
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient synthesis of antiviral agent uprifosbuvir enabled by new synthetic methods†
Artis
Klapars
 *a,
John Y. L.
Chung
*a,
John
Limanto
a,
Ralph
Calabria
a,
Louis-Charles
Campeau
a,
Kevin R.
Campos
a,
Wenyong
Chen
a,
Stephen M.
Dalby
a,
Tyler A.
Davis
a,
Daniel A.
DiRocco
a,
Alan M.
Hyde
a,
Amude M.
Kassim
a,
Mona Utne
Larsen
a,
Guiquan
Liu
b,
Peter E.
Maligres
a,
Aaron
Moment
a,
Feng
Peng
a,
Rebecca T.
Ruck
a,
Michael
Shevlin
a,
Bryon L.
Simmons
a,
Zhiguo Jake
Song
a,
Lushi
Tan
a,
Timothy J.
Wright
a and
Susan L.
Zultanski
a
*a,
John Y. L.
Chung
*a,
John
Limanto
a,
Ralph
Calabria
a,
Louis-Charles
Campeau
a,
Kevin R.
Campos
a,
Wenyong
Chen
a,
Stephen M.
Dalby
a,
Tyler A.
Davis
a,
Daniel A.
DiRocco
a,
Alan M.
Hyde
a,
Amude M.
Kassim
a,
Mona Utne
Larsen
a,
Guiquan
Liu
b,
Peter E.
Maligres
a,
Aaron
Moment
a,
Feng
Peng
a,
Rebecca T.
Ruck
a,
Michael
Shevlin
a,
Bryon L.
Simmons
a,
Zhiguo Jake
Song
a,
Lushi
Tan
a,
Timothy J.
Wright
a and
Susan L.
Zultanski
a
aDepartment of Process Research and Development, Merck & Co., Inc., Rahway, New Jersey 07065, USA. E-mail: artis_klapars@merck.com
bWuXi STA, 90 Delin Road, Waigaoqiao Free Trade Zone, Shanghai 200131, China
First published on 19th May 2021
Abstract
An efficient route to the HCV antiviral agent uprifosbuvir was developed in 5 steps from readily available uridine in 50% overall yield. This concise synthesis was achieved by development of several synthetic methods: (1) complexation-driven selective acyl migration/oxidation; (2) BSA-mediated cyclization to anhydrouridine; (3) hydrochlorination using FeCl3/TMDSO; (4) dynamic stereoselective phosphoramidation using a chiral nucleophilic catalyst. The new route improves the yield of uprifosbuvir 50-fold over the previous manufacturing process and expands the tool set available for synthesis of antiviral nucleotides.
Introduction
As highlighted by the COVID-19 pandemic, availability of efficient antiviral treatments remains a starkly unmet medical need for most viral infections. Among existing antiviral drugs, several have been approved for the treatment of more than one viral disease1 and already approved antiviral drugs can sometimes be repurposed as treatments for emerging infectious diseases.2 This potential for emergency use underscores the need for a large and diverse stockpile of antiviral agents and the importance of efficient manufacturing processes to enable a rapid response to a potentially massive increase in demand.Chronic hepatitis C virus (HCV) infection affects an estimated 71 million people globally, and the WHO has estimated that in 2016 approximately 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people died from HCV-related complications.3 Recently, encouraging progress has been achieved in the treatment of HCV with cure rates now exceeding 95%.4 Uprifosbuvir 1 is an NS5b inhibitor developed for the treatment of HCV,5 representing a class of 2′-branched nucleosides modified with a ProTide sidechain.6 Many other nucleoside antivirals also contain 2′, 3′, or 4′-modifications of the ribose core, presenting formidable synthetic challenges that have been approached through de novo synthesis.5,7 The alternative and more direct strategy to functionalize preexisting nucleosides is less developed.8 Here, we report a highly efficient synthesis of uprifosbuvir 1via direct functionalization of uridine enabled by development of new synthetic methods to overcome limitations of the existing tool set available for the synthesis of nucleotides.
000 people died from HCV-related complications.3 Recently, encouraging progress has been achieved in the treatment of HCV with cure rates now exceeding 95%.4 Uprifosbuvir 1 is an NS5b inhibitor developed for the treatment of HCV,5 representing a class of 2′-branched nucleosides modified with a ProTide sidechain.6 Many other nucleoside antivirals also contain 2′, 3′, or 4′-modifications of the ribose core, presenting formidable synthetic challenges that have been approached through de novo synthesis.5,7 The alternative and more direct strategy to functionalize preexisting nucleosides is less developed.8 Here, we report a highly efficient synthesis of uprifosbuvir 1via direct functionalization of uridine enabled by development of new synthetic methods to overcome limitations of the existing tool set available for the synthesis of nucleotides.
Results and discussion
The original multi-kilogram route to uprifosbuvir proceeded in 12 linear steps and only 1% overall yield (Scheme 1). Commencing with D-glucose 2, the route utilized an ingenious albeit low-yielding (19%) rearrangement cascade to reach lactone 3 containing the tertiary alkyl group at the 2′-position.9 Further elaboration of lactone 3 over 10 steps provided uprifosbuvir 1. In addition to the challenges in stereoselective installation of the chiral tertiary alkyl chloride, the route suffered from low yields and poor regio- and diastereoselectivity in phosphoramidation of intermediate 4 to install the ProTide sidechain (Scheme 1). A more efficient synthesis was urgently required to address these two key synthetic challenges and to meet the antiviral therapy demand.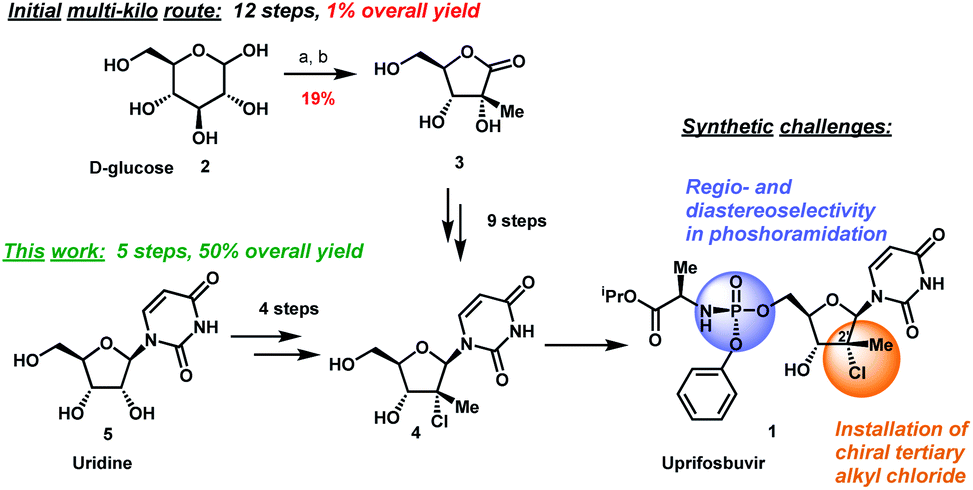 | ||
| Scheme 1 Synthetic approaches to uprifosbuvir 1 with the two main challenges highlighted. (a) Me2NH, AcOH, EtOH/MeOH, 80 °C, 1.5 h; (b) Ca(OH)2, water, 70 °C, 24 h, 19% over 2 steps.9 | ||
We envisioned a shorter and more efficient route to uprifosbuvir starting from the inexpensive and readily available uridine 5,10 which already contains all of the atoms in the nucleoside portion of the molecule, with the glaring – and challenging – exception of the tertiary alkyl chloride in the 2′-position. We started out by exploring selective functionalization in the 2′-position in uridine. All chemical11 or biocatalytic efforts to directly oxidize uridine failed due to poor selectivity or instability of 2′-ketouridine 6. Uracil 9 was the main product observable by HPLC in the biocatalytic oxidation using ketoreductase (KRED) enzymes in the reverse direction, presumably formed via fast enolization of 2′-ketouridine 6 (or the isomeric 3′-ketouridine) to 7 followed by elimination of uracil (Scheme 2).12
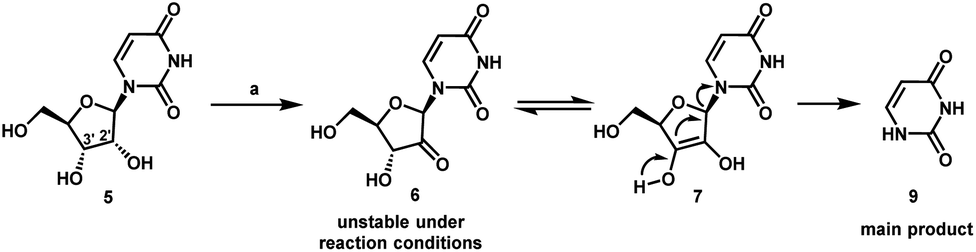 | ||
| Scheme 2 Decomposition during attempted direct biocatalytic oxidation of uridine 5. (a) General screening conditions: KRED enzyme variants, NAD or NADP cofactor, acetone-water, 30 °C. The reactions were screened using a set of buffers with pH of 6.5–7 (potassium phosphate) and pH of 5–6 (sodium borate). | ||
It became clear that a minimal protecting group strategy for uridine would be required to address the aforementioned instability, as well as solubility, issues. Protection of the 3′,5′-diol moiety in riboses is traditionally achieved with 1,3-dichloro-1,1,3,3-tetraisopropyl-disiloxane (TIPDSCl2)8,13 or di-tert-butylsilyl ditriflate (DTBS(OTf)2).14 Although these protecting reagents are selective for 3′,5′-diprotection, they are expensive and have high molecular weights, rendering them impractical for commercial manufacturing. In order to better accomplish our goals for the synthesis of uprifosbuvir, we decided to evaluate simple acyl chlorides as far more practical protecting groups.
Unsurprisingly, acylation of uridine provided mixtures of products, including a series of mono-, bis-, and tri-acylated products. Of several simple acyl chlorides that were evaluated,15 pivaloyl chloride (PivCl) delivered the highest selectivity for diacylation (>90%) vs. mono- and tri-acylation; therefore, PivCl was selected as the acylating agent for further optimization. Although the kinetic ratio favoured the undesired 2′,5′-isomer 10 (87:13 of 10/11 at 0 °C), the two isomers could be equilibrated to a 1:3.3 thermodynamic ratio of 10/11 (Scheme 3). We then investigated the strategy of influencing this equilibrium through selective complexation of 11 with a Lewis acid. This could be followed by oxidation to the desired ketone 12 in a complexation-driven selective acyl migration/oxidation process. Carrying out a high throughput screen of 96 Lewis acidic additives led to rapid identification of BF3·OEt2 as a unique additive that afforded >99:1 selectivity of 11/10 in toluene solvent. The likely driving force for this isomerization is formation of the crystalline 11·BF3 complex.16 This hypothesis is supported by our observations that selective isomerization was not observed in alternative solvents, such as THF, MeCN, EtOAc that are more polar and Lewis basic towards BF3 than toluene and could potentially compete with 11 or dissolve the 11·BF3 complex. The slurry of crystalline 11·BF3 complex (>50:1) in toluene could be treated with water to dissolve the crystals and provide a slightly acidic organic layer (pH = 2) that maintained its enrichment in 11 (>50:1) due to very slow rate of equilibration of 10 and 11 under mildly acidic conditions. For the subsequent oxidation step, the improved stability of both alcohol 11 and ketone 12 at low pH led us to select the mildly acidic TEMPO/Bu4NBr/AcOOH conditions. Direct isolation of crystalline ketone 12 from the reaction mixture provided 83% yield from uridine 5 with >50:1 selectivity (Scheme 3). In this way, the unfavorable kinetic selectivity of 13:87 (11/10) in the pivaloylation of 5 could be inverted to >50:1 selectivity in the formation of ketone 12 using a novel complexation-driven selective acyl migration/oxidation.
