
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe underlying mechanism for reduction stability of organic electrolytes in lithium secondary batteries†
Xiaohui
Shen‡
,
Peng
Li‡
,
Xingwei
Liu
,
Shengli
Chen
 ,
Xinping
Ai
,
Hanxi
Yang
and
Yuliang
Cao
*
,
Xinping
Ai
,
Hanxi
Yang
and
Yuliang
Cao
*
College of Chemistry and Molecular Sciences, Hubei Key Laboratory of Electrochemical Power Sources, Wuhan University, Wuhan 430072, China. E-mail: ylcao@whu.edu.cn
First published on 1st June 2021
Abstract
Many organic solvents have very desirable solution properties, such as wide temperature range, high solubility of Li salts and nonflammability, and should be able but fail in reality to serve as electrolyte solvents for Li-ion or -metal batteries due to their reduction instability. The origin of this interfacial instability remains unsolved and disputed so far. Here, we reveal for the first time the origin of the reduction stability of organic carbonate electrolytes by combining ab initio molecular dynamics (AIMD) simulations, density functional theory (DFT) calculations and electrochemical stability experiments. It is found that with the increase of the molar ratio (MR) of salt to solvent, the anion progressively enters into the solvation shell of Li+ to form an anion-induced ion–solvent-coordinated (AI-ISC) structure, leading to a “V-shaped” change of the LUMO energy level of coordinated solvent molecules, whose interfacial stability first decreases and then increases with the increased MRs of salt to solvent. This mechanism perfectly explains the long-standing puzzle about the interfacial compatibility of organic electrolytes with Li or similar low potential anodes and provides a basic understanding and new insights into the rational design of the advanced electrolytes for next generation lithium secondary batteries.
The state-of-the-art electrolytes in Li-ion batteries (LIBs) are mostly based on 1.0 mol L−1 LiPF6/ethylene carbonate (EC)-based carbonate due to the surface passivation of the graphite anode by forming a stable solid electrolyte interphase (SEI). However, these electrolytes cannot operate well for new electrode materials and battery systems that are expected to have higher voltage, better safety and wider temperature range than current commercial LIBs.1–3 For example, EC-based carbonate electrolytes are easily oxidized on a high voltage cathode at or above 4.3 V, resulting in depletion of electrolytes, gas evolution and low coulombic efficiency, which reduce the cycle life and create safety hazards for LIBs.4 These problems of the conventional electrolyte significantly hinder the development of new generation lithium batteries and limit these batteries for high voltage and/or high capacity applications and operation in a wide temperature range.
To overcome these problems, great efforts have been devoted in recent years to the development of new electrolytes, such as solid state electrolytes,5 ionic liquids,6–8 highly-concentrated electrolytes (HCEs),9 electrolyte stabilizing additive,10–13 and so on. Among them, the HCEs or high-molar-ratio electrolytes (HMREs) of salt to solvent have received particular attention, owing to their unusual electrochemical stability, nonflammability, and good compatibility with a wide range of anode and cathode materials.14–17 These desirable properties are apparently attributed to the solution structure of HCEs, where there exist almost no free solvent molecules, and the parasitic side reactions of solvents are thereby greatly reduced. Due to the lack of solvent molecules in HCEs, anions have to enter into the solvation shell of Li+, in order to meet the Li+ coordination number of 4–6, to form an ion–solvent-coordinated (ISC) structure.18 Several studies have shown that the unique ISC structure of HCEs leads to the shift of the lowest unoccupied molecular orbital (LUMO) from solvent to salt, which makes anions preferentially reduced or decomposed to produce a robust anion-derived SEI.14,19 In recent years, the anion-derived SEI structure has been regarded as the “holy grail” of electrolyte chemistry for understanding the interfacial stability and compatibility of HCEs. However, recent studies have showed that some HCEs containing non-film-forming salts and solvents can still achieve excellent reversible Li+ insertion reactions.20 Therefore, an intrinsic origin for the interfacial stability of HCEs still remains unrevealed. In our previous studies on HCEs or HMREs, their interfacial stability was found to depend predominately on the molar ratio (MR) of salt to solvent rather than the molar concentration.2,21,22 Thus, the HMREs instead of the HCEs in the following study could more clearly describe the nature of electrolyte stability.
In this work, we reveal the correlation between the solvation microstructures and the LUMO energy levels of typical ISC structures in the electrolytes at various MRs with non-film-forming lithium salt (LiClO4) and organic carbonate solvents (PC, DMC, EMC and DEC) by ab initio molecular dynamics (AIMD) simulations and density functional theory (DFT) calculations. The choice of non-film-forming lithium salt and solvent in this study was aimed to exclude the contribution of the formation of the SEI film to the interfacial stability of the electrolytes. It is found from this study that the LUMO energy level of the ISC structure formed at a low MR is lower than that of pure solvent. With the increase of the MR, anions gradually enter into the first solvation shell of Li+ to form the anion-induced ISC (AI-ISC) structure, resulting in the increase of the LUMO energy level that enhances the reduction stability of the electrolyte. Also, it is revealed that the LUMO levels of ISC structures at different MRs are always situated at the coordinated solvent molecules, i.e., the strong reduction stability of HMREs is dominated by the modulation of solvent molecules rather than only the formation of the anion-derived SEI. Such a theoretical insight is further unequivocally evidenced by chemical compatibility experiments in this work. These findings reveal the origin of the greatly improved interfacial stability of HMREs and provide a mechanistic insight into the rational design of stable electrolytes for new generation alkali or alkaline metal based batteries.
To investigate the specific ISC microstructures of the electrolytes with different MRs, AIMD simulations were first performed (see computational details in the ESI†). Taking non-film-forming DEC solvent as an example, three types of electrolytes with MRs of LiClO4 to DEC = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 1:5 and 1:2 are considered (Table S1†). After long-time AIMD simulation, the representative images of the equilibrium structures are shown in Fig. 1a–c. To characterize the solution structures, the radial distribution function g(r) of the electrolyte with different MRs is analyzed (Fig. 1e–g), and the changes in the Li+ coordination number with the O atoms of solvents and anions are listed in Table 1, where the g(r) profiles display one distinct peak at around 2.0 Å for all the electrolytes, representing the first solvation shell of Li+ ions. It is obvious from Table 1 that with the increase of the MR from 1:10 to 1:2, the average coordination number of Li+ with the O atom of solvents decreases from 3.1 to 1.6, whereas the average coordination number of Li+ contributed by the O atom in anions increases from 1.0 to 2.4, indicating that the anions gradually invade into the first solvation shell of Li+ and then form the AI-ISC structure, as illustrated in Fig. 1d. In addition, it should be noted that the total coordination number of Li+ always remains around 4, which implies that the stable tetragonal solvation shell structure of Li+ does not change in the different MR electrolytes; meanwhile, both the coordination numbers of Li+ contributed by the solvent and anion change oppositely. This phenomenon can be corroborated experimentally through infrared spectroscopy (IR) because the C
:10, 1:5 and 1:2 are considered (Table S1†). After long-time AIMD simulation, the representative images of the equilibrium structures are shown in Fig. 1a–c. To characterize the solution structures, the radial distribution function g(r) of the electrolyte with different MRs is analyzed (Fig. 1e–g), and the changes in the Li+ coordination number with the O atoms of solvents and anions are listed in Table 1, where the g(r) profiles display one distinct peak at around 2.0 Å for all the electrolytes, representing the first solvation shell of Li+ ions. It is obvious from Table 1 that with the increase of the MR from 1:10 to 1:2, the average coordination number of Li+ with the O atom of solvents decreases from 3.1 to 1.6, whereas the average coordination number of Li+ contributed by the O atom in anions increases from 1.0 to 2.4, indicating that the anions gradually invade into the first solvation shell of Li+ and then form the AI-ISC structure, as illustrated in Fig. 1d. In addition, it should be noted that the total coordination number of Li+ always remains around 4, which implies that the stable tetragonal solvation shell structure of Li+ does not change in the different MR electrolytes; meanwhile, both the coordination numbers of Li+ contributed by the solvent and anion change oppositely. This phenomenon can be corroborated experimentally through infrared spectroscopy (IR) because the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the carbonate group has a strong IR absorption in the carbonyl region (1650–1850 cm−1) and its IR peak position shifts sensitively with its coordination environment. As shown in Fig. 1h, the IR band of carbonyl groups in pure DEC is located at ∼1741 cm−1, which is shifted to ∼1710 cm−1 in a LiClO4/DEC (MR = 1:10) electrolyte due to the coordination of the O atom in CO with Li+. With the increase of the MR of Li+/DEC, its IR peak at ∼1741 cm−1 gradually disappears, reflecting a gradual decrease in the number of free DEC molecules. In addition, the IR band of free ClO4− in a LiClO4/DEC (MR = 1:10) electrolyte is located at ∼931 cm−1, which is shifted to ∼942 cm−1 in the 1:2 LiClO4/DEC electrolyte due to the ionic association of Li+ and ClO4− (Fig. S1†). Combining AIMD simulations and IR experiments, it can be concluded that with the increase of the MR of the electrolyte, the anions gradually enter into the solvation shell of Li+, which modulates the chemical stability of the electrolyte.
O bond of the carbonate group has a strong IR absorption in the carbonyl region (1650–1850 cm−1) and its IR peak position shifts sensitively with its coordination environment. As shown in Fig. 1h, the IR band of carbonyl groups in pure DEC is located at ∼1741 cm−1, which is shifted to ∼1710 cm−1 in a LiClO4/DEC (MR = 1:10) electrolyte due to the coordination of the O atom in CO with Li+. With the increase of the MR of Li+/DEC, its IR peak at ∼1741 cm−1 gradually disappears, reflecting a gradual decrease in the number of free DEC molecules. In addition, the IR band of free ClO4− in a LiClO4/DEC (MR = 1:10) electrolyte is located at ∼931 cm−1, which is shifted to ∼942 cm−1 in the 1:2 LiClO4/DEC electrolyte due to the ionic association of Li+ and ClO4− (Fig. S1†). Combining AIMD simulations and IR experiments, it can be concluded that with the increase of the MR of the electrolyte, the anions gradually enter into the solvation shell of Li+, which modulates the chemical stability of the electrolyte.
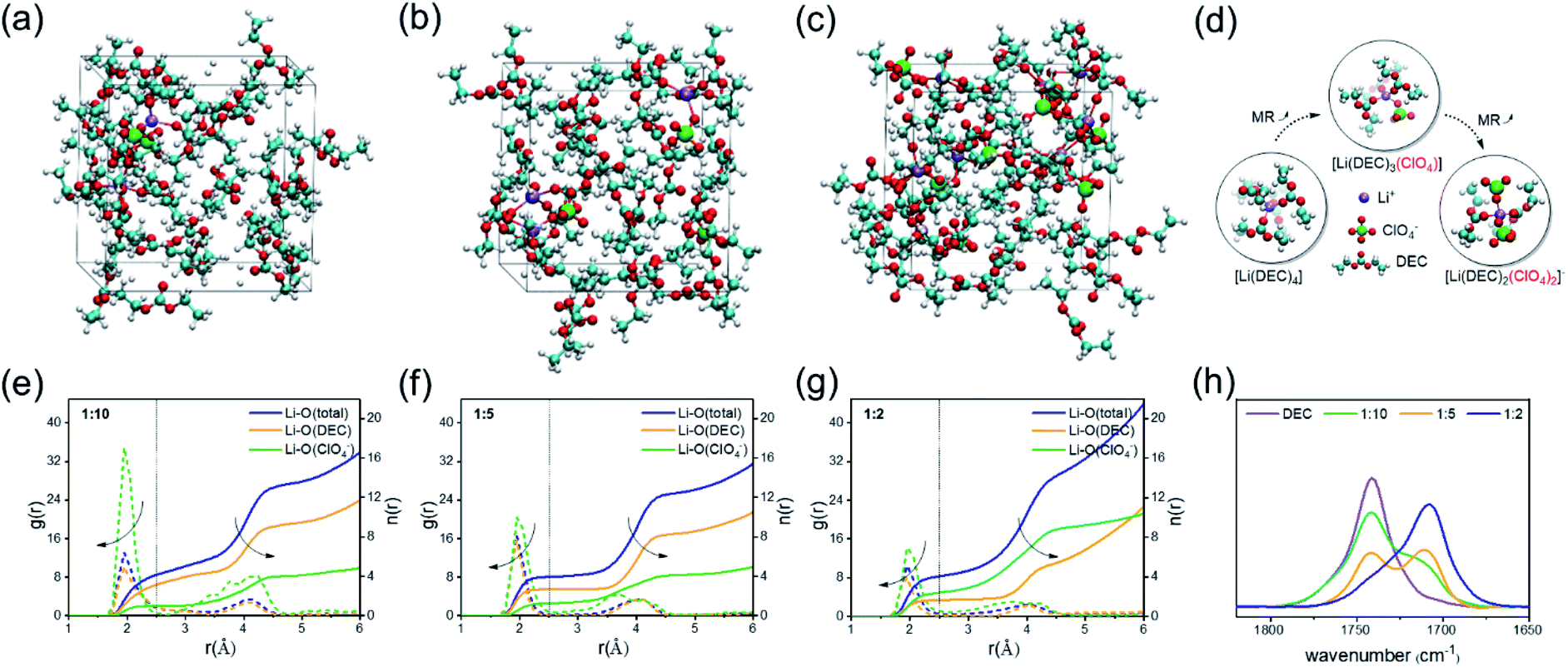 | ||
| Fig. 1 Snapshots of typical equilibrium trajectories from DFT-MD simulations: (a) 1:10 LiClO4/DEC solution (2-LiClO4/20-DEC), (b) 1:5 LiClO4/DEC solution (3-LiClO4/15-DEC) and (c) 1:2 LiClO4/DEC solution (7-LiClO4/14-DEC). (d) Typical ISC structure extracted from DFT-MD. (e–g) Radial distribution function of lithium–oxygen interaction (short dashed lines) and relationship between the coordination number and bond distances (full lines). (h) FTIR spectra of the carbonyl group in LiClO4/DEC solution. Atom color: H, white; Li, purple; C, cyan; O, red; Cl, green. | ||
| Molar ratio | Li–O(DEC) | Li–O(ClO4−) | Total |
|---|---|---|---|
| 1:10 |
3.1 | 1.0 | 4.1 |
| 1:5 |
2.7 | 1.3 | 4.0 |
| 1:2 |
1.6 | 2.4 | 4.0 |
To further understand how the coordination of anions with Li+ can modulate the reduction stability of the electrolyte, DFT calculations were performed to evaluate the lowest unoccupied molecular orbital (LUMO) of the ISC structures in electrolytes with different MRs. The optimized configuration of DEC molecules and associated ISC structures are shown in Fig. S2.