
The odyssey of cobaloximes for catalytic H2 production and their recent revival with enzyme-inspired design
Dependu
Dolui
 a,
Shikha
Khandelwal
a,
Piyali
Majumder
b and
Arnab
Dutta
*c
a,
Shikha
Khandelwal
a,
Piyali
Majumder
b and
Arnab
Dutta
*c
aChemistry Discipline, Indian Institute of Technology Gandhinagar, Palaj 382355, India
bBiological Engineering Discipline, Indian Institute of Technology Gandhinagar, Palaj 382355, India
cChemistry Department, Indian Institute of Technology Bombay, Powai 400076, India. E-mail: arnabdutta@chem.iitb.ac.in
First published on 5th June 2020
Abstract
Cobaloxime complexes gained attention for their intrinsic ability of catalytic H2 production despite their initial emergence as a vitamin B12 model. The simple, robust, and synthetically manoeuvrable cobaloxime core represents a model catalyst molecule for the investigation of optimal conditions for both photo- and electrocatalytic H2 production catalytic assemblies. Cobaloxime is one of the rare catalysts that finds equal applications in the analysis of homogeneous and heterogeneous catalytic conditions. However, the poor aqueous solubility and long-term instability of cobaloximes have severely impeded their growth. Lately, interest in the cobaloxime-based catalysts has been resuscitated with the rational use of extended enzymatic features. This unique enzyme-inspired catalyst design strategy has instigated the formation of a new genre of cobaloxime molecules that exhibit enhanced photo- and electrocatalytic H2 evolution with improved aqueous and air stability.
 Dependu Dolui | Dependu Dolui obtained his Master's degree with a specialization in Inorganic Chemistry from the Central University of Punjab (CUPB) (2017). After receiving the DST-Inspire Fellowship for Doctoral Research in India, he joined the bioinorganic research group of Dr Arnab Dutta at the Indian Institute of Technology, Gandhinagar (IITGN). His major research interests focus on designing bio-inspired molecular catalysts for energy-relevant small molecule activation and chemical transformation. In his free time, he loves to travel. |
 Shikha Khandelwal | Shikha Khandelwal received her Master's degree in Chemistry from the School of Chemical Sciences, DAVV, Indore, India. She joined IIT Gandhinagar to pursue a PhD in inorganic chemistry under the supervision of Dr Arnab Dutta. Her research focuses on the development of a cobalt-based synthetic catalyst inspired by natural enzymes for the hydrogen evolution reaction (HER). |
 Piyali Majumder | Piyali Majumder completed her Bachelor's degree in Biochemistry from Presidency College (2010), followed by a Master's degree in Biotechnology from the University of Calcutta (CU) (2012). Then, she joined the Saha Institute of Nuclear Physics (SINP) under the guidance of Dr Debashis Mukhopadhyay. After her PhD, she was awarded a National Post-doctorate Fellowship to work with Dr Bhaskar Datta at the Indian Institute of Technology Gandhinagar (IITGN). She specializes in the biochemistry of neurodegenerative diseases and metalloenzyme activity. |
 Arnab Dutta | The intricate framework of natural metalloenzymes has always fascinated Arnab Dutta from the beginning. He had an opportunity to study those structural marvels of biology during his PhD with Dr Anne Jones, where he developed metallopetide-based candid models of hydrogenase (Arizona State University, 2012). In the next phase of his career, he mastered the finer details of enzyme-mimicry with Dr Wendy Shaw. This time, he generated versatile molecular catalysts, boasting minimal but essential enzymatic traits in their periphery (Pacific Northwest National Laboratory, 2015). As an independent researcher, he is trying to develop sustainable and all-weather ready catalysts that can boost the use of renewable energy. His quest to bring life into poorly active catalysts with the Midas touch of enzyme-derived features continues at IIT Bombay. |
Introduction
The structural elucidation of vitamin B12 sprung a surprise in the bioinorganic community with the presence of a rare organometallic cobalt–carbon bond in biology.1–3 Vitamin B12 performs an array of metabolically vital chemical reactions such as methyl transfer, dehalogenation, and isomerization. Hence, the basic structure of vitamin B12 soon became the source of inspiration for developing several biomimetic model complexes for executing versatile industry-relevant chemical reactions, including alkene coupling, ring-opening metathesis, hydrogenation, and rearrangement.4 A stable organometallic cobalt complex, 5-deoxyadenosyl(5,6-dimethylbenzimidazolyl)-cobinamide ((1), Fig. 1A), resides in the core of vitamin B12, where the equatorial corrin ring plays a crucial role in tuning its reactivity.5 The requirement of such a bulky and synthetically challenging tetradentate corrin ring can be avoided by innovative substitution with two molecules of bidentate dimethyl glyoximes while retaining the versatile edge of a Co–N4 square planar core structure.6,7 Schrauzer and Kohnle prepared one of the first examples of such square planar cobalt dimethylglyoxime complexes, where the axial coordination sites were occupied by cyanide and pyridine groups.8 Following up on this work, Wagner and Bernhauer replaced the axial cyanide group with a Grignard reagent to generate an air-stable cobalt-alkyl derivative, imitating the vitamin B12 coenzyme core.9 Later, the term “cobaloxime” was coined to describe the cobalt dimethylglyoxime [Co(DH)2] moiety. The unique structure of these vitamin B12-inspired cobaloximes has instigated a series of attempts to probe their activity even as a functional model for the hydrogenase enzyme. The excellent reactivity of hydrogenases for H2 production (2H+ + 2e−![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) H2) is attributed to the synchronized conglomeration of a redox-active metal cofactor, electron-transporting [FeS] cluster proteins, and proton-relaying polar amino acid side-chains. Among these, the redox-active metal core and closely lying polar functionalities (oximes) are already present in cobaloximes. Hence, an appropriate electron supplement (either from a photo- or potential-driven source) can activate the cobaloximes for the catalytic hydrogen evolution reaction (HER) (Fig. 1B).
H2) is attributed to the synchronized conglomeration of a redox-active metal cofactor, electron-transporting [FeS] cluster proteins, and proton-relaying polar amino acid side-chains. Among these, the redox-active metal core and closely lying polar functionalities (oximes) are already present in cobaloximes. Hence, an appropriate electron supplement (either from a photo- or potential-driven source) can activate the cobaloximes for the catalytic hydrogen evolution reaction (HER) (Fig. 1B).
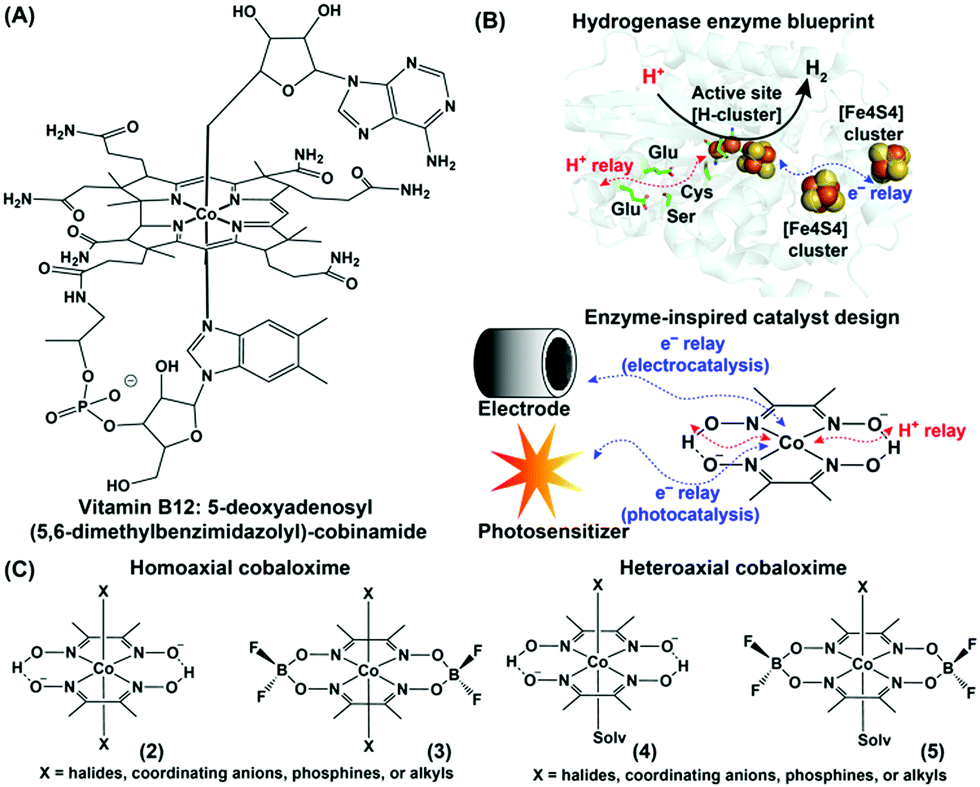 | ||
| Fig. 1 (A) Chemical structures of the vitamin B12 core (cobinamide). (B) Inclusion of the H+ relay and electron channel around the metal active site in the hydrogenase enzyme (PDB: 1HFE) and synthetic cobaloxime. The electron for the synthetic cobaloxime is transported from the electrode (electrocatalysis) or excited photosensitizer (photocatalysis). (C) Comparative structures of homo- and hetero-axial cobaloximes. | ||
This first proof of this “enzyme-inspired catalyst design” hypothesis was provided by Ziessel and co-workers, who observed photocatalytic H2 production by cobaloximes in the presence of [Ru(bpy)3]2+ and an organic amine as the photosensitizer and electron donor, respectively.10 Here, the Co(II) centre of cobaloximes readily coverts to Co(I) following the reductive quenching of photo-excited [Ru(bpy)3]2+. Next, the subsequent protonation generates a highly reactive Co(III)-hydride intermediate, which ultimately decomposes to display H2 production. The intrinsic O2 tolerance of cobaloximes allowed the occurrence of this catalytic H2 production even in aerobic conditions. Despite exhibiting such unique catalytic HER activity, cobaloximes failed to achieve prominence as HER catalysts due to their long-term instability.11,12 In biology, the axial ligands are believed to bring the stability for the cobalt-core in vitamin B12.13 Following this cue, several ligands (halides, coordinating mono-anions, phosphines, alkyls, and even coordinating solvent molecules) were screened in the pursuit of stable cobaloxime [XCo(DH)2B] motifs. The internal hydrogen-bonding network of oxime (–N–OH⋯O−–N–) groups were further difluoroborylated (BF2) to generate derivatives of cobaloximes [XCo(DBF2)2B], while the identity of the axial ligands was utilized to classify them into homo- or hetero-axial cobaloximes ((2–5), Fig. 1C).14 Introduction of these electron-withdrawing difluoroboryl (BF2)-functionality shrinks the energy barrier for catalytically vital cobalt reduction steps.15 Espenson et al. was able to utilize this thermodynamic advantage, and reported on the chemical H2 production from hydrochloric acid by [XCo(DBF2)2B] in the presence of Cr2+ as an external reducing agent.16 Peters, Artero, and Reisner independently investigated the electrocatalytic H2 evolution by cobaloxime derivatives in an acidic organic medium.17–19 Their success revived the research on oxygen-tolerant cobaloxime-based HER activity on both photocatalytic and electrocatalytic fronts. However, the limited catalytic efficiency, long-term aqueous instability, and loss of catalytic activity under acidic aqueous conditions have raised serious questions on the potential of these cobaloximes for large-scale applications. A similar roadblock was also observed for Ni-bis-(diphosphine)-based Dubois catalysts, which was resolved by the rational incorporation of extended features of enzyme architecture into their periphery of the Ni-core.20,21 Buoyed by this success of such unconventional enzyme-inspired catalyst design, researchers have also amended outer coordination sphere features around the cobaloxime core. This innovative track of catalyst design technique was the key to the formation of a new genre of cobaloxime catalysts that resuscitates the cobaloxime as a viable option for renewable energy research. The simple, robust, and synthetically maneuverable structure of the cobaloxime core has allowed researchers to optimize the combinations of ingredients for photocatalytic HER, analyze the catalytic pathways during the electrocatalytic H2 evolution, and explore the effect of extended enzyme-inspired features, which are discussed in detail in the following sections.
Photocatalytic HER activity by cobaloximes
The photocatalytic HER activity is typically driven by collaboration between three independent ingredients: (i) the photosensitizer (PS), (ii) the catalyst, and (iii) the sacrificial electron donor (ED). Here, our discussion will be limited to those systems where a cobaloxime derivative plays the catalytic role. Depending on the nature of the photosensitizer (PS), we have further fragmented our discussion into sections: (i) inorganic noble-metal-based PS, (ii) inorganic non-noble-metal based PS, (iii) organic PS, and (iv) nanomaterial-based PS. The robust nature, synthetic flexibility, and chemical property tunability of the cobaloxime core made it a popular choice for screening the photocatalytic efficiency of an array of photosensitizers under a wide chemical space.Noble metal-based photosensitizer
As mentioned earlier, photocatalytic hydrogen production can be obtained by using these cobaloximes in the presence of a photosensitizer (PS) and a sacrificial electron donor.22 The majority of such early studies reported the utilization of expensive Ru/Pt/Ir-based photosensitizers for light-harvesting (Fig. 2 and Table 1).23–26 Due to the limited water solubility of typical cobaloximes, organic solvents remained the primary choice of medium for photocatalytic HER investigation. The photocatalytic activity is conventionally reported with the parameter turnover number (TON), where the amount of H2 produced during the experiment is measured against the concentration of PS or cobaloxime catalyst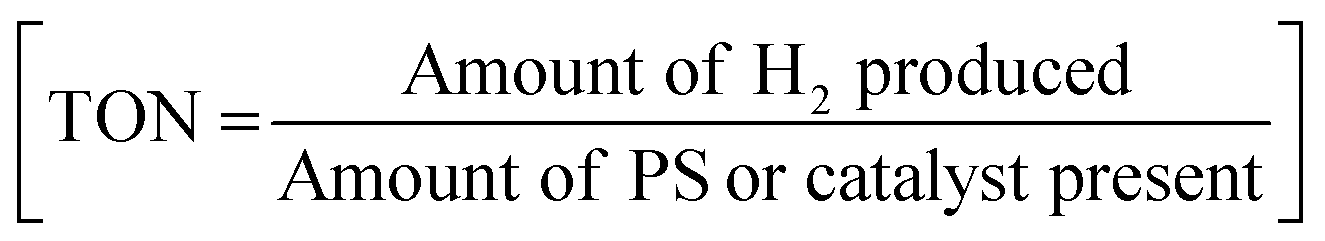 . In one of the first examples, considerable photocatalytic H2 production was observed for the [Co(DH)2] complex in the presence of [Ru(bpy)3]2+-PS (7) following 1 hour of irradiation (TON ∼ 38 vs. PS). The aero-sensitivity and catalytic performance were improved with specific functionalization of the bipyridyl moiety at the 4,4′-position by methyl, nitrile27 or carbene28 groups.25 Platinum (Pt)-based PS and Pt-acetylide (8) allowed for the prolonged irradiation and presence of external water during an analogous experimental setup, which significantly improved the TON.29 The improvement in TON was continued with the use of an extended π-conjugated Pt-based PS (9).24 One of the highest initial-stage photocatalytic HER rates has been observed for [Ru(bpy)3]2+ among the cobaloxime-noble-metal PS dyad, although its long-term stability is poor in contrast to [ReBr(CO)3(bpy)] (10). Probst et al. reported one of the first rhenium-based complexes for photocatalytic HER using triethanolamine (TEOA) as a sacrificial electron donor. After 9 h of irradiation in DMF solution, [Co(DH)2] displayed 150 TON (250 W, λ > 400 nm) in the presence of Re-based PS.30 However, the use of such Re-based PS was futile as it exhibited significant degradation over time.30 The substitution of the labile Br− with NCS− improved the stability of this Re-complex that resulted in better photocatalytic hydrogen production.31 Castellano and co-workers developed a cyclometalated Ir-based photosensitizer (11), along with cobaloxime, for photocatalytic H2 production in near neutral condition.23
. In one of the first examples, considerable photocatalytic H2 production was observed for the [Co(DH)2] complex in the presence of [Ru(bpy)3]2+-PS (7) following 1 hour of irradiation (TON ∼ 38 vs. PS). The aero-sensitivity and catalytic performance were improved with specific functionalization of the bipyridyl moiety at the 4,4′-position by methyl, nitrile27 or carbene28 groups.25 Platinum (Pt)-based PS and Pt-acetylide (8) allowed for the prolonged irradiation and presence of external water during an analogous experimental setup, which significantly improved the TON.29 The improvement in TON was continued with the use of an extended π-conjugated Pt-based PS (9).24 One of the highest initial-stage photocatalytic HER rates has been observed for [Ru(bpy)3]2+ among the cobaloxime-noble-metal PS dyad, although its long-term stability is poor in contrast to [ReBr(CO)3(bpy)] (10). Probst et al. reported one of the first rhenium-based complexes for photocatalytic HER using triethanolamine (TEOA) as a sacrificial electron donor. After 9 h of irradiation in DMF solution, [Co(DH)2] displayed 150 TON (250 W, λ > 400 nm) in the presence of Re-based PS.30 However, the use of such Re-based PS was futile as it exhibited significant degradation over time.30 The substitution of the labile Br− with NCS− improved the stability of this Re-complex that resulted in better photocatalytic hydrogen production.31 Castellano and co-workers developed a cyclometalated Ir-based photosensitizer (11), along with cobaloxime, for photocatalytic H2 production in near neutral condition.23
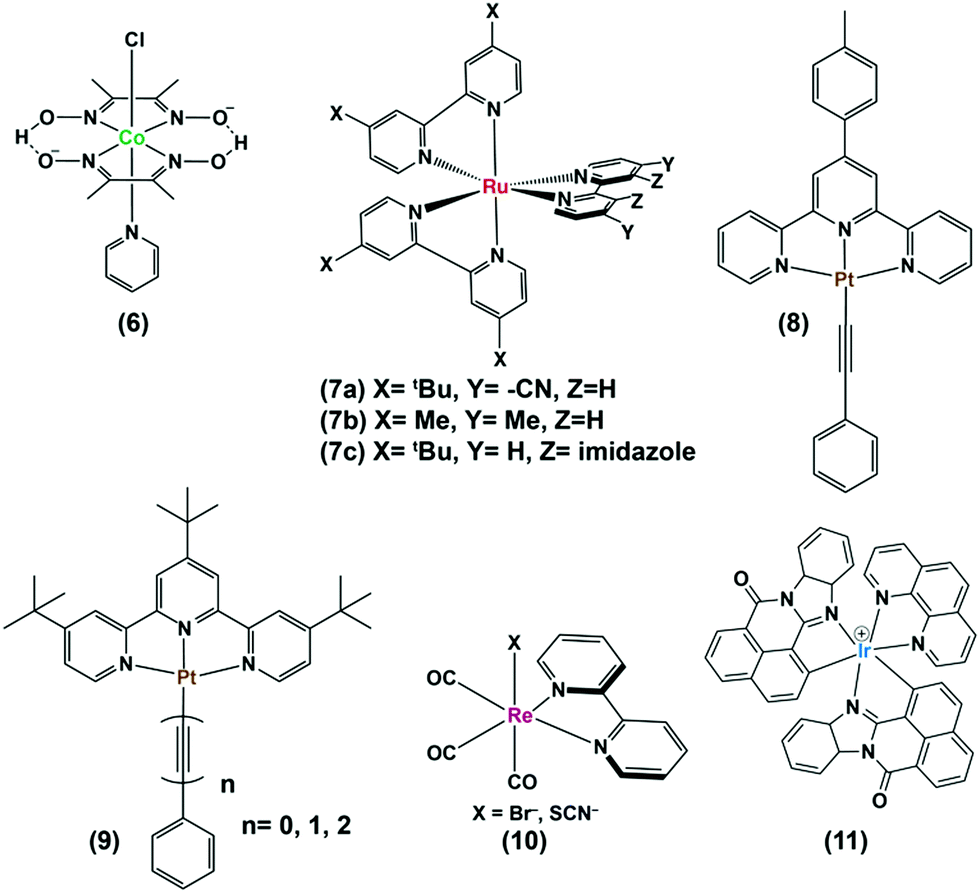 | ||
| Fig. 2 Structures of various noble metal-based photosensitizers (7–11) used with cobaloxime derivatives, such as (6) for photocatalytic hydrogen evolution in homogeneous condition. | ||
| Cbxa | PSb | Solvent/proton source | EDc | λ ex cut off (nm) | Irr. time (h) | TON (vs. PS) | Ref. |
|---|---|---|---|---|---|---|---|
| a Cbx: cobaloxime derivative. b PS: photosensitizer. c ED: electron donor. d DMT: N,N′-dimethyl-p-toluidine. e TEA: triethylamine. f AscH: ascorbic acid. g TEOA: triethanolamine. | |||||||
| (6) | (7) | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN/H2O :1 CH3CN/H2O |
DMTd | 452 | 10 | 91 | 23 |
| (2) | (11) | 2:1 CH3CN/H2O |
DMT | 452 | 10 | 352 | 23 |
| (2) | (7a) | Methanol | TEAe | 470 | 20 | 470 | 27 |
| (2) | (7b) | 9:1 CH3CN/H2O |
AscHf | 470 | 24 | 792 | 28 |
| (6) | (7c) | 1:1 CH3CN/H2O |
AscH | 400 | 1 | 214 | 25 |
| (6) | (8) | 3:2 CH3CN/H2O |
TEOAg | 410 | 10 | 400 | 29 |
| (6) | (9) | 1:1 CH3CN/H2O, pH 8.5 |
TEOA | 420 | 3.5 | 789 | 24 |
| (2) | ((10), X = Br) | DMF/AcOH | TEOA | 415 | 9 | 150 | 30 |
| (2) | ((10), X = NCS) | DMF/HBF4 | TEOA | 476 | 120 | 2000 | 31 |
Continuing the search for a stable photocatalytic system, the axial pyridine ligand was used as a linker to covalently connect noble metal-based photosensitizers.32–35 Fihri et al. prepared a supramolecular assembly, where a ruthenium-tris-di-imine PS was coupled to a cobaloxime core ((12–13), Fig. 3) through the axial pyridine. This modification positively shifted the CoII/I reduction potential by ∼80 mV, but it eventually slowed down in the longer run.36 After 15 hours of irradiation (λ > 350 nm), a TON of 103 (measured vs. cobaloxime catalyst) was observed in the presence of 100 equivalents of Et3N (electron donor) and Et3NHBF4 (proton source).33
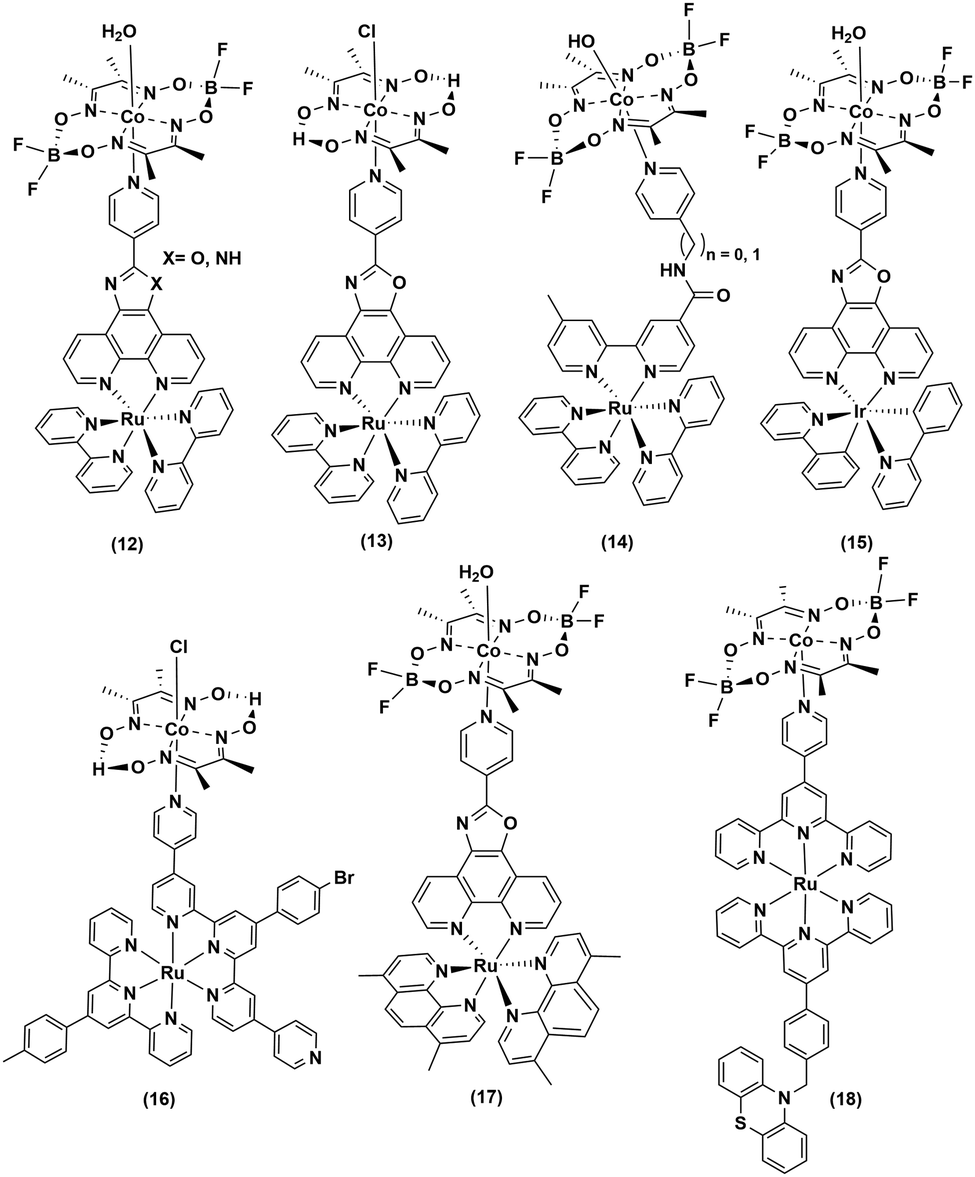 | ||
| Fig. 3 Noble metal-based photosensitizer–cobaloxime dyads for photocatalytic hydrogen evolution. | ||
Sun and co-workers improvised this strategy to incorporate an amide linker at a variable length between the axial-pyridine and PS ((14), Fig. 3). The presence of a methylene spacer between the linker and pyridine turned out to be a critical stabilizing factor, as it amplified the TON by ∼1.5 times compared to the control complex during a 4 h irradiation (Xe lamp, 500 W, λ > 400 nm) in acetone. It is believed that this unconjugated methylene spacer possibly impedes the back electron transfer from the reduced metal to the PS to improve the overall photocatalytic efficiency.34
The Ir-polypyridyl photosensitizer-based supramolecular PS–cobaloxime dyad (15) yielded a comparatively higher TON (∼140; after 8 h of irradiation in an acetone solvent with a Hg lamp (150 W, λ > 380 nm)).32 Analogous heteroleptic Ru(II)-photosensitizer-bound cobaloxime assemblies ((16–19), Fig. 3) were also developed to harvest the longer wavelengths due to their broad red-shifted emission.37 However, they exhibited poor photocatalytic HER (4 h irradiation, TON: 9) that was attributed to the low quantum yield and short-lived excited states of those heteroleptic PS-catalyst dyads.38 Further studies with such Ir-containing multi-component PS–cobaloxime assemblies ((20), Fig. 4) revealed low efficiency, albeit a higher catalytic turnover with prolonged irradiation.39,40 In a different approach, Mulfort and co-workers targeted the equatorial sites of the cobaloximes to append the light-harvesting Ru-PS component ((21–22), Fig. 4). Through this transformation, they were even able to identify the key photosynthetic charge-separated intermediate (CoI) for ∼26 ps.41
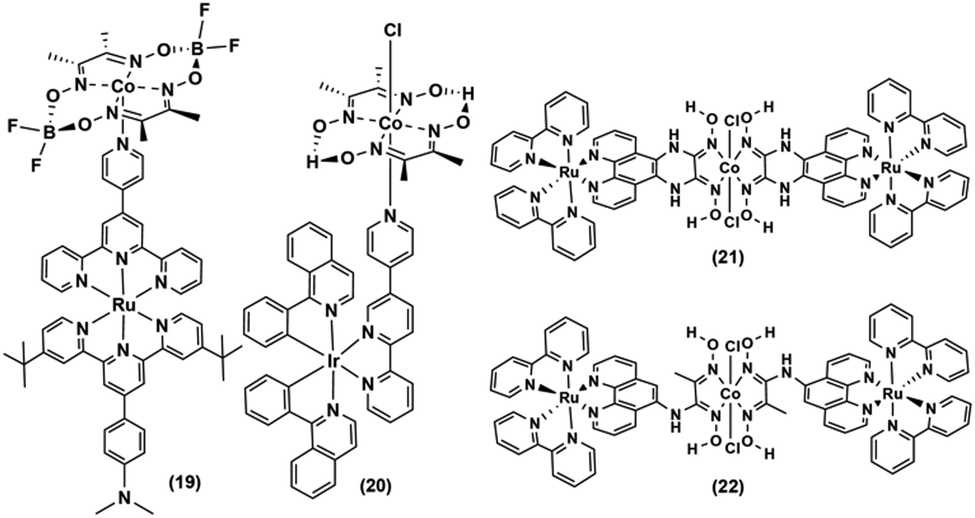 | ||
| Fig. 4 Various noble metal-based PS-axial- (19–20) and -equatorial-linked (21–22) cobaloximes. | ||
Most of the above-mentioned photocatalytic H2 production reactions, containing a noble-metal-based PS and cobaloxime catalyst, suffered a setback when water was added to the reaction medium. Eisenberg and his team utilized a mixture of Pt-based PS [Pt(ttpy)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)]ClO4 (ttpy = 4′-p-tolyterpyridine) and cobaloxime derivative [ClCo(DH)2(methylisonicotinate)] to demonstrate effective H2 production even in a 3:2 acetonitrile:water mixture (200 W mercury xenon lamp, λ > 410 nm).42
CPh)]ClO4 (ttpy = 4′-p-tolyterpyridine) and cobaloxime derivative [ClCo(DH)2(methylisonicotinate)] to demonstrate effective H2 production even in a 3:2 acetonitrile:water mixture (200 W mercury xenon lamp, λ > 410 nm).42
It can be summarized here that Ru- and Ir-based photosensitizers exhibited better photocatalytic HER in partnership with the cobaloxime catalyst, and the reactivity improved significantly when both PS and catalyst modules were connected via covalent linker (Table 2).