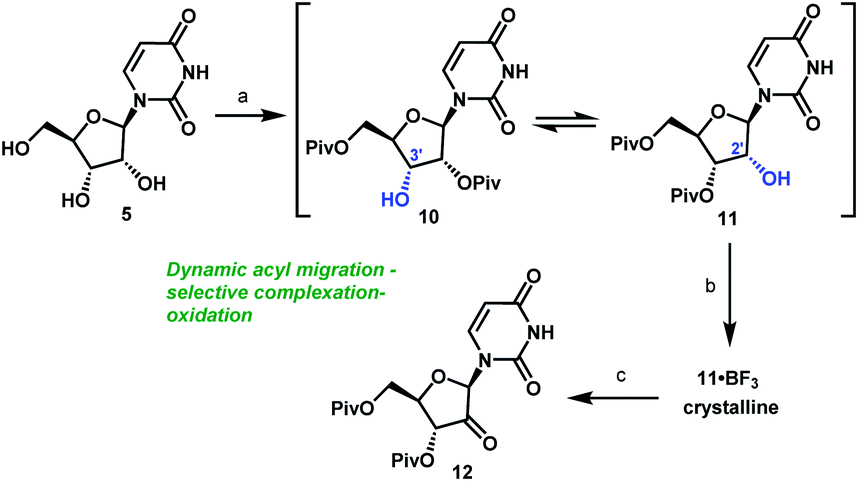 | ||
| Scheme 3 Complexation-driven selective acyl migration/oxidation to access 12. (a) PivCl, pyridine, 0 °C, 16 h; (b) BF3·OEt2, PhMe, 40 °C, 10 h; (c) TEMPO, Bu4NBr, AcOOH, dioctyl sulphide, PhMe, −10 °C to 20 °C, 24 h, 83% from 5. | ||
With a practical process for the preparation of 2′-ketone 12 achieved, we explored means to introduce the methyl and chloro substituents in the 2′-position. First, we investigated a sequence of olefination/hydrochlorination. Although Wittig olefination had been reported for a related ketone where TIPDS protecting group was used instead of Piv protecting groups,17 only low yields of olefin 14 were obtained from ketone 12 under a variety of conditions. We consequently moved on to explore Peterson olefination (Scheme 4). The β-hydroxysilane 13 could be formed in high yield upon treatment of ketone 12 with Me3SiCH2MgCl. Elimination to olefin 14 was challenging under either basic or acidic conditions; nevertheless, we were able to achieve it through activation of the tertiary alcohol as a trifluoroacetate ester followed by fluoride-mediated desilylation/elimination to provide the desired olefin 15. Unfortunately, direct hydrochlorination of olefin 15 with various HCl sources failed to afford the desired tertiary alkyl chloride 4. Instead, decomposition of 15 was observed, as evidenced by the formation of uracil 9. In our search for milder hydrochlorination conditions, we were intrigued by a single example in a publication by Boger and co-workers where Fe(III) oxalate, 4-AcNHC6H4SO2Cl, and NaBH4 were used to effect the hydrochlorination of a simple alkene.18 Applying these conditions to 15 led to formation of the desired alkyl chloride 4 in 6% yield. Further development revealed FeCl3 and PhSiH3 as much more efficient reagents; moreover, a sulfonyl chloride was not required. Under these improved and streamlined conditions, olefin 15 could be converted into chloride 4 at rt with high diastereoselectivity (>50:1), 95% assay and 82% isolated yield. This novel olefination/hydrochlorination sequence allowed us to install the two critical methyl and chloro functionalities in 3 steps starting from ketone 12.
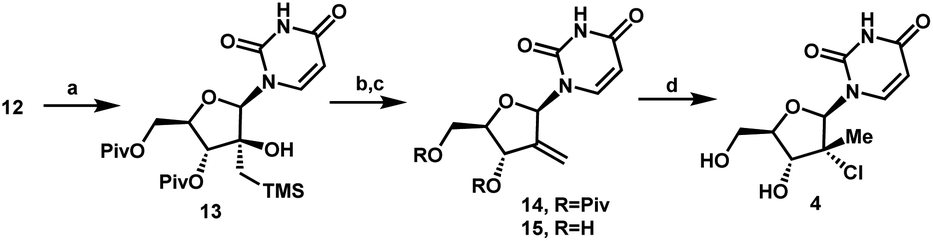 | ||
| Scheme 4 Olefination/hydrochlorination of the ketone 12. (a) TMSCH2MgCl, CPME, 0 °C to rt, 24 h, 91%; (b) (CF3CO)2O, pyridine, DMAP, MeCN, rt, 15 h, then KF, 70 °C, 24 h, 88%; (c) K2CO3, MeOH/THF, 40 °C, 15 h, 94%; (d) FeCl3, PhSiH3, rt, 48 h, 82%. | ||
Although the olefination/hydrochlorination route was a major improvement over the previous synthesis, we were not satisfied with the relatively high cost and poor availability of the Me3SiCH2MgCl reagent and unfavorable process mass intensity inherent to most olefination reactions. Therefore, we desired to define an alternative route with comparable step count. For the introduction of the methyl group, leveraging methylmagnesium reagents was particularly attractive due to their low cost and excellent availability. Under all conditions evaluated, methyl addition to ketone 12 favoured the α-facial attack, although the selectivity was modest (Table 1, entries 1 and 2). With three equivalents of the Grignard reagent, >94% conversion was achieved, but the reactions suffered from poor yields of the desired tertiary alcohol 16 due to competitive cleavage of the pivaloyl ester. In order to suppress these side reactions, we evaluated milder methyl nucleophiles. Using 3.3 equiv. of Me2AlCl, the reaction proceeded to 98% conversion with 49:1 dr and 86% yield and completely avoided the de-Piv side reaction (Table 1, entry 3). However, due to the highly hazardous nature of alkylaluminums, we continued to look for a more practical reagent for this transformation. Given the attractiveness of utilizing a Grignard reagent from a cost, safety and availability perspective, we hypothesized that the presence of Lewis acid additives could attenuate the reactivity of MeMgBr and offset the competitive undesired reactions. Favourable results were obtained in the presence of ZnCl2, which afforded 91% yield with 21:1 dr; however, 4 equiv. of MeMgBr and 2 equiv. of ZnCl2 were required to drive the reaction to 98% conversion, leading to cost and environmental concerns (entry 4). We ultimately identified that using the slightly more reactive organomanganese reagent,19 prepared from 2.5 equivalents each of MeMgBr and MnCl2, afforded high yield and high dr of the desired methylated product. Changing the solvent to anisole/toluene led to further improvement in the dr (entries 5 and 6). These conditions provide the ideal solution for installation of the methyl moiety of uprifosbuvir.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Methyl nucleophile | Equiv. | Solvent | d.r. (16:17) |
De-Piva (%) | AY (%) of 16b |
| a Deprotection of Piv group, as indicated by the sum of mono-Piv derivatives of 16 and 17. b Assay yield after work-up. | ||||||
| 1 | MeMgBr | 3.0 | CH2Cl2 | 10:1 |
14 | 62 |
| 2 | MeMgCl | 3.0 | CH2Cl2 | 5:1 |
40 | — |
| 3 | Me2AlCl | 3.3 | CH2Cl2 | 49:1 |
— | 86 |
| 4 | MeMgBr/ZnCl2 | 4/2 | PhMe/2-MeTHF | 21:1 |
— | 91 |
| 5 | MeMgBr/MnCl2 | 2.5/2.5 | PhMe/2-MeTHF | 12:1 |
— | 92 |
| 6 | MeMgBr/MnCl2 | 2.5/2.5 | PhMe/anisole | 32:1 |
— | 93 |
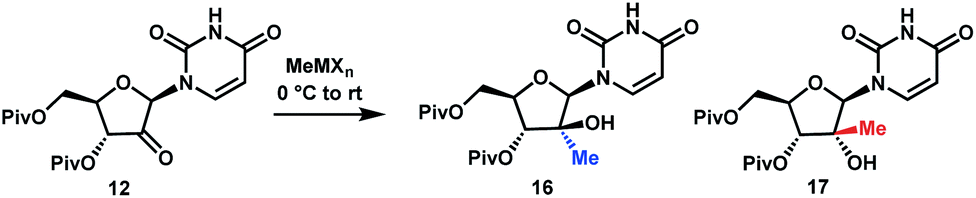
Having introduced the methyl group in the 2′-position, it was necessary to convert the tertiary alcohol 16 into the corresponding tertiary chloride 4 with concomitant inversion of configuration. Carrying out this transformation on such densely functionalized molecule as 16 presents a series of challenges: (1) selectivity among the OH and ester groups, (2) risk of elimination from the tertiary alcohol, (3) diastereoselectivity of displacement at the 2′-position, and (4) intramolecular reactivity of neighbouring groups. Attempts at direct conversion of alcohol 16 into chlorouridine 4 resulted in complex mixtures with low yields of the desired product 4. Therefore, we decided to take advantage of the neighbouring uracil ring to form the desired chloride 4via anhydrouridine 1920,21 with overall inversion of configuration at the 2′-position (Scheme 5). First, we needed to form anhydrouridine 18 with retention of configuration at 2′-position of alcohol 16, which was unprecedented and required selective activation of the uracil ring in the presence of the 2′-alcohol.22 We screened various activating agents and found that bis-trimethylsilylacetamide (BSA) promoted cyclodehydration of 16 to provide 18. Unfortunately, a significant amount of O-TMS derivative 20 was also formed as an impurity (>20% assay yield) that failed to convert to the desired 18 under the reaction conditions. We speculated that the formation of TMS ether impurity 20 was likely base-catalyzed and were delighted to find that addition of 1 mol% HCl suppressed the TMS ether impurity 20 to <3%. Following cyclodehydration, the pivalate protecting groups were conveniently removed in the same reaction vessel by addition of DBU and MeOH, and the crystalline deprotected anhydrouridine 19 was directly isolated from the reaction mixture in 87% isolated yield, thereby positioning us for installation of the key chloro functionality.
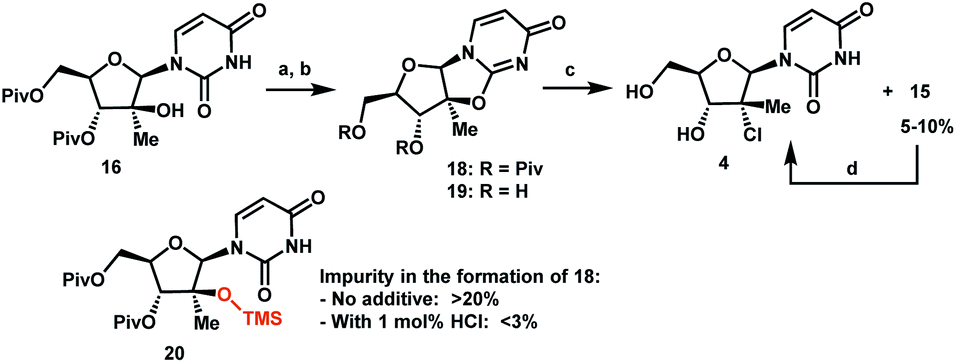 | ||
| Scheme 5 Formation of tertiary chloride 4. (a) BSA, 1 mol% of 37% aq HCl, anisole, 75 °C, 8 h; (b) DBU, MeOH, 60 °C, 10 h, 87% yield from 16; (c) Me2SiCl2, DMF, 1,2-dimethoxyethane (DME), 70 °C, 10 h; (d) FeCl3–6H2O, (Me2SiH)2O, 25 °C, 12 h, 85% from 19. | ||
Formation of the tertiary chloride 4 from 19 is challenging due to the presence of considerable steric hindrance and vicinal electron-withdrawing substituents. The initial conditions developed were highly hazardous (HCl gas, AcOH, 1,4-dioxane, under pressure at 100 °C) and provided only 56% yield of 4 from 19. Milder and more convenient conditions were established by using dichlorodimethylsilane to form a more soluble silyl ether intermediate and generate HCl in situ.23 However, under these and many other conditions explored, 5–10% of olefin 15 was consistently formed as an impurity. This outcome not only served to reduce the yield of the desired chloride 4 but also complicated the isolation and purification of 4 by crystallization due to low solubility of the olefin impurity 15. Having previously developed a highly efficient Fe-mediated hydrochlorination of olefin 15 (Scheme 4), we employed this innovation and were delighted to find that, simply adding the inexpensive reagents FeCl3·6H2O and tetramethyldisiloxane (TMDSO) at the end of the dichlorodimethylsilane-mediated hydrochlorination of 19 cleanly funnelled the olefin impurity 15 into tertiary alkyl chloride 4 with high diastereoselectivity (>100:1 overall dr of 4).24 In so doing, we carried out a two-step sequence to successfully convert a sterically encumbered tertiary alcohol into the corresponding inverted tertiary chloride and reveal the full nucleoside architecture of uprifosbuvir.
To complete the synthesis of uprifosbuvir, it remained to install the phosphoramidate side chain, presenting a significant synthetic challenge. The initial kilo-scale synthesis proceeded through the diastereopure pentafluorophenyl ester 2325,26 and utilized tBuMgCl as base to provide uprifosbuvir 1 with poor 3′/5′ regioselectivity and only 50% isolated yield. We found that introducing Me2AlCl as an additive led to a significant improvement in the phosphoramidation regioselectivity, resulting in an 81% isolated yield of uprifosbuvir.27 Nevertheless, this approach still required preparation of the enantio and diastereopure phosphorylating agent 23. An even more efficient route would bypass 23 altogether and would directly introduce the phosphoramidate sidechain in 4 using chlorophosphoramidate 22, which can be readily prepared from alanine ester 21b and conveniently used as a solution in iPrOAc.28 However, chlorophosphoramidate 22 exists as a ∼1:1 mixture of diastereomers and uncatalyzed phosphoramidation of 4 with 22 was shown to proceed to uprifosbuvir in poor diastereoselectivity (55:45 dr at phosphorus). In an earlier communication,29,30 we disclosed a class of dimeric chiral imidazole carbamate catalysts that was highly effective in overcoming the unfavourable inherent diastereoselectivity in dynamic stereoselective phosphoramidation of nucleosides. Upon further development, we identified 24 as the optimal catalyst for 1 and found that the crude chlorophosphoramidate mixture 22 (1:1 dr) could be coupled with 4 in 97:3 dr and 94% assay yield using only 3 mol% of catalyst 24, providing 88% isolated yield of uprifosbuvir 1 after crystallization. This highly efficient dynamic stereoselective phosphoramidation step concluded the 5-step synthesis of uprifosbuvir in 50% overall yield from uridine (Scheme 6).
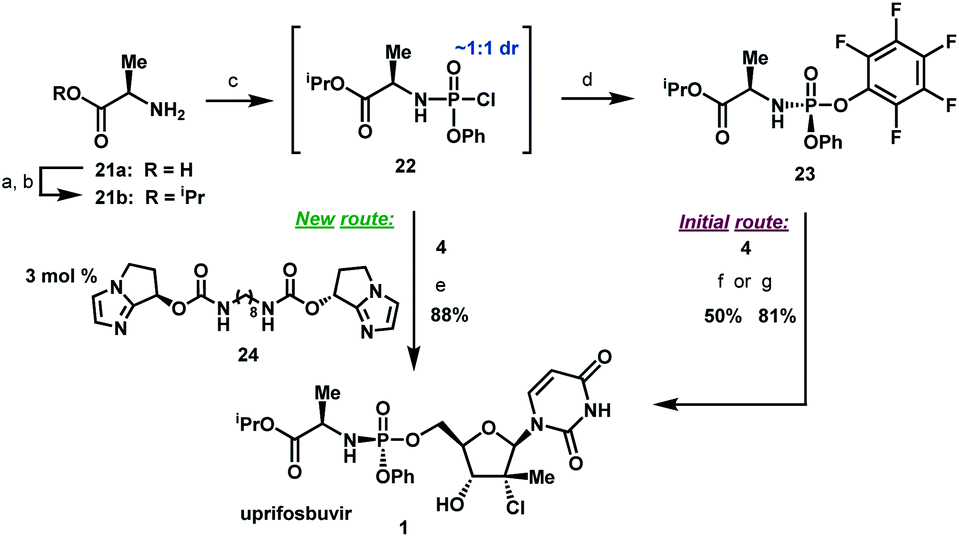 | ||
| Scheme 6 Completion of uprifosbuvir synthesis. (a) TMS-Cl, iPrOH, 70 °C, 12 h; (b) NEt3, iPrOAc, wiped film evaporation, 80%; (c) PhOP(O)Cl2, NEt3, iPrOAc, −20 °C, 2 h, 90%; (d) C6F5OH, NEt3, iPrOAc, −5 °C to 10 °C, 18 h, 76%;26 (e) 4, 3 mol% 24, 2,6-lutidine, 1,3-dioxolane, −10 °C, 24 h, 88%; (f) 4, tBuMgCl, THF, −5 °C to 5 °C, 15 h, 50%;27 (g) 4, Me2AlCl, 2,6-lutidine, THF, 35 °C, 16 h, 81%.27 | ||
Conclusions
In summary, we have developed a highly efficient route to HCV antiviral uprifosbuvir 1 in five easily scalable steps from the readily available raw material uridine 5 in 50% overall yield, which represents a 50-fold yield improvement over the initial multi-kilo route. This achievement was made possible by the development of several synthetic methods: (1) complexation-driven selective acyl migration/oxidation; (2) BSA-mediated cyclization to anhydrouridine; (3) hydrochlorination using Me2SiCl2 and FeCl3/TMDSO; (4) dynamic stereoselective phosphoramidation using a chiral nucleophilic imidazole carbamate catalyst (Scheme 7).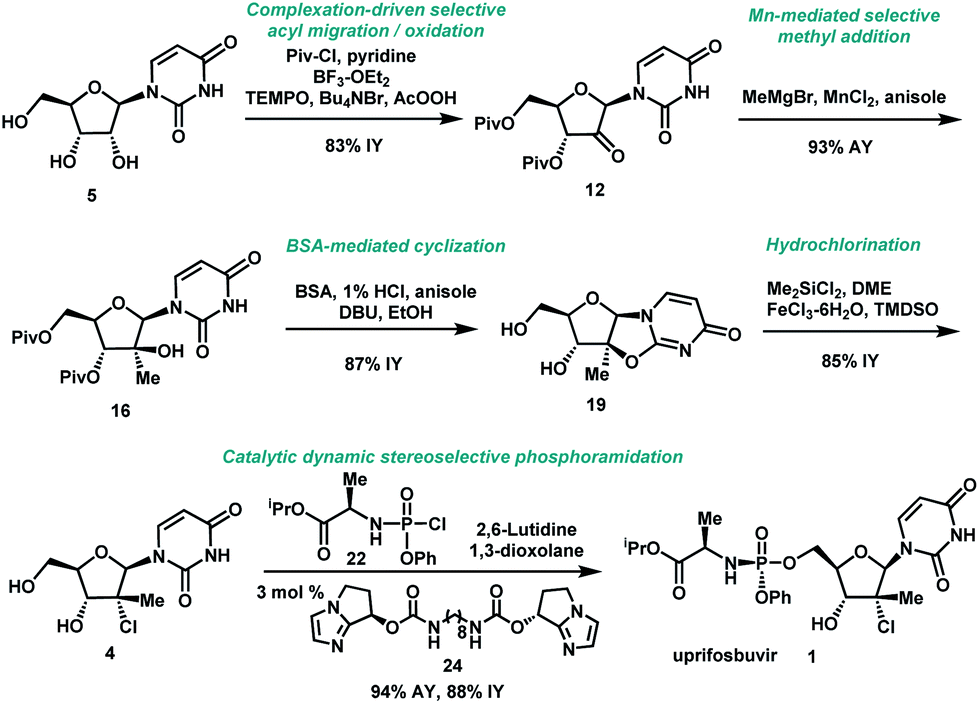 | ||
| Scheme 7 Summary of uprifosbuvir synthesis. AY = assay yield; IY = isolated yield. | ||
Nucleoside antiviral agents for various indications often share common structural features, such as ribose modifications and the ProTide moiety. We believe that the methods disclosed here for the synthesis of uprifosbuvir should facilitate more efficient preparation of other nucleoside antivirals to enable access to these life-saving medicines.31
Author contributions
A. K., J. Y. L. C., J. L., R. C., W. C., S. M. D., T. A. D., D. A. D., A. M. H., A. M. K., M. U. L., P. E. M., A. M., F. P., M. S., B. L. S., T. J. W. and S. L. Z. conceived and performed the experiments; A. K., J. L., L.-C. C., K. R. C., M. U. L., G. L., A. M., R. T. R. and L. T. led the project and conceived ideas; A.K. and J. Y. L. C. wrote the original draft; L.-C. C., A. M. H. and R. T. R. reviewed and edited the manuscript; all authors contributed to the discussion of the results and the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Cyndi Q. He and Edward C. Sherer for computational support, Frank Bernardoni, Kristine L. Cappuccio, Ryan D. Cohen, Peter G. Dormer, Holst M. Halsey, Robert Hartman, Yining Ji, Alfred Y. Lee, Ian Mangion, Justin A. Newman, Timothy Nowak, Erik L. Regalado, Mikhail Reibarkh, Weidong Tong, and Michael A. Zompa for analytical support, Daniel M. Bishara, Darryl Chang, Meredith Green, Shane T. Grosser, Alexander Judge, David Lashen, Ivan Lee, Keith A. Mattern, Charles Orella, Megan Roth, Eric Sirota, James Ulis, and Ralph Zhao for engineering support, Birgit Kosjek, Hongmei Li, Rose Mathew, Sylvie Pailloux, Xianghui Wen, Honglin Ye, and Guofeng Xu for experimental contributions and discussions.Notes and references
- E. De Clercq and G. Li, Clin. Microbiol. Rev., 2016, 29, 695–747 Search PubMed.
- G. Li and E. De Clercq, Nat. Rev. Drug Discovery, 2020, 19, 149–150 Search PubMed; P. N. Batalha, L. S. M. Forezi, C. G. S. Lima, F. P. Pauli, F. C. S. Boechat, M. C. B. V. de Souza, A. C. Cunha, V. F. Ferreira and F. de C. da Silva, Bioorg. Chem., 2021, 106, 104488 Search PubMed.
- https://www.who.int/news-room/fact-sheets/detail/hepatitis-c .
- HCV: The Journey from Discovery to a Cure, ed. M. J. Sofia, Top. Med. Chem., 2019, vol. 32 Search PubMed.
- F.-R. Alexandre, E. Badaroux, J. P. Bilello, S. Bot, T. Bouisset, G. Brandt, S. Cappelle, C. Chapron, D. Chaves, T. Convard, C. Counor, D. Da Costa, D. Dukhan, M. Gay, G. Gosselin, J.-F. Griffon, K. Gupta, B. Hernandez-Santiago, M. La Colla, M.-P. Lioure, J. Milhau, J.-L. Paparin, J. Peyronnet, C. Parsy, C. P. Rouvière, H. Rahali, R. Rahali, A. Salanson, M. Seifer, I. Serra, D. Standring, D. Surleraux and C. B. Dousson, Bioorg. Med. Chem. Lett., 2017, 27, 4323–4330 Search PubMed.
- C. McGuigan, R. N. Pathirana, N. Mahmood, K. G. Devine and A. J. Hay, Antiviral Res., 1992, 17, 311–321 Search PubMed.
- For selected examples, see: M. Peifer, R. Berger, V. W. Shurtleff, J. C. Conrad and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5900–5903 Search PubMed; M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 Search PubMed; M. Meanwell, S. M. Silverman, J. Lehmann, B. Adluri, Y. Wang, R. Cohen, L.-C. Campeau and R. Britton, Science, 2020, 369, 725–730 Search PubMed.
- For an example, see: J. L. Clark, L. Hollecker, J. C. Mason, L. J. Stuyver, P. M. Tharnish, S. Lostia, T. R. McBrayer, R. F. Schinazi, K. A. Watanabe, M. J. Otto, P. A. Furman, W. J. Stec, S. E. Patterson and K. W. Pankiewicz, J. Med. Chem., 2005, 48, 5504–5508 Search PubMed.
- R. Storer, A. Moussa, N. Chaudhuri and F. Waligora, WO 2004/052899 A2, 24 June, 2004; D. J. Hotchkiss, S. F. Jenkinson, R. Storer, T. Heinz and G. W. J. Fleet, Tetrahedron Lett., 2006, 47, 315–318 Search PubMed.
- For a one-step biocatalytic synthesis of protected uridine from ribose and uracil, see: T. Benkovics, J. A. McIntosh, S. M. Silverman, J. Kong, P. Maligres, T. Itoh, H. Yang, M. Huffman, D. Verma, W. Pan, H.-I. Ho, J. Vroom, A. Knight, J. Hurtak, W. Morris, N. A. Strotman, G. Murphy, K. M. Maloney and P. S. Fier, DOI: DOI:10.26434/chemrxiv.13472373.v1.
- The following set of conditions, which have been used for selective oxidation of other secondary alcohols, did not provide any of the desired ketone 6: N-bromosuccinimide-NaHCO3; H2O2-HBr; H2O2-Bu4NCl-ammonium molybdate; NaOCl-AcOH.
- 2′-Ketouridine 6 has been prepared through trityl deprotection under acidic conditions. Rapid decomposition of 6via elimination of uracil was observed under basic conditions, see: A. F. Cook and J. G. Moffatt, J. Am. Chem. Soc., 1967, 89, 2697–2705 Search PubMed.
- W. T. Markiewicz, J. Chem. Res., Synop., 1979, 24–25 Search PubMed.
- K. Furusawa, K. Ueno and T. Katsura, Chem. Lett., 1990, 19, 97–100 Search PubMed.
- Acetyl, benzoyl, 2,4,6-trimethylbenzoyl, 2-toluoyl, and 4-chlorobenzoyl chlorides provided inferior selectivity for bis-acylation.
- See ESI† for XRPD and HRMS data of complex 11·BF3.
- A. Matsuda, K. Takenuki, M. Tanaka, T. Sasaki and T. Ueda, J. Med. Chem., 1991, 34, 812–819 Search PubMed; V. Samano and M. J. Robins, Synthesis, 1991, 283–288 Search PubMed.
- E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428–1431 Search PubMed.
- G. Cahiez, C. Duplais and J. Buendia, Chem. Rev., 2009, 109, 1434–1476 Search PubMed.
- For a completely different route to anhydrouridine 19, which was utilized in the original kilo route, see: B. L. Simmons, K. R. Campos, A. Klapars, A. J. Stewart, B. A. Mayes, P. E. Maligres, A. Hyde, S. M. Silverman, Y. L. Zhong, A. M. Moussa, K. Baker and K. Van Valkenburg, WO 2016/064797 A1 April 28, 2016. A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Org. Process Res. Dev., 2019, 23, 1860–1871 Search PubMed.
- For another route to anhydrouridine 19via the Holy reaction, see: S. F. Jenkinson, N. A. Jones, A. Moussa, A. J. Stewart, T. Heinz and G. W. J. Fleet, Tetrahedron Lett., 2007, 48, 4441–4444 Search PubMed.
- The nucleophilic attack of the tertiary alcohol on the activated uracil ring, as opposed to uracil acting as a nucleophile, is supported by 18O labeling, see the Supporting Information.†.
- Although we have communicated preliminary results using dichlorodimethylsilane, the iron-mediated hydrochlorination of the olefin 15 has not been reported previously, see: A. M. Hyde, S. L. Zultanski, J. H. Waldman, Y. L. Zhong, M. Shevlin and F. Peng, Org. Process Res. Dev., 2017, 21, 1355–1370 Search PubMed.
- If pure olefin 15 was used in the Fe-mediated hydrochlorination reaction under similar conditions, a 20:1 dr of the tertiary alkyl chloride 4 was obtained. Since only 5-10% of the olefin 15 is present at the end of Me2SiCl2-mediated hydrochlorination of anhydride 19, the 20:1 selectivity in Fe-mediated olefin hydrochlorination translated into >100:1 overall dr of compound 4.
- B. S. Ross, P. G. Reddy, H.-R. Zhang, S. Rachakonda and M. J. Sofia, J. Org. Chem., 2011, 76, 8311–8319 Search PubMed.
- For an improved preparation of 23, see: R. K. Orr, J. M. McCabe Dunn, A. Nolting, A. M. Hyde, E. R. Ashley, J. Leone, E. Sirota, J. A. Jurica, A. Gibson, C. Wise, S. Oliver and R. T. Ruck, Green Chem., 2018, 20, 2519–2525 Search PubMed.
- Z. Liu, A. Klapars, B. Simmons, A. Bellomo, A. Kalinin, M. Weisel, J. Hill and S. M. Silverman, Org. Process Res. Dev., 2021, 25, 661–667 Search PubMed.
- A slight excess of triethylamine improved the rate and selectivity in the formation of 22. Presumably, triethylamine acts not only as a base but also as a nucleophilic catalyst; for a precedent, see: F. Ramirez, J. F. Marecek and H. Okazaki, J. Am. Chem. Soc., 1975, 97, 7181–7182 Search PubMed.
- D. A. DiRocco, Y. Ji, E. C. Sherer, A. Klapars, M. Reibarkh, J. Dropinski, R. Mathew, P. Maligres, A. M. Hyde, J. Limanto, A. Brunskill, R. T. Ruck, L.-C. Campeau and I. W. Davies, Science, 2017, 356, 426–430 Search PubMed.
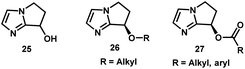
- The imidazole carbamate catalyst 24 is based on the bicyclic imidazole alcohol scaffold 25, which was first prepared by Weintraub, see: P. M. Weintraub, P. L. Tiernan and J. C. Huffman, J. Heterocycl. Chem., 1987, 24, 561–563 Search PubMed . Chiral ether 26 and ester 27 derivatives of this scaffold have been utilized as chiral nucleophilic catalysts for acyl transfer reactions, see: Z. Zhang, F. Xie, J. Jia and W. Zhang, J. Am. Chem. Soc., 2010, 132, 15939–15941 Search PubMed; M. Wang, Z. Zhang, S. Liu, F. Xie and W. Zhang, Chem. Commun., 2014, 50, 1227–1230 Search PubMed . Although enantioselective N-phosphorylation of lactams had been reported using the imidazole ether catalysts 26, the stereoselectivity was modest (<40% ee), see: S. Liu, Z. Zhang, F. Xie, N. A. Butt, L. Sun and W. Zhang, Tetrahedron: Asymmetry, 2012, 23, 329–332
 .
. - A recent synthesis of the anti-COVID treatment remdesivir leveraged the chiral imidazole carbamate catalysts closely related to 24, that we had developed for regio- and stereoselective phosphoramidation of 4 and other nucleosides, see: M. Wang, L. Zhang, X. Huo, Z. Zhang, Q. Yuan, P. Li, J. Chen, Y. Zou, Z. Wu and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20814–20819 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopy data for new compounds. See DOI: 10.1039/d1sc01978c |
| This journal is © The Royal Society of Chemistry 2021 |