† Table S2† gives the calculated reaction energies for several different modes. It is found that the formation of all ISC structures is strongly favourable and enthalpy driven. The corresponding LUMO and energy levels of ISC structures are shown in Fig. 2. Firstly, our theoretical result clearly demonstrates that with the increase of the MR, the LUMOs of all ISC structures are invariably located on DEC molecules but have never shifted from the solvent to the salt as described in previous reports.14,19 This suggests that the reductive decomposition of the anion (ClO4−) won't easily occur at low potentials. The discrepancy between this study and previous reports is mainly because we deliberately chose reduction-tolerant and non-film-forming LiClO4 as the electrolyte salt to avoid the influence of stable SEI film formation on the electrolyte stability, whereas the use of film-forming LiFSI or LiPF6 may contain the contribution of the SEI for interfacial stabilization, as shown in previous reports. Secondly, our results reveal that the change of the LUMO energy level of ISC structures appears to be a “V-shaped” profile with the increase of the MR, as shown in Fig. 2. At a low MR, the LUMO of [Li(DEC)4]+ is much lower than that of pure DEC molecules, owing to the coordination with cations to decrease the reduction stability of DEC, which is in line with Zhang's results.23,24 Nevertheless, when the anions gradually enter into the first solvation shell of Li+ with the increase of the MR, the LUMO of the ISC structure notably rise up to a much higher level than that of pure DEC molecules, indicating that introducing anions into the ISC structure to coordinate with Li+ can increase the LUMO located on solvents and thereby intensify the reduction stability of the electrolyte. In addition, we have analyzed the projected density of states (PDOS) averaged over the AIMD trajectories for electrolytes with different MRs (Fig. S3†). It is found that in LiClO4/DEC electrolytes with any MR, the DEC molecules coordinated with Li+ dominate the LUMO, which agrees well with our above DFT results and highlights that the anions (ClO4−) will not be reduced to form the SEI film in this system. Thus, the reduction stability of HRMEs is controlled by the position of the LUMO of coordinated solvent molecules, which is shifted by the participation of anions into the first solvation shell of Li+ to form the anion-induced ISC structure. This mechanism is markedly different from the traditional and general understanding, in which the high reduction stability of HRMEs is attributed to the formation of the anion-derived SEI film. In other words, the interfacial stability of the electrolyte can be adjusted through the modulation of the AI-ISC structure and not necessarily the anion-derived SEI film.
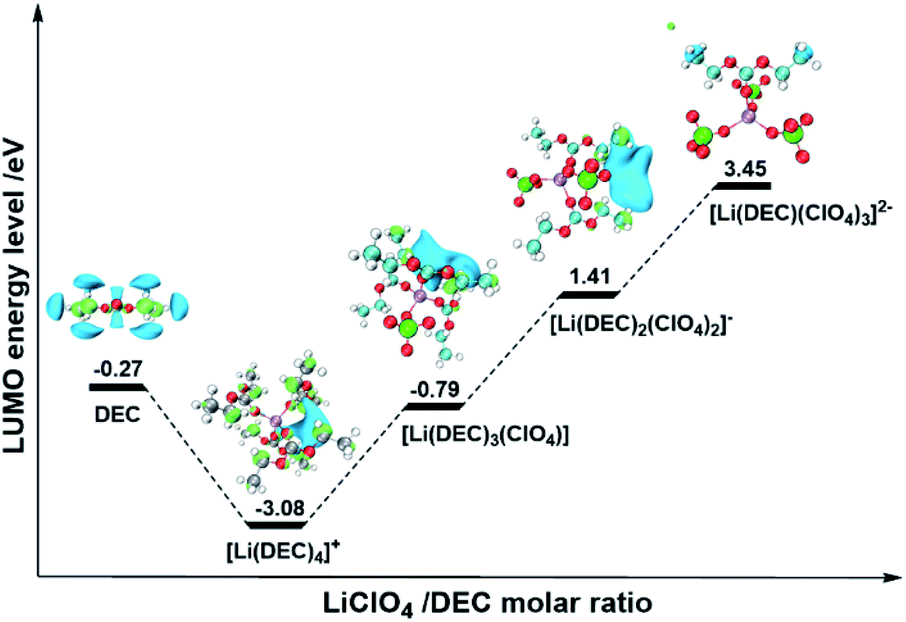 | ||
| Fig. 2 The visual LUMOs and energy level of ISC structures. The hydrogen, lithium, carbon, oxygen, and chlorine atoms are marked with white, purple, gray, red, and green, respectively. The light green and light blue regions of LUMOs represent the positive and negative parts of the orbitals, respectively (isovalue = 0.02). | ||
In order to verify the above theoretical analysis, we performed a series of chemical stability experiments of the electrolytes. The change trend of the reduction stability of electrolytes with various MRs can be visually observed by immersing Li pieces in the LiClO4/DEC electrolyte. Fig. 3a shows the photos of these electrolytes and lithium pieces before (as-prepared) and after storage for 5 h, 12 h and 24 h, respectively. It is clear that the LiClO4/DEC electrolyte with a MR of 1:10 and 1:5 turned yellow after only 5 hours. However, in pure DEC and LiClO4/DEC with a MR of 1:2, there is still no obvious color change for lithium foil and solution even after 24 h. Such an experimental phenomenon indicates that the reduction stability of the electrolyte decreases distinctly from the pure solvent to the LiClO4/DEC electrolyte with a low MR (1:10 and 1:5) due to the presence of the anion-free ISC structure that decreases the LUMO (Fig. 2). In addition, we can note that the electrolyte with a MR of 1:5 has a stronger reactivity with lithium piece than the electrolyte with a MR of 1:10, mainly because there exist more anion-free ISC structures in the 1:5 MR electrolyte (Fig. 3e). As the MR increases to 1:2, there is an increased number of ISC structures containing more anions to form the AI-ISC structure (Fig. 3f), which promotes the elevation of the LUMO so as to increase the reduction tolerance. To further confirm this mechanism, we also investigated the interfacial stability of the LiClO4/DEC electrolyte on a graphite (Gr) electrode at different MRs of LiClO4/DEC. As shown in Fig. 3b, the reversible capacity of the Gr electrode decreases from 130 mA h g−1 to 105 mA h g−1 and increases to 260 mA h g−1 with the increase of the MR of LiClO4/DEC from 1:10 to 1:5 and 1:2 in the electrolyte, consistent with the trend in Fig. 3a. Also, the Gr electrode demonstrates similar three-staged Li+ insertion reactions in the LiClO4/DEC (1:2) electrolyte as in conventional 1 M LiPF6 EC–EMC electrolyte (Fig. 3c), indicating that the interfacial compatibility of the electrolyte with Gr can also be achieved by introducing an AI-ISC structure (Fig. 3f). Encouragingly, such experimental phenomena are completely in line with our DFT calculations and AIMD simulations (Fig. 2), in which the LUMO energy level of the ISC structure formed in the electrolyte with a low MR (<1:4) is lower than that of pure solvent, while with the increase of the MR, the AI-ISC structure elevates the LUMO energy level of the electrolyte. It is worth mentioning that for the electrolytes with reduction-stable anions (e.g. ClO4−), their LUMO is still located on the coordinated solvent molecules even with a high MR (>1:4), demonstrating that the increase of reduction stability is controlled by the coordinated solvent molecules in ISC structures, rather than the formation of the anion-derived SEI film.
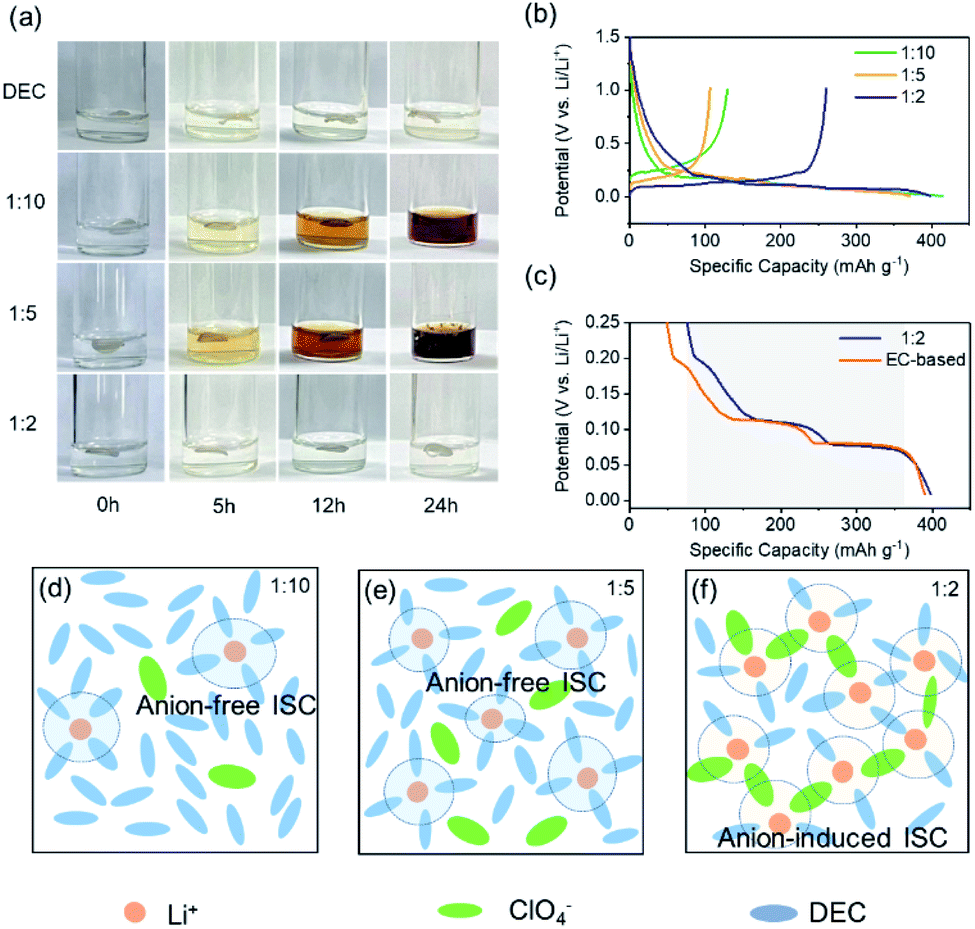 | ||
| Fig. 3 (a) Reactivity of lithium metal foil and LiClO4/DEC solution at room temperature. (b) Charge–discharge curves of the Gr anode in LiClO4/DEC electrolytes with different MRs. (c) Magnified discharge curves of the Gr anode in 1:2 MR LiClO4/DEC and traditional EC-based (1 M LiPF6 EC–EMC) electrolytes. Schematics of LiClO4/DEC electrolyte with a MR of (d) 1:10, (e) 1:5 and (f) 1:2. | ||
In order to further confirm the universality of the above conclusion, DFT calculations were also carried out for three other electrolyte systems, which are composed of non-film-forming PC, DMC or EMC solvents and non-film-forming LiClO4 salt. The optimized structures of pure solvent molecules and ISC structures with various MRs are shown in Fig. S2,† and their corresponding LUMO levels are shown in Fig. 4. It can be clearly seen that the LUMO levels are still situated at the solvent molecules for all pure solvents and ISC structures. In addition, as the MR increases, the LUMO energy levels change to a V-shape, which are in line with the case of LiClO4/DEC electrolyte and confirms the rationality and correctness of our proposed mechanism.
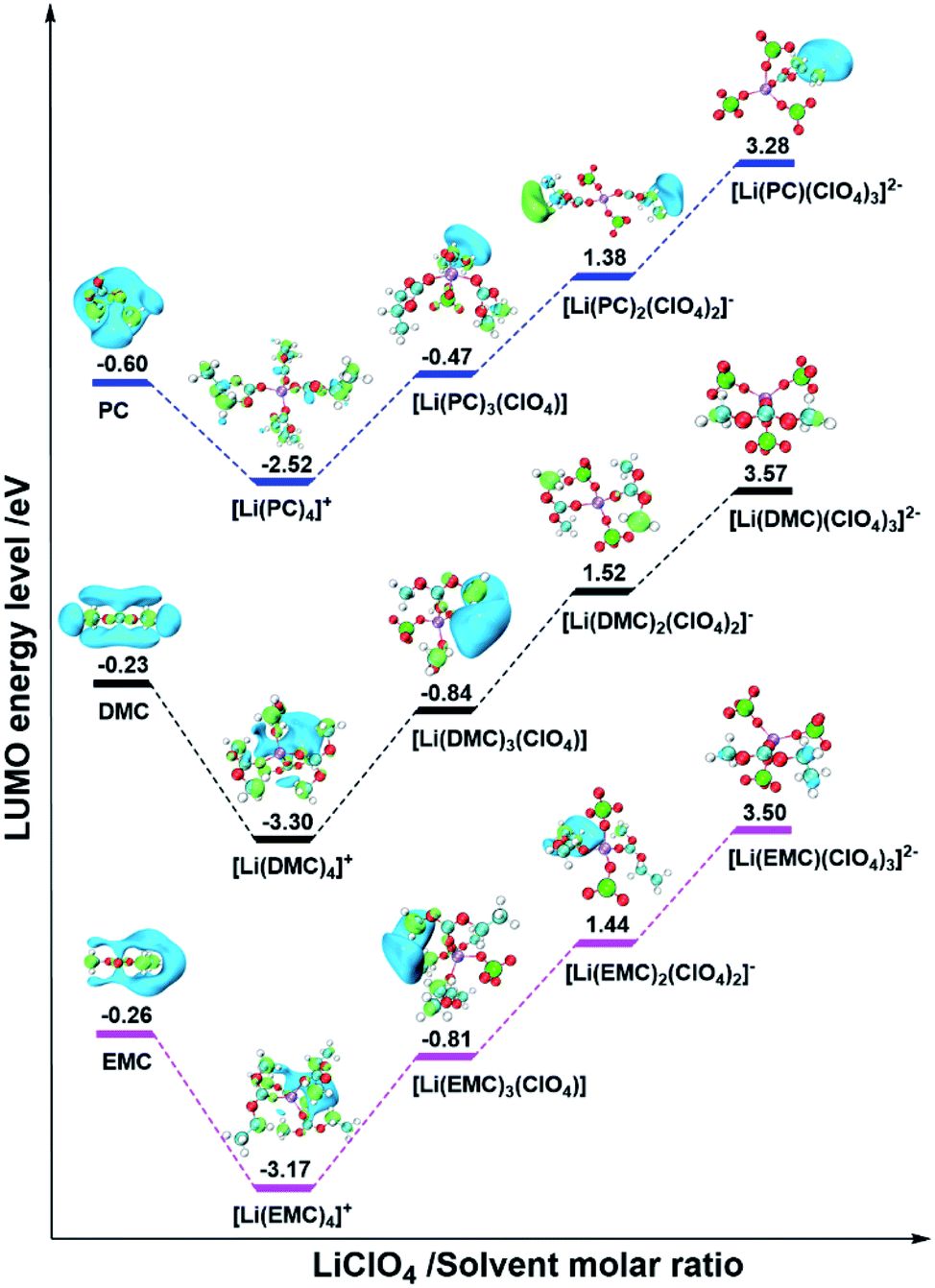 | ||
| Fig. 4 The visual LUMOs and energy level of solvents and ISCs. The hydrogen, lithium, carbon, oxygen, and chlorine atoms are marked with white, purple, gray, red, and green, respectively. Besides, the light green and light blue regions of LUMOs represent the positive and negative parts of the orbitals, respectively (isovalue = 0.02). | ||
In conclusion, by combining AIMD simulations and DFT calculations with chemical stability experiments, we establish a close correlation between ISC structures and reduction stability of commonly used carbonate electrolytes with different MRs. The LUMO energy level of the anion-free ISC structure formed in the electrolyte with a low MR (<1:4) is lower than that of pure solvent, suggesting a decreased interfacial stability of less concentrated electrolytes. With the increase of the MR, anions gradually coordinated with Li+ to form AI-ISC structures, leading to the increase of the LUMO energy level of ISC structures and therefore an enhanced reduction stability. The change of the LUMO energy level with ISC structures appears as a “V-shaped” profile with the increase of the MR. It is worth noting that at any MR, the LUMO is always located on the coordinated solvent molecules without shifting from the solvent towards the salt (only stable anion). Consequently, the underlying origin of the enhanced reduction stability for electrolytes with higher MRs is revealed to arise from the entry of anions into the first solvation shell of Li+ for the formation of the AI-ISC structure. Our findings provide a novel and molecular level understanding of the stability mechanism of HMREs (or HCEs) and a new insight into the rational design of highly stable and multifunctional electrolytes for new generation rechargeable batteries.
Data availability
All the data supporting this article have been included in the main text and the supplementary material.Author contributions
Xiaohui Shen and Peng Li completed the theoretical calculation together. Xingwei Liu designed and completed the experiment. All authors contributed to the analysis and discussion of the results. Xiaohui Shen, Peng Li and Yuliang Cao wrote the manuscript. Yuliang Cao put forward this idea and supervised this research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support by the Regional Innovation and Development Joint Fund, National Natural Science Foundation of China (No. U20A20249), National Natural Science Foundation of China (No. 21972108) and the Key Research Program of Hubei Province (2020BAA030). The numerical calculations in this paper have been done on the supercomputing system in the Supercomputing Center of Wuhan University.Notes and references
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 Search PubMed.
- Z. Zeng, V. Murugesan, K. S. Han, X. Jiang, Y. Cao, L. Xiao, X. Ai, H. Yang, J.-G. Zhang, M. L. Sushko and J. Liu, Nat. Energy, 2018, 3, 674–681 Search PubMed.
- X. Dong, Y. Lin, P. Li, Y. Ma, J. Huang, D. Bin, Y. Wang, Y. Qi and Y. Xia, Angew. Chem., 2019, 58, 5623–5627 Search PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2017, 3, 22–29 Search PubMed.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 Search PubMed.
- K. Dokko, N. Tachikawa, K. Yamauchi, M. Tsuchiya, A. Yamazaki, E. Takashima, J.-W. Park, K. Ueno, S. Seki and N. Serizawa, J. Electrochem. Soc., 2013, 160, A1304 Search PubMed.
- J. B. Haskins, H. Yildirim, C. W. Bauschlicher and J. W. Lawson, J. Phys. Chem. C, 2017, 121, 28235–28248 Search PubMed.
- H. Yildirim, J. B. Haskins, C. W. Bauschlicher and J. W. Lawson, J. Phys. Chem. C, 2017, 121, 28214–28234 Search PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 Search PubMed.
- X. Chen, X. Shen, T.-Z. Hou, R. Zhang, H.-J. Peng and Q. Zhang, Chem, 2020, 6, 2242–2256 Search PubMed.
- C. Yan, Y.-X. Yao, X. Chen, X.-B. Cheng, X.-Q. Zhang, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 14055–14059 Search PubMed.
- X.-Q. Zhang, X. Chen, L.-P. Hou, B.-Q. Li, X.-B. Cheng, J.-Q. Huang and Q. Zhang, ACS Energy Lett., 2019, 4, 411–416 Search PubMed.
- X. Q. Zhang, X. Chen, X. B. Cheng, B. Q. Li, X. Shen, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., 2018, 130, 5399–5403 Search PubMed.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 Search PubMed.
- Y. Yamada, K. Usui, C. H. Chiang, K. Kikuchi, K. Furukawa and A. Yamada, ACS Appl. Mater. Interfaces, 2014, 6, 10892–10899 Search PubMed.
- K. Fujii, H. Wakamatsu, Y. Todorov, N. Yoshimoto and M. Morita, J. Phys. Chem. C, 2016, 120, 17196–17204 Search PubMed.
- M. He, K. C. Lau, X. Ren, N. Xiao, W. D. McCulloch, L. A. Curtiss and Y. Wu, Angew. Chem., Int. Ed., 2016, 55, 15310–15314 Search PubMed.
- L. Xiao, Z. Zeng, X. Liu, Y. Fang, X. Jiang, Y. Shao, L. Zhuang, X. Ai, H. Yang, Y. Cao and J. Liu, ACS Energy Lett., 2019, 4, 483–488 Search PubMed.
- K. Sodeyama, Y. Yamada, K. Aikawa, A. Yamada and Y. Tateyama, J. Phys. Chem. C, 2014, 118, 14091–14097 Search PubMed.
- X. Liu, X. Jiang, Z. Zeng, X. Ai, H. Yang, F. Zhong, Y. Xia and Y. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 38141–38150 Search PubMed.
- X. Jiang, X. Liu, Z. Zeng, L. Xiao, X. Ai, H. Yang and Y. Cao, Adv. Energy Mater., 2018, 8, 1802176 Search PubMed.
- X. Liu, X. Shen, F. Zhong, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Chem. Commun., 2020, 56, 6559–6562 Search PubMed.
- X. Chen, H. R. Li, X. Shen and Q. Zhang, Angew. Chem., Int. Ed. Engl., 2018, 57, 16643–16647 Search PubMed.
- X. Chen, X. Shen, B. Li, H. J. Peng, X. B. Cheng, B. Q. Li, X. Q. Zhang, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed. Engl., 2018, 57, 734–737 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01363g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |