
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA visible-light mediated three-component radical process using dithiocarbamate anion catalysis†
Sara
Cuadros‡
a,
Matthew A.
Horwitz‡
a,
Bertrand
Schweitzer-Chaput
 a and
Paolo
Melchiorre
*abc
a and
Paolo
Melchiorre
*abc
aICIQ – Institute of Chemical Research of Catalonia, The Barcelona Institute of Science and Technology, Avenida Països Catalans 16, 43007, Tarragona, Spain. E-mail: pmelchiorre@iciq.es; Web: http://www.iciq.org/research/research_group/prof-paolo-melchiorre/
bIIT – Istituto Italiano di Tecnologia, Laboratory of Asymmetric Catalysis and Photochemistry, Via Morego 30, 16163, Genoa, Italy
cICREA – Catalan Institution for Research and Advanced Studies, Passeig Lluís Companys 23, 08010, Barcelona, Spain
First published on 30th April 2019
Abstract
We report a photoinduced three-component radical process, which couples readily available alkyl chlorides, maleimides, and heteroaromatic fragments to rapidly generate complex chiral products with high diastereocontrol. This method generates radicals via an SN2-based photochemical catalytic mechanism, which is not reliant on the redox properties of the precursors. It therefore grants access to open-shell intermediates from substrates that would be incompatible with or inert to classical radical-generating strategies. The redox-neutral conditions of this process make it tolerant of redox-sensitive substrates and allow the installation of multiple biologically relevant heterocycles within the cascade products.
Introduction
Multicomponent reactions (MCRs) are important in synthetic chemistry because they allow the rapid construction of complex molecules from simple substrates and in a single step.1 While chemists have predominantly developed methods based on polar pathways, radical reactivity provides a powerful alternative.2,3 However, methods to initiate radical MCRs have traditionally required unstable initiators or stoichiometric oxidants or reductants (Fig. 1a).4 These relatively harsh conditions for generating open-shell intermediates (high temperatures, toxic reagents or strong redox-active reagents) significantly affect the selectivity and the functional group tolerance of the overall radical cascade processes. Recently, the field of photoredox catalysis5 has overcome some of these limitations, offering effective tools to trigger radical MCRs under mild conditions while avoiding the need for stoichiometric metals or harsh atom abstractors.6 However, this radical generation strategy mainly activates substrates via single-electron transfer (SET) pathways, thus relying on their redox properties.7 This intrinsic feature poses a voltage-gated limit to the generality of the radical precursors that can be used.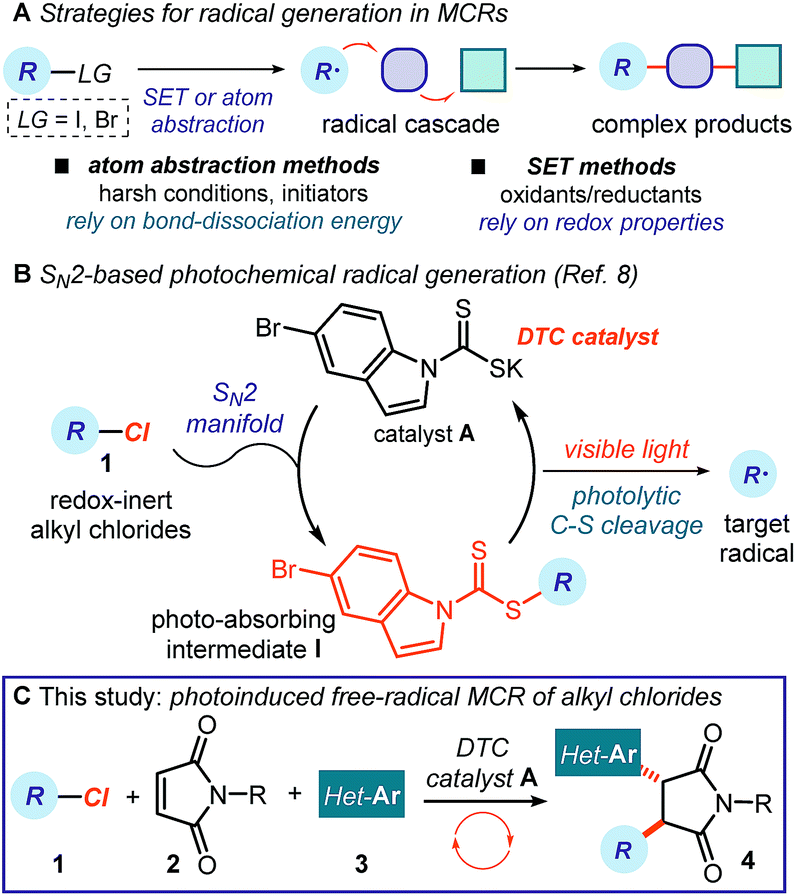 | ||
| Fig. 1 (A) Classical radical-generating strategies to initiate multicomponent reactions require precursors with suitable bond-dissociation energies (BDE) or redox properties. (B) Our recently developed SN2-based method to photochemically generate radicals is not reliant on the redox properties of substrates. (C) Proposed photoinduced three-component radical reaction using redox-inert alkyl chlorides 1, mediated by dithiocarbamate (DTC) anion catalysis. | ||
In this context, our laboratory recently reported a unique photochemical catalytic strategy, which harnesses different physical properties of the substrates to generate radicals (Fig. 1b).8 Specifically, we designed the readily available and air- and moisture-stable dithiocarbamate (DTC)9 anion catalyst A, which is adorned with an indole chromophoric unit. This organic catalyst is nucleophilic enough10 to activate alkyl electrophiles by displacing a variety of leaving groups via an SN2 pathway. The resulting photon-absorbing intermediates I afford radicals upon excitation by visible light and homolytic cleavage of the weak C–S bond.11 This catalytic SN2-based strategy, which is not reliant on the redox properties of the radical precursor, grants access to open-shell intermediates from substrates that would be incompatible with or inert to classical radical-generating strategies, including photoredox catalysis.
Here, we demonstrate how this photochemical strategy can be translated into the realm of radical MCRs to design cascades that would be difficult to implement with other methods. Specifically, we report a three-component reaction mediated by the DTC potassium salt catalyst A that uses difficult-to-reduce alkyl chlorides 1 as radical precursors and couples them with readily available maleimides 2 and heteroaromatic fragments 3 (Fig. 1c). This carbo-difunctionalization of maleimides12 combines two sequential intermolecular radical-based bond-forming processes to afford complex products 4 containing a succinimide moiety. This structural element is found in numerous biologically active molecules.13 Importantly, the method's redox neutral conditions and high functional group tolerance allow us to easily adorn products 4 with a variety of biologically relevant N-heterocycles,14 which would not be compatible with strong redox-active reagents.
Mechanistic plan
The specific mechanistic details of our proposed photochemical radical MCR are outlined in Fig. 2. We envisioned a catalytic cycle where the DTC catalyst A would activate alkyl chlorides 1 upon SN2 attack.10 The resulting intermediate I possesses a weak C–S bond, which would be cleaved by low-energy photons (blue LEDs) to generate the target carbon radical II and the dithiocarbonyl radical III.11 The nucleophilic open-shell intermediate II would initiate the radical cascade upon interception from maleimide 2, which forges a new C–C bond. The emerging electrophilic radical IV would in turn participate in a second trapping event with an electron-rich heteroaromatic compound, affording the open-shell intermediate V. This crucial intermediate would act as a reductant to turn over catalyst A upon SET reduction of the dithiocarbonyl radical III.15 Overall, this net redox-neutral transformation would deliver the functionalized cascade product 4.8 | ||
| Fig. 2 Proposed mechanism for the visible-light-driven three-component radical reaction; Z: chromophore. | ||
Results and discussion
To test the feasibility of our strategy, we selected the commercially available N-(chloromethyl)phthalimide 1a as precursor of a nucleophilic radical, N-methylmaleimide 2a as the radical trap, and N-methylpyrrole 3a as the terminal electron-rich heteroaromatic trap (Table 1). The experiments were conducted at 60 °C in DCE using blue LEDs emitting at 465 nm, 20 mol% of the DTC catalyst A and a large excess of 3a. These conditions furnished the cascade product trans-4a in good chemical yield and with excellent diastereoselectivity (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, entry 1). Under these conditions, we did not observe any byproduct arising from either a pyrrole alkylation radical path with 1a or a polar addition of 3a to maleimide 2a. Variations of the initial conditions, including a lower catalyst amount and temperature (entries 3 and 4), led to poorer results. Control experiments established that both catalyst A and light are necessary to trigger the three-component radical process (entries 5 and 6), thus excluding an alternative polar pathway for the formation of 4a. Interestingly, no product formation was observed when replacing catalyst A with the strongly reducing fac-Ir(ppy)3 photoredox catalyst (E1/2 Ir(IV)/Ir(III*) = −1.73 vs. SCE),16 which highlights the inability of photoredox catalysis to trigger the radical MCR process (entry 7).17