| Cbx–PSa | EDb | λ ex cut off (nm) | Irr. time | TON vs. PS | Ref. |
|---|---|---|---|---|---|
| a Cbx–PS: cobaloxime–photosensitizer conjugate. b ED: electron donor; solvents. c DMF. d Acetone. e Acetonitrile. | |||||
| (12) | TEAc | 350 | 4 h | 56 | 33 |
| (13) | TEAc | 350 | 4 h | 17 | 33 |
| ((14), n = 0) | TEAd | 400 | 8 h | 38 | 34 |
| ((14), n = 1) | TEAd | 400 | 8 h | 48 | 34 |
| (16) | TEOAc | 450 | 15 days | 764 | 38 |
| (15) | TEAd | 380 | 15 h | 210 | 32 |
| (17) | TEAd | 380 | 15 h | 103 | 32 |
| (19) | TEOAc | 525 | 2.5 h | 14 | 37 |
| (20) | TEOAe | 595 | 18 h | 180 | 39 |
Non-noble metal-based photosensitizers
The low abundance and high expense of noble metals compelled the researchers to explore the possibility of employing non-noble metal-based photosensitizers for photocatalytic H2 production with cobaloximes (Fig. 5 and 6). The presence of Mg-based chlorophylls and their active participation in the photo-harvesting process inspired the synthesis of magnesium (Mg)-based synthetic porphyrins. Following the similar synthetic methodology, zinc (Zn)-based porphyrin analogues were also prepared. Zinc porphyrins illustrated higher photocatalytic efficiency compared to the magnesium counterpart when they were appended with the cobaloxime via axial ligation ((23), Fig. 5). This higher efficiency is attributed to the possible pre-coordination of trimethylamine (TEA) to the zinc porphyrin that facilitates inner sphere electron transfer (ET) from TEA to PS.43 In a follow-up study, an amide spacer was included in the linker that quenched the electron transfer rate, causing the irradiation to slow down the overall photocatalytic HER activity by this modified Zn-porphyrin–PS–cobaloxime assembly ((24), Fig. 5).44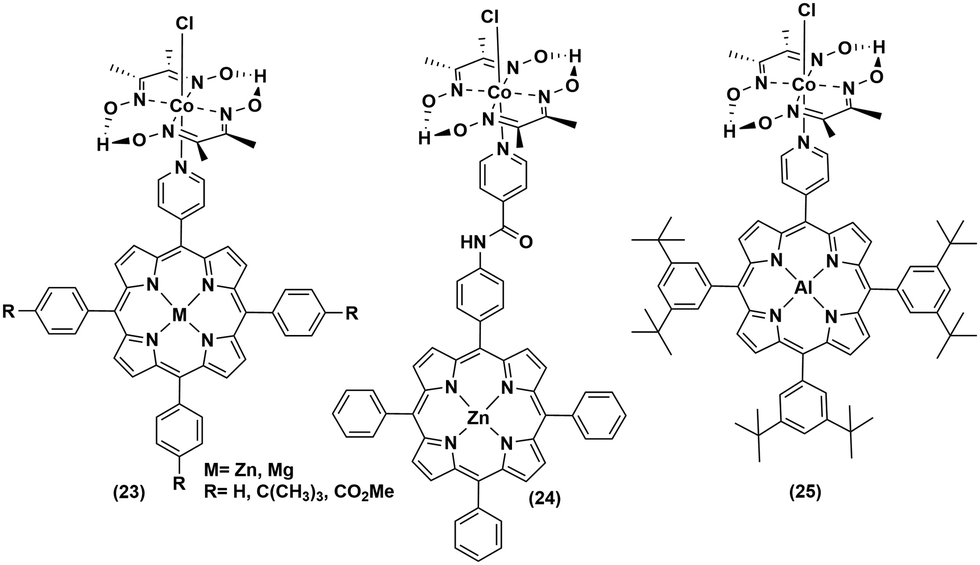 | ||
| Fig. 5 Non-noble metalloporphyrin-linked cobaloximes for photocatalytic hydrogen evolution. | ||
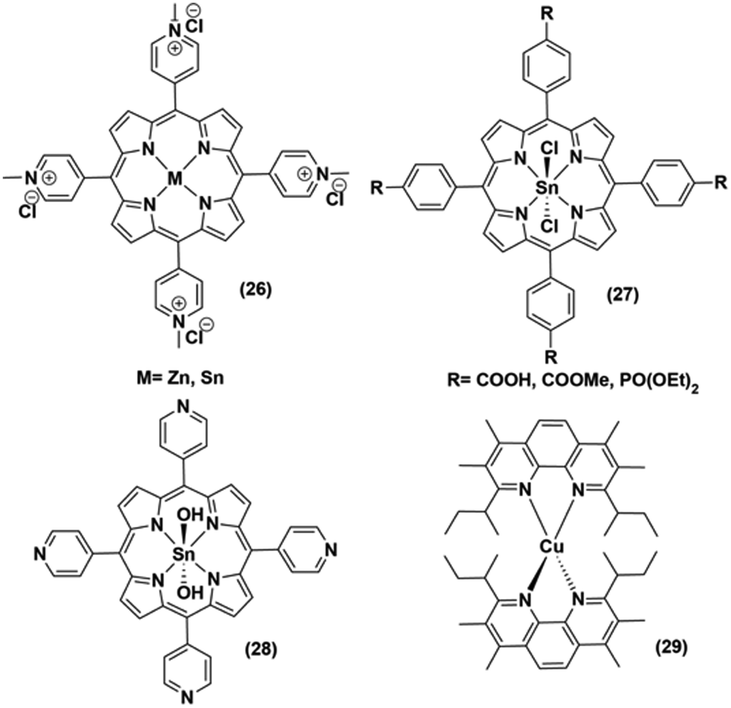 | ||
| Fig. 6 Non-noble metal-based PS used with various cobaloximes for photocatalytic hydrogen evolution. | ||
It is noteworthy that axial imidazole-linked cobaloximes display superior photocatalytic HER activity in comparison to axial pyridine-linked cobaloximes.45 Natali and co-workers developed in situ Al-porphyrin linked to the hydrogen-bridged cobaloximes ((25), Fig. 5) through the pyridyl group, which exhibited limited photocatalytic HER [TONmax = 352 (vs. PS, 175 W Xe-arc, λ > 400 nm).46 In a parallel study, tin-based porphyrins ((26–28), Fig. 6) were also probed for the role of PS under similar conditions. Interestingly, photocatalytic H2 production was observed only in the presence of the water-insoluble tin-porphyrin derivative [SnTPyP(OH)2] ((28), Fig. 6), but not with the water-soluble tin-porphyrin chloride [SnTmPyP] ((26), Fig. 6).47 Probst et al. reported a water-soluble Sn-porphyrin system ((27), Fig. 6) that was active as a PS for photocatalytic H2 production. However, its short-lived excited state reduced the overall catalytic efficiency.48 Later, several other zinc-porphyrin or -corrole chromophores were screened with cobaloximes in aqueous or aqueous–organic solvent mixtures.49,50 In recent years, the earth-abundant copper-based [Cu(I)(dsbtmp)2]+ (dsbtmp = 2,9-di(sec-butyl)-3,4,7,8-tetramethyl-1,10-phenanthroline) PS ((29), Fig. 6) was also developed. This Cu(I)–PS exhibited only moderate photocatalytic H2 production in the presence of cobaloxime (TON ∼ 35 vs. cobalt catalyst, λex 450 nm), although the photosensitizer retained activity over five days and six cycles of experiments under visible light.51 Analogously, tellurium (Te)-linked cobaloximes were also generated, but it showed negligible hydrogen evolution even in the presence of multiple light-harvesting components.52
Hence, it can be mentioned here that despite the evolution of a variety of non-noble metal-based photosensitizers, they were unable to match the excellent photo-driven cobaloxime-catalysed HER activity showcased by noble metal (especially Ru and Ir)-based PS assemblies (Table 3). However, the photo-bleaching of these inorganic photosensitizers under prolonged irradiation has cast serious doubts on their long-term applications.
| Catalyst | PS | λ ex cut off (nm) | Irr. time (h) | TON vs. PS | Ref. |
|---|---|---|---|---|---|
| Solvent/electron donor.a THF–H2O (8:2)/TEA.b 1:1 CH3CN–H2O/TEOA.c 7:3 acetone–H2O/ascorbic acid.d 1:1 CH3CN–H2O/DMT.e pH 8.5/TEOA. |
|||||
| ((23), M = Zn)a | 400 | 5 | 22 | 43 | |
| ((23), M = Mg)a | 400 | 3 | 2 | 43 | |
| (24) | 400 | 5 | 5 | 44 | |
| [ClCo(DH2)2(pyrCN)] | ((26), M = Zn)b | 440 | 11 | 77 | 22 |
| [ClCo(DH2)2(pyrCooH)] | [Zn(TmPyP)]b | 440 | 26 | 40 | 22 |
| [ClCo(DH2)2(pyr)] | [Zn(TmPyP)]b | 440 | 63 | 320 | 22 |
| [ClCo(DH2)2(pyrSH)] | [Zn(TmPyP)]b | 440 | 43 | 425 | 22 |
| [ClCo(DH2)2(pyrMe)] | [Zn(TmPyP)]b | 440 | 40 | 443 | 22 |
| [ClCo(DH2)2(Im)] | [Zn(TmPyP)]b | 440 | 50 | 565 | 22 |
| [ClCo(DH2)2(ImMe)] | [Zn(TmPyP)]b | 440 | 50 | 1135 | 22 |
| (2) | [Zn(TmPyP)]b | 440 | 23 | 290 | 22 |
| (6) | (28) | 440 | 140 | 150 | 47 |
| [ClCo(DH2)2(pyrCOOH)] | [Sn(TPyP(OH)2)]b | 440 | 100 | 40 | 47 |
| [ClCo(DH2)2(ImMe)] | (27, R = COOMe)b | 440 | 48 | 128 | 58 |
| [ClCo(DH2)2(ImMe)] | [27, R = PO(OEt)2]b | 440 | 48 | 48 | 58 |
| (25) | 400 | 2 | 280 | 46 | |
| (6) | (29) | 452 | 20 | 70 | 51 |
| (6) | ((27), R = COOH)e | 390 | 20 | 20 | 48 |
Organic photosensitizer
The plausible role of organic dyes as a photosensitizer was also probed in detail in the pursuit of an artificial photosynthetic assembly. Various organic dye molecules containing diverse categories of functionalities, such as boron dipyrromethane (BODIPY),53 Rose Bengal,54 Rhodamine,55 Acriflavin,56 and Eosin,57 represent the face of organic dye instigated photocatalytic HER research using cobaloxime derivatives as a diagnostic tool (Fig. 7). Analogous to their metal-based PS counterpart, the organic dye molecules were also utilized in two distinct formats for cobaloxime-derived photocatalytic HER: (i) untethered free organic dye PS and cobaloxime in solution, and (ii) covalently linked organic dye–cobaloxime dyads. We have reviewed the consistent growth in either of those approaches in the following sections.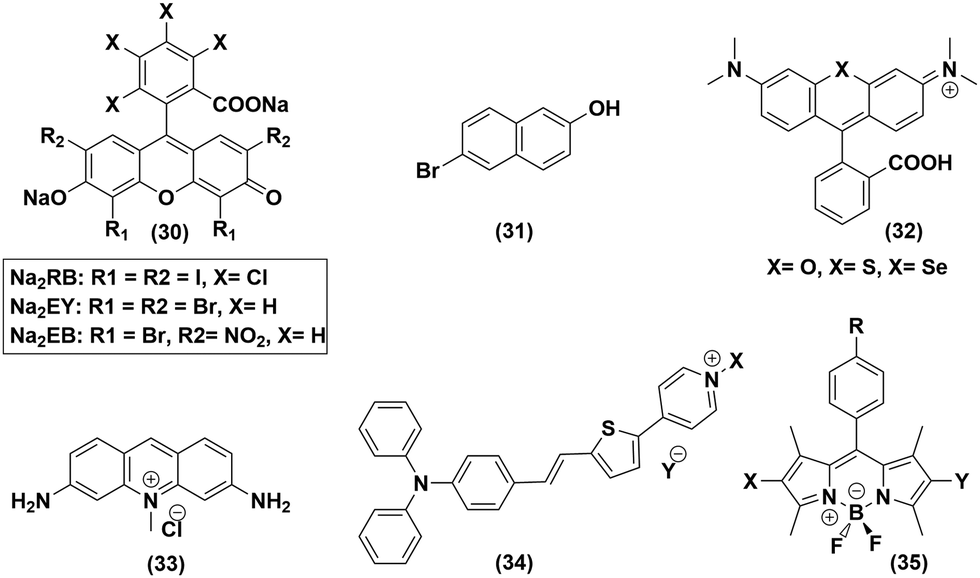 | ||
| Fig. 7 Various organic dye molecules used in the homogenous solution phase, along with cobaloximes, for photocatalytic hydrogen evolution. | ||
Untethered free organic dye as photosensitizer
Xanthene-based dyes are one of the widely explored photoactive molecules, and were chosen for the initial photocatalytic studies to play the role of a PS. When the basic xanthene skeleton was mixed along with cobaloxime in solution, it failed to produce any H2 under illumination. A close look into this failed experiment revealed that the excited state of the PS should be a long-lived one to support such a bimolecular photocatalytic reaction. The photophysical properties of xanthene molecules suggest that the lifetime of the photo-excited state can be prolonged by facilitating the intersystem crossing (ISC) process with the introduction of heavy atoms like halogens. With the use of Eosin and Rose Bengal (RB) as PS ((30), Fig. 7), the successful photocatalytic H2 evolution validated that hypothesis.59 Complex 6 exhibited TON = 900 (vs. catalyst, pH 7, 5% TEOA, 1:1 CH3CN/H2O, λ > 410 nm, 12 h irradiation, excess ligand) in the presence of Eosin-Y (EY2−). However, this photocatalytic activity was hampered whether in acidic or basic medium.57 The BF2-bridged cobaloximes [CoII(DBF2)2] exhibited better stability compared to the hydrogen-bonded cobaloximes when these xanthene derivatives were used as PS. The annulated cobaloxime [CoII(DBF2)2] displayed a TON of 32 with maximum activity in basic medium (pH 10) in the presence of Rose Bengal PS (500 W Xe lamp, 5 h irradiation, 10% TEOA, λ > 400 nm).54 The photo-activated proton movement in 6-bromo-2-napthol (31) was also employed for cobaloxime-generated photocatalytic H2 production, which highlights the versatility of the cobaloxime core in screening different PS sources.60 The photocatalytic HER activity was boosted when the O atom in the heterocycle of xanthine framework was replaced with heavier chalcogens S and Se ((32), Fig. 7). The S- and Se-substituted Rhodamine (RB) exhibited excellent cobaloxime-catalysed photo-driven H2 production with TON values of 1700 and 5500, respectively (5% TEOA, 1:1 CH3CN/H2O, pH 7, 520 nm LED, 0.15 W).55 The use of acriflavine (33) and pyridinium-based donor–π–acceptor assemblies (34) as the photosensitizer, along with cobaloxime, also exhibited photocatalytic H2 production.56,61
The variable combinations of cobaloxime and xanthine-based dyes allowed the researchers to have a glimpse of the importance of the sacrificial electron donor on the photocatalytic HER (Table 4). It was found that, in the presence of triethanolamine (TEOA), the efficiency of photo-generated H2 production follows the trend: EY2− (alcohol) > EY2− (water) > HF (halogenated fluorescein) > EB2− (Eosin-Blue) >RB2−,62 while it was altered when triethylamine (TEA) donated an electron: EY2− > RB2− > EB2−.63 Hence, Eosin-Y (EY2−) and TEOA turned out to be one of the best PS-sacrificial electron donor recipes for cobaloxime-derived photocatalytic HER, where a TON of 562 (5% TEOA, λ > 420 nm, 1:1 CH3CN/H2O) was observed. The halogenated aza-/meso-BODIPY dyes ((35), Fig. 7) represent another section of organic dyes that were extensively examined with cobaloximes. BODIPY PS typically operates in neutral or near basic-neutral media with a maximum photocatalytic efficiency at pH 8.5.64,65 Recently, Sun and co-workers incorporated an 8-hydroxyl-quinoline group in the meso-position of iodinated BODIPY, which was able to reduce the proton, even under acidic medium, in the presence of complex (6) and ascorbic acid as the catalyst and electron donor, respectively.66
| Catalyst | PS | λ ex cut off (nm) | Irr. time (h) | TON vs. PS | Ref. |
|---|---|---|---|---|---|
| Solvent/electron donor.a 2:1 CH3CN–H2O/TEA.b 1:1 CH3CN–H2O/TEOA.c 3:1 DMF–H2O/TEOA.d 3:2 CH3CN–H2O/TEOA. |
|||||
| (6) | RB2−a |
400 | 5 | 321 | 54 |
| (6) | ((32), X = Se)b | 520 | 5 | 3737 | 55 |
| (6) | (33) | 420 | 2 | 110 | 56 |
| (6) | EY2−b |
450 | 30 | 220 | 57 |
| (6) | RB2−b |
450 | 5 | 540 | 59 |
| (3) |
(31)
|
226 | 0.75 | — | 60 |
| (6) | ((35), X = Y = I, R = COOH)d | 420 | 9 | 132 | 64 |
| Bis-pyridyl-propane-[Co(DH2)2]2 | EY2−b |
420 | 2 | 562.5 | 62 |
| 4-4′-bpy-[Co(DH2)2]2 | EY2−b |
420 | 10 | 160 | 63 and 69 |
| [Cl-Co(DH2)2(pyrm-EtCOOH)] | EY2−b |
400 | 5 | 444 | 67 |
In a similar manner, the cobaloxime motif was also varied to enhance the photo-driven H2 production in the presence of an identical PS (EY2−). Initially, carboxylic acid groups (–COOH) were appended at the variable position of the axial pyridine ligand of the complex (6) molecule. The detailed studies displayed a better catalytic response from a meta-carboxylic acid bound complex compared to the para-substituted analog. On the other hand, the inclusion of ester functionality reduced the photocatalytic HER to emphasize the possible role of these peripheral functionalities in proton movement and electron distribution during photocatalysis.67 Recently, our group also observed analogous results when such protic group substitution was performed on the axial imidazole (Im) ligand of the [ClCo(DH2)2(Im)] molecule. The presence of multiple interactive basic functionalities and their fluxional dynamics around the cobalt core transpired to be an essential modulating factor for the photocatalytic activity of cobaloximes.68 Additionally, the use of a self-assembled multinuclear cobaloxime62 or inclusion of a 4,4′-bipyridine ligand to the cobaloxime core exhibited improved photocatalytic efficiency in comparison to the unsubstituted mononuclear cobaloxime motif.69
However, none of these untethered combinations of organic dye and cobaloxime derivatives were primed for inclusion in large-scale usage photo-harvesting procedures due to their long-term instability, especially in water-blended media.
Covalently linked organic dye–cobaloxime dyad
In an attempt to improve the long-term stability and raise the photocatalytic efficiency, the organic dyes were covalently anchored with cobaloxime to generate a series of organic PS-catalyst assemblies (Fig. 8 and Table 5). First, BODIPY dyes were tethered to the axial position of [Co(DH2)2] through the meso-positioned pyridyl group ((36–38), Fig. 8). Even with the variation of the electron-donating and -withdrawing substituents on the BODIPY motif, these adducts demonstrated poor reactivity.70 However, the inclusion of heavy atoms like halides was able to enhance the photocatalytic H2 production to reach a TON of 85 (5 h Irr, 4:1 CH3CN/H2O, pH 8.5).71 The efficiency of such light-activated HER improved further with the inclusion of thienyl functionality to the BODIPY–cobaloxime dyad.72 Despite this initial success, the instability of these adducts in aqueous blended solvents continued to impede their durability.73 The reactive Co–N(pyridyl) bond is believed to be the primary reason for the instability of the BODIPY–cobaloxime adducts, especially under irradiation.74
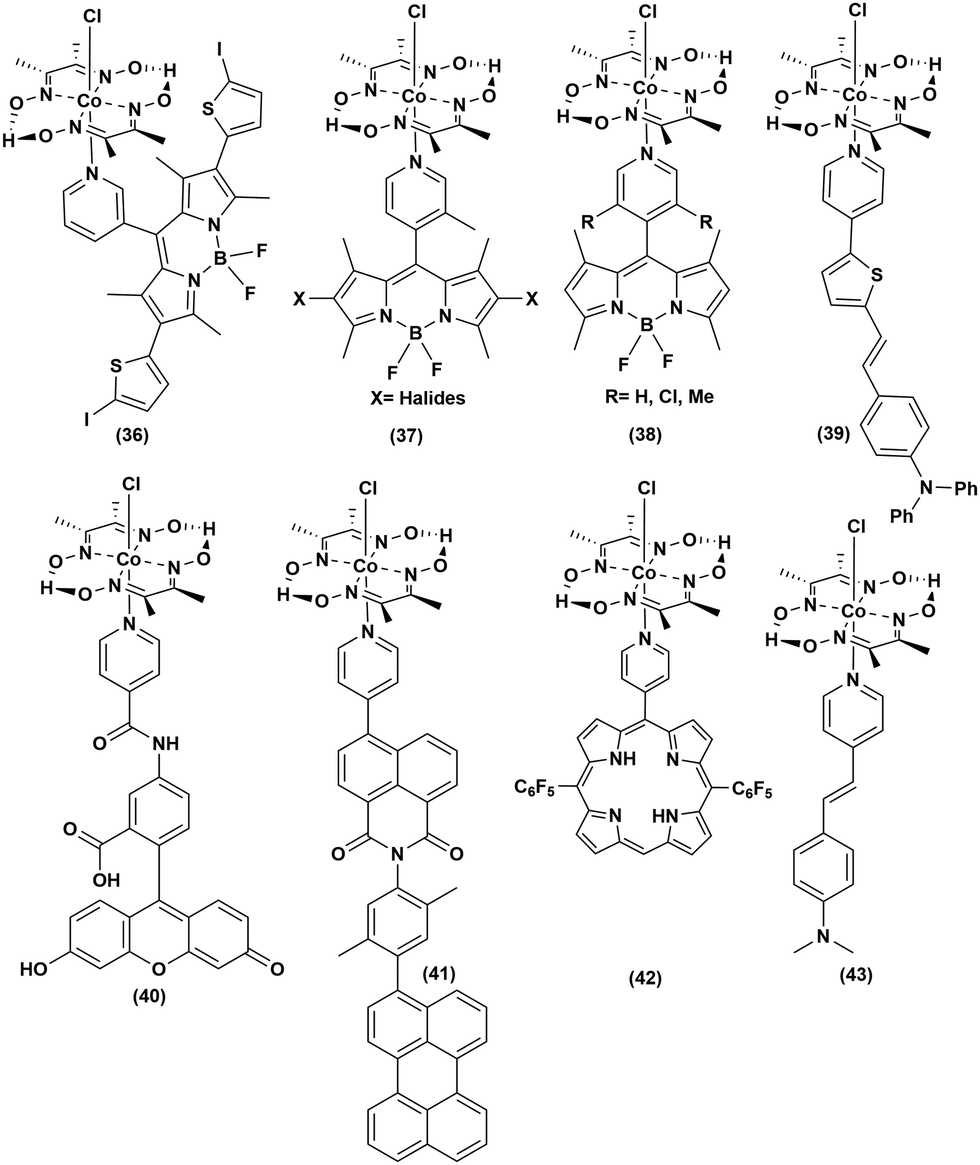 | ||
| Fig. 8 Metal-free organic dye-linked photosensitizer–cobaloxime conjugates for photocatalytic hydrogen evolution. | ||
| Catalyst–PS conjugates | Solvent | EDa | λ ex cut off (nm) | Irr. time | TON vs. PS | Ref. |
|---|---|---|---|---|---|---|
| a ED: electron donor. b DCM: dichloromethane. | ||||||
| Bis-thienyl-BODIPY-Co(DH2)2 | 8:2 CH3CN–H2O |
TEA | 420 | 8 h | 5.6 | 53 |
| ((37), X = I) | 3:2 CH3CN–H2O |
TEOA | 525 | 17 h | 30.9 | 71 |
| (36) | 4:1 CH3CN–H2O, pH 8.5 |
TEOA | 420 | 10 h | 73 | 72 |
| 3-pyr-meso-BODIPYIodine-Co(DH2)2 | 24:1 CH3CN–H2O |
TEOA | 420 | 5 h | 85 | 73 |
| (39) | 3:1 CH3CN–MeOH |
TEOA | 455 | 9 days | 122 | 61 |
| (40) | 24:1 CH3CN–H2O |
TEOA | 520 | 28 h | 32 | 75 |
| (41) | DCMb | TEA | 400 | 2 h | — | 76 |
The activity was improved slightly when the triphenylamine-vinylthiophene-pyridine chain ((39), Fig. 8) was used as a donor–π-linker–acceptor triad.61 Following the footsteps of BODIPY, Eisenberg et al. linked amido-fluorescein to the axial position of cobaloxime ((40), Fig. 8). This amido-fluorescein–cobaloxime adduct exhibited approximately three-fold improvement in photocatalytic HER activity compared to the untethered components under analogous reaction conditions.75 The Wasielewski group developed a rational model of a donor–linker–acceptor-[Co(DBF2)2] cobaloxime assembly framework, without the presence of any heavy atom, to improve the transfer of the photoactivated electron. In this strategy, they synthesized a molecule containing perylene, xylene and pyridyl-substituted 1,8-naphthalimide as a donor, bridge and acceptor, respectively, along with a [Co(DBF2)2]cobaloxime core to achieve moderate photocatalytic HER activity ((41–42), Fig. 8).76 In a recent study, our group simplified the donor–linker–acceptor chain by using a pyridinyl derivative of a stilbene motif ((43), Fig. 8). Although this [ClCo(DH2)2pyridnyl-stillbene] adduct was able to exhibit photocatalytic H2 evolution even under natural sunlight, the overall photocatalytic efficiency and long-term aqueous stability remained poor.77
Nanomaterial-based photosensitizer
An ideal photocatalytic assembly should exhibit the following properties to fulfill the demands of harsh practical applications: (i) the PS component should be stable under prolonged irradiation in aerobic conditions, (ii) the catalyst module should be able to produce H2 from the ubiquitous source of protons, i.e., water. Hence, the generation of a robust photocatalytic assembly, operational in pure aqueous media, remains the final frontier for their practical applications. As mentioned in the earlier sections, the rational utilization of the cobaloxime motif allowed the scientists to screen an array of photosensitizers, ranging from inorganic to organic dye molecules, in the pursuit of an optimized photocatalytic H2 production assembly. Although some of those dye molecules exhibited considerable H2 evolution in tandem with cobaloxime, the overall effectivity of such combinations (in either tethered or untethered forms), especially in the presence of water, were low due to the long-term instability of the dye molecules. In this scenario, the semiconductor materials emerged as a possible solution for this conundrum owing to their robust photophysical properties, where it can play the role of a charge mediator between PS and catalyst, while stabilizing the dye molecules under photocatalytic conditions.Reisner et al. employed the resilient TiO2 nanoparticle (TiO2 nP) as a mediator for immobilizing both cobaloxime [ClCo(DH)2(pyridyl-4-hydrophosphonate)] (CoP) and [Ru(bpy)2(bpy-phosphonate)2+] (RuP) PS via phosphonate–TiO2 binding. The CoP–TiO2 nP–RuP combination exhibited significant photocatalytic H2 production [400 mmol H2 h−1 (g TiO2)−1] in neutral aqueous conditions (pH 7, in the presence of TEOA) (Fig. 9A). Here, the conduction band of the TiO2 nP was believed to facilitate the fast movement of charge carriers from RuP to the CoP motif. Despite expanding the reactivity in pure aqueous solution, the photocatalytic H2 evolution by this assembly slows down after 2 hours, and completely ceases after 8 hours under continuous irradiation.78
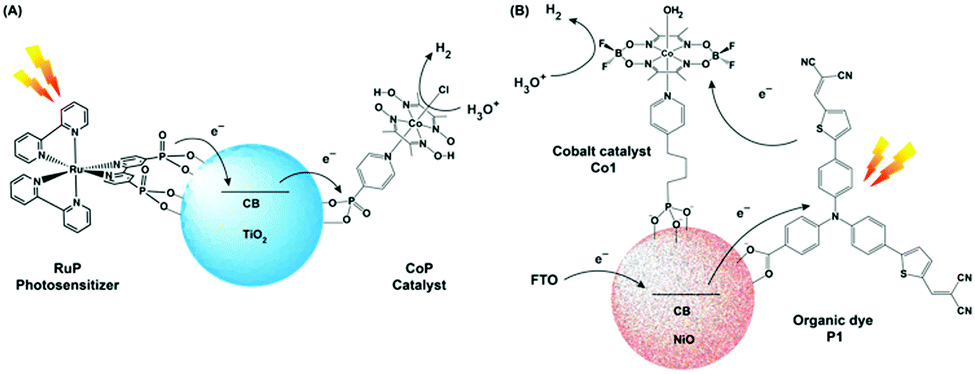 | ||
| Fig. 9 (A) Schematic representation of photocatalytic H2 production TiO2 NPs containing a covalently attached cobalt catalyst (CoP) and a ruthenium photosensitiser (RuP). (B) Representative figure of the photocathode containing organic dye P1 and cobaloxime catalyst Co1 on the NiO surface. The figures are redrawn from ref. 78 and 79 respectively. | ||
Licheng Sun and team designed an organic dye-sensitized photoelectrochemical (PEC) cell that can perform water splitting under neutral aqueous (pH 7) conditions using visible light. This PEC cell consisted of two parts: (i) photoanode (for water splitting), and (ii) photocathode (for hydrogen generation). A cobalt catalyst Co1 [Co(DBF2)2(H2O)2] was directly connected to the semiconductor-based photocathode (p-type NiO) surface with phosphoric acid linker along with an organic dye P1, which acted as a photosensitizer (Fig. 9B). This photocathode produced a decent amount of hydrogen upon irradiation.79 However, scaling up such NiO film is an issue, which prevented a higher amount of catalyst and dye loading for improved H2 production. The poor charge carrier property of the NiO surface also negates their long-term usage.80 Recently, Artero and co-workers have improved the charge transfer between the semiconductor and catalyst by introducing a Ru-diacetylide-spacer between them.81 In a parallel set of studies, photoactive nanomaterials were used that can directly instigate light-driven HER with the cobaloxime catalyst. For this purpose, a range of materials were used, including graphite-like C3N4 (cobaloxime bound via π–π stacking through a pyrene tail),82 CdS quantum dot (containing cobaloxime film on its surface),83 and the azine-linked N2-based covalent organic framework (COF) (cobaloxime present in the cavity).84 All of these materials exhibited moderate photocatalytic HER activity mostly in the organic/water mixture.
Turner and co-workers further modified the photoelectrode design, and used the atomic layer deposited (ALD) TiO2-modified p-GaInP2 for this purpose. In a sequential synthetic step, cobaloxime [ClCo(DH)2(pyr-COO−)] was immobilized on the TiO2 terminal of the photoelectrode surface via carboxylate functionality. This modified electrode illustrated excellent photo-electrocatalytic H2 production activity (TON 139000 and average TOF of 1.9 s−1) during a 20 h period of 1 sun illumination in basic aqueous media (pH 13). As mentioned earlier, this system required the push of electrons from the electrode surface (which was held at 0 V vs. RHE), and its reactivity was limited to strong alkaline conditions. The photocurrent density for this assembly decreased rapidly (∼50%) over the first 4 hours of the experiment to raise its long-term instability issue.85 The detachment of the cobalt catalyst from the TiO2 surface is believed to be the possible reason for this instability.86
In this section, we have established that the presence of a semiconductor surface can play two crucial roles for cobaloxime-based photocatalytic H2 production reactions. First, the surface immobilization of PS and catalyst moieties ensure that they remain in close distance, which is essential for the rapid charge transfer process following photo-excitation. Second, the electroactive semiconductor surface can be utilized for modulating the potential of the surface to drive the photocatalytic reactions in the desired direction. The second factor even allowed us to probe the otherwise unexplored photocatalytic water oxidation pathway by a cobaloxime derivative [ClCo(DH)2(pyr-cysteamine)], which was immobilized on the TiO2–gold nanomaterial surface.87
Electrocatalytic HER activity by cobaloximes
As mentioned earlier, cobaloxime can act as a functional model for the hydrogenase enzyme, where the electron-transporting [Fe4S4] clusters can be mimicked by an electrode surface (Fig. 1B). The catalytic efficiency of the cobaloxime derivatives can be monitored as a function of the applied potential on the electrode surface (i.e., the energy of the electron) and the resultant catalytic current (that is directly proportional to the catalytic rate).88–90 Utilizing the unique structure of the cobaloxime core, researchers have probed the effect of electron-donating/-withdrawing substituents present in the primary coordination sphere and the influence of peripheral proton network via electrochemistry experiments. Such electrocatalytic studies have been performed on cobaloxime molecules in two different conditions: homogeneous and heterogeneous. In homogeneous conditions, the catalytic molecule and substrate (H+ in the case of H2 production) remain in the solution phase, and it interacts with the solid electrode surface in its electrochemical double layer. On the other hand, the catalyst is immobilized on the electrode surface during the heterogeneous study, while only the substrate stays in the solvent phase. In the following sections, the results from either set of experiments have been analyzed. It is worth mentioning that analogous nickel dimethylglyoximato complexes exhibited moderate HER activity in organic solvents, while they degraded into nanoparticles in aqueous solution under a reducing potential.91Homogeneous electrocatalytic HER
Instability of the basic [Co(DH2)2] framework led to the development of an air- and moisture-stable N-axial pyridine cobaloxime system. [ClCo(DH)2(pyr)] typically displays a reversible (CoII/I) and an irreversible (CoIII/II) redox reduction feature in dry organic media. Irreversible CoIII/II is related to the loss of an axial ligand in the CoII state. The addition of an acid (Et3NH+Cl−) in the solution transforms the reversible CoII/I signal into an irreversible one, and the reductive current continues to increase with an increasing amount of acid.17 Complementary bulk electrolysis (i.e., chronocoulometry) and gas chromatography experiments identified the enhanced reductive response as catalytic hydrogen generation. Complex (6) exhibited 85–100% faradaic efficiency (FE) during this electrocatalytic HER during the 2.5 h of bulk electrolysis in dichloromethane (graphite electrode, −0.9 V vs. Ag/AgCl).92The initial modifications to the cobaloxime core were executed on the equatorial motif. The substitution of the methyl groups of the dimethylglyoxime moiety with phenyl groups not only reduced the catalytic HER response, but also increased the overpotential requirement (the difference between the applied potential and standard reduction potential for H2 evolution at the experimental conditions; 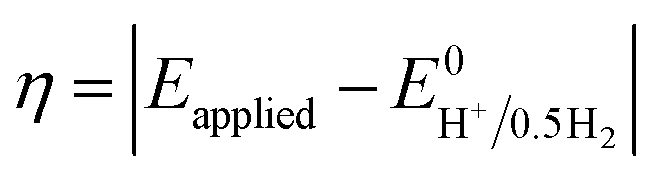 ).93 Interestingly, the overpotential requirement value was lowered when the hydrogen-bonded oxime network was replaced by the BF2 groups in [(CH3CN)2CoII(DBF2)2] (−0.28 V vs. SCE in CH3CN).18 However, this difluoroboryl annulated complex demonstrated reduced HER activity, and it required the presence of a stronger acid for electrocatalytic H2 production.94 As the axial halide ligands are labile in nature, especially in the reduced oxidation states of cobalt, the axial pyridine gained more attention for axial ligand modification. A series of diverse functionalities (such as amine, alcohol, ester, carboxylic acid, thiol, phosphate, alkyl, halide) have been introduced in the axial pyridyl motif to generate a library of axial-pyridine modified cobaloximes (Fig. 10). Reisner and co-workers have developed an electrochemical method, known as “Electrochemical Molecular Catalyst Screening” or EMOCS, for studying the electrocatalytic behaviour of such in situ generated pyridyl cobaloximes prior to their actual synthesis. This EMOCS method turns out to be a user-friendly screening method for comparing the axial ligand effect on the HER activity of cobaloxime core in analogous conditions.95 The detailed electrochemical results from these studies revealed a unique structure–function relationship for the axial-pyridine modified cobaloximes. The electron-donating substituents improved the electrocatalytic rate for the cobaloximes, but at the expense of the catalytic efficiency (overpotential also increases). In contrast, the electrocatalytic H2 production rate was reduced in the presence of an electron-withdrawing group in the axial pyridine, while making it an efficient catalyst (overpotential also decreases). Inclusion of the phosphate derivative in the axial ligand further improves the oxygen-tolerance of the cobaloxime core. However, it exhibited an acute CO poisoning effect.96,97 The bimolecular cobaloximes, containing an equatorial octamethylene unit, were also prepared via the equatorial BF2 groups as the bridging ligand. However, no significant enhancement in the electrocatalytic performance was noticed in comparison to the mononuclear cobaloxime.88
).93 Interestingly, the overpotential requirement value was lowered when the hydrogen-bonded oxime network was replaced by the BF2 groups in [(CH3CN)2CoII(DBF2)2] (−0.28 V vs. SCE in CH3CN).18 However, this difluoroboryl annulated complex demonstrated reduced HER activity, and it required the presence of a stronger acid for electrocatalytic H2 production.94 As the axial halide ligands are labile in nature, especially in the reduced oxidation states of cobalt, the axial pyridine gained more attention for axial ligand modification. A series of diverse functionalities (such as amine, alcohol, ester, carboxylic acid, thiol, phosphate, alkyl, halide) have been introduced in the axial pyridyl motif to generate a library of axial-pyridine modified cobaloximes (Fig. 10). Reisner and co-workers have developed an electrochemical method, known as “Electrochemical Molecular Catalyst Screening” or EMOCS, for studying the electrocatalytic behaviour of such in situ generated pyridyl cobaloximes prior to their actual synthesis. This EMOCS method turns out to be a user-friendly screening method for comparing the axial ligand effect on the HER activity of cobaloxime core in analogous conditions.95 The detailed electrochemical results from these studies revealed a unique structure–function relationship for the axial-pyridine modified cobaloximes. The electron-donating substituents improved the electrocatalytic rate for the cobaloximes, but at the expense of the catalytic efficiency (overpotential also increases). In contrast, the electrocatalytic H2 production rate was reduced in the presence of an electron-withdrawing group in the axial pyridine, while making it an efficient catalyst (overpotential also decreases). Inclusion of the phosphate derivative in the axial ligand further improves the oxygen-tolerance of the cobaloxime core. However, it exhibited an acute CO poisoning effect.96,97 The bimolecular cobaloximes, containing an equatorial octamethylene unit, were also prepared via the equatorial BF2 groups as the bridging ligand. However, no significant enhancement in the electrocatalytic performance was noticed in comparison to the mononuclear cobaloxime.88
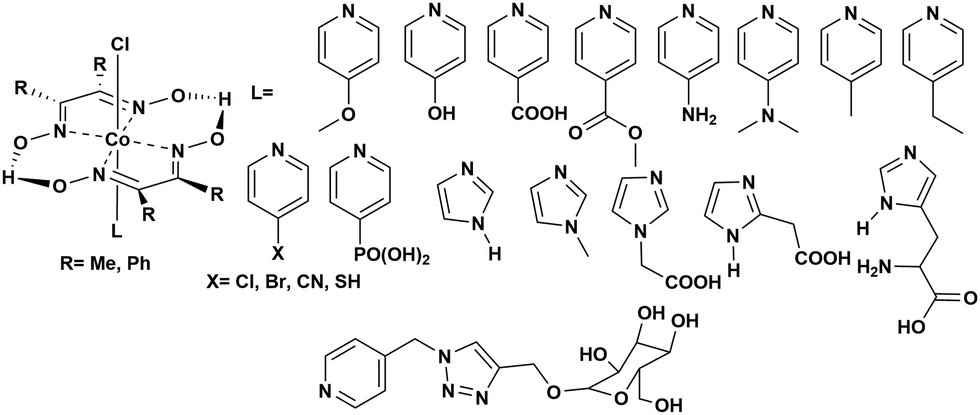 | ||
| Fig. 10 N-Axial pyridine cobaloxime derivatives used for the study of the electrocatalytic hydrogen evolution reaction. | ||
All of the cobaloxime derivatives discussed so far in this section were screened in an organic solvent due to their poor aqueous solubility. Zhang et al. introduced a glucose-functionalized pyridine motif to the cobaloximes to improve the water solubility. This modification produced water-soluble cobaloximes that operated under a neutral aqueous medium. However, they lost their catalytic properties as the acidity of the solution increased (pH ≤ 5).98 This loss of activity under acidic conditions is attributed to the cleavage of the oxime-derived hydrogen-bonding network, which acts as the primary protonation site during the electrocatalytic cycle. Hence, the activity of cobaloximes, even in acidic conditions, can be revived with the inclusion of an alternative protonation site. The introduction of an enzyme-inspired outer coordination sphere around the cobaloxime core allowed the presence of such a secondary protonation site that will be discussed in detail in a following section.99
Heterogeneous electrocatalytic HER
The cobaloxime motif was immobilized on variable electroactive surfaces in an attempt to improve its electrocatalytic performance compared to the homogeneous conditions. Reisner and co-workers led the research in this front and developed a cobaloxime-meso-ITO film (via phosphonate group) on an ITO electrode, which exhibited H2 production at a moderate overpotential. However, the cobaloxime bound on this mesoporous electrode degraded over time.100 They improved the stability of the surface-bound cobaloxime by using multiwall carbon nanotube (mw-CNT) material. Multiple cobaloxime molecules containing pyrene tails were anchored on the mw-CNT surface through π–π interactions.101 Artero et al. further improvised this approach and prepared a nanohybrid of mw-CNT and cobaloxime using a self-assembled polymeric amphiphile. This amphiphile contained diacetylene (DA) as a hydrophobic tail and a pyridine (pyr) moiety as the polar head, which was directly coordinated to the cobaloxime core (Fig. 11A). A glassy carbon electrode surface was modified with this cobaloxime–CNT hybrid that exhibited 110–120 turnovers for electrocatalytic HER in a neutral aqueous medium. However, this surface-modified catalyst suffered from degradation after 30 min under electrolytic conditions as the unstable cobaloxime leached out from the CNT surface.102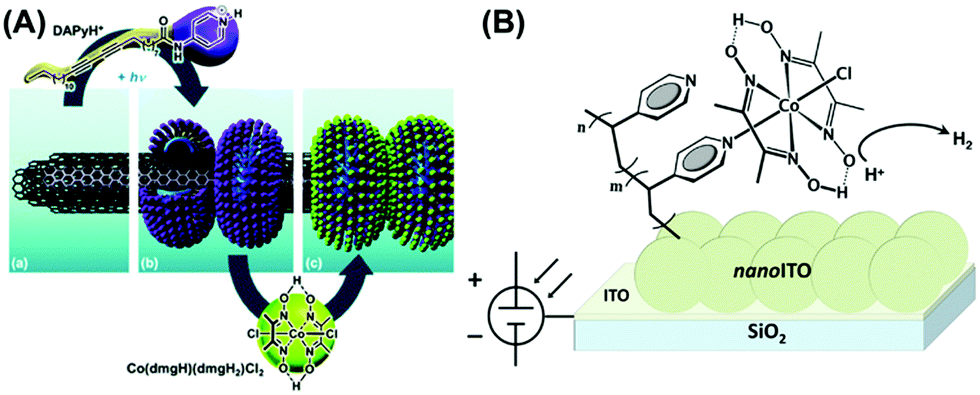 | ||
| Fig. 11 (A) The self-assembled structure of polymeric amphiphile (DApyr)–cobaloxime–CNT nanohybrid, exhibiting (a) the layers of multi-walled carbon nanotube (CNT), (b) nano-rings generated by the polymeric amphiphile, and (c) the cobaloxime-functionalized nano-hybrid. Reproduced from ref. 102 with permission from The Royal Society of Chemistry.102 (B) The cobaloxime–PVP polymer-immobilized nano-ITO Electrode surface. Reproduced from ref. 103 with permission from © American Chemical Society, 2016.103 | ||
Moore and co-workers studied the photoelectrocatalytic behaviour of a cobaloxime catalyst grafted on a p-type gallium phosphide (100) semiconductor via polyvinylpyridine (PVP) ligands. This assembly was active for photocatalytic HER activity, which was sensed via electrochemistry.104 They developed the next generation of such modified electrodes by immobilizing cobaloximes on the nanostructured indium tin oxide (nano-ITO) surface via in-built polyvinylpyridine (PVP) ligands (Fig. 11B). This nano-ITO was employed as a working electrode for the hydrogen evolution reaction in phosphate buffer (pH 7). This heterogeneous assembly displayed a hydrogen production rate of 3 μL min−1 cm−2 with a relatively lower faradaic efficiency of 50 ± 3%. The calculated TOF for this electrocatalytic HER was 1200 ± 200 h−1 per cobalt centre, which is orders of magnitude lower in contrast to analogous cobaloximes in homogeneous conditions.103 Recently, the Artero group was also able to achieve electrocatalytic HER activity by using a modified electrode containing CuFexOy semiconductor–TiO2–cobaloxime layers.105
Ott and co-workers immobilized a cobaloxime derivative on a metal–organic framework (MOF), and studied its electrochemical behaviour in acetate buffer (pH 4). This catalyst consisted of a 3D MFO UU-100 Co and cobaloxime catalyst with carboxylates as the anchoring groups (Fig. 12). This cobaloxime-grafted MOF generated hydrogen with a faradaic efficiency of 79%, with an average TOF of 1171 h−1. The overall TON of 20875 was obtained for this catalyst after 18 h of electrocatalysis.106 The MOF backbone ensured that the cobaloxime motif could be active in a 3D structure, while the robust nature of MOF provided the acid-stability. The porous structure of the catalyst-modified MOF also allowed access to the internally buried catalytic side, while instigating a facile diffusion of the electrolyte and substrates during electrocatalysis. However, the poor charge transfer properties and non-conducting nature of MOF have raised questions on its long-term implementation.107,108
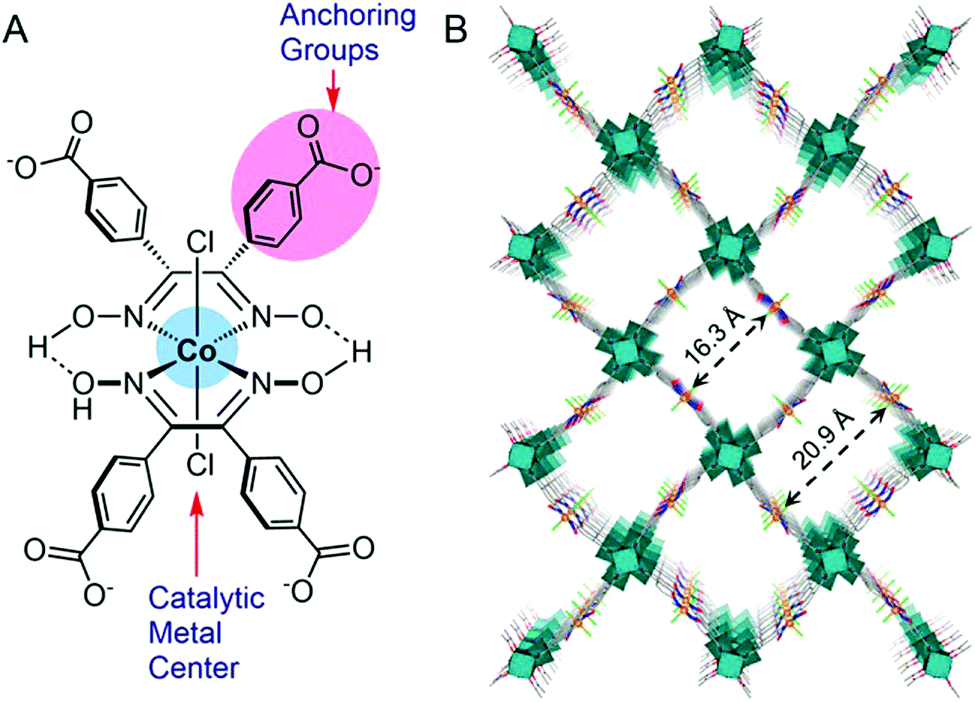 | ||
| Fig. 12 (A) The basic structure of the cobaloxime derivative containing benzoate substituents that is used as linker in UU-100(Co), and (B) the 3D structural model of the UU-100(Co) MOF with a view along the [001] plane. Reproduced from ref. 106 with permission from © American Chemical Society, 2019. Further permissions related to this figure should be directed to the American Chemical Society.106 | ||
Thus, the homogeneous electrocatalytic experiments with cobaloxime derivatives provided us with a deep insight into the structural and electronic factors that regulate their electrocatalytic behaviour. However, the overall catalytic performance (both catalytic rate and overpotential) was inferior compared to the state of the art Ni-bis-(di-phosphine)-based Dubois catalysts.109 On the other hand, cobaloximes were immobilized on different solid surfaces, including carbon nanotubes, semiconductor nanomaterials, and metal organic frameworks. Although these heterogeneous materials replicated the electrocatalytic HER activity of cobaloximes in heterogeneous conditions, they were unable to improve its long-term durability and catalytic efficiency under practical conditions.
The limited success with the current approaches towards cobaloxime-promoted photocatalytic and electrocatalytic HER activities for extensive industrial-scale usage necessitated a new direction in this research area. The successful evolution of Dubois catalysts indicates that the unconventional enzyme-inspired catalyst design strategy can be a way forward to invigorate the basic cobaloxime core in the first place. In the next step, the improvised cobaloxime derivatives can be investigated for efficient and robust photocatalytic and electrocatalytic H2 evolution under the most practical conditions.
Enzyme-inspired modifications of cobaloximes
Naturally evolved hydrogenase enzyme has been known to play a critical role in H2-based metabolism in various life forms for millions of years. All of the different variants of this enzyme contain naturally abundant Fe and Ni in its active site (referred to as [FeFe]- and [NiFe]-hydrogenase). They catalyze H2 production from water at a rapid rate, while maintaining a low overpotential requirement.110,111 All of these features have prompted the hydrogenase active site as a popular choice for the development of biomimetic model complexes for artificial hydrogenases.112 Interestingly, most of the active site models fail to imitate the enzymatic reactivity under practical conditions, which led to the hypothesis that the presence of the protein scaffold around the active site has a significant role in attaining the catalytic excellence.20 Consequently, several research groups have extended their bio-mimetic design beyond the active site to the secondary and outer coordination sphere features.20,113 The HER activity by the cobaloxime core is sensitive to its surrounding environment. Hence, it was rationally combined with variable supramolecular scaffolds (proteins/polymers) to probe the effect of secondary and outer coordination sphere features on the cobaloxime-derived HER activity.A research group led by Utschig and Tiede inserted a cobaloxime motif [Co(DH)2(pyr)Cl, (6)] with a natural Photosystem-I (PSI) protein to fabricate a PSI–cobaloxime hybrid (Fig. 13A). This hybrid construct was active for photocatalytic H2 production in a neutral aqueous solution in the presence of an electron mediator cytochrome c6. This particular construct was able to produce 170 mol H2 per mol of PSI per min that is equivalent to the 50% catalytic rate of an analogous PSI–Pt nanoparticle system, while it outperformed the PSI–hydrogenase hybrid.114,115 Later, they simplified their construct and removed the bulky PSI protein. In this new assembly, they utilized an electron-transporting ferredoxin (Fd) protein to bridge a synthetic photosensitizer [Ru(bpy)32+] with the catalytic cobaloxime core [Co(DBF2)2] (Fig. 13B). This [Ru(bpy)32+]–Fd–[Co(DBF2)2] hybrid exhibited the production of 60 mol H2 per mol of [Ru(bpy)32+] per hour in a neutral aqueous solution upon photo-irradiation, which was improved by ∼3 times in the presence of the axial pyridine bound cobaloxime.116,117 In a follow-up study, another [Ru(bpy)32+]–Fld–[Co(DBF2)2] biohybrid was prepared by replacing the Fd protein with a flavodoxin (Fld) scaffold, which afforded HER activity via a different pathway.117
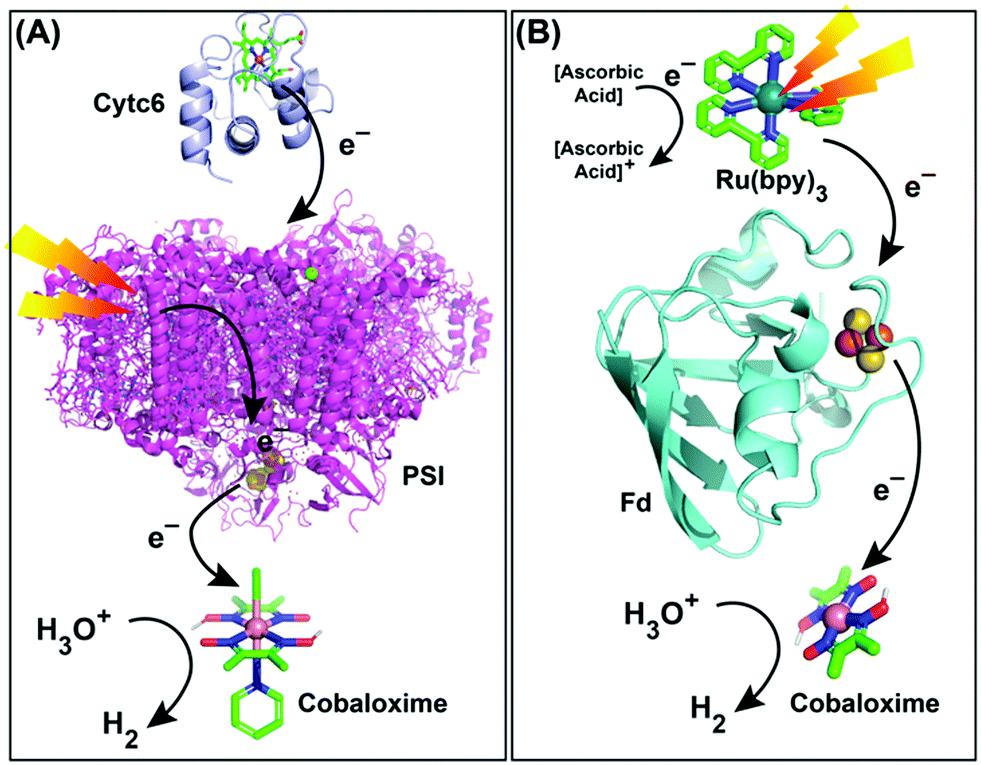 | ||
| Fig. 13 Photocatalytic HER activity exhibited by (A) the PSI–cobaloxime [Co(dmgH)2(pyridine)Cl] hybrid in the presence of cytochrome c6 (Cytc6) as an electron transfer partner, and (B) the [Ru(bpy)32+]–ferredoxin (Fd)–cobaloxime [Co(dmgBF2)2] assembly in the presence of ascorbic acid as a sacrificial electron donor. These figures are redrawn from ref. 114 and 116. The molecules and proteins are not drawn to scale for clarity. | ||
As discussed earlier, the inclusion of pyridine or imidazole axial ligands can positively influence the catalytic H2 production by cobaloxime-based complexes. On the other hand, biology has widely employed the imidazole side chain from the histidine residue for anchoring metal cofactors, especially in hemoproteins.118,119 Following this lead, Artero et al. have rationally inserted two different variants of cobaloxime motifs (hydrogen-bonded [Co(DH)2] and difluoroboryl annulated [Co(DBF2)2]) into the heme-binding pocket of apo-Sperm-whale myoglobin (SwMb) (Fig. 14A). These synthetic metalloproteins displayed photocatalytic activity for HER, although at a considerably reduced efficiency (TON ∼ 3–4, calculated vs. [catalyst] in the presence of [Ru(bpy)32+]PS) compared to the corresponding free cobaloximes. This drop in the catalytic activity was attributed to the strained flexibility experienced by the histidine-ligated cobaloxime in the heme-binding cavity, which in turn terminated the competitive binuclear pathway for H2 production (Fig. 14B).120 In a follow-up study, they tuned the reactivity of the cobaloxime-bound biohybrids by altering the heme-binding pocket. For this purpose, the cobaloxime cores were installed in the heme-binding cavity of two strains of heme oxygenase proteins: rat heme oxygenase 1 (HO1) and Corynebacterium diphteriae heme oxygenase (HmuO). The resultant synthetic metalloproteins demonstrated an increase of ∼3 times in the photocatalytic HER activity (TON ∼ 6.3–15.3, calculated vs. [catalyst] in the presence of deazaflavin PS) compared to either free or SwMb-supported cobaloxime [Co(DH)2].121
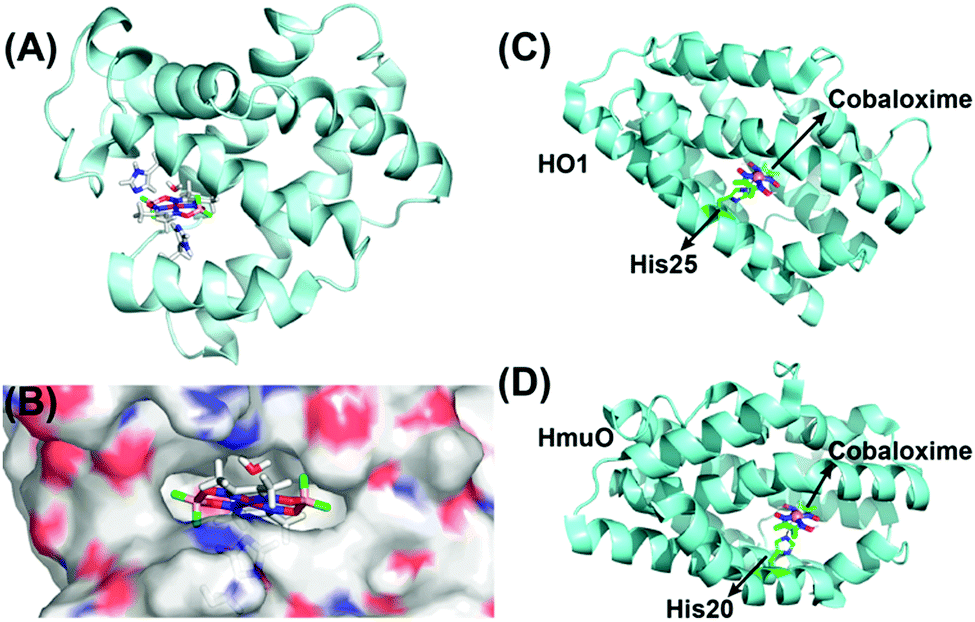 | ||
| Fig. 14 (A) The QC/MM calculated structure of SwMb-cobaloxime [Co(dmgBF2)2], and (B) its electrostatic environment around the cobaloxime core. The hydrophilic regions are depicted in red (O atoms) and blue (N atoms), while the hydrophobic regions are displayed in grey. This figure is adapted from ref. 120. Permission was obtained from the © American Chemical Society, 2014. The heme oxygenase-bound cobaloxime structures of (C) HO1–cobaloxime [Co(dmgH2)2], and (D) HmuO–cobaloxime [Co(dmgH2)2]. The black arrows direct the position of the cobaloxime and the protein-derived axial histidine ligands. The cobalt atom is depicted in purple, boron in pink, carbon in green and fluorine in fluorescent green. These figures were redrawn from ref. 121. | ||
Kato and Yagi explored cyclodextrins (cyclic oligosaccharides of glucose) as an alternative for protein structures to induce a supramolecular effect on the reactivity of cobaloximes. For this purpose, they have designed a 1:1 adduct between the sulfonated-cobaloxime [Co(DH)2(pyridine-4-sulfonate)Cl] and β- and γ-cyclodextrin via host–guest interaction (Fig. 15A). Among them, only the γ-cyclodextrin bound cobaloxime exhibited a ∼10% increase in photocatalytic HER activity in the presence of the Eosin-Y photosensitizer in aqueous solution (TON ∼ 47, calculated vs. [catalyst] in the presence of Eosin-Y PS). The best-fitted insertion of the sulfonate group into the cyclodextrin cavity occurred in the case of γ-cyclodextrin, presumably due to the optimal size-matching. During the formation of this host–guest adduct, the core Co–N4 motif of cobaloxime approached within the hydrogen-bonding distance with the hydroxyl groups present in the rim of the cyclodextrin (Fig. 15B). The dynamic proton exchange interaction between those groups possibly played a crucial role to induce the kinetic effect on the cobaloxime HER activity.122
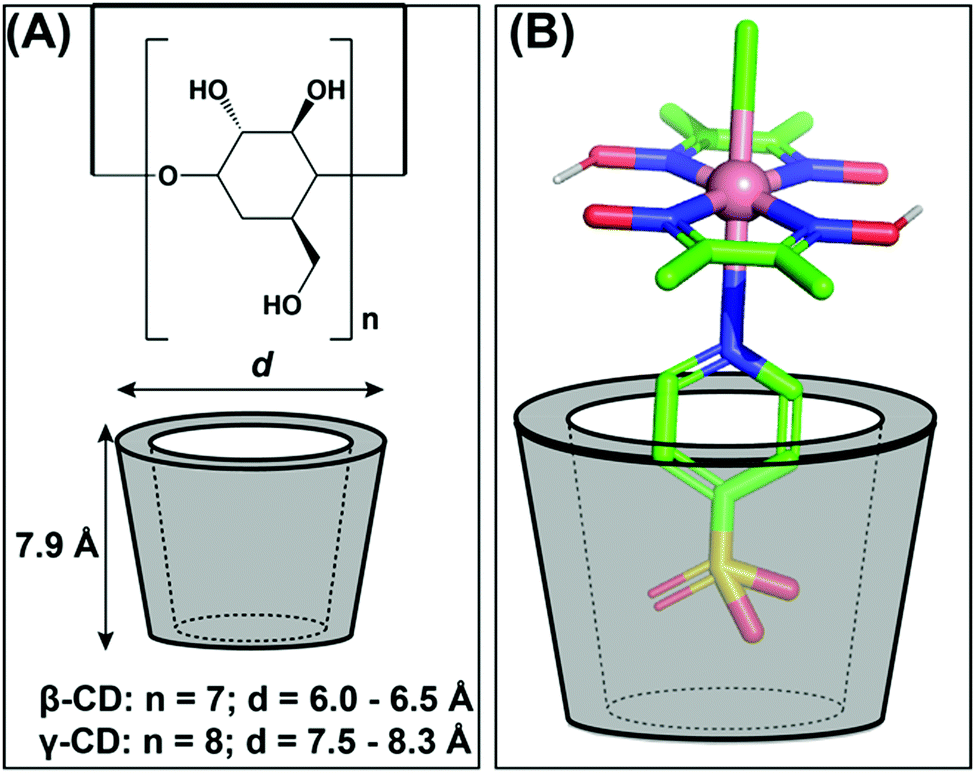 | ||
| Fig. 15 (A) The basic structure of β- and γ-cyclodextrins (cyclic oligosaccharides of glucose) and their corresponding sizes. (B) The possible interaction between the sulfonate group from cobaloxime [Co(dmgH)2(pyridine-4-sulfonate)Cl] and the γ-cyclodextrin. These figures were redrawn from ref. 122. | ||
Interestingly, the increment in the catalytic HER activity for these biohybrid constructs was either inconsequential (or even negative) compared to the free cobaloxime analogues. This observation suggested that the inclusion of cobaloxime inside the robust supramolecular structure may severely restrict the fluxionality of the cobaloxime motif to affect the overall reaction rate. A significant increase in the catalytic H2 evolution was noticed only for a few constructs where the correctly oriented hydrogen-bonding interactions induced facile H+ movement. The results from these earlier studies indicate that “a minimally constrained cobaloxime motif containing a peripheral proton relay” possibly holds the key for generating the second generation of enzyme-inspired cobaloxime-based H2 production catalysts. Following this queue, a series of four cobaloxime complexes were created, where only the para-position of the axially coordinated pyridine ligand was altered, around an otherwise identical cobaloxime core [Co(DH)2(pyridine-derivative)Cl]. Variable combinations of the secondary amine (–NH–), carboxylic acid group (–COOH), and phenolic (–Ph–OH) groups were utilized to fabricate one, two, and three-component proton relays in the cobaloxime periphery (Fig. 16).99 The effect of the incorporation of the minimal (but essential) outer coordination sphere features was imminent as these cobaloxime derivatives were found to be active for electrocatalytic H2 evolution in 100% aqueous solution. The three-component H+-relay containing [ClCo(DH)2(pyridine–methylene–tyrosine)] recorded the fastest TOF for HER (∼8830 s−1, pH 7) among these complexes, which is a ∼10-fold increase in the catalytic rate compared to the original [ClCo(DH)2(pyridine)]. The catalytic rate for H2 evolution for these complexes followed the following trend with varying pyridine substituents, tyrosine > tyramine ≈ phenylalanine > tyramine, to establish a direct connection between the extent of the outer coordination sphere feature and catalytic rate. This hypothesis was further supported by the two-dimensional Nuclear Overhauser effect spectroscopy (NOESY) NMR experiment, where a dynamic proton exchange among those peripheral basic groups, the oxime hydrogen-bonded network, and the solvent water molecules were observed. Typically, the cobaloxime motif becomes inactive for homogeneous H2 production with increasing acidity of the solution (pH ≤ 5.0). The disruption of the oxime-derived hydrogen-bonding network, which acts as the protonation site during HER catalysis, is believed to be the prime reason for this behaviour. However, the outer coordination sphere functionalities present in the cobaloxime derivatives provides an alternate protonation spot, even in the acidic condition, to allow homogeneous H2 production by those complexes even at pH 3–5. This particular aspect highlights that the incorporation of enzyme-inspired features can even impact the overall stability of the complexes. However, the effect of this distant protic groups is inferior in comparison to the closely lying oxime network, which is reflected by a contrasting change in the HER catalytic rate with a decrease in pH for all of those cobaloximes (Fig. 16).99
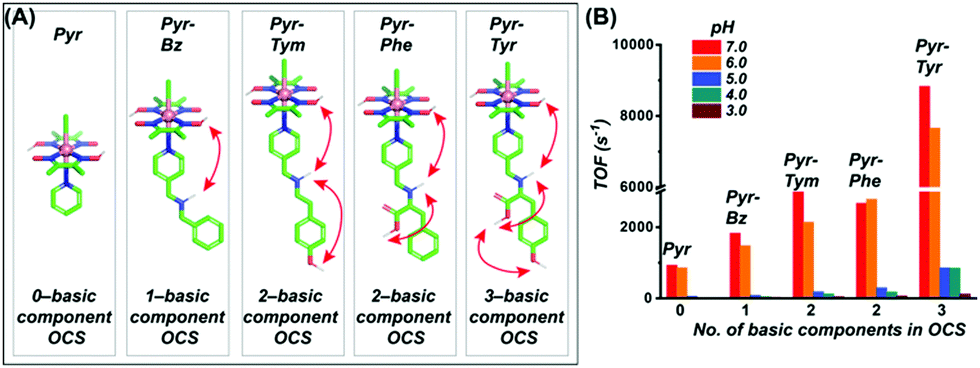 | ||
| Fig. 16 (A) The basic structures of the [ClCo(dmgH)2(pyridine-derivative)] complexes, along with the number of basic components present in their corresponding outer coordination sphere (OCS): (from left to right) only pyridine (Pyr), zero component; pyridine–methylene–benzylamine (Pyr–Bz), one component (–NH); pyridine–methylene–tyramine (Pyr–Tym), two components (–NH– and –Ph–OH); pyridine–methylene–phenylalanine (Pyr–Phe), two components (–NH– and –COOH); pyridine–methylene–tyrosine (Pyr–Tyr), three components (–NH–, –COOH, and –Ph–OH). (B) The experimentally tabulated electrocatalytic H2 evolution reaction rate (TOF) variation for each complex in aqueous solution (pH 3.0–7.0). These figures were adapted from ref. 99. | ||
Next, the effect of such neighbouring basic groups on the cobaloxime motif was studied on the [Co(DH)2(imidazole-derivative)Cl] platform, with an alternating axially ligated imidazole scaffold (Fig. 17).68 The presence of multiple protic functionalities and their spatial orientation tunes the HER activity of these complexes. As expected, the natural amino acid histidine (which contains an imidazole side chain along with carboxylic acid and primary amine groups)-bound cobaloxime exhibited the highest kinetic rate (TOF ∼ 4500 s−1) for electrolytic H2 production in a neutral aqueous solution. These complexes were probed even in the photocatalytic conditions to monitor the effect of the outer coordination sphere features beyond the standard electrochemical settings. In the presence of the Eosin-Y dye, the overall photocatalytic H2 production by [Co(DH)2(histidine)Cl] was enhanced by 1.7 times (TON ∼ 250, calculated vs. [catalyst] in the presence of Eosin-Y PS), compared to the rudimentary [Co(DH)2(imidazole)Cl] in 1:1 water/acetonitrile media, to underline the impact of the additional protic groups appended from the imidazole. Again, the facile proton dynamics among these peripheral groups, along with the oxime and solvent water molecules, are believed to be the prime reason for this amplified HER performance. The presence of these enzyme-stimulated outer coordination sphere features even allowed us to obtain the first generation of fully water-soluble [ClCo(DH)2(imidazole-derivative)] complexes. This unique trait allowed us to achieve the first natural sunlight-irradiated photocatalytic H2-producing cobaloxime complex that is active in an aerobic, neutral (pH 7.0) aqueous solution (TON ∼ 1000, calculated vs. [catalyst] in the presence of Eosin-Y PS).68 Hence, this enzyme-inspired complex represents an ideal candidate for investigating solar-driven H2 production from water under the most practical circumstances.
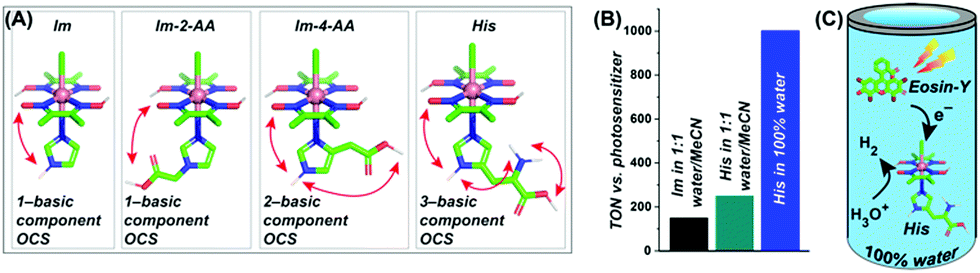 | ||
| Fig. 17 (A) The basic structures of the [ClCo(DH)2(imidazole-derivative)] complexes along with the number of basic components present in their corresponding outer coordination sphere (OCS): (from left to right) only imidazole (Im), one component (imidazole –NH–); imidazole-2-acetic acid (Im-2-AA), one component (–COOH); imidazole-4-acetic acid (Im-4-AA), two components (imidazole–NH– and –COOH); histidine (His), three components (imidazole–NH–, –COOH, and –NH2). (B) The comparative TON (measured vs. photosensitizer) for photocatalytic H2 evolution for Im (black bar) and His (cyan bar) in 1:1 water/acetonitrile, and His in 100% water (blue bar). (C) A schematic diagram of His activity in 100% water in the presence of Eosin-Y as a photosensitizer. These figures were adapted from ref. 68. | ||
Conclusions
Although cobaloxime was developed as a structural model for vitamin B12, it has recently gained more attention due to its catalytic properties for H2 evolution reactions. The structural flexibility of this cobaloxime framework enabled the generation of a myriad of compounds with varying physiochemical electronic and solubility properties. The HER activity of the cobaloxime derivatives across a wide variety of conditions has established it as a diagnostic tool for optimizing photosensitizers for photocatalytic H2 production. The display of better HER activity by the covalently-linked cobaloxime–photosensitizer adduct compared to independent segments established the significance of the directly coordinated photocatalytic assemblies for photo-driven H2 production reactions. Later, the electrochemical investigation of cobaloximes provided insight into the intricacy of the structure–function relationship at the molecular level. Again, the versatility of the cobaloxime core shines as a critical element for probing its HER activity in both aqueous and non-aqueous solvent media. Despite all these advantages, the acid-instability of the primary coordination sphere-bound oxime motifs has narrowed the active HER catalytic space of cobaloximes to near-neutral conditions. Additionally, the poor H+ movement around the core has limited the HER activity of cobaloximes to moderate compared to contemporary first-row transition metal-based catalysts. As a result, the cobaloxime-based HER research faced a lag period.The architectural blueprint of metalloenzymes suggested that the inclusion of enzymatic hallmarks, such as an artificial proton relay, can influence the HER activity by cobaloxime. The simple yet robust nature of the cobaloxime core provided ample opportunity to either include cobaloxime into an already existing protein pocket, or rationally design a multi-component proton relay with precisely positioned peripheral functionalities around the core. This bio-mimetic catalyst design strategy has evolved a new genre of widely stable cobaloxime complexes that can photo-/electro-chemically produce H2 at an unprecedented rate. Hence, the cobaloxime core is utilized as a model indicator for a broad spectrum of catalytic H2 production functionalities, including optimizing the photocatalytic assembly, probing the electrocatalytic conditions, and exploring the effects of the peripheral enzyme-inspired features. In this review, we have covered the revival of the cobaloxime motif in the pursuit of a durable, efficient, and user-friendly H2-producing catalyst, a key ingredient for the development of the H2-based renewable energy infrastructure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the support provided by the Indian Institute of Technology Gandhinagar (IITGN) and Indian Institute of Technology Bombay (IITB). D. D. would like to thank the support of the DST-Inspire fellowship provided by the Department of Science and Technology (DST), India (DST/INSPIRE Fellowship/2017/IF170385). A. D. acknowledges the Ramanujan Fellowship (SB/S2/RJN-112/2015). P. M. would like to thank DST-SERB national post-doctoral fellowship (PDF/2018/000837).Notes and references
- D. C. Hodgkin, J. Kamper, M. Mackay, J. Pickworth, K. N. Trueblood and J. G. White, Nature, 1956, 178, 64–66 CrossRef CAS PubMed.
- D. M. C. Hodgkin, J. Kamper, J. Lindsey, M. MacKay, J. Pickworth, J. H. Robertson, C. Brink-Shoemaker, J. G. White, R. J. Prosen and K. N. Trueblood, Proc. R. Soc. London, Ser. A, 1957, 242, 228–263 CAS.
- D. Lindsay and W. Kerr, Nat. Chem., 2011, 3, 494 CrossRef CAS PubMed.
- M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391–3404 RSC.
- G. N. Schrauzer, Acc. Chem. Res., 1968, 1, 97–103 CrossRef CAS.
- R. Dreos, S. Geremia, L. Randaccio and P. Siega, PATAI’S Chemistry of Functional Groups, American Cancer Society, 2010 Search PubMed.
- G. N. Schrauzer, G. W. Parshall and E. R. Wonchoba, Inorganic Syntheses, John Wiley & Sons, Ltd, 2007, pp. 61–70 Search PubMed.
- G. N. Schrauzer and J. Kohnle, Chem. Ber., 1964, 97, 3056–3064 CrossRef CAS.
- F. Wagner and K. Bernhauer, Ann. N. Y. Acad. Sci., 1964, 112, 580–589 CrossRef CAS PubMed.
- J. Hawecker, J. M. Lehn and R. Ziessel, Nouv. J. Chim., 1983, 7, 271 CAS.
- A. Gerli and L. G. Marzilli, Inorg. Chem., 1992, 31, 1152–1160 CrossRef CAS.
- L. G. Marzilli, A. Gerli and A. M. Calafat, Inorg. Chem., 1992, 31, 4617–4627 CrossRef CAS.
- S. W. Ragsdale, P. A. Lindahl and E. Münck, J. Biol. Chem., 1987, 262, 14289–14297 CAS.
- B. S. Tovrog, D. J. Kitko and R. S. Drago, J. Am. Chem. Soc., 1976, 98, 5144–5153 CrossRef CAS.
- A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1984, 106, 5197–5202 CrossRef CAS.
- P. Connolly and J. H. Espenson, Inorg. Chem., 1986, 25, 2684–2688 CrossRef CAS.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS PubMed.
- X. Hu, B. M. Cossairt, B. S. Brunschwig, N. S. Lewis and J. C. Peters, Chem. Commun., 2005, 4723–4725 RSC.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS PubMed.
- A. Dutta, A. M. Appel and W. J. Shaw, Nat. Rev. Chem., 2018, 2, 244–252 CrossRef CAS.
- A. Panagiotopoulos, K. Ladomenou, D. Sun, V. Artero and A. G. Coutsolelos, Dalton Trans., 2016, 45, 6732–6738 RSC.
- M. Yang, J. E. Yarnell, K. El Roz and F. N. Castellano, ACS Appl. Energy Mater., 2020, 3, 1842–1853 CrossRef CAS.
- X. Wang, S. Goeb, Z. Ji, N. A. Pogulaichenko and F. N. Castellano, Inorg. Chem., 2011, 50, 705–707 CrossRef CAS PubMed.
- E. Deponti and M. Natali, Dalton Trans., 2016, 45, 9136–9147 RSC.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- R. Staehle, S. Losse, M. R. Filipovic, I. Ivanović-Burmazović, J. G. Vos and S. Rau, ChemPlusChem, 2014, 79, 1614–1621 CrossRef CAS.
- L. Petermann, R. Staehle, M. Pfeifer, C. Reichardt, D. Sorsche, M. Wächtler, J. Popp, B. Dietzek and S. Rau, Chem. – Eur. J., 2016, 22, 8240–8253 CrossRef CAS PubMed.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed.
- B. Probst, C. Kolano, P. Hamm and R. Alberto, Inorg. Chem., 2009, 48, 1836–1843 CrossRef CAS PubMed.
- B. Probst, A. Rodenberg, M. Guttentag, P. Hamm and R. Alberto, Inorg. Chem., 2010, 49, 6453–6460 CrossRef CAS PubMed.
- A. Fihri, V. Artero, A. Pereira and M. Fontecave, Dalton Trans., 2008, 5567–5569 RSC.
- A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564–567 CrossRef CAS PubMed.
- C. Li, M. Wang, J. Pan, P. Zhang, R. Zhang and L. Sun, J. Organomet. Chem., 2009, 694, 2814–2819 CrossRef CAS.
- K. L. Mulfort and D. M. Tiede, J. Phys. Chem. B, 2010, 114, 14572–14581 CrossRef CAS PubMed.
- D. M. Cropek, A. Metz, A. M. Müller, H. B. Gray, T. Horne, D. C. Horton, O. Poluektov, D. M. Tiede, R. T. Weber, W. L. Jarrett, J. D. Phillips and A. A. Holder, Dalton Trans., 2012, 41, 13060–13073 RSC.
- T. Auvray, R. Sahoo, D. Deschênes and G. S. Hanan, Dalton Trans., 2019, 48, 15136–15143 RSC.
- M. Rupp, T. Auvray, E. Rousset, G. M. Mercier, V. Marvaud, D. G. Kurth and G. S. Hanan, Inorg. Chem., 2019, 58, 9127–9134 CrossRef CAS PubMed.
- C. Lentz, O. Schott, T. Auvray, G. Hanan and B. Elias, Inorg. Chem., 2017, 56, 10875–10881 CrossRef CAS PubMed.
- C. Lentz, O. Schott, T. Auvray, G. S. Hanan and B. Elias, Dalton Trans., 2019, 48, 15567–15576 RSC.
- A. Mukherjee, O. Kokhan, J. Huang, J. Niklas, L. X. Chen, D. M. Tiede and K. L. Mulfort, Phys. Chem. Chem. Phys., 2013, 15, 21070–21076 RSC.
- P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS PubMed.
- P. Zhang, M. Wang, C. Li, X. Li, J. Dong and L. Sun, Chem. Commun., 2009, 8806–8808 Search PubMed.
- P. Zhang, M. Wang, X. Li, H. Cui, J. Dong and L. Sun, Sci. China: Chem., 2012, 55, 1274–1282 CrossRef CAS.
- T. Lazarides, M. Delor, I. V. Sazanovich, T. M. McCormick, I. Georgakaki, G. Charalambidis, J. A. Weinstein and A. G. Coutsolelos, Chem. Commun., 2013, 50, 521–523 RSC.
- M. Natali, R. Argazzi, C. Chiorboli, E. Iengo and F. Scandola, Chem. – Eur. J., 2013, 19, 9261–9271 CrossRef CAS PubMed.
- G. Landrou, A. A. Panagiotopoulos, K. Ladomenou and A. G. Coutsolelos, J. Porphyrins phthalocyanines, 2016, 20, 534–541 CrossRef CAS.
- L. Mintrop, J. Windisch, C. Gotzmann, R. Alberto, B. Probst and P. Kurz, J. Phys. Chem. B, 2015, 119, 13698–13706 CrossRef CAS PubMed.
- K. Peuntinger, T. Lazarides, D. Dafnomili, G. Charalambidis, G. Landrou, A. Kahnt, R. P. Sabatini, D. W. McCamant, D. T. Gryko, A. G. Coutsolelos and D. M. Guldi, J. Phys. Chem. C, 2013, 117, 1647–1655 CrossRef CAS.
- J. Manono, P. A. Marzilli, F. R. Fronczek and L. G. Marzilli, Inorg. Chem., 2009, 48, 5626–5635 CrossRef CAS PubMed.
- R. S. Khnayzer, C. E. McCusker, B. S. Olaiya and F. N. Castellano, J. Am. Chem. Soc., 2013, 135, 14068–14070 CrossRef CAS PubMed.
- G. García-Herbosa, W. R. McNamara, W. W. Brennessel, J. V. Cuevas, S. Sur and R. Eisenberg, Polyhedron, 2013, 58, 39–46 CrossRef.
- W.-C. Xiao, Y.-W. Tao, Y. Zhao, J.-X. Luo and W.-Z. Lai, Inorg. Chem. Commun., 2020, 113, 107800 CrossRef CAS.
- P. Zhang, M. Wang, J. Dong, X. Li, F. Wang, L. Wu and L. Sun, J. Phys. Chem. C, 2010, 114, 15868–15874 CrossRef CAS.
- T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480–15483 CrossRef CAS PubMed.
- L. Gong, J. Wang, H. Li, L. Wang, J. Zhao and Z. Zhu, Catal. Commun., 2011, 12, 1099–1103 CrossRef CAS.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- E. Giannoudis, E. Benazzi, J. Karlsson, G. Copley, S. Panagiotakis, G. Landrou, P. Angaridis, V. Nikolaou, C. Matthaiaki, G. Charalambidis, E. A. Gibson and A. G. Coutsolelos, Inorg. Chem., 2020, 59, 1611–1621 CrossRef CAS PubMed.
- J. Dong, M. Wang, P. Zhang, S. Yang, J. Liu, X. Li and L. Sun, J. Phys. Chem. C, 2011, 115, 15089–15096 CrossRef CAS.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 16774–16776 CrossRef CAS PubMed.
- D. D. Konieczna, H. Biller, M. Witte, W. G. Schmidt, A. Neuba and R. Wilhelm, Tetrahedron, 2018, 74, 142–149 CrossRef CAS.
- Z.-Y. Wang, H. Rao, M.-F. Deng, Y.-T. Fan and H.-W. Hou, Phys. Chem. Chem. Phys., 2013, 15, 16665–16671 RSC.
- H.-Q. Zheng, H. Rao, X.-Z. Hu, X.-H. Li, Y.-T. Fan and H.-W. Hou, Sol. Energy, 2014, 105, 648–655 CrossRef CAS.
- G.-G. Luo, H. Lu, X.-L. Zhang, J.-C. Dai, J.-H. Wu and J.-J. Wu, Phys. Chem. Chem. Phys., 2015, 17, 9716–9729 RSC.
- X. Li, G. Luo, K. Fang, J. Zhou, Q. Zhao and R. Wu, Sci. Sin.: Chim., 2015, 45, 843–854 CrossRef CAS.
- A. Xie, Z.-H. Pan, M. Yu, G.-G. Luo and D. Sun, Chin. Chem. Lett., 2019, 30, 225–228 CrossRef CAS.
- J. Wang, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and X. Wang, Dalton Trans., 2015, 44, 17704–17711 RSC.
- D. Dolui, S. Das, J. Bharti, S. Kumar, P. Kumar and A. Dutta, Cell Rep. Phys. Sci., 2020, 1, 100007 CrossRef.
- X.-F. Liu, Y.-X. Zhang and J. Yan, Transition Met. Chem., 2015, 40, 305–311 CrossRef CAS.
- J. Bartelmess, W. W. Weare and R. D. Sommer, Dalton Trans., 2013, 42, 14883–14891 RSC.
- J. Bartelmess, A. J. Francis, K. A. El Roz, F. N. Castellano, W. W. Weare and R. D. Sommer, Inorg. Chem., 2014, 53, 4527–4534 CrossRef CAS PubMed.
- G.-G. Luo, K. Fang, J.-H. Wu and J. Mo, Chem. Commun., 2015, 51, 12361–12364 RSC.
- G.-G. Luo, K. Fang, J.-H. Wu, J.-C. Dai and Q.-H. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 23884–23894 RSC.
- J. C. Manton, C. Long, J. G. Vos and M. T. Pryce, Phys. Chem. Chem. Phys., 2014, 16, 5229–5236 RSC.
- T. M. McCormick, Z. Han, D. J. Weinberg, W. W. Brennessel, P. L. Holland and R. Eisenberg, Inorg. Chem., 2011, 50, 10660–10666 CrossRef CAS PubMed.
- B. S. Veldkamp, W.-S. Han, S. M. Dyar, S. W. Eaton, M. A. Ratner and M. R. Wasielewski, Energy Environ. Sci., 2013, 6, 1917–1928 RSC.
- A. Q. Mir, D. Dolui, S. Khandelwal, H. Bhatt, B. Kumari, S. Barman, S. Kanvah and A. Dutta, JoVE, 2019, e60231 Search PubMed.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- A. Morandeira, G. Boschloo, A. Hagfeldt and L. Hammarström, J. Phys. Chem. B, 2005, 109, 19403–19410 CrossRef CAS PubMed.
- S. Lyu, J. Massin, M. Pavone, A. B. Muñoz-García, C. Labrugère, T. Toupance, M. Chavarot-Kerlidou, V. Artero and C. Olivier, ACS Appl. Energy Mater., 2019, 2, 4971–4980 CrossRef CAS.
- X.-W. Song, H.-M. Wen, C.-B. Ma, H.-H. Cui, H. Chen and C.-N. Chen, RSC Adv., 2014, 4, 18853–18861 RSC.
- F. Niu, S. Shen, N. Zhang, J. Chen and L. Guo, Appl. Catal., B, 2016, 199, 134–141 CrossRef CAS.
- T. Banerjee, F. Haase, G. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228–16234 CrossRef CAS PubMed.
- J. Gu, Y. Yan, J. L. Young, K. X. Steirer, N. R. Neale and J. A. Turner, Nat. Mater., 2016, 15, 456–460 CrossRef CAS PubMed.
- N. R. Neale, N. Kopidakis, J. van de Lagemaat, M. Grätzel and A. J. Frank, J. Phys. Chem. B, 2005, 109, 23183–23189 CrossRef CAS PubMed.
- A. Q. Mir, G. Joshi, P. Ghosh, S. Khandelwal, A. Kar, R. Hegde, S. Khatua and A. Dutta, ACS Energy Lett., 2019, 4, 2428–2435 CrossRef CAS.
- C. N. Valdez, J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15589–15593 CrossRef CAS PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- D. Dolui, S. Ghorai and A. Dutta, Coord. Chem. Rev., 2020, 416, 213335 CrossRef CAS.
- S. Cherdo, S. E. Ghachtouli, M. Sircoglou, F. Brisset, M. Orio and A. Aukauloo, Chem. Commun., 2014, 50, 13514–13516 RSC.
- V. Artero and M. Fontecave, Coord. Chem. Rev., 2005, 249, 1518–1535 CrossRef CAS.
- J. A. S. Roberts and R. M. Bullock, Inorg. Chem., 2013, 52, 3823–3835 CrossRef CAS PubMed.
- X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed.
- D. W. Wakerley and E. Reisner, Phys. Chem. Chem. Phys., 2014, 16, 5739–5746 RSC.
- D. W. Wakerley, M. A. Gross and E. Reisner, Chem. Commun., 2014, 50, 15995–15998 RSC.
- D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2015, 8, 2283–2295 RSC.
- X. Yin, C. Liu, S. Zhuo, Y. Xu and B. Zhang, Dalton Trans., 2015, 44, 1526–1529 RSC.
- D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, ACS Catal., 2019, 9, 10115–10125 CrossRef CAS.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- B. Reuillard, J. Warnan, J. J. Leung, D. W. Wakerley and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 3952–3957 CrossRef CAS PubMed.
- S. Donck, J. Fize, E. Gravel, E. Doris and V. Artero, Chem. Commun., 2016, 52, 11783–11786 RSC.
- B. L. Wadsworth, A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, ACS Catal., 2016, 6, 8048–8057 CrossRef CAS.
- A. Krawicz, J. Yang, E. Anzenberg, J. Yano, I. D. Sharp and G. F. Moore, J. Am. Chem. Soc., 2013, 135, 11861–11868 CrossRef CAS PubMed.
- C. Tapia, E. Bellet-Amalric, D. Aldakov, F. Boudoire, K. Sivula, L. Cagnon and V. Artero, Green Chem., 2020, 22, 3141–3149 RSC.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- E. Mijangos, S. Roy, S. Pullen, R. Lomoth and S. Ott, Dalton Trans., 2017, 46, 4907–4911 RSC.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- R. Cammack, M. Frey, R. Robson, M. Frey and R. Robson, Hydrogen as a Fuel: Learning from Nature, CRC Press, 2001 Search PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS PubMed.
- L. M. Utschig, S. C. Silver, K. L. Mulfort and D. M. Tiede, J. Am. Chem. Soc., 2011, 133, 16334–16337 CrossRef CAS PubMed.
- L. M. Utschig, S. R. Soltau and D. M. Tiede, Curr. Opin. Chem. Biol., 2015, 25, 1–8 CrossRef CAS PubMed.
- S. R. Soltau, J. Niklas, P. D. Dahlberg, O. G. Poluektov, D. M. Tiede, K. L. Mulfort and L. M. Utschig, Chem. Commun., 2015, 51, 10628–10631 RSC.
- S. R. Soltau, P. D. Dahlberg, J. Niklas, O. G. Poluektov, K. L. Mulfort and L. M. Utschig, Chem. Sci., 2016, 7, 7068–7078 RSC.
- C. J. Reedy and B. R. Gibney, Chem. Rev., 2004, 104, 617–650 CrossRef CAS PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- M. Bacchi, G. Berggren, J. Niklas, E. Veinberg, M. W. Mara, M. L. Shelby, O. G. Poluektov, L. X. Chen, D. M. Tiede, C. Cavazza, M. J. Field, M. Fontecave and V. Artero, Inorg. Chem., 2014, 53, 8071–8082 CrossRef CAS PubMed.
- M. Bacchi, E. Veinberg, M. J. Field, J. Niklas, T. Matsui, D. M. Tiede, O. G. Poluektov, M. Ikeda-Saito, M. Fontecave and V. Artero, ChemPlusChem, 2016, 81, 1083–1089 CrossRef CAS PubMed.
- M. Kato, K. Kon, J. Hirayama and I. Yagi, New J. Chem., 2019, 43, 10087–10092 RSC.
| This journal is © The Royal Society of Chemistry 2020 |