
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isothiourea catalysed enantioselective generation of point and axially chiral iminothia- and iminoselenazinanones†
Alastair J.
Nimmo
 ,
Alister S.
Goodfellow
,
Jacob T.
Guntley
,
Aidan P.
McKay
,
David B.
Cordes
,
Michael
Bühl
* and
Andrew D.
Smith
*
,
Alister S.
Goodfellow
,
Jacob T.
Guntley
,
Aidan P.
McKay
,
David B.
Cordes
,
Michael
Bühl
* and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, Fife, KY16 9ST, UK. E-mail: buehl@st-andrews.ac.uk; ads10@st-andrews.ac.uk
First published on 30th April 2025
Abstract
Symmetrical and unsymmetrical thioureas, as well as unsymmetrical selenoureas, are used in an isothiourea-catalysed Michael addition-lactamisation protocol using α,β-unsaturated pentafluorophenyl esters to generate iminothia- and iminoselenazinanone heterocycles with high enantioselectivity (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er). The scope and limitations of this process have been widely investigated (40 examples in total) with unsymmetrical thio- and selenoureas containing ortho-substituted N-aryl substituents giving atropisomeric products, leading to an effective process for iminothia- and iminoselenazinanones heterocyclic products containing both point and axially chiral stereogenic elements with excellent stereocontrol (up to >95:5 dr and 98:2 er). Mechanistic investigation showed that (i) the catalytically liberated aryloxide could deprotonate an electron-deficient thiourea; (ii) in the absence of an isothiourea catalyst, this leads to formation of racemic product; (iii) a crossover experiment indicates the reversibility of the thia-Michael addition. Computational analysis has identified the factors leading to enantioselectivity within this process, with stereocontrol arising from the lactamisation step within the catalytic cycle.
:1 er). The scope and limitations of this process have been widely investigated (40 examples in total) with unsymmetrical thio- and selenoureas containing ortho-substituted N-aryl substituents giving atropisomeric products, leading to an effective process for iminothia- and iminoselenazinanones heterocyclic products containing both point and axially chiral stereogenic elements with excellent stereocontrol (up to >95:5 dr and 98:2 er). Mechanistic investigation showed that (i) the catalytically liberated aryloxide could deprotonate an electron-deficient thiourea; (ii) in the absence of an isothiourea catalyst, this leads to formation of racemic product; (iii) a crossover experiment indicates the reversibility of the thia-Michael addition. Computational analysis has identified the factors leading to enantioselectivity within this process, with stereocontrol arising from the lactamisation step within the catalytic cycle.
1. Introduction
Since the introduction of isothioureas as catalysts for the acylative kinetic resolution of alcohols by Birman,1 these versatile Lewis bases have been widely developed and applied in enantioselective processes. Their simple and scalable synthesis, combined with their ability to generate multiple reactive intermediates 1–5 (acyl ammonium,2 α,β-unsaturated acyl ammonium,3 C(1)-ammonium enolate,4 betaine5 and silyl ammonium6 species) from simple starting materials has led to their widespread popularity (Scheme 1A). Isothiourea-catalysed Michael additions to in situ generated α,β-unsaturated acyl ammonium species have been used extensively to achieve enantioselective C–C bond formation.7 However, their use to selectively generate C-heteroatom bonds through conjugate addition of non-carbon centred nucleophiles is limited to relatively few C–S and C–N bond-forming processes. Within this area, Matsubara has reported a thia-Michael addition-lactamisation strategy for the synthesis of 1,5-benzothiazepines 9 using N-tosylated aminothiophenols 6 and α,β-unsaturated mixed anhydrides 7 in the presence of (R)-BTM 8 (Scheme 1B).8 The products were isolated in good to excellent yields (70 to 99%) and with excellent enantioselectivity (94:6 to 99:1 er). Mechanistic investigations revealed that the high enantioselectivity is a result of a dynamic kinetic process determined by the relative lactamisation rates of the diastereomeric thia-Michael addition products rather than the thia-Michael addition, which is readily reversible. This methodology was extended to 3-substituted and 2,3-disubstituted benzothiazepines with excellent stereoselectivity (79:21 to 94:6 er).9 Birman subsequently reported a route to enantioenriched thiochromenes 13 using thioesters 10 as α,β-unsaturated acyl ammonium precursors (Scheme 1C).10
 | ||
| Scheme 1 (A) Isothiourea derived catalytic intermediate. (B) & (C) Enantioselective C–S bond forming processes using α,β-unsaturated acyl ammonium species. | ||
Enantioselective C–S bond formation via thia-Michael addition is followed by an aldol-lactonisation cascade forming β-lactones 12, which readily decarboxylate, giving the corresponding thiochromene 13 in good to excellent yields (53 to 99%) and with excellent enantiocontrol (97:3 to >99:1 er). Alongside these advances, in previous work we developed the aza-Michael addition of 2-hydroxybenzophenone imines to α,β-unsaturated esters allowing access to β-amino amides in excellent yield (95%) and enantioselectivity (96:4 er).11
In addition, in recent years there has been an explosion of interest concerning the development of selective synthetic methods for the preparation of atropisomeric species.12 While axially chiral biaryl (and heterobiaryl) species containing a C–C axis have been most widely studied, recent work has shown that the generation of axially chiral and configurationally stable C–N atropisomers is possible.13 Given these precedents, we considered alternative ways of developing enantioselective C–S and C–Se bond formation via an α,β-unsaturated acyl ammonium intermediate that could lead to heterocycles bearing both point and axially chiral stereogenic elements. In recent work Jin, Chi and coworkers used thioureas as dinucleophiles in an atropselective synthesis of iminothiazinones 18 using the NHC derived from precatalyst 17 (20 mol%, Scheme 2A).14 Thia-Michael addition-lactamisation of the thiourea 14 with an in situ generated acyl azolium intermediate gives iminothiazinones 18 in moderate to high yields (29 to 71%) with excellent enantioselectivity (91:9 to >99:1 er) with the formation of a C–N stereogenic axis. While effective, stoichiometric quantities (3 equiv.) of oxidant 16 was required to achieve oxidation of the Breslow intermediate in this protocol and furan was required as a solvent. This manuscript describes an alternative methodology to access related heterocyclic products that utilises α,β-unsaturated ester substrates and so avoids the need for in situ oxidation. Reaction via an in situ generated α,β-unsaturated acyl ammonium species derived from an α,β-unsaturated aryl ester and an isothiourea was postulated.15 In such processes, the aryloxide liberated upon isothiourea acylation can fulfil multiple roles, including that of Brønsted base.10,15,16 The hydrogen bond donor ability of electron-deficient thioureas, commonly exploited in catalytic applications, is concomitant with a pKa that facilitates deprotonation by the catalytically liberated aryloxide,17 activating this towards thia-Michael addition with an α,β-unsaturated acyl ammonium intermediate 23 (Scheme 2B). This simple methodology would require no additional reagents and allow access to iminothiazinanone heterocycles 25 that possess a stereogenic centre. Furthermore, the incorporation of a 2-substituted aryl substituent on the thiourea would generate atropisomeric products 24 that also contain a stereogenic centre, while extension to selenoureas would give access to chiral iminoselenazinanones for the first time.
 | ||
| Scheme 2 (A) NHC-catalysed synthesis of atropisomeric iminothiazinones. (B) This work: isothiourea catalysed preparation of point and axially chiral heterocycles. | ||
2. Results and discussion
2.1 Development of model process with symmetrical thioureas
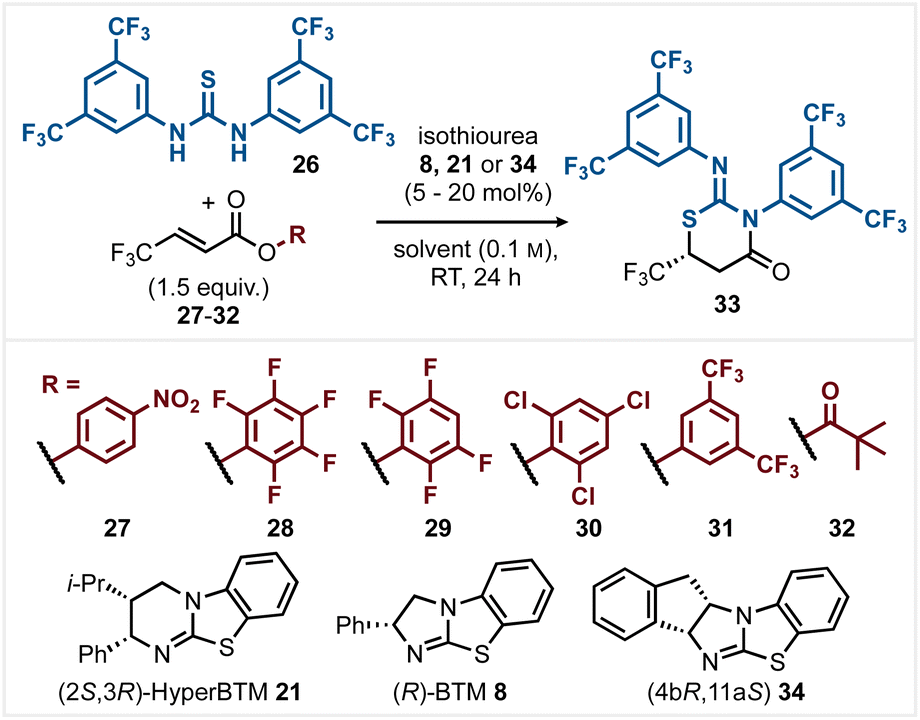
Optimisation began with the reaction of α,β-unsaturated para-nitrophenyl (PNP) ester 27 with Schreiner's thiourea 26 and (2S,3R)-HyperBTM 21 (20 mol%) in MeCN (0.1 M) (Table 1, entry 1) giving product 33 in quantitative yield with 78:22 er. Variation of the reaction solvent (see ESI† for full range of solvents trialled) indicated that most common solvents gave >95% conversion to product. CH2Cl2 (entry 2) gave reduced enantioselectivity (66:34 er), while EtOAc, DMF, and THF (entries 3–5) all gave improved enantioselectivity. The use of 2-MeTHF and i-PrOAc also led to high product enantioselectivity (entries 6 and 7) with further optimisation using THF. Catalyst variation and loading were next tested. While HyperBTM 20 (5 mol%) gave 33 in quantitative yield and 91:9 er, the use of alternative isothioureas 8 and 34 led to only moderate selectivity (entries 8–10). Variation of the nucleofuge within the α,β-unsaturated ester or anhydride reaction component was next trialled. Among the range of aryl esters tested (entries 11–14) the observed product enantioselectivity correlated with the pKa of the corresponding phenol.18 Pentafluorophenyl (PFP) ester 28 (entry 11) with the most acidic phenol (pKa 5.53 in H2O) gave the greatest enantioselectivity (93:7 er) at 90% conversion, while 3,5-bis(trifluoromethyl)phenyl ester 31 (entry 14) with the least acidic phenol (pKa 8.26 in H2O) gave the lowest observed product enantioselectivity (90:10 er). The use of in situ generated pivalic anhydride 32 did not lead to improved selectivity (entry 15, 83%, 90:10 er). The use of PFP ester 28 at 0 °C led to optimal reaction selectivity, giving product 33 in 95% isolated yield and 95:5 er (entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acyl donor | Catalyst (mol%) | Solvent | Yielda | erb |
| a Yield determined by 1H NMR analysis relative to internal standard 1,3,5-trimethoxybenzene (isolated yield in parentheses). b er measured by HPLC analysis on a chiral stationary phase. c Performed at 0 °C. | |||||
| 1 | 27 | 21 (20) | MeCN | Quant. | 78:22 |
| 2 | 27 | 21 (20) | CH2Cl2 | Quant. | 66:34 |
| 3 | 27 | 21 (20) | EtOAc | Quant. | 90:10 |
| 4 | 27 | 21 (20) | DMF | Quant. | 84:16 |
| 5 | 27 | 21 (20) | THF | Quant. | 91:9 |
| 6 | 27 | 21 (20) | 2-MeTHF | Quant. | 91:9 |
| 7 | 27 | 21 (20) | i-PrOAc | 98 | 90:10 |
| 8 | 27 | 21 (5) | THF | Quant. | 91:9 |
| 9 | 27 | 8 (5) | THF | 96 | 84:16 |
| 10 | 27 | 34 (5) | THF | 98 | 33:67 |
| 11 | 28 | 21 (5) | THF | 90 | 93:7 |
| 12 | 29 | 21 (5) | THF | Quant. | 92:8 |
| 13 | 30 | 21 (5) | THF | 96 | 92:8 |
| 14 | 31 | 21 (5) | THF | 95 | 90:10 |
| 15 | 32 | 21 (5) | THF | 83 | 90:10 |
| 16c | 28 | 21 (5) | THF | (95) | 95:5 |
2.2 Scope of the developed process with symmetrical thioureas
With optimised conditions for the generation of model heterocycle 33 developed, the scope and limitations of this process were investigated through variation of the N-aryl substituents within the thiourea and the β-substituent within a range of α,β-unsaturated esters (Scheme 3). Using β-trifluoromethyl α,β-unsaturated PFP ester 28 as standard, the incorporation of 4-F3CC6H4 substituents within the bis-N-arylthiourea led to reduced product conversion at 0 °C, but at RT gave 35 in 79% yield with excellent enantioselectivity (95:5 er). The incorporation of strongly electron-withdrawing 4-O2NC6H4 and 4-NCC6H4N-aryl substituents within the thiourea showed similar reactivity to that of the model substrate, giving products 36 and 37 in excellent yields (95% and 83%) and enantioselectivity (98:2 er and 97:3 er respectively). The absolute configuration within (6R,Z)-37 was confirmed by single crystal X-ray analysis, with the configuration within all other products assigned by analogy.19 Further work probed the scope of the α,β-unsaturated ester Michael acceptors using bis-4-O2NC6H4 substituted thiourea. The incorporation of perhalogenated β-CF2Cl- and perfluoroethyl β-substituents gave products 38 and 39 in good yield (78% and 59%) with excellent enantioselectivity (both 98:2 er).20 While β-CF2H and β-CH2F substituents within the ester were tolerated giving products 40 and 41 respectively, reduced enantioselectivity was observed in both cases (80:20 er and 83:17 er respectively). To further test this methodology the use of β-alkyl substituted esters was probed as these generally show moderate reactivity in reactions involving the corresponding α,β-unsaturated acyl ammonium species. In this context, 42–45 were generated in excellent yields and promising enantioselectivity (86:14 er to 94:6 er) although these reactions were carried out at room temperature and required higher catalyst loadings (20–30 mol%). Unfortunately the use of β-aryl substituted PFP esters (β-aryl = Ph, 4-NO2C6H4) were unreactive in this process using bis-4-O2NC6H4 substituted thiourea and represent a limitation of this methodology.
 | ||
| Scheme 3 All yields are isolated; all er ratios determined by HPLC analysis on a chiral stationary phase; [a] 10 mol% 21 used at 0 °C; [b] 20 mol% 21 used at RT; [c] 30 mol% 21 used at RT. | ||
2.3 Application to unsymmetrical thio- and selenoureas
Further work extended this process to the use of unsymmetrical thio- and selenoureas (Scheme 4). In principle these starting materials could lead to the formation of regioisomeric products, and so to bias regioselectivity variation in the electronic properties of the nitrogen substituents was used to dictate reactivity. Initial studies considered the utility of a range of thioureas bearing electronically distinct N-substituents with PFP ester 28 (Scheme 4A). In each case a single regioisomeric product was observed but with varying levels of enantioselectivity. Reaction to generate 46 required 20 mol% catalyst and 48 h at RT to reach completion, giving 46 in 77% yield but in racemic form. Replacing the N-4-nitrophenyl substituent with an N-tosyl substituent led to improved reactivity, giving 47 in high yield (88%) using 5 mol% catalyst at 0 °C but with moderate enantioselectivity (67:33 er). Using an N-benzyl substituted derivative gave 48 in 98% yield with improved enantioselectivity (78:22 er), while N-aryl variants gave 49–51 in high yields (79 to 91%) and good enantioselectivity (88:12 to 91:9 er). Variation within the electron-withdrawing substituents was also tolerated, with N-4-methoxybenzenesulfonyl and N-trifluoroacetyl variants giving 52 and 53 with high enantioselectivity (91:9 er and 96:4 er). As a further test, it was postulated that regioselectivity could be achieved through a steric bias within two electron-withdrawing N-aryl substituents. Using the sterically demanding N-2,4,6-trichlorophenyl substituent to disfavour cyclisation led to a single regioisomer of 54 in excellent yield (92%) with excellent enantioselectivity (98:2 er). The constitution of 46 and 54 were confirmed by 1H–15N HMBC (see ESI† for further information).
 | ||
| Scheme 4 All yields are isolated; all er ratios determined by HPLC analysis on a chiral stationary phase; [a] 20 mol% 21 used at RT; [b] 10 mol% 21 used at 0 °C; [c] 10 mol% 21 used at RT; [d] 30 mol% 21 used at RT. | ||
Subsequent work extended this process to the use of unsymmetrical selenoureas to give iminoselenazinanones (Scheme 4B). Pleasingly, N-Bz,N-Ph-substituted selenourea led to 55 in excellent yield and enantioselectivity (97%, 99:1 er). In this series, alternative N-aryl substitution was investigated, with the incorporation of the antibiotic sulfamethoxazole within a selenourea giving 56 in good yield (61%) with excellent 98:2 er. 4-MeOC6H4 substitution gave 57 with excellent yield (97%) and enantioselectivity (98:2 er). Variation within the N-benzoyl substitution was also well tolerated, with 4-MeOC6H4, 4-BrC6H4, and 4-O2NC6H4 substituents giving products 58, 59, and 60 respectively in high yields (80 to 92%) and excellent enantioselectivity (97:3 to 99:1 er). To demonstrate that the selenoureas were reactive with alternative Michael acceptors, two representative examples were chosen. Reaction with ethyl ester substituted Michael acceptor at RT gave product 61 in 71% yield with high enantioselectivity (93:7 er). The reactivity of selenoureas was pushed to a limit with the β-methyl substituted Michael acceptor with the use of 30 mol% 21 at RT for 48 h giving 62 in low 29% yield (87:13 er) and so represents a limit of this methodology. Further work investigated the use of thioureas and a selenourea bearing ortho-substituted aryl substituents to give atropisomeric products (Scheme 4C). Initial studies showed that chalcogenoureas bearing an ortho-substituted aryl substituent give atropisomeric products 63–66 upon treatment with pentafluorophenyl acrylate and HyperBTM. Although the isolated yields of products 63–66 were good to excellent (71 to 98%), the enantioselectivities were only moderate (73:27 er to 87:13 er). The configurational stability of 63–66 was determined as described by Armstrong,21 with increasing barriers to rotation observed with increased steric hindrance of the 2-aryl substituent (Ph < i-Pr < t-Bu), and with Se-containing 66 possessing a lower barrier than its sulfur analogue. Intrigued by these observations, application to products containing both a stereogenic centre and a stereogenic axis were investigated. Using the previously developed conditions, treatment of a thiourea bearing an ortho-iso-propyl substituent with PFP ester 28 and HyperBTM gave 67 in excellent yield (94%) and stereoselectivity (92:8 dr, 98:2 er). Similarly, thioureas bearing ortho-phenyl and ortho-tert-butyl substituents gave the corresponding products 68 and 69 with excellent stereoselectivity. Using a selenourea bearing an ortho-iso-propyl substituent gave 70 in 86% yield and excellent stereoselectivity (>95:5 dr, 98:2 er). The generality of the enantio- and atropselective methodology was further investigated by reaction of an ortho-iso-propyl substituted thiourea with three alternative β-substituted α,β-unsaturated esters. CF2Cl substitution gave 71 in excellent yield (89%) and stereoselectivity (>95:5 dr, 98:2 er), while pleasingly β-alkyl substituted Michael acceptors were also tolerated, giving 72 and 73 with high yields (82% and 86%) and stereoselectivity (88:12 dr, 92:8 er and 92:8 dr, 95:5 er). The absolute configuration of (6R,Ra,Z)-67 was confirmed by single crystal X-ray crystallography, with the relative configuration within 72 confirmed by 1H NOESY NMR analysis (see ESI Section 13† for further information). The configuration within all other products was assigned by analogy.
2.4 Mechanistic analysis and control studies
 | ||
| Scheme 5 (A) Determining role of aryloxide by 1H NMR titration. (B) Aryloxide catalysed racemic background reaction. (C) Probing reversibility of the thia-Michael addition by crossover experiment. | ||
2.5 Computational analysis and proposed mechanism
Based upon these observations a catalytic cycle for this process can be outlined, with the origins of stereocontrol probed by DFT calculations performed at the M06- 2XSMD(THF)/def2-TZVP//M06-2XSMD(THF)/def2-SVP level of theory using Gaussian16 (Scheme 6). Using the symmetric bis-4-O2NC6H4 substituted thiourea and PFP ester 28 as a model, N-acylation of the PFP ester 28 by the Lewis base (2R,3S)-HyperBTM 21 generates the corresponding acyl ammonium ion pair I with a stabilising 1,5-O⋯S chalcogen bonding interaction (nO → σ*S–C,E(2) = 6.0 kcal mol−1, described by NBO second-order perturbation analysis).23–32 This ensures coplanarity between the 1,5-O- and S-atoms and provides a conformational bias.26 Deprotonation of the thiourea promotes thia-Michael addition viaTS1 to give II. Subsequent proton transfer gives III, with intramolecular cyclisation viaTS2 generating IV. Catalyst release is promoted by collapse of the tetrahedral intermediate IVviaTS3 generating the heterocyclic product 36 in high enantioselectivity (Scheme 6A). | ||
| Scheme 6 (A) Proposed catalytic cycle. (B) DFT analysis of the stereodetermining transition state. (C) Computed reaction profile leading to enantiomeric products. M06-2XSMD(THF)/def2-TZVP//M06-2XSMD(THF)/def2-SVP Gibbs free energies (ΔG273) shown in kcal mol−1. | ||
Stereoselectivity in the initial Michael addition considered the anionic thiourea nucleophile approaching either the Re- or Si-face of acyl ammonium intermediate I (Scheme 6B and C). Approach to the Si-face is hindered by the stereodirecting phenyl group within (2S,3R)-HyperBTM leading to favoured addition to the Re-face (TS1, ΔΔG273‡ = 1.3 kcal mol; 92:8 er (R)). After proton transfer, the pendant nitrogen nucleophile can initiate lactamisation (TS2) to generate the tetrahedral cyclised product adduct, before catalyst turnover (TS3) to generate the product. These diastereomeric lactamisation transition states (TS2) were computed to be higher in energy than the thia-Michael addition transition states, TS1 (2.5 kcal mol−1 higher for the (R)-pathway), due to the geometric constraints of ring-closure. While the stereogenic centre is formed in the Michael addition (TS1), computation suggested that the observed enantioselectivity arises from the energetic difference between the higher energy lactamisation transition states (TS2). Turnover of the catalyst (TS3) is strongly exergonic and proceeds through a lower energy transition state compared to TS2. Every step of the reaction (Scheme 6C) was computed to be reversible up until catalyst turnover (TS3), in agreement with the experimentally observed crossover and retro-Michael addition in Scheme 5C. Lactamisation (TS2) was computed to be enantiodetermining with the favoured (R)-pathway proceeding through nucleophilic addition from the less hindered Si-face of the acyl ammonium. The disfavoured (S)-pathway is initially positioned to lactamise from the hindered Re-face of the acyl ammonium (TS2-(S)′, ΔΔG273‡ = +5.3 kcal mol−1). However, rotation allows lactamisation from the less hindered Si-face (TS2-(S), ΔΔG273‡ = +2.7 kcal mol−1).
3. Conclusion
In conclusion, a range of symmetrical and unsymmetrical thioureas and selenoureas can be utilised in an isothiourea-catalysed protocol to generate iminothia- and iminoselenazinanone heterocycles with high enantioselectivity (up to 99:1 er). Notably, unsymmetrical thio- and selenoureas containing ortho-substituted N-aryl substituents generate atropisomeric products. This allows the generation of iminothia- and iminoselenazinanone heterocyclic products containing both point and axially chiral stereogenic elements with excellent stereocontrol (up to >95:5 dr and 98:2 er) for the first time. Mechanistic investigations indicate that catalytically liberated aryloxide can deprotonate an electron-deficient thiourea, while a crossover experiment indicates the reversibility of the thia-Michael addition. Extensive computational analysis has identified the factors leading to enantioselectivity within this process, with stereocontrol shown to arise from the lactamisation step within the catalytic cycle.
Data availability
All data (experimental procedures, characterisation data and cartesian coordinates for all DFT calculations) that support the findings of this study are available within the article and its ESI.† The research data supporting this publication can be accessed from “Isothiourea Catalysed Enantioselective Generation of Point and Axially Chiral Iminothia- and Iminoselenazinanones”. Pure ID: 314763684. University of St Andrews Research Portal “PURE” [https://doi.org/10.17630/3ffd33a9-50ae-441d-bd88-6a3343351c63].Author contributions
Alastair J. Nimmo – conceptualization, investigation, writing. Alister S. Goodfellow – formal analysis, investigation writing-review and editing. Jacob T. Guntley – investigation. Aidan P. McKay, David. B. Cordes, investigation (X-ray analysis). Michael Bühl – investigation, funding acquisition, writing-review and editing. Andrew D. Smith – conceptualization, funding acquisition, project administration writing-review and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the EaSICAT Centre for Doctoral Training (AJN, ASG) for funding. ADS thanks the EPSRC Programme Grant “Boron: Beyond the Reagent” (EP/W007517) for support. MB thanks EaStCHEM and the School of Chemistry for support. Computations were performed on a local HPC cluster maintained by Dr H. Früchtl. To meet institutional and research funder open access requirements, any accepted manuscript arising shall be open access under a Creative Commons Attribution (CC BY) reuse licence with zero embargo.Notes and references
- V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS.
- For reviews see: (a) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org Chem., 2016, 2016, 5589–5610 CrossRef CAS; (b) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC; (c) V. B. Birman, Aldrichimica Acta, 2016, 49, 23–41 CAS.
- For reviews see: (a) S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2016, 55, 13934–13943 CrossRef CAS; (b) J. Bitai, M. T. Westwood and A. D. Smith, Org. Biomol. Chem., 2021, 19, 2366–2384 RSC.
- For reviews see (a) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC; (b) C. McLaughlin and A. D. Smith, Chem.–Eur. J., 2021, 27, 1533–1555 CrossRef CAS PubMed.
- (a) L. S. Vogl, P. Mayer, R. Robiette and M. Waser, Angew. Chem., Int. Ed., 2024, 63, e202315345 CrossRef CAS; (b) M. Piringer, M. Hofer, L. S. Vogl, P. Mayer and M. Waser, Adv. Synth. Catal., 2024, 366, 2115–2122 CrossRef CAS PubMed.
- (a) C. I. Sheppard, J. L. Taylor and S. L. Wiskur, Org. Lett., 2011, 13, 3794–3797 CrossRef CAS PubMed; (b) R. W. Cark, T. M. Deaton, Y. Zhang, M. I. Moore and S. L. Wiskur, Org. Lett., 2013, 15, 6132–6135 CrossRef; (c) T. Zhang, B. K. Redden and S. L. Wiskur, Eur. J. Org Chem., 2019, 30, 4827–4831 CrossRef; (d) L. Wang, R. K. Akhani and S. L. Wiskur, Org. Lett., 2015, 17, 2408–2411 CrossRef CAS; (e) T. Hu, Y. Zhang, W. Wang, Q. Li, L. Huang, J. Gao, Y. Kuang, C. Zhao, S. Zhou, Z. Su and Z. Song, J. Am. Chem. Soc., 2024, 146, 23092–23102 CrossRef CAS PubMed.
- (a) A. Matviitsuk, M. D. Greenhalgh, D.-J. B. Antúnez, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 12282–12287 CrossRef CAS; (b) E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC; (c) G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049–1057 CrossRef CAS; (d) E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193–2200 RSC; (e) A. Matviitsuk, J. E. Taylor, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Chem.–Eur. J., 2016, 22, 17748–17757 CrossRef CAS PubMed; (f) E. R. T. Robinson, A. B. Frost, P. Elías-Rodríguez and A. D. Smith, Synthesis, 2017, 49, 409–423 CAS.
- Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320–5323 CrossRef CAS PubMed.
- Y. Fukata, K. Yao, R. Miyaji, K. Asano and S. Matsubara, J. Org. Chem., 2017, 82, 12655–12668 CrossRef CAS PubMed.
- N. A. Ahlemeyer and V. B. Birman, Org. Lett., 2016, 18, 3454–3457 CrossRef CAS PubMed.
- J. E. Lapetaje, C. M. Young, C. Shu and A. D. Smith, Chem. Commun., 2022, 58, 6886–6889 RSC.
- For selected review articles, see: (a) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed; (b) B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981–2988 CrossRef CAS; (c) V. Corti and G. Bertuzzi, Synthesis, 2020, 52, 2450–2468 CrossRef CAS; (d) T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem.–Eur. J., 2020, 26, 15779–15792 CrossRef CAS; (e) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed; (f) R. Song, Y. Xie, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2021, 60, 26026–26037 CrossRef CAS; (g) G. Hedouin, S. Hazra, F. Gallou and S. Handa, ACS Catal., 2022, 12, 4918–4937 CrossRef CAS; (h) X. Zhang, K. Zhao and Z. Gu, Acc. Chem. Res., 2022, 55, 1620–1633 CrossRef CAS; (i) O. F. B. Watts, J. Berreur, B. S. L. Collins and J. Clayden, Acc. Chem. Res., 2022, 55, 3362–3375 CrossRef CAS PubMed.
- For selected reviews on C–N atropisomerism, see: (a) I. Takahashi, Y. Suzuki and O. Kitagawa, Org. Prep. Proced. Int., 2014, 46, 1–23 CrossRef CAS; (b) J. Frey, S. Choppin, F. Colobert and J. Wencel-Delord, Chimia, 2020, 74, 883–889 CrossRef CAS; (c) O. Kitagawa, Acc. Chem. Res., 2021, 54, 719–730 CrossRef CAS PubMed; (d) Y.-J. Wu, G. Liao and B.-F. Shi, Green Synth. Catal., 2022, 3, 117–136 CrossRef; (e) P. Rodríguez-Salamanca, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem.–Eur. J., 2022, 28, e202104442 CrossRef; (f) J. S. Sweet and P. C. Knipe, Synthesis, 2022, 54, 2119–2132 CrossRef CAS; (g) G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1855–1893 CrossRef CAS; (h) X. Xiao, B. Chen, Y.-P. Yao, H.-J. Zhou, X. Wang, N.-Z. Wang and F.-E. Chen, Molecules, 2022, 27, 6583 CrossRef CAS; (i) A. D. G. Campbell and R. J. Armstrong, Synthesis, 2023, 55, 2427–2438 CrossRef CAS.
- T. Li, C. Mou, P. Qi, X. Peng, S. Jiang, G. Hao, W. Xue, S. Yang, L. Hao, Y. R. Chi and Z. Jin, Angew. Chem., Int. Ed., 2021, 60, 9362–9367 CrossRef CAS.
- (a) J. Wu, C. M. Young, A. A. Watts, A. M. Z. Slawin, G. R. Boyce, M. Bühl and A. D. Smith, Org. Lett., 2022, 24, 4040–4045 CrossRef CAS PubMed; (b) J. Bitai, A. J. Nimmo, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2022, 61, e202202621 CrossRef CAS; (c) M. D. Greenhalgh, S. Qu, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2018, 9, 4909–4918 RSC; (d) D. Yuan, A. S. Goodfellow, K. Kasten, Z. Duan, T. Kang, D. B. Cordes, A. P. McKay, M. Bühl, G. R. Boyce and A. D. Smith, Chem. Sci., 2023, 14, 14146–14156 RSC.
- C. McLaughlin, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2019, 58, 15111–15119 CrossRef CAS PubMed.
- G. Jakab, C. Tancon, Z. Zhang, K. M. Lippert and P. R. Schreiner, Org. Lett., 2012, 14, 1724–1727 CrossRef CAS.
- (a) A. Kütt, V. Movchun, T. Rodima, T. Dansauer, E. B. Rusanov, I. Leito, I. Kaljurand, J. Koppel, V. Pihl, I. Koppel, G. Ovsjannikov, L. Toom, M. Mishima, M. Medebielle, E. Lork, G.-V. Röschenthaler, I. A. Koppel and A. A. Kolomeitsev, J. Org. Chem., 2008, 73, 2607–2620 CrossRef; (b) S. Espinosa, E. Bosch and M. Roses, J. Chromatogr. A, 2002, 964, 55–66 CrossRef CAS; (c) J. Han and F. M. Tao, J. Phys. Chem. A, 2006, 110, 257–263 CrossRef CAS.
- Crystallographic data for compounds 37, 55 and 67 have been deposited with the Cambridge Crystallographic Data Centre under deposition numbers 2410987, 2410988 and 2410989 respectively..
- In the perfluoroethyl case a regioisomeric product was isolated in 10% yield as a racemate. For further information and structural assignment see ESI† Section 10..
- J.-P. Heeb, J. Clayden, M. D. Smith and R. J. Armstrong, Nat. Protoc., 2023, 18, 2745–2771 CrossRef CAS.
- C. Pérez-Casas and A. K. Yatsimirsky, J. Org. Chem., 2008, 73, 2275–2284 CrossRef PubMed.
- V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed.
- P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS.
- M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS.
- E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC.
- M. D. Greenhalgh, S. M. Smith, D. M. Walden, J. E. Taylor, Z. Brice, E. R. T. Robinson, C. Fallan, D. B. Cordes, A. M. Z. Slawin, H. C. Richardson, M. A. Grove, P. H.-Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2018, 57, 3200–3206 CrossRef CAS PubMed.
- C. M. Young, A. Elmi, D. J. Pascoe, R. K. Morris, C. McLaughlin, A. M. Woods, A. B. Frost, A. de la Houpliere, K. B. Ling, T. K. Smith, A. M. Z. Slawin, P. H. Willoughby, S. L. Cockroft and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 3705–3710 CrossRef CAS.
- Y. Nagao, S. Miyamoto, M. Miyamoto, H. Takeshige, K. Hayashi, S. Sano, M. Shiro, K. Yamaguchi and Y. Sei, J. Am. Chem. Soc., 2006, 128, 9722–9729 CrossRef CAS PubMed.
- B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed.
- D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS.
- For a selection of other enantioselective processes that involve chalcogen bonding interactions see: (a) M. Breugst and J. J. Koenig, Eur. J. Org Chem., 2020, 2020, 5473–5487 CrossRef CAS; (b) A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357–4416 CrossRef CAS; (c) K.-i. Fujita, M. Iwaoka and S. Tomoda, Chem. Lett., 2006, 23, 923–926 CrossRef; (d) K.-i. Fujita, K. Murata, M. Iwaoka and S. Tomoda, J. Chem. Soc., Chem. Commun., 1995, 1995, 1641–1642 RSC; (e) K.-i. Fujita, K. Murata, M. Iwaoka and S. Tomoda, Tetrahedron Lett., 1995, 36, 5219–5222 CAS; (f) T. Wirth, Angew. Chem., Int. Ed., 1995, 34, 1726–1728 CrossRef CAS; (g) C. Bleiholder, R. Gleiter, D. B. Werz and H. Köppel, Inorg. Chem., 2007, 46, 2249–2260 CrossRef CAS PubMed; (h) S. Kolb, G. A. Oliver and D. B. Werz, Angew. Chem., Int. Ed., 2020, 59, 22306–22310 CrossRef CAS PubMed; (i) S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef CAS; (j) P. Wonner, L. Vogel, M. Düser, L. Gomes, F. Kniep, B. Mallick, D. B. Werz and S. M. Huber, Angew. Chem., Int. Ed., 2017, 56, 12009–12012 CrossRef CAS; (k) W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2410987–2410989. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02435h |
| This journal is © The Royal Society of Chemistry 2025 |