:1, entry 1). Under these conditions, we did not observe any byproduct arising from either a pyrrole alkylation radical path with 1a or a polar addition of 3a to maleimide 2a. Variations of the initial conditions, including a lower catalyst amount and temperature (entries 3 and 4), led to poorer results. Control experiments established that both catalyst A and light are necessary to trigger the three-component radical process (entries 5 and 6), thus excluding an alternative polar pathway for the formation of 4a. Interestingly, no product formation was observed when replacing catalyst A with the strongly reducing fac-Ir(ppy)3 photoredox catalyst (E1/2 Ir(IV)/Ir(III*) = −1.73 vs. SCE),16 which highlights the inability of photoredox catalysis to trigger the radical MCR process (entry 7).17
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yield 4ab (%) |
| a Reactions performed on a 0.1 mmol scale at 60 °C for 20 h using 0.2 mL of solvent under illumination by blue LED strip (λmax = 465 nm) and using catalyst A (20 mol%), 1.5 equiv. of 1a, and 10 equiv. of 3a. b Yield of 4a determined by 1H NMR analysis of the crude mixture using trichloroethylene as the internal standard; value in parenthesis refers to the yield of isolated 4a. DCE: 1,2-dichloroethane; AcOEt: ethyl acetate; NPhth: phthalimide. | ||
| 1 | None | 70 (57) |
| 2 | AcOEt instead of DCE | 45 |
| 3 | 40 °C | 23 |
| 4 | 10 mol% catalyst A | 47 |
| 5 | No light | 0 |
| 6 | No catalyst A | 0 |
| 7 | fac-Ir(ppy)3 (3 mol%); no catalyst A | 0 |

Using the optimized conditions described in Table 1, entry 1, we turned our attention to exploring the generality of the photoinduced radical cascade process (Fig. 3). The method is amenable to synthetically useful purposes, since a high efficiency was maintained when running the reaction on a 5 mmol scale. This experiment did not require any modification of the standard experimental set-up (see ESI† for details), and it afforded product 4a in good yield and with complete diastereoselectivity (0.91 g, 52% yield, >20:1 dr). We then explored the scope of the radical precursors 1 that could be activated by the DTC catalyst A. Our approach displayed a high degree of tolerance towards N-heterocycles, as the cascade products 4 could be readily adorned with triazole (4b), pyrazole (4c), isoxazole (4d), benzothiazole (4e), and thiazole (4f) scaffolds. Other heterocyclic moieties, such as furan (4g) and thiophene (4h), were also tolerated. These common motifs are typically found in drug molecules. However, they generally represent a significant tolerability challenge for synthetic methods.14a The corresponding MCR products 4 were obtained with good yields and complete diastereoselectivity. Importantly, the reduction potentials of the heterocyclic-containing substrates 1, evaluated by cyclic voltammetry, are sufficiently negative (Ered < −2.0 V vs. Ag/AgCl, see ESI†) to remain out of reach of most commonly used photoredox catalysts.5 Therefore, SET-based reduction methods to generate the target radicals and trigger this MCR process would require strong reductants, greatly limiting functional group compatibility.
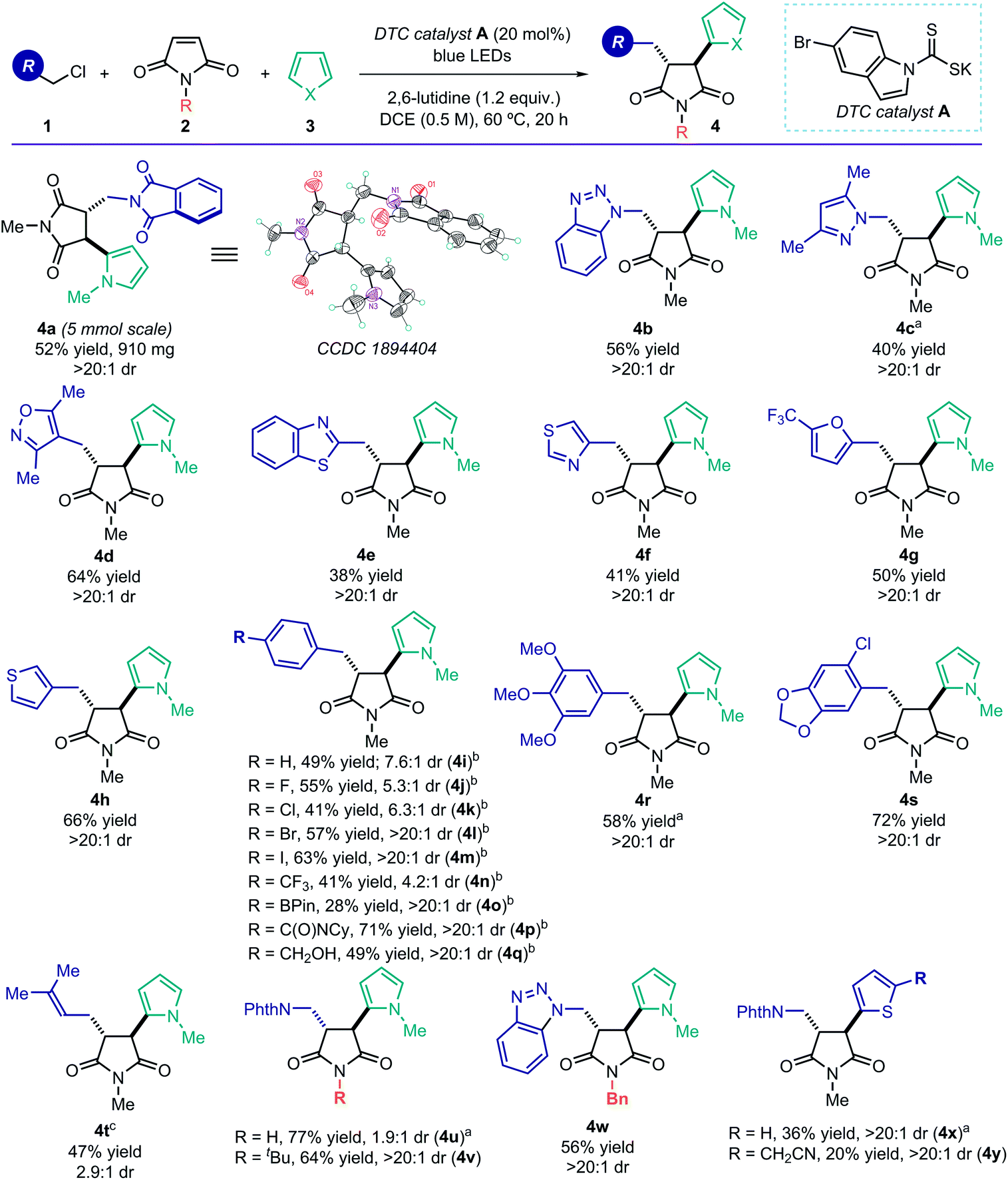 | ||
| Fig. 3 Reaction scope: reactions performed on 0.5 mmol scale using 1.5 equiv. of 1, 10 equiv. of 3, and 1.0 mL of 1,2-dichloroethane; yields of products 4 refer to isolated material after purification. aNMR yield determined using 1,1,2-trichloroethylene as the internal standard. bThe corresponding bromides 1 were used as radical precursors. c3 equiv. of 1; NPhth: phthalimide. | ||
Benzylic radical precursors, bearing both electron-rich and electron-poor aryl substituents, were also competent substrates, affording the complex products in moderate to high chemical yields (4i–4s, 28–72% yield). Reactive functional groups, including an aryl iodide (4m), a boron ester (4o), an amide (4p), and a free alcohol (4q), were all tolerated and activated exclusively at the desired benzylic position. Regarding the diastereoselectivity, full control was achieved with electron-rich benzyl systems, while the presence of electron-withdrawing moieties affected the relative stereocontrol (4i–k, 4n). In addition, prenyl chloride, which delivered an allylic radical upon activation, could be successfully used in the MCR (product 4t), albeit with reduced diastereoselectivity. The N-protecting group of the maleimide 2 was found to be important for stereocontrol, as unprotected maleimide resulted in low diastereoselectivity (product 4u). Substrates 2 bearing more encumbered protecting groups were well tolerated (adducts 4v, 4w), and thiophene was found to be an alternative terminal radical trap (4x and 4y). A complete list of moderately successful and unsuccessful substrates for this radical MCR strategy is reported in Section D, Fig. S5 of the ESI.†
To showcase the system's synthetic utility, we used this strategy to prepare highly functionalized pyrroles, which are relevant motifs found in natural products, pharmaceuticals, and materials.18 Specifically, we envisioned an assembly line protocol that combines two sequential radical processes, governed by the DTC catalyst A and visible light, to achieve a 2,5-difunctionalization of a commercial pyrrole building block (Scheme 1a). First, the photochemical activation of chloroacetonitrile 5 by A generates an electrophilic radical, which is readily intercepted by N-methyl pyrrole to deliver the functionalized intermediate 6 in 92% yield.8 By exploiting the residual nucleophilic character of C5, the pyrrole 6 was then used in the MCR process with N-(chloromethyl)phthalimide 1a and N-methylmaleimide 2a to rapidly assemble the difunctionalized pyrrole 7 from commercially available substrates.
 | ||
| Scheme 1 (A) Photochemical radical assembly line and (B) derivatization of MCR products. | ||
Finally, we demonstrated that the succinimide moiety within the MCR products 4 can be readily transformed into a pyrrolidine, which is a synthetically and biologically relevant framework.19 As highlighted in Scheme 1b, the reduction of adduct 4w with LiAlH4 affords the corresponding trans-3,4-disubstituted pyrrolidine 8. This transformation further testifies to the potential of this MCR strategy to cover biologically relevant chemical space.
Conclusions
We have developed a visible-light-mediated three-component radical strategy, which couples readily available alkyl chlorides, maleimides, and heteroaromatic fragments to rapidly generate complex products. The MCR is founded upon an SN2-based radical-generation strategy, which is not reliant on the redox properties of the radical precursors and therefore overcomes the limitations of photoredox chemistry. The redox neutral conditions of this process make it tolerant of redox-sensitive substrates and allow the installation of multiple biologically relevant heterocycles within the products. These findings, along with the experimental simplicity and the low cost of the catalyst, suggest that this method can inspire the design of other voltage-independent radical applications and would be amenable to interrogating new chemical space in medicinal chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by MICIU (CTQ2016-75520-P), the AGAUR (Grant 2017 SGR 981), and the European Research Council (ERC-2015-CoG 681840 - CATA-LUX). S. C. thanks the MEFP for an FPU predoctoral fellowship (ref. FPU14/06541).Notes and references
- (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed; (b) L. F. Tietze, G. Brasche and K. M. Gericke, in Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006 CrossRef.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed; (b) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- (a) V. Liautard and Y. Landais, ‘Free-radical Multicomponent Processes’, in Multicomponent Reactions in Organic Synthesis, ed. J. Zhu, Q. Wang and M.-X. Wang, Wiley-VCH, 2015, ch. 14, pp. 401–438 Search PubMed; (b) E. Godineau and Y. Landais, Chem.–Eur. J., 2009, 15, 3044–3055 CrossRef CAS PubMed.
- J. Lalevée and J. P. Fouassier, ‘Overview of Radical Initiation’, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, 2012, vol. 1, pp. 37–56 Search PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed; (c) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 Search PubMed.
- (a) M. P. Plesniak, H.-M. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1, 0077 CrossRef; (b) L. J. Sebren, J. J. Devery and C. R. J. Stephenson, ACS Catal., 2014, 4, 703–716 CrossRef CAS PubMed; (c) S. Garbino, D. Ravelli, S. Protti and A. Basso, Angew. Chem., Int. Ed., 2016, 55, 15476–15484 CrossRef PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed.
- Substrates bearing thio functions, including xanthates, have been extensively used in stoichiometric amounts to efficiently generate radicals, see: (a) D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC; (b) D. H. R. Barton and S. Z. Zard, Pure Appl. Chem., 1986, 58, 675–684 CAS; (c) P. Delduc, C. Tailhan and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1988, 308–310 RSC; (d) S. Z. Zard, Angew. Chem., Int. Ed., 1997, 36, 672–685 CrossRef.
- For a study discussing the high nucleophilicity of dithiocarbonate and dithiocarbamate anions of type A, see: X.-H. Duan, B. Maji and H. Mayr, Org. Biomol. Chem., 2011, 9, 8046–8050 RSC.
- For studies discussing the tendency of thiocarbonyl compounds of type I to generate radicals upon photolytic cleavage, see: (a) D. H. R. Barton, M. V. George and M. Tomoeda, J. Chem. Soc., 1962, 1967–1974 RSC; (b) J. Lalevée, N. Blanchard, M. El-Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2347–2352 CrossRef.
- Few examples of carbo-difunctionalization of maleimides via radical MCRs have been reported, see: (a) C. Cadot, P. I. Dalko, J. Cossy, C. Ollivier, R. Chuard and P. Renaud, J. Org. Chem., 2002, 67, 7193–7202 CrossRef CAS PubMed; (b) A. P. Schaffner, K. Sarkunam and P. Renaud, Helv. Chim. Acta, 2006, 89, 2450–2461 CrossRef CAS; (c) Q. Huang and S. Z. Zard, Org. Lett., 2017, 19, 3895–3898 CrossRef CAS PubMed; (d) Q. Huang and S. Z. Zard, Org. Lett., 2018, 20, 5304–5308 CrossRef CAS PubMed. For the use of N-protected pyrroles, see: (e) J. Tang, J.-J. Yue, F.-F. Tao, G. Grampp, B.-X. Wang, F. Li, X.-Z. Liang, Y.-M. Shen and J.-H. Xu, J. Org. Chem., 2014, 79, 7572–7582 CrossRef CAS PubMed . For a pioneering carbo-heteroatom functionalization of maleimides via radical paths, see: ; (f) D. H. R. Barton and M. Ramesh, J. Am. Chem. Soc., 1990, 112, 891–892 CrossRef CAS.
- (a) M. L. Curtin, R. B. Garland, H. R. Heyman, R. R. Frey, M. R. Michaelides, J. Li, L. J. Pease, K. B. Glaser, P. A. Marcotte and S. K. Davidsen, Bioorg. Med. Chem. Lett., 2002, 12, 2919–2923 CrossRef CAS PubMed; (b) M. J. Kornet, A. M. Crider and E. O. Magarian, J. Med. Chem., 1977, 20, 405–409 CrossRef CAS PubMed; (c) A. M. Crider, T. M. Kolczynski and K. M. Yates, J. Med. Chem., 1980, 23, 324–326 CrossRef CAS PubMed.
- (a) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (b) W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, J. Med. Chem., 2009, 52, 2952–2963 CrossRef CAS PubMed.
- The parent ethyl xanthogenate anion, where the indole moiety in A is replaced by OEt, has an oxidation potential as low as +0.04 V vs. SCE, see: Ö. Dag, S. Ö. Yaman, A. M. Onal and H. Isci, J. Chem. Soc., Dalton Trans., 2001, 2819–2824 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- Previous approaches to generate the phthalimidomethyl radical required atom abstraction mechanisms or the use of stoichiometric xanthates; for examples see: (a) K. Yamada, Y. Matsumoto, S. Fujii, T. Konishi, Y. Yamaoka and K. Takasu, J. Org. Chem., 2016, 81, 3809–3817 CrossRef CAS PubMed; (b) B. Quiclet-Sire and S. Z. Zard, Org. Lett., 2008, 10, 3279–3282 CrossRef CAS PubMed.
- (a) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed; (b) C. Schmuck and D. Rupprecht, Synthesis, 2007, 3095–3110 CrossRef CAS.
- (a) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC; (b) J. M. Schomaker, S. Bhatacharjee, J. Yan and B. Borhan, J. Am. Chem. Soc., 2007, 129, 1996–2003 CrossRef CAS PubMed , and references therein.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and full compound characterization. CCDC 1894404 (4a) contains the supplementary crystallographic data for this paper. CCDC 1894404. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00833k |
| ‡ S. C. and M. A. H. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |