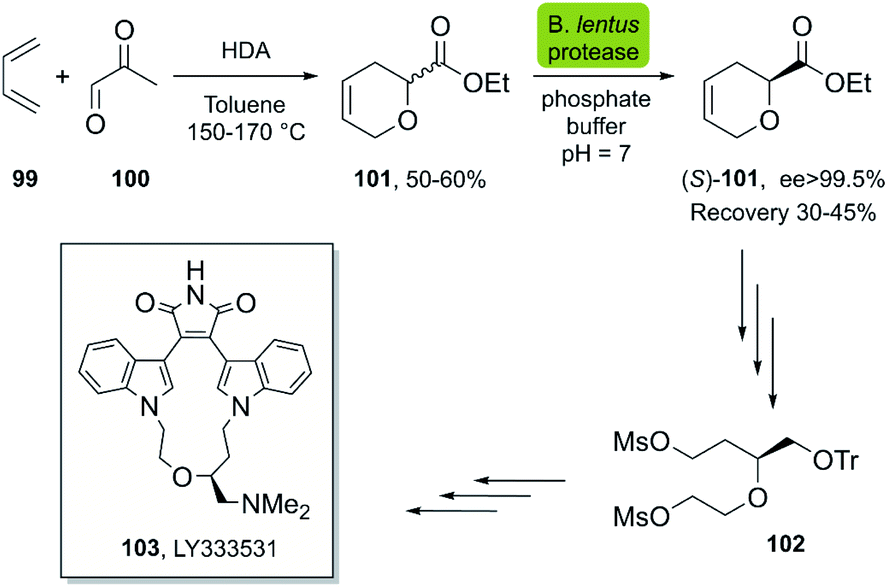 | ||
| Scheme 14 Chemoenzymatic synthesis of (S)-101, a key intermediate in the synthesis of LY333531, a protein kinase C inhibitor (PKC β). | ||
In 2010, Wirz et al. reported the scalable enantioselective synthesis of a benzothiazole derivative 109, which was evaluated as an A2 receptor antagonist for the treatment of major depression.57 The developed chemoenzymatic process consists of the DA-reaction of furan 104 with acrylonitrile 105, activated by ZnCl2, furnishing bicyclic product 106. The following steps gave rise to the racemic product 107, which was hydrolyzed by CALA lipase in a stereoselective fashion, furnishing the (S)-enantiomer 108 with excellent enantioselectivity (Scheme 15).
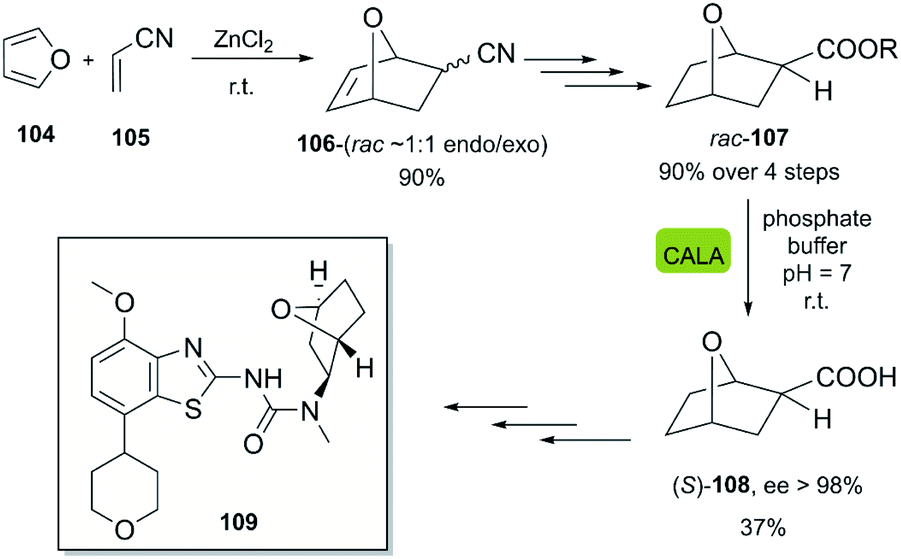 | ||
| Scheme 15 Bio-click approach for the synthesis of (S)-108. Precursor in the synthesis of A2 receptor antagonist 109. | ||
Abacavir is a carbocyclic nucleoside with antiviral activity, acting as a reverse transcriptase inhibitor (see Scheme 16). It is used in combinations with other nucleoside analogues, such as lamivudine, for the treatment of HIV-infection.58 Crimmins et al. described an asymmetric synthesis of abacavir in 1996, making use of a first-generation Grubbs catalyst.59 An alternative synthetic route (Scheme 16a), used the Vince lactam 111, which can be obtained by the DA reaction of cyclopentadiene 110 and tosyl cyanide.60 The subsequent enzymatic kinetic resolution of the racemic Vince lactam with lactamase furnished the corresponding amino acid 113 (with (−) lactamase) or the (−) Vince lactam 112 (using (+) lactamase), which was then hydrolyzed into the desired amino acid 113.61 This key intermediate can then be converted into the desired product abacavir.62
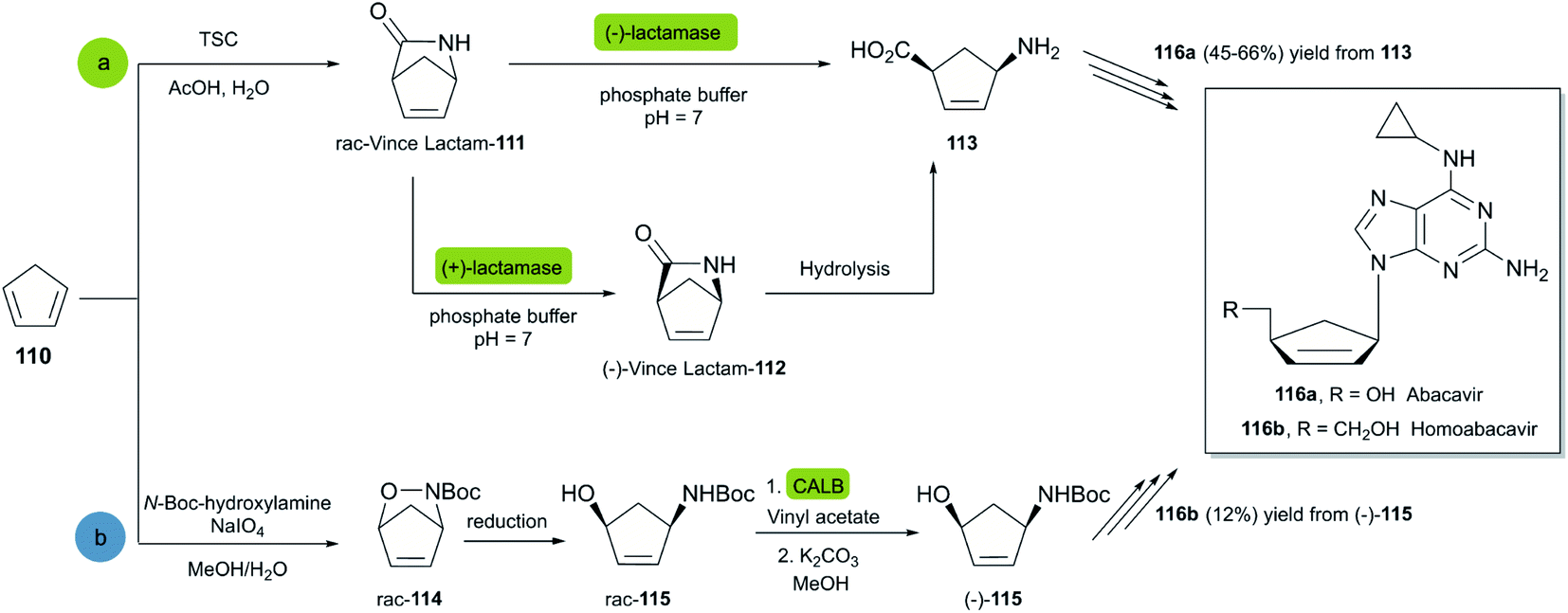 | ||
| Scheme 16 Chemoenzymatic synthesis of abacavir. (a) Using a lactamase by kinetic resolution of rac-Vince lactam and (b) using a CALB lipase in the kinetic resolution of aminocyclopentenol derivate. | ||
The Vince lactam is not only a useful intermediate in the synthesis of abacavir, but also in the synthesis of peramivir and carbovir. In fact, numerous groups have reported the application of different lactamases for the resolution of the Vince lactam, en route to these powerful antiviral agents.63 A work carried out by Tardibono et al.64 presents a bio-click methodology towards homoabacavir 116b (Scheme 16b). In this approach, the hetero Diels Alder-reaction between cyclopentadiene and the transient nitroso-compound of N-Boc-hydroxylamine is used for the formation of intermediate 114. The subsequent reduction of the N–O bond furnished the racemic aminocyclopentenol derivative 115, which was then successfully subjected to a kinetic resolution with CALB lipase, using vinyl acetate with posterior basic hydrolysis, furnishing 115-(−) (80% ee) as a key intermediate for the further syntheses of homoabacavir 116b and homocarbovir.64
3.3 Epoxide-opening and biocatalysis
Epoxides are valuable and versatile building blocks and synthetic intermediates common in nature and synthesis.65,66 The outstanding reactivity of the three-membered ring is mainly caused by the special geometry and angle strain, endowing the ring opening reaction with a favourable thermodynamic driving force (24 to 28 kcal mol−1).67,68 The SN2 ring opening of epoxides shows a variety of desirable characteristics, such as reliability, stereospecificity and regioselectivity.4 The congener aziridines present similar characteristics, however with the advantage of the presence of a nitrogen atom, allowing subsequent chemical transformations in a somewhat easier way.69,70 Nevertheless, fewer efficient methods for the direct synthesis of aziridines are available,71 also reflected in the scarce number of respective bio-click approaches incorporating these substrates. The recent development of methods for the enzyme-catalyzed olefin aziridination72 could change these shortcomings and foster the development of a greater number of bio-click approaches incorporating aziridines.The ring-opening of epoxides has been extensively applied for the synthesis of complex molecules.73 In fact, the synthesis of numerous Active Pharmaceutical Ingredients (APIs) uses ring-opening of epoxides as a key step.74,75 The bio-click approach towards the ring-opening reaction of epoxides is a field of increasing interest.
Generally, the bio-click approach incorporating epoxides is applied in two ways: (a) the selective enzymatic oxidation of alkenes, furnishing epoxides in green conditions for the subsequent ring opening; (b) the oxidation of alkenes, a subsequent enzymatic resolution of the racemic epoxides and the ring-opening of the desired candidate (Scheme 17).
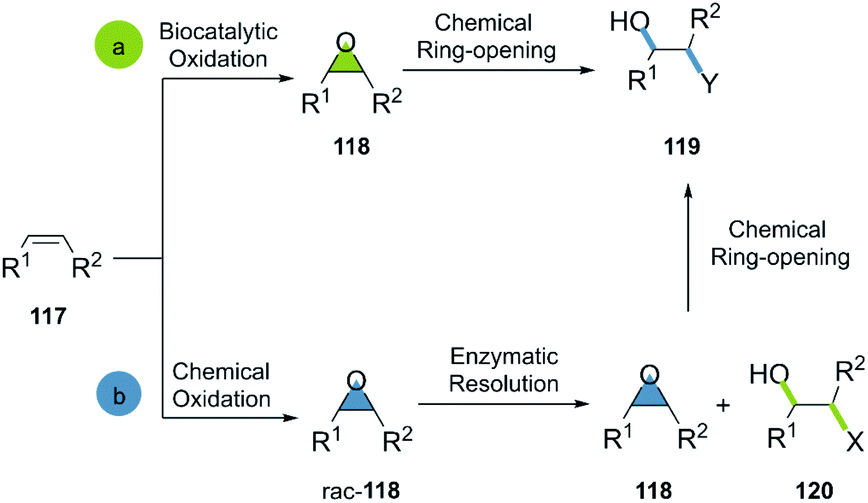 | ||
| Scheme 17 General bio-click approach in epoxide-opening reactions. | ||
cis-1,2-Dihydrocatechols 46 are obtained by the enzymatic dihydroxylation of arenes 45 and represent an excellent platform for the synthesis of alkaloids, sugars, cyclitols, prostaglandins, terpenes, polymers and others (Scheme 18a).81 As an example, the enantioselective synthesis of cis-1,2-dihydrocatechols is carried out in a whole-cell fermentation, which provides an efficient cofactor regeneration system.82 E. coli JM109 (pDT601) which contains the genes from Pseudomonas putida F1 for the overexpression of the toluene dioxygenase system (TDO),83 is the most common biocatalyst applied for the enantioselective synthesis of cis-1,2-dihydrocatechol derivates. Careful optimization of the reaction conditions in a biphasic system can achieve yields as high as 35 g L−1.84 This is quite remarkable, considering that no efficient chemical method is known for the production of this class of compounds at scale.79,81
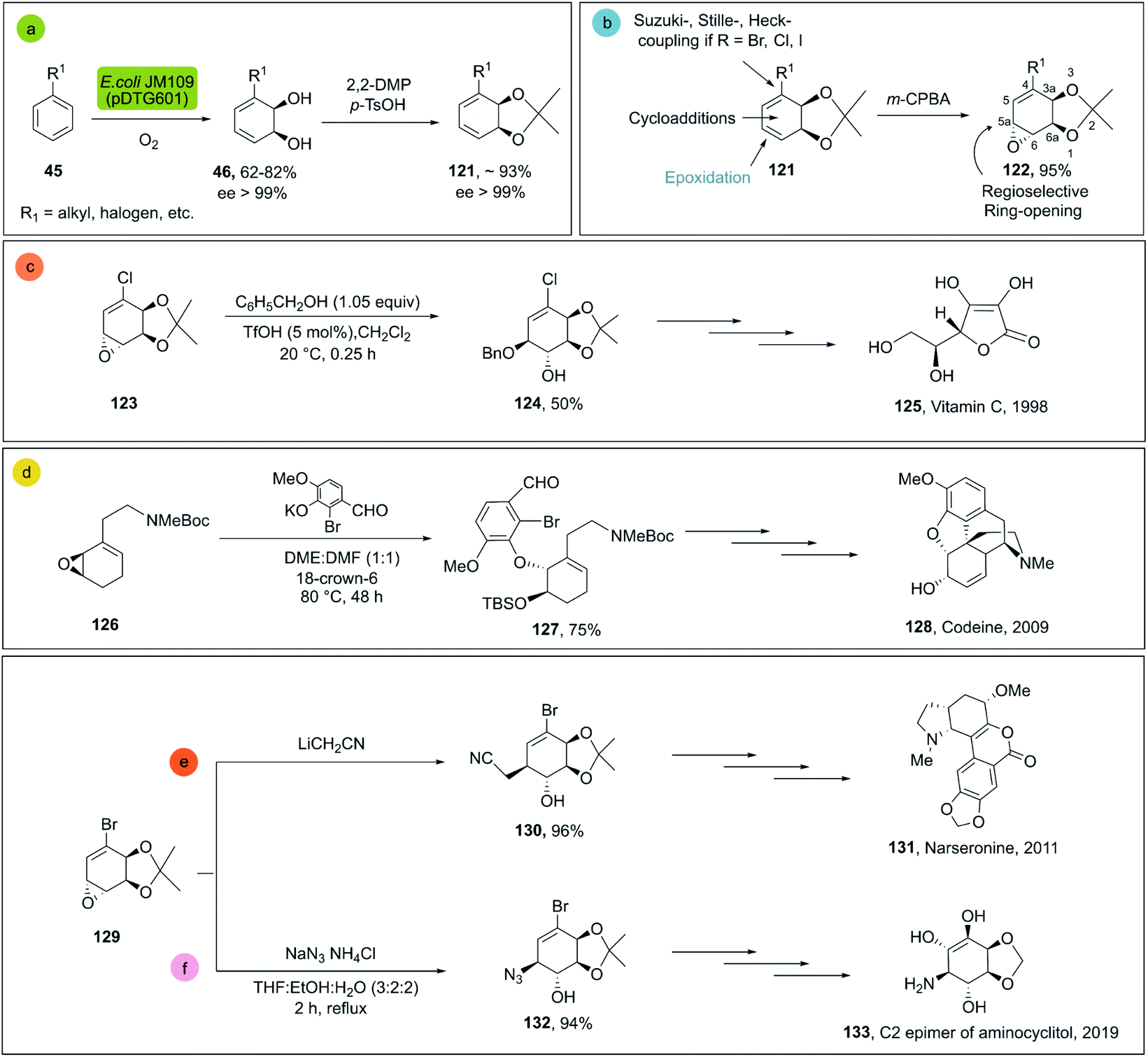 | ||
| Scheme 18 (a) Biocatalytic dihydroxylation of arenes by E. coli JM109 (pDT601), (b) chemical reactivity of cis-1,2-dihydrocatechols. Representative molecules obtained from bio-click methodologies (TDO-ring opening). (c) Vitamin C 1998 by Longmore et al.;87 (d) codeine 2009 by Leisch;88 (e) narseronine 2011 by Schwartz et al.;89 (f) C2 epimer of aminocyclitol 2019 by Carrau.90 | ||
The cis-1,2-dihydrocatechols are highly versatile and a great platform for enantioselective synthesis85 (Scheme 18b). Monoepoxidation, for example, is a transformation that can be easily applied to cis-1,2-dihydrocatechols 121 and combined with enantio- and regioselective 122 ring-openings, which can be efficiently applied in total synthesis.86
For the ring opening of the epoxides on dihydrocatechols 122 the general preference is towards the cleavage in the 5a position (see Scheme 18b), mainly associated to two factors: (a) the greater stabilization of the transient carbocation in the allylic position; (b) less steric hindrance at position 5a as compared to 6a. In this way, the cleavage of epoxides is generally achieved with high regio- and enantioselectivity, providing the corresponding products of the trans-1,2 addition efficiently.86
Representative examples for the opening of monoepoxides 122 (obtained from biocatalytic enzymatic cis-dihydroxylation of benzene derivates 45) with different nucleophiles are ubiquitous (see Scheme 18, vitamin C,87 codeine,88 narseronine,89 C2 epimer of aminocyclitol90) and crucial in the synthesis of natural products.
The indirect epoxidation of α-haloketones using ketoreductases (KREDs) has become a common strategy to obtain chiral epoxides, also reflected in the stereoselective synthesis of different APIs obtained on a kg-scale. The development of new variants of KREDs through protein engineering has widely enhanced scope, robustness, and selectivity of these biocatalysts. The fact that they are an excellent green alternative to conventional catalysts is further underscored by the easy workup, generally avoiding chromatographic product purifications.
In 2017, Hou et al. reported a chemo-enzymatic synthesis of BMS960, a powerful S1P1-receptor agonist.91 Its synthesis involved the enzymatic reduction of α-bromoketone 134 (100 g in 5 h at 40 °C) to the corresponding alcohol 135 using the commercially available KRED-NADH-110 (substrate enzyme ratio 200:1). The chiral alcohol was then extracted with MTBE and reacted with sodium tert-butoxide towards the epoxide with 93% overall yield and 100% ee (S-enantiomer). Subsequently, a regio- and stereospecific ring opening of the epoxide 136 with (S)-ethyl piperidine-3-carboxilate in the presence of a catalytic amount of DMAP at 50 °C, provided the product 137 in up to 77% yield and with an ee of 99.6% after recrystallization. Four successive chemical transformations furnished BMS-960 in 23–33% overall yield (Scheme 19a).
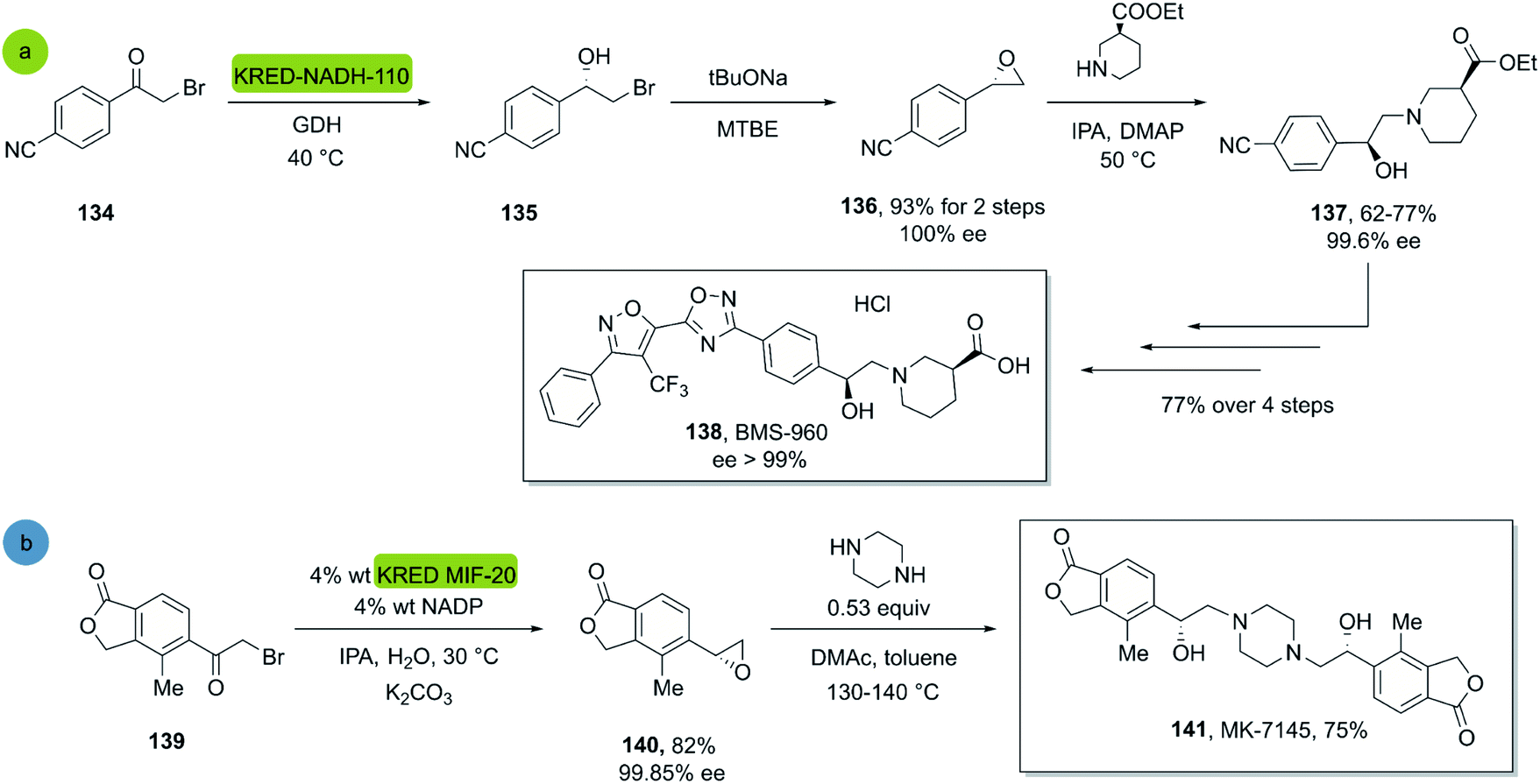 | ||
| Scheme 19 KRED mediated epoxidation of alfa-bromo ketones and ring opening: (a) synthesis of BMS-960, (b) synthesis of MK7145. | ||
In 2020, Ruck et al. reported a kg-scale synthesis of MK-7145, a clinical candidate for the treatment of hypertension and associated heart failures.92 The synthesis involved the enzymatic reduction of a bromo-ketone 139 to the corresponding bromohydrin as a key step. The commercial KRED-MIF-20-induced reduction and a subsequent ring closure furnished the corresponding enantiopure epoxide in 82% yield and excellent enantiopurity. Subsequently the target molecule MK-7145 141 was obtained through a bis-epoxide opening with piperazine at 140 °C, furnishing the product in 75% yield (Scheme 19b).
Similar to the abovementioned biocatalytic cis-dihydroxylation of arenes 142, the Hollmann group recently described the direct biocatalytic epoxidation of naphthalene derivatives.93 The methodology described is based on the recombinantly evolved peroxygenase variant PaDa-I from Agrocybe aegerita (rAaeUPO), a self-sufficient enzyme. Careful control of the reaction time of the nucleophilic addition allows for the generation of the ring-opening products 144 using nucleophiles such as sodium azide (Scheme 20).
 | ||
| Scheme 20 Bio-click synthesis of trans-disubstituted cyclohexadiene derivatives using peroxygenase PaDa-I, and chemical ring-opening with different nucleophiles. | ||
In this way, the synthesis of eleven derivatives in yields ranging from 6 to 73% was reported. These arene oxides are excellent building blocks for the synthesis of trans-disubstituted cyclohexadienes and can be further functionalized towards triazoles, amino alcohols, and other valuable entities. Epichlorohydrin 145 is a versatile molecule with multiple applications,94 and a useful starting material for the synthesis of different APIs.
The synthesis of rivaroxaban, an anticoagulant widely used in the prophylaxis of cardiovascular diseases,95 has been described by numerous groups using (R)-epichlorohydrin 145 as a building block.96 In a bio-click approach, rivaroxaban was synthesized via (R)-145, which can be obtained either by an enantioselective biooxidation of 3-chloropropene 146 using chloroperoxidase CPO from Caldariomyces fumago,97 or by resolution of racemic 145 using epoxyhydrolase EH ArEH98 from Agrobacterium radiobacter (Scheme 21). Further functionalisation of the desired epoxide with phthalimide under basic conditions using a phase transfer agent and a subsequent regio- and enantiospecific ring opening of epoxide 148 with arylamine 149, furnished the product 150 smoothly en route to rivaroxaban.99
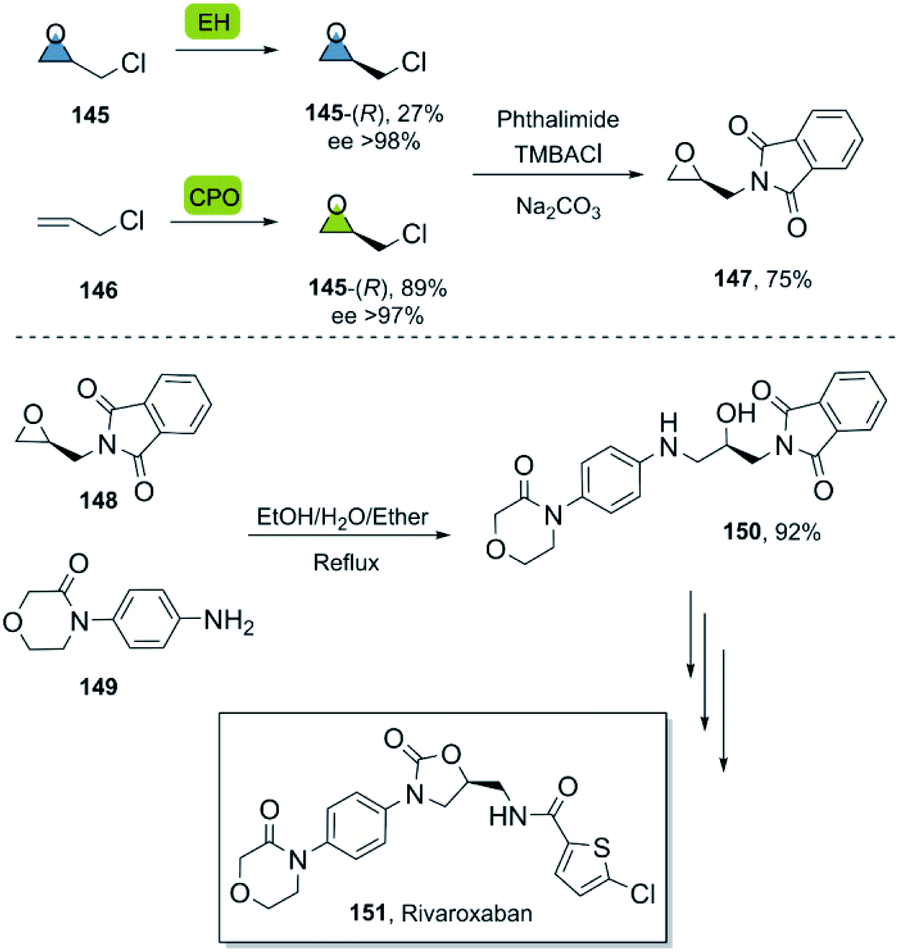 | ||
| Scheme 21 Bio-click approach for the synthesis of rivaroxaban. | ||
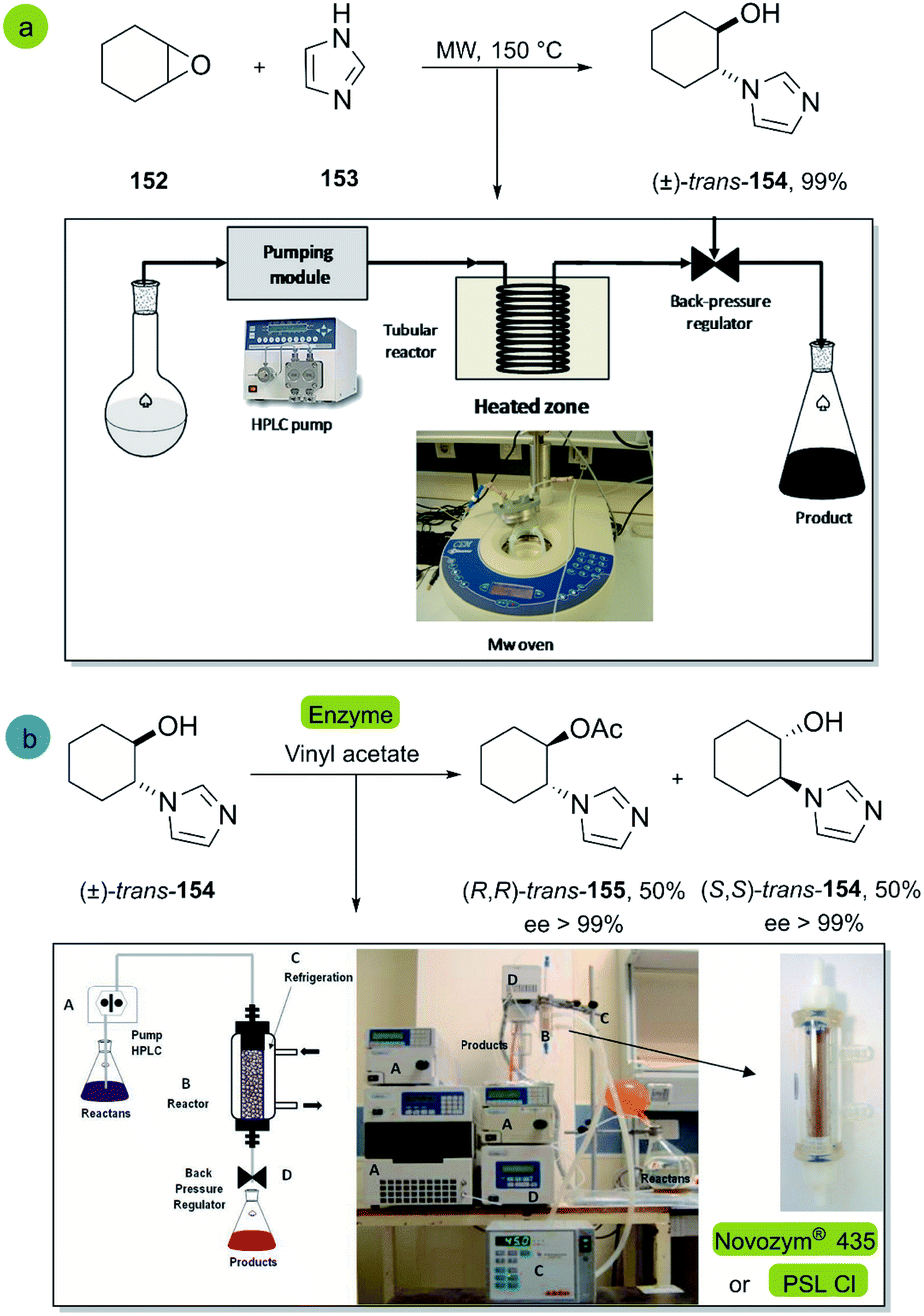 | ||
| Scheme 22 Synthesis of trans-2-(1H-imidazol-1-yl)cyclohexanol (b) enantioselective acylation of (±)-trans-2-(1H-imidazol-1-yl)cyclohexanol 154 with vinyl acetate catalyzed by Novozym® 435 or PSL-CI. This figure was used with permission.100 | ||
In 2015, Villar et al. reported the chemoenzymatic synthesis of optically pure, orthogonally protected trans-3-amino-4-hydroxypiperidines, potential molecules for the synthesis of chiral bioactive compounds (Scheme 23).102 The developed bio-click methodology developed, starts with the regioselective epoxide-opening with diallylamine, providing access to the racemic trans-cyclohexanol 157. The subsequent enzymatic kinetic resolution via transesterification with vinyl acetate was performed with Novozym® 435, which gave the desired product 158 with the highest conversion (47%) and enantioselectivity (ee > 99%) among the enzymes tested.
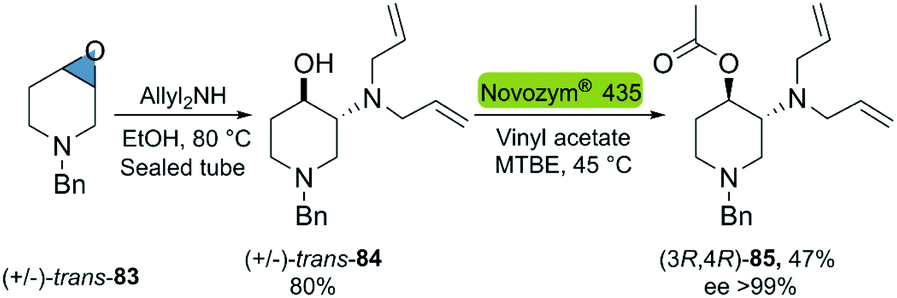 | ||
| Scheme 23 Chemoenzymatic synthesis of optically pure orthogonally protected trans-3-amino-4-hydroxypiperidines. | ||
Propranolol is a versatile beta-adrenergic receptor antagonist used in the treatment of several cardiovascular such as hypertension, cardiac arrhythmias, etc.103 Quite remarkably, (S)-propranolol is 100 times more potent β-adrenergic receptor blocker than its enantiomer.104 In 2015, Dong et al.105 reported the chemoenzymatic two-step synthesis of (S)-propranolol (Scheme 24). The enzymatic kinetic resolution of racemic epoxide 159 with the hydrolase BmEH128T (from Bacillus megaterium), smoothly furnished the (S)-configured product 159 smoothly after centrifugation of the undesired enantiomer. The subsequent regioselective ring-opening of (S)-159 with isopropylamine under reflux conditions gave (S)-propanolol 160 in an overall yield of 42% and excellent enantioselectivity after recrystallization (ee > 99%).
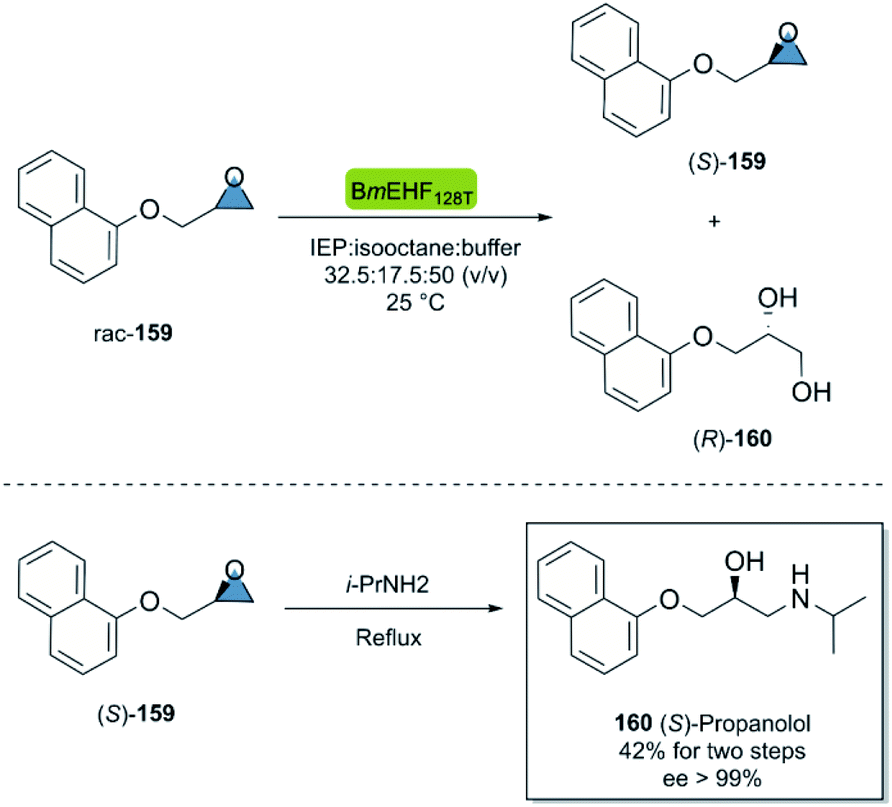 | ||
| Scheme 24 Chemoenzymatic synthesis of (S)-propranolol. | ||
3.4 Thiol-Michael reactions and biocatalysis
Thiol-Michael click reactions, either via free-radical, or catalyzed Michael-additions, are valuable transformations widely recognized in polymer science and bioconjugation chemistry, since they allow for the efficient formation of C–S bonds from thiols and alkenes (Scheme 25).106–108 They are usually highly efficient, work under mild reaction conditions, are regioselective and atom-economic, and only generate few or no by-products which are generally easily separated.109,110 Bio-click reactions approaches featuring thiol-Michael chemistry are scarce to date, despite the advantages of chemoenzymatic reactions in this context. | ||
| Scheme 25 Thiol-Michael click chemistry. | ||
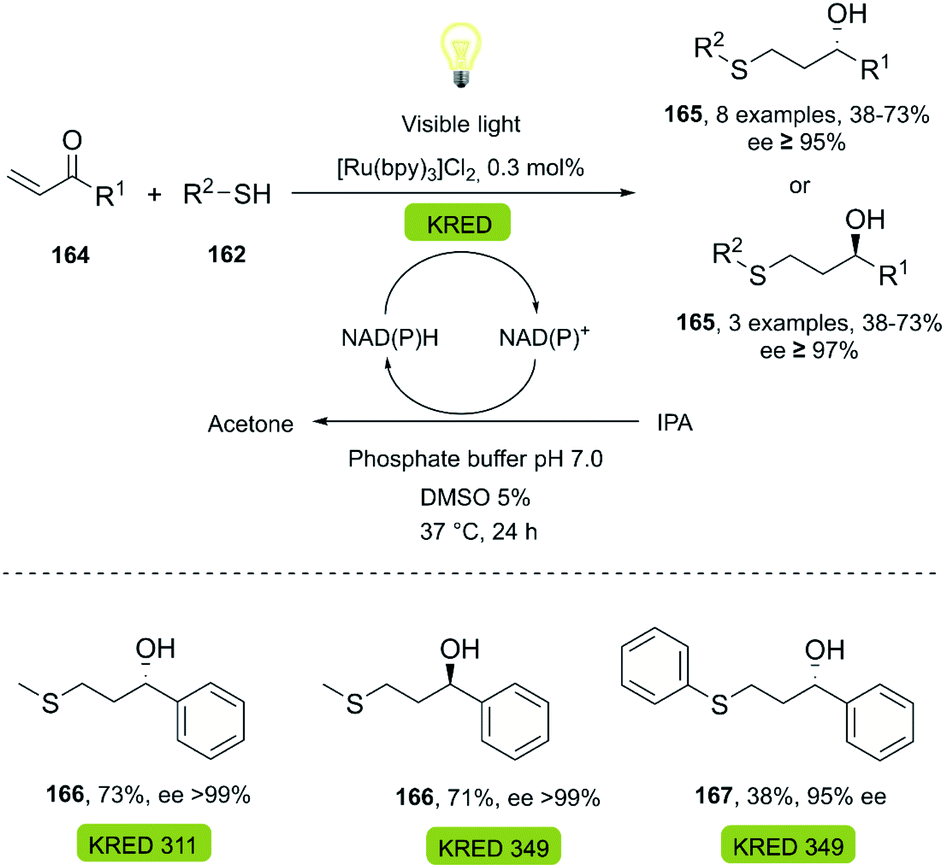 | ||
| Scheme 26 One-pot photo-biocatalytic (thiol-Michael/KRED reduction). | ||
4. Summary and outlook
This review summarizes developments in a field of research that combines biocatalytic transformations and click reactions. The proposal of the term bio-click chemistry comes quite naturally if one considers the varied and efficient syntheses of complex molecules explored. A clear classification of the different chemoenzymatic transformations will undoubtedly promote the development of more sustainable and efficient processes in this area (see Table 1).| Click reaction | Enzyme or microorganism | Structure | Function | Reference |
|---|---|---|---|---|
| CuAAC | Carbonyl reductase from Candida magnoliae (CMCR) |  |
β-Adrenergic receptor blocker analogue | 29 |
| Diels–Alder or hetero-Diels–Alder | (+)-γ-Lactamase, (−)-γ-lactamase from Bradyrhizobium japonicum USDA 6 | 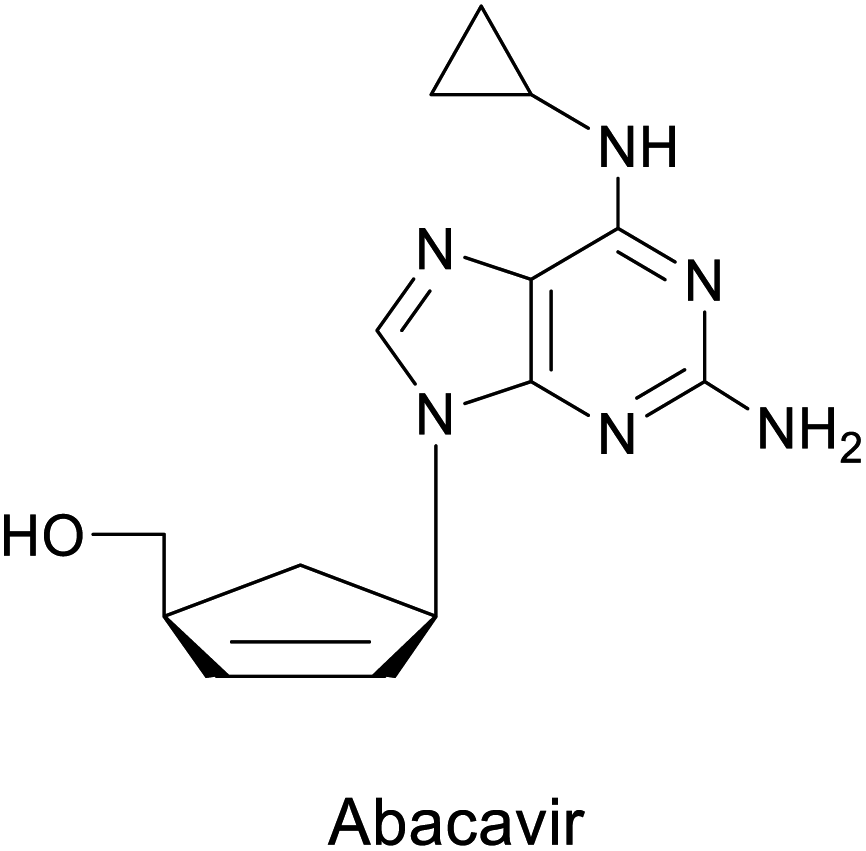 |
Carbocyclic nucleoside with antiviral activity | 58, 60, 61 and 112 |
| Candida antarctica B lipase (CALB) | 64 | |||
| Oxidoreductase SorbC | 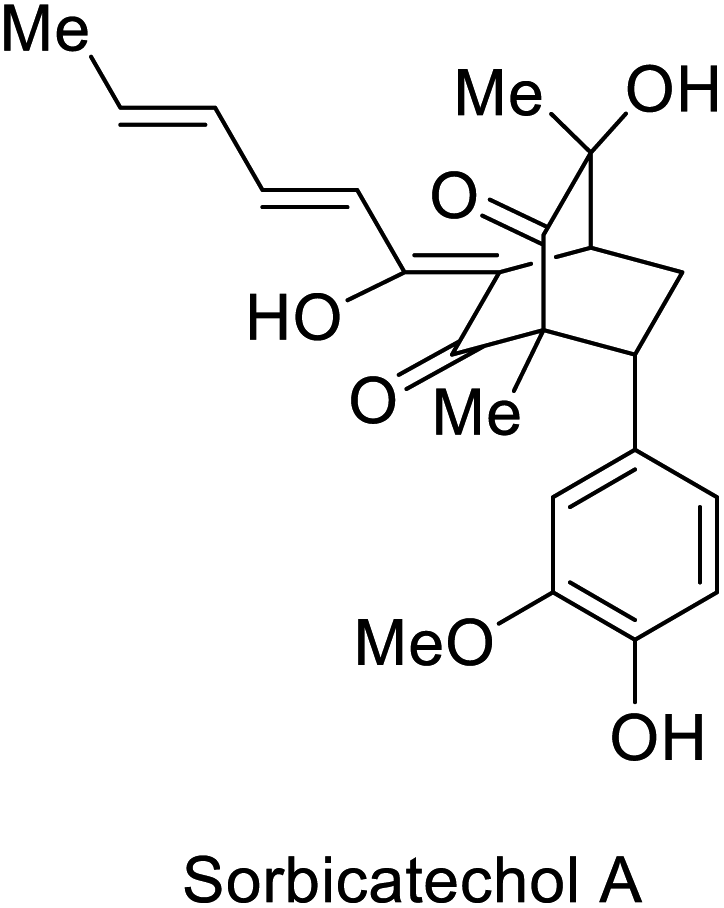 |
Antiviral activity against H1N1 | 53, 54 and 113 | |
| Epoxide-opening | E. coli JM109 (pDTG601) | 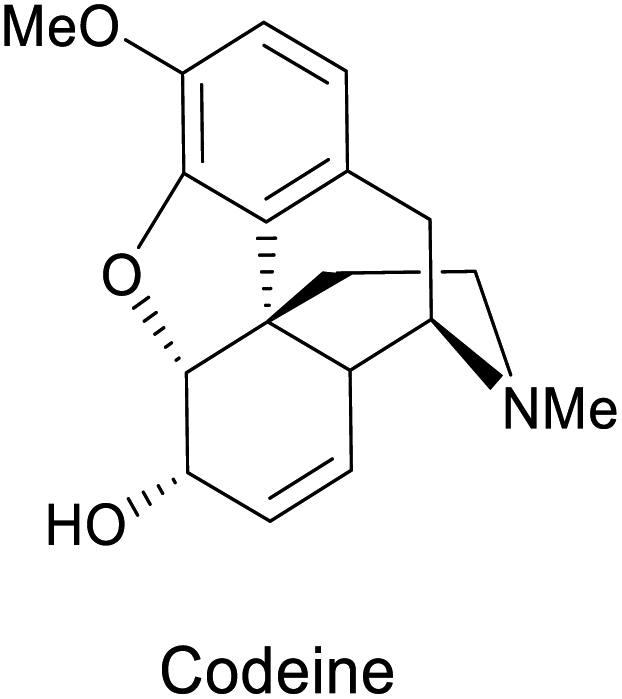 |
Painkiller | 88 |
| Ketoreductase, KRED-NADH-110 |  |
S1P1 receptor agonist | 91 | |
| Thiol-Michael | Ketoreductase, KRED-311 |  |
Volatile sulfur compound | 111 |
Bio-click chemistry reactions are demonstrated to be remarkably complementary, as is the case of click chemical reactions, which are regiospecific by nature but not necessarily enantioselective. The joint use of enzymes/microorganisms allows to overcome these limitations, introducing chirality without hampering the overall efficiency of the processes. The global need of, and tendency towards more sustainable and green processes is thoroughly reflected by the complementary use of efficient chemical reactions and green biocatalysis. It can be envisioned, that the development of more robust and active biocatalysts with wider substrate ranges will facilitate the broadening of efficient bio-click processes foreseeable future. A very promising example are ketoreductases (KREDs), which are by now widely commercially available in great variety. The use of KREDs has proven to be a competitive alternative to conventional enantioselective oxidations, even on an industrial scale.
It is essential to emphasize, that click reactions, biocatalysis or their combination do not generate greener processes per se, and it is always necessary to carefully evaluate the exact methodology, the implementation in a reaction sequence and the expected overall efficiency and sustainability for future improvements.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
FZ is grateful to ANID/CONICYT/FONDECYT Regular No. 1210763. DR thanks CONICYT-PCHA/Doctorado Nacional/2018-21180422. CARS is supported by ANID Millennium Science Initiative Program ICN17_022. We also want to thank Lars Ratjen PhD for the English revision of this manuscript.Notes and references
- M. Hönig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942–8973 CrossRef PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185 CrossRef CAS PubMed.
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J.-Y. Berthon and B. H. Lipshutz, Nat. Commun., 2019, 10, 2169 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- E. Oueis, C. Sabot and P.-Y. Renard, Chem. Commun., 2015, 51, 12158–12169 RSC.
- J. M. Palomo, Curr. Org. Chem., 2013, 17, 691–700 CrossRef CAS.
- J. L. Brennan, N. S. Hatzakis, T. R. Tshikhudo, V. Razumas, S. Patkar, J. Vind, A. Svendsen, R. J. M. Nolte, A. E. Rowan and M. Brust, Bioconjugate Chem., 2006, 17, 1373–1375 CrossRef CAS PubMed.
- J. M. Palomo, Org. Biomol. Chem., 2012, 10, 9309–9318 RSC.
- R. B. Woodward, W. A. Ayer, J. M. Beaton, F. Bickelhaupt, R. Bonnett, P. Buchschacher, G. L. Closs, H. Dutler, J. Hannah, F. P. Hauck, S. Itô, A. Langemann, E. Le Goff, W. Leimgruber, W. Lwowski, J. Sauer, Z. Valenta and H. Volz, J. Am. Chem. Soc., 1960, 82, 3800–3802 CrossRef CAS.
- K. C. Nicolaou, Z. Yang, J. J. Liu, H. Ueno, P. G. Nantermet, R. K. Guy, C. F. Claiborne, J. Renaud, E. A. Couladouros, K. Paulvannan and E. J. Sorensen, Nature, 1994, 367, 630–634 CrossRef CAS PubMed.
- R. A. Holton, H. B. Kim, C. Somoza, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith and S. Kim, J. Am. Chem. Soc., 1994, 116, 1599–1600 CrossRef CAS.
- T. Durek, V. Y. Torbeev and S. B. H. Kent, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4846–4851 CrossRef CAS PubMed.
- R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- P. Prasher and M. Sharma, Medchemcomm, 2019, 10, 1302–1328 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- M. A. Tasdelen, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2016, 52, 19–78 CrossRef CAS.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- C. A. Martinez, S. Hu, Y. Dumond, J. Tao, P. Kelleher and L. Tully, Org. Process Res. Dev., 2008, 12, 392–398 CrossRef CAS.
- R. A. Sheldon, Green Biocatal., 2016, 1–13 Search PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- M. K. M. Leung, C. E. Hagemeyer, A. P. R. Johnston, C. Gonzales, M. M. J. Kamphuis, K. Ardipradja, G. K. Such, K. Peter and F. Caruso, Angew. Chem., Int. Ed., 2012, 51, 7132–7136 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Nature, 2019, 574, 86–89 CrossRef CAS PubMed.
- N. M. Meghani, H. H. Amin and B.-J. Lee, Drug Discovery Today, 2017, 22, 1604–1619 CrossRef CAS PubMed.
- A. Brik, J. Alexandratos, Y.-C. Lin, J. H. Elder, A. J. Olson, A. Wlodawer, D. S. Goodsell and C.-H. Wong, ChemBioChem, 2005, 6, 1167–1169 CrossRef CAS PubMed.
- H. Ankati, Y. Yang, D. Zhu, E. R. Biehl and L. Hua, J. Org. Chem., 2008, 73, 6433–6436 CrossRef CAS PubMed.
- C. Aguirre-Pranzoni, R. D. Tosso, F. R. Bisogno, M. Kurina-Sanz and A. A. Orden, Process Biochem., 2019, 79, 114–117 CrossRef CAS.
- P. Sivaguru, Y. Ning and X. Bi, Chem. Rev., 2021, 121, 4253–4307 CrossRef CAS PubMed.
- W. Szymanski, C. P. Postema, C. Tarabiono, F. Berthiol, L. Campbell-Verduyn, S. de Wildeman, J. G. de Vries, L. Feringa and D. B. Janssen, Adv. Synth. Catal., 2010, 352, 2111–2115 CrossRef CAS.
- L. S. Campbell-Verduyn, L. Mirfeizi, R. A. Dierckx, P. H. Elsinga and L. Feringa, Chem. Commun., 2009, 2139–2141 RSC.
- T. N. Nguyen, P.-A. Chen, K. Setthakarn and J. A. May, Molecules, 2018, 23 CAS.
- K. Leijondahl, L. Borén, R. Braun and J.-E. Bäckvall, Org. Lett., 2008, 10, 2027–2030 CrossRef CAS PubMed.
- A. Cuetos, F. R. Bisogno, I. Lavandera and V. Gotor, Chem. Commun., 2013, 49, 2625–2627 RSC.
- C. de Souza de Oliveira, K. T. de Andrade and A. T. Omori, J. Mol. Catal. B: Enzym., 2013, 91, 93–97 CrossRef CAS.
- V. de la Sovera, A. Bellomo, J. M. Pena, D. Gonzalez and H. A. Stefani, Mol. Diversity, 2011, 15, 163–172 CrossRef PubMed.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- D. S. Pedersen and A. Abell, Eur. J. Org. Chem., 2011, 2011, 2399–2411 CrossRef.
- S. Hassan, R. Tschersich and T. J. J. Müller, Tetrahedron Lett., 2013, 54, 4641–4644 CrossRef CAS.
- P. Gesse and T. J. J. Müller, Eur. J. Org. Chem., 2019, 2019, 2150–2157 CrossRef CAS.
- N. N. Büyükadali, S. Seven, N. Aslan, D. Yenidede and A. Gümüş, Tetrahedron: Asymmetry, 2015, 26, 1285–1291 CrossRef.
- M. E. Moisă, L. Poppe, C. A. Gal, L. C. Bencze, F. D. Irimie, C. Paizs, F. Peter and M. I. Toşa, React. Chem. Eng., 2018, 3, 790–798 RSC.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- S. Otto, W. Blokzijl and J. B. F. N. Engberts, J. Org. Chem., 1994, 59, 5372–5376 CrossRef CAS.
- A. Wassermann, J. Chem. Soc., 1942, 618–621 RSC.
- S. Otto and J. B. F. N. Engberts, Tetrahedron Lett., 1995, 36, 2645–2648 CrossRef CAS.
- P. Vermeeren, T. A. Hamlin, I. Fernández and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2020, 59, 6201–6206 CrossRef CAS PubMed.
- J. Meng, X. Wang, D. Xu, X. Fu, X. Zhang, D. Lai, L. Zhou and G. Zhang, Molecules, 2016, 21, 715 CrossRef PubMed.
- A. Sib and T. A. M. Gulder, Angew. Chem., Int. Ed., 2017, 56, 12888–12891 CrossRef CAS PubMed.
- R. Hong, Y. Chen and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 3478–3481 CrossRef CAS PubMed.
- A. Sib and T. A. M. Gulder, Angew. Chem., Int. Ed., 2018, 57, 14650–14653 CrossRef CAS PubMed.
- S. A. Baker Dockrey, A. L. Lukowski, M. R. Becker and A. R. H. Narayan, Nat. Chem., 2018, 10, 119–125 CrossRef CAS PubMed.
- J.-C. Caille, C. K. Govindan, H. Junga, J. Lalonde and Y. Yao, Org. Process Res. Dev., 2002, 6, 471–476 CrossRef CAS.
- M. R. Jirousek, J. R. Gillig, C. M. Gonzalez, W. F. Heath, J. H. McDonald, D. A. Neel, C. J. Rito, U. Singh, L. E. Stramm, A. Melikian-Badalian, M. Baevsky, L. M. Ballas, S. E. Hall, L. L. Winneroski and M. M. Faul, J. Med. Chem., 1996, 39, 2664–2671 CrossRef CAS PubMed.
- B. Wirz, P. Spurr and C. Pfleger, Tetrahedron: Asymmetry, 2010, 21, 159–161 CrossRef CAS.
- G. J. Moyle, E. DeJesus, P. Cahn, S. A. Castillo, H. Zhao, D. N. Gordon, C. Craig and T. R. Scott, JAIDS, J. Acquired Immune Defic. Syndr., 2005, 38, 417–425 CrossRef CAS PubMed.
- M. T. Crimmins and B. W. King, J. Org. Chem., 1996, 61, 4192–4193 CrossRef CAS PubMed.
- R. Singh and R. Vince, Chem. Rev., 2012, 112, 4642–4686 CrossRef CAS PubMed.
- S. Zhu, L. Ren, S. Yu, C. Gong, D. Song and G. Zheng, Bioorg. Med. Chem. Lett., 2014, 24, 4899–4902 CrossRef CAS PubMed.
- S. M. Daluge, M. T. Martin, B. R. Sickles and D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 297–327 CrossRef CAS PubMed.
- S. Slagman and W.-D. Fessner, Chem. Soc. Rev., 2021, 50, 1968–2009 RSC.
- L. P. Tardibono, M. J. Miller and J. Balzarini, Tetrahedron, 2011, 67, 825–829 CrossRef CAS PubMed.
- Y. Zou, M. Garcia-Borràs, M. C. Tang, Y. Hirayama, D. H. Li, L. Li, K. Watanabe, K. N. Houk and Y. Tang, Nat. Chem. Biol., 2017, 13, 325–332 CrossRef CAS PubMed.
- C. Bonini and G. Righi, Tetrahedron, 2002, 58, 4981–5021 CrossRef CAS.
- H. Pellissier, A. Lattanzi and R. Dalpozzo, in Asymmetric Synthesis of Three-Membered Rings, Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 1st edn, 2017, ch. 3, pp. 379–538 Search PubMed.
- K. M. Morgan, J. A. Ellis, J. Lee, A. Fulton, S. L. Wilson, P. S. Dupart and R. Dastoori, J. Org. Chem., 2013, 78, 4303–4311 CrossRef CAS PubMed.
- X. E. Hu, Tetrahedron, 2004, 60, 2701–2743 CrossRef CAS.
- P. Lu, Tetrahedron, 2010, 66, 2549–2560 CrossRef CAS.
- H. Pellissier, A. Lattanzi and R. Dalpozzo, in Asymmetric Synthesis of Three-Membered Rings, Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 1st edn, 2017, ch. 2, pp. 205–378 Search PubMed.
- C. C. Farwell, R. K. Zhang, J. A. McIntosh, T. K. Hyster and F. H. Arnold, ACS Cent. Sci., 2015, 1, 89–93 CrossRef CAS PubMed.
- P. Crotti and M. Pineschi, in Aziridines Epoxides Organic Synthesis, ed. A.K. Yudin, Wiley-VCH, Weinheim, 1st edn. 2006, pp. 271–313 Search PubMed.
- M. G. Russell and T. F. Jamison, Angew. Chem., Int. Ed., 2019, 58, 7678–7681 CrossRef CAS PubMed.
- F. Moschona, I. Savvopoulou, M. Tsitopoulou, D. Tataraki and G. Rassias, Catal., 2020, 10, 1117–1182 CrossRef CAS.
- A. Archelas and R. Furstoss, Annu. Rev. Microbiol., 1997, 51, 491–525 CrossRef CAS PubMed.
- A. Archelas and R. Furstoss, Tetrahedron Lett., 1992, 33, 5241–5242 CrossRef CAS.
- F. J. Lakner and L. P. Hager, J. Org. Chem., 1996, 61, 3923–3925 CrossRef CAS PubMed.
- A. T. Martínez, F. J. Ruiz-Dueñas, S. Camarero, A. Serrano, D. Linde, H. Lund, J. Vind, M. Tovborg, O. M. Herold-Majumdar, M. Hofrichter, C. Liers, R. Ullrich, K. Scheibner, G. Sannia, A. Piscitelli, C. Pezzella, M. E. Sener, S. Kılıç, W. J. H. van Berkel, V. Guallar, M. F. Lucas, R. Zuhse, R. Ludwig, F. Hollmann, E. Fernández-Fueyo, E. Record, C. B. Faulds, M. Tortajada, I. Winckelmann, J.-A. Rasmussen, M. Gelo-Pujic, A. Gutiérrez, J. C. del Río, J. Rencoret and M. Alcalde, Biotechnol. Adv., 2017, 35, 815–831 CrossRef PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- T. Hudlicky, ACS Omega, 2018, 3, 17326–17340 CrossRef CAS.
- F. F. Özgen and S. Schmidt, in Biocatalysis. Enzymatic Basics and Applications, ed. Q. Husain and M. F. Ullah, Springer International Publishing, New York, 1st edn, 2019, ch. 4, pp. 57–82 Search PubMed.
- G. J. Zylstra, W. R. McCombie, D. T. Gibson and B. A. Finette, Appl. Environ. Microbiol., 1988, 54, 1498–1503 CrossRef CAS PubMed.
- M. A. Vila, M. Brovetto, D. Gamenara, P. Bracco, G. Zinola, G. Seoane, S. Rodríguez and I. Carrera, J. Mol. Catal. B: Enzym., 2013, 96, 14–20 CrossRef CAS.
- T. Hudlicky, J. D. Price, F. Rulin and T. Tsunoda, J. Am. Chem. Soc., 1990, 112, 9439–9440 CrossRef CAS.
- M. G. Banwell, N. Haddad, T. Hudlicky, T. C. Nugent, M. F. Mackay and S. L. Richards, J. Chem. Soc., Perkin Trans. 1, 1997, 1779–1792 RSC.
- M. Banwell, S. Blakey, G. Harfoot and R. Longmore, J. Chem. Soc., Perkin Trans. 1, 1998, 3141–3142 RSC.
- H. Leisch, A. T. Omori, K. J. Finn, J. Gilmet, T. Bissett, D. Ilceski and T. Hudlický, Tetrahedron, 2009, 65, 9862–9875 CrossRef CAS.
- B. D. Schwartz, M. G. Banwell and I. A. Cade, Tetrahedron Lett., 2011, 52, 4526–4528 CrossRef CAS.
- G. Carrau, A. I. Bellomo, L. Suescun and D. Gonzalez, Eur. J. Org. Chem., 2019, 2019, 788–802 CrossRef CAS.
- X. Hou, H. Zhang, B.-C. Chen, Z. Guo, A. Singh, A. Goswami, J. L. Gilmore, J. E. Sheppeck, A. J. Dyckman, P. H. Carter and A. Mathur, Org. Process Res. Dev., 2017, 21, 200–207 CrossRef CAS.
- R. T. Ruck, Q. Chen, N. Rivera, J. Kong, I. K. Mangion, L. Tan and F. J. Fleitz, Org. Process Res. Dev., 2021, 25, 405–410 CrossRef CAS.
- W. Zhang, H. Li, S. H. H. Younes, P. Gómez de Santos, F. Tieves, G. Grogan, M. Pabst, M. Alcalde, A. C. Whitwood and F. Hollmann, ACS Catal., 2021, 11, 2644–2649 CrossRef CAS PubMed.
- N. Kasai, T. Suzuki and Y. Furukawa, J. Mol. Catal. B: Enzym., 1998, 4, 237–252 CrossRef CAS.
- E. Perzborn, S. Roehrig, A. Straub, D. Kubitza and F. Misselwitz, Nat. Rev. Drug Discovery, 2010, 10, 61 CrossRef.
- T. A. Fattah and A. Saeed, Tetrahedron: Asymmetry, 2017, 28, 485–504 CrossRef.
- J. Wu, C. Liu, Y. Jiang, M. Hu, S. Li and Q. Zhai, Catal. Commun., 2010, 11, 727–731 CrossRef CAS.
- H.-X. Jin, Z.-Q. Liu, Z.-C. Hu and Y.-G. Zheng, Eng. Life Sci., 2013, 13, 385–392 CrossRef CAS.
- A. C. Mali, D. G. Deshmukh, D. R. Joshi, H. D. Lad, P. I. Patel, V. J. Medhane and V. T. Mathad, Sustainable Chem. Processes, 2015, 3, 11 CrossRef.
- R. Porcar, V. Sans, N. Ríos-Lombardía, V. Gotor-Fernández, V. Gotor, M. I. Burguete, E. García-Verdugo and S. V Luis, ACS Catal., 2012, 2, 1976–1983 CrossRef CAS.
- E. Busto, V. Gotor-Fernández, N. Ríos-Lombardía, E. García-Verdugo, I. Alfonso, S. García-Granda, A. Menéndez-Velázquez, M. I. Burguete, S. V Luis and V. Gotor, Tetrahedron Lett., 2007, 48, 5251–5254 CrossRef CAS.
- Á. Villar-Barro, V. Gotor and R. Brieva, Tetrahedron, 2015, 71, 6907–6912 CrossRef.
- A. V Srinivasan, Ann. Indian Acad. Neurol., 2019, 22, 21–26 CrossRef PubMed.
- B. Silber, N. H. G. Holford and S. Riegelman, J. Pharm. Sci., 1982, 71, 699–704 CrossRef CAS PubMed.
- X.-D. Kong, H.-L. Yu, S. Yang, J. Zhou, B.-B. Zeng and J.-H. Xu, J. Mol. Catal. B: Enzym., 2015, 122, 275–281 CrossRef CAS.
- E. J. Park, T. N. Gevrek, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2014, 25, 2004–2011 CrossRef CAS PubMed.
- S. S. Ghosh, P. M. Kao, A. W. McCue and H. L. Chappelle, Bioconjugate Chem., 1990, 1, 71–76 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- E. K. Skinner, F. M. Whiffin and G. J. Price, Chem. Commun., 2012, 48, 6800–6802 RSC.
- K. Lauder, A. Toscani, Y. Qi, J. Lim, S. J. Charnock, K. Korah and D. Castagnolo, Angew. Chem., Int. Ed., 2018, 57, 5803–5807 CrossRef CAS PubMed.
- R. Vince and M. Hua, Curr. Protoc. Nucleic Acid Chem., 2006, 25, 14.4.1–14.4.8 Search PubMed.
- J. Peng, X. Zhang, L. Du, W. Wang, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2014, 77, 424–428 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |