
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen chemical precipitation of manganese, cobalt, and nickel from acid mine drainage using ozone: mechanism and chemical kinetics
Younes
Shekarian
,
Mohammad
Rezaee
 * and
Sarma
Pisupati
* and
Sarma
Pisupati
John and Willie Leone Family Department of Energy and Mineral Engineering, Center for Critical Minerals, EMS Energy Institute, College of Earth and Mineral Sciences, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: m.rezaee@psu.edu
First published on 1st July 2025
Abstract
Manganese (Mn), cobalt (Co), and nickel (Ni) are designated as critical elements by the U.S. Department of the Interior. Acid mine drainage (AMD) is a viable secondary source for these metals. Conventional AMD treatment processes necessitate high pH levels (∼pH 9) or costly oxidants to recover these elements. Building upon prior work, this study utilizes an ozone oxidative precipitation method, currently patent-pending, to reduce chemical use and recover Mn, Co, and Ni from AMD. Saturation index calculations and Pourbaix diagram analyses demonstrated that ozone could recover these elements across a broad pH range (2–8). The effects of process parameters, particularly gas flow rate, stirring rate, and temperature, on the precipitation of these elements from AMD were investigated. It was found that the recovery of Mn–Co–Ni is enhanced when there is an increase in these parameters to a certain level, below which no statistically significant differences were observed. Additionally, a kinetic study on the oxidative precipitation of Mn–Co–Ni was conducted employing the pseudo-homogeneous model, and the activation energies were calculated. The effect of the process parameters, along with the calculated activation energy values (Ea(Mn) = −13.9 kJ mol−1; Ea(Co) = 16.3 kJ mol−1; Ea(Ni) = 14.5 kJ mol−1), collectively suggests that the ozone oxidative precipitation process of Mn–Co–Ni is diffusion-controlled.
1. Introduction
The recognition of manganese (Mn), cobalt (Co), and nickel (Ni) as critical elements by the U.S. Department of the Interior highlights the pressing issues over the U.S. dependency on imported resources for these metals.1,2 This reliance is particularly concerning as the demand for Mn–Co–Ni continues to increase due to the transition to electric vehicles and renewable energy technologies.2,3 Given the scarcity of primary resources for Mn–Co–Ni in the U.S., harnessing these critical elements from various viable secondary sources is crucial. One promising alternative is acid mine drainage (AMD), an effluent associated with coal and sulfide mineral mining activities, which has recently been of environmental concern but recently recognized as a viable source of multiple critical elements, including aluminum (Al), rare earth elements (REEs), Mn, Co, and Ni.4–6AMD forms when pyrite in coal and sulfide mineral waste streams reacts with oxygen and water, releasing ferric iron, sulfate, and hydrogen ions, creating an acidic effluent. This process involves pyrite oxidation, ferrous iron oxidation, and subsequent further oxidation of pyrite by ferric iron.4,7,8 Under the Clean Water Act (Act 33 U.S.C. §1251), these effluents must be neutralized and treated prior to release into the environment, incurring significant costs influenced by AMD's acidity and flow rate. However, recovering critical elements during the environmental treatment process could mitigate costs and enhance sustainability by transforming these waste streams into valuable resources.3,9,10
Several chemical processes have been used for extracting heavy metal ions like Mn–Co–Ni from aqueous solutions, including chemical precipitation (oxidative, hydroxide, sulfide, carbonate, etc.), ion-exchange, membranes, solvent extraction, electrochemical methods (electrodeposition, electrosorption, and surface e-precipitation), and adsorption.9,11–16 The effectiveness of these processes depends on various factors such as solution pH, temperature, the presence of other ions, and redox potential. The choice of the most effective chemical process for Mn, Co, and Ni recovery depends on the application specifics, solution composition, and desired product purity.11,17 While conventional methods for metal recovery from aqueous solutions, such as chemical precipitation, ion-exchange, membranes, solvent extraction, electrochemical techniques, and adsorption, are widely applied, there is continued interest in developing more efficient and environmentally sustainable alternatives for the recovery of valuable metals from complex secondary resources. Recent studies have demonstrated various hydrometallurgical strategies to address the challenges associated with waste streams. For example, Weshahy et al.18 reported a multi-stage hydrometallurgical process for the selective recovery of high-purity cadmium, cobalt, and nickel from spent Ni–Cd batteries, involving sulfuric acid leaching, followed by solvent extraction and adsorption using a mesoporous silica-based material. This approach showcases the potential of combining diverse separation methods for comprehensive resource recovery from waste.18 In another case, ultrasound-assisted oxidative acid leaching was applied to enhance zinc recovery from low-grade residues, significantly improving leaching kinetics and overall efficiency.19 Similarly, surfactant-assisted HCl and L-tartaric acid mixed leaching has been used to enhance impurity removal from diamond wire saw silicon powder by weakening electrostatic adsorption of metal ions.20 Parallel to these applied developments, mechanistic studies such as those employing the surface complexation model (SCM) have improved the understanding of solid–liquid interface behavior and protonation reactions on mineral surfaces.21 These recent advancements highlight the need for further development of intensified and fundamentally understood techniques that are grounded in a clear understanding of reaction mechanisms and interfacial processes. Such approaches are particularly important for extracting valuable elements from diverse and complex secondary waste streams, where recovery often relies on advanced metal precipitation methods, including oxidative pathways.
Among various methods, metal precipitation has been widely used in water treatment and metal recovery and involves adding chemicals to react with dissolved metal ions, forming insoluble compounds that precipitate as solid particles.22,23 This process forms a solid phase in a saturated solution, driven by supersaturation, and includes sub-processes like crystal nucleation, growth, ripening, and agglomeration, though these are not always distinct in practical applications.24 Precipitation has several defining characteristics. Firstly, the resulting precipitates are often scarcely soluble, forming under conditions of substantial supersaturation, and rapid precipitation usually results from homogeneous or heterogeneous nucleation without secondary nucleation. Due to high supersaturation, nucleation dominates, producing a multitude of small crystals, typically ranging from 0.1 to 10 micrometers in size and with particle concentrations between 1011 and 1016 particles per cm3. Second, the high particle concentration and small crystal size facilitate secondary processes like Ostwald ripening and aggregation, which can significantly affect precipitate properties. Lastly, supersaturation—often triggered by a chemical reaction—is generally required to initiate precipitation.22
Staged precipitation has emerged as a promising technique for selective recovery of elements from AMD. A patented staged carbonate precipitation process, developed by the authors, effectively recovers Fe, Al, and REEs from AMD at pH levels up to the conventional treatment threshold of pH 7.25 However, the recovery of Mn–Co–Ni presents challenges due to the high pH requirements or the expensive oxidants needed for their precipitation. Conventional hydroxide or ammoniacal AMD treatments initiate the precipitation of these metals around a pH of 9, with optimal recovery often requiring pH levels as high as 10.5.9,26 Alternatively, oxidative precipitation has proven highly effective in the hydrometallurgical processing of Mn–Co–Ni, employing various oxidants, such as hydrogen peroxide, SO2/O2 mixture, ozone, Caro's acid, peroxydisulfuric acid, hypochlorite and chlorite, sodium persulfate and potassium permanganate, to recover these elements from aqueous solutions.9,11–13,17,27
Among these oxidants, ozone is one of the most effective oxidizing agents.28–30 Due to its high reactivity, ozone facilitates rapid treatment processes with shorter contact times compared to other oxidants. The double-film theory, also known as gas–liquid mass transfer theory, is a conceptual framework used to describe the mass transfer of ozone from the gas phase to the liquid phase during ozone–water reactions. This theory proposes that ozone transfer across the gas–liquid interface occurs in two distinct steps: mass transfer through the gas phase and subsequent chemical reaction within the liquid phase.31–33 In the context of Mn–Co–Ni precipitation, once ozone molecules diffuse into the bulk liquid, they undergo an irreversible reaction to the dissolved metal ions, forming insoluble oxides or oxyhydroxides. Calzado et al. investigated the feasibility of using ozone for nickel recovery from dilute aqueous solutions, identifying three main steps: ozone transfer from gas to liquid, ozone decomposition by hydroxyl ions, and oxidative-precipitation of nickel. X-ray diffraction analysis identified the resulting nickel precipitates as Ni(OH)2, NiOOH, Ni3O2(OH)4, and Ni2O2(OH)4, suggesting a reaction sequence of hydrolysis–precipitation–oxidation of nickel in aqueous sulfate solutions.11 Additionally, research by Tian et al. highlighted that variables such as flow rate, ion concentration, ozone partial pressure, and temperature significantly affect Co recovery in chloride solutions when using ozone.34 Previous research by the authors9 identified ozone as an exceptionally effective oxidant for extracting Co and Mn from AMD at neutral pH, offering a greener and more chemical-efficient extraction method. Ozone also facilitates the recovery of other critical transition elements such as Pb, Cu, Ni, Ag, and Pd from aqueous solutions, including AMD and brine sources.9,11
To effectively control the oxidative precipitation process, both thermodynamics and kinetics of the process should be understood.22 However, the impact of various process parameters such as flow rate, stirring rate, and temperature on the oxidative precipitation of Mn–Co–Ni by ozone remains understudied, with limited kinetic studies available.32,34–38 Consequently, a comprehensive kinetic study is crucial to advance the understanding of ozone-based recovery mechanisms.
Therefore, this study investigates the effects of oxygen flow rate, stirring rate, and temperature on the ozone oxidative precipitation of Mn–Co–Ni to identify the significant parameters and optimize them to maximize the elemental recovery. Kinetic analyses were conducted to assess precipitation rates, applying various models—linear, Higbie, and pseudo-homogeneous—to fit kinetic data and calculate activation energies. The results of this research have substantial implications for the development, design, and scale-up the ozone oxidative precipitation process to recover Mn–Co–Ni from aqueous solutions including AMD.
2. Materials and methods
2.1. Materials
Approximately 800 L of AMD was collected at the feed point of a treatment facility in Pennsylvania operated by the Pennsylvania Department of Environmental Protection (PADEP). The AMD, sourced from the Lower Kittanning coal seam, contained an elevated concentration of critical elements, including Al, REEs, Mn, Co, and Ni with a pH and Eh of 3.5 and 589 mV, respectively (Table 1). These findings highlight the potential of AMD as a valuable secondary source of multiple critical elements.2.2. Methods
 according to reaction (1) and (2), therefore the formation of ozone through a three-body reaction (reaction (3)).39–41
according to reaction (1) and (2), therefore the formation of ozone through a three-body reaction (reaction (3)).39–41 | (1) |
 | (2) |
 | (3) |
 denotes the initial transient excited state of ozone. The ozone generation (reaction (3)) competes with other reactions (reaction (4)–(6)) that also involve oxygen atoms.39,42
denotes the initial transient excited state of ozone. The ozone generation (reaction (3)) competes with other reactions (reaction (4)–(6)) that also involve oxygen atoms.39,42| O + O + M → O2 + M | (4) |
| O + O3 + M → 2O2 + M | (5) |
 | (6) |
The experimental setup involved a 1000 mg h−1 ozone generator (T-king Enaly model), utilizing 99% pure oxygen as the feed gas to generate ozone. High-purity, dry oxygen was used to ensure efficient ozone generation and stability, as impurities such as moisture and nitrogen in air-fed systems promote ozone decomposition and side reactions that reduce ozone yield, impairing oxidative precipitation efficiency.41 The ozone was sparged into the solution through a porous bubble sparger, with the oxygen flow rate precisely controlled by a flowmeter. The system configuration included potential measurement, as illustrated in Fig. 1. Oxygen flow to the generators was manually controlled by a flowmeter, and the O2/O3 mixture was then introduced into a 500 ml reactor through the diffuser.
 | ||
| Fig. 1 Schematic experimental setup for ozone oxidative precipitation of Mn–Co–Ni from AMD. | ||
Throughout the experiment, the pH and redox potential were continuously monitored within the reactor. 5 ml samples were collected at various intervals—0 seconds, 5 seconds, 15 seconds, 30 seconds, 45 seconds, 60 seconds, 90 seconds, 120 seconds, 5 minutes, 10 minutes, 15 minutes, 20 minutes, and 30 minutes—to assess the concentrations of Mn–Co–Ni. Each 5 ml sample was immediately filtered through EZFlow® syringe filters with a 0.22 μm pore, and then quickly acidified using 70% HNO3 to achieve a final concentration of 5% HNO3 in the solutions, preventing further precipitation.
The experiments were conducted in triplicate, and the average results are presented with a 95% confidence interval. Each experiment was conducted using a 500 ml solution at pH 7. The methodology, including selecting process parameters and reagents, was based on previous research9,24 and a patented method.43 The experimental setup is shown schematically in Fig. 1.
2.2.3.1. Kinetic models. In this study, oxidative precipitation experiments using ozone were conducted by varying the temperature while collecting incremental samples over time and precipitation kinetics were modeled using the following three kinetic models:
• Linear model: the linear model is a simple kinetic model that assumes the reaction rate is proportional to the concentration of a reactant (e.g., Mn, Co, or Ni). The linear model neglects any potential mass transfer limitations and any fluctuations in ozone concentration, assuming a uniform concentration instead. The linear model serves as an approximation and provides a baseline to understand the primary behavior of the system.38 The reaction rate in the linear model is calculated using eqn (1).
| Ct/C0 = k × t | (1) |
• Higbie model: the Higbie model, based on film theory, is primarily used to describe mass transfer processes. This model assumes that the boundary layer thickness controls the kinetic reaction.45 This model was initially developed to describe the rate at which a solute in a gas is mixed with a solvent in a liquid, where the process is controlled by diffusion through a stagnant film at the gas–liquid interface.38 By considering the effect of concentration gradients, the Higbie model (described using eqn (2)) can provide a more accurate description of the mass transfer-controlled reaction kinetics at the gas–liquid interface compared to the linear model.38
| ln(Ct) = 2k′t0.5 + ln(C0) | (2) |
• Pseudo-homogeneous model: the pseudo-homogeneous model is a more complex model that attempts to bridge the linear and Higbie models. It considers the reaction kinetics (similar to the linear model) and mass transfer limitations (as in the Higbie model). In this model, the resistance to mass transfer from the gas phase to the liquid phase is considered negligible, and the amount of ozone dissolved in the liquid is assumed to be in ample supply.37,38Eqn (3) describes the corresponding rate equation.
| ln(Ct/C0) = −k′′ × t | (3) |
kg = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Eg/RT) exp(−Eg/RT) | (4) |
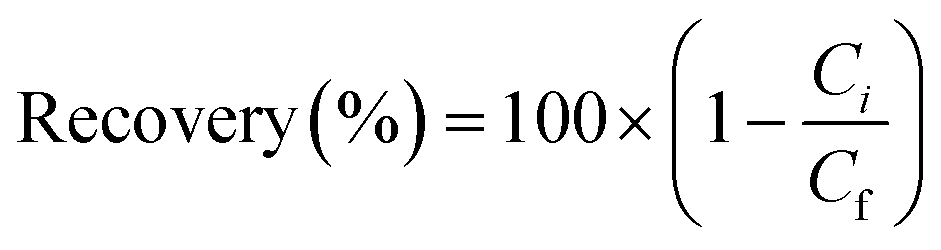 | (5) |
3. Results and discussion
3.1. Solution chemistry study
The saturation index (SI) was utilized to determine the likelihood of precipitation for Mn–Co–Ni hydroxides or oxides. The formula for calculating the saturation index is as follows:| SI = log(IAP/Ksp) | (6) |
• IAP is the ion activity product, representing the product of the dissolved metal and oxide/hydroxide ions' activities (or concentrations) in the solution.
• Ksp is the solubility product constant for the metal hydroxide or oxide at the given temperature.
The oxidation of manganese(II), cobalt(II), and nickel(II) by ozone in water, resulting in the formation of manganese(III) oxide, cobalt(III) oxide, and nickel(III) oxide can be expressed by the following reactions (reaction (7)–(9)):
| Mn2+(aq) + 1/2O3(aq) + H2O(l) → 1/2Mn2O3(s) + 2H+(aq) + 1/2O2(g) | (7) |
| 2Co2+(aq) + O3(aq) + H2O(l) → Co2O3(s) + 2H+(aq) + 1/2O2(g) | (8) |
| 2Ni2+(aq) + O3(aq) + 3H2O(l) → 2NiOOH(s) + 4H+(aq) + O2(g) | (9) |
Calculated saturation indices based on the concentrations of Mn–Co–Ni in AMD (shown in Fig. 2) showed positive SI values for Co and Mn oxides at a pH of around 2 when ozone was incorporated, while it was around pH 7 for NiOOH, suggesting favorable precipitation conditions. Notably, precipitation of these elements with ligands required a pH above 9. Across all tested pH levels, SI values for Mn(OH)2, Co(OH)2, and Ni(OH)2 remained lower than those for Mn2O3, Co2O3, and NiOOH, reflecting the higher solubility of Mn–Co–Ni(II) hydroxides in aqueous solutions. These saturation index results corroborate the findings from previous experiments, which demonstrated that over 95% of Mn–Co–Ni could be recovered from AMD using ozone as an oxidizing agent under either acidic or neutral pH conditions.9,44
 | ||
| Fig. 2 Saturation indices of Mn–Co–Ni as a function of solution pH. | ||
These pH–Eh diagrams provide baselines to determine the solution parameters (such as potential, pH, and temperature) for precipitation specific elements of interest. Mn predominantly exists in oxidation states II, III, and IV in nature, while Co and Ni occur in states II and III. The stability of each oxidation state in aqueous solutions is significantly governed by the oxidation–reduction potential (ORP) and pH, as illustrated in Fig. 3. Changes in Eh–pH conditions facilitate the transformation of Mn–Co–Ni into their most stable phase under new conditions, provided that thermodynamically favorable pathways exist (Fig. 3 and Table 2). Kinetic analyses are essential to predict these transformation rates.48,49
 | ||
| Fig. 3 Eh–pH diagrams for (a) Mn (1 mM), (b) Co (0.1 mM), and (c) Ni (0.1 mM), showing the predominant species in the H2O system at 25 °C. | ||
| Pathway | Reaction | Precipitation method | ΔG0 (kJ) |
|---|---|---|---|
| A1 | Mn2+ + 2OH− = Mn(OH)2 | Ligand | −74.165 |
| A2 | Mn2+ + O3 + H2O = 0.3Mn3O4 + 2H+ + 1.3O2 | Oxidative | −636.322 |
| A3 | Mn2+ + 0.5O3 + H2O = 0.5Mn2O3 + 2H+ + 0.5O2 | Oxidative | −470.491 |
| Mn2+ + 1.5O3 + 1.5H2O = MnO*OH + 2H+ + 2O2 | −243.440 | ||
| A4 | Mn2+ + O3 + H2O = MnO2 + 2H+ + O2 | Oxidative | −173.493 |
| B1 | Co2+ + 2OH− = Co(OH)2 | Ligand | −83.085 |
| B2 | Co2+ + O3 + H2O = 0.3Co3O4 + 2H+ + 1.3O2 | Oxidative | −136.514 |
| C1 | Ni2+ + 2OH− = Ni(OH)2 | Ligand | −86.328 |
| C2 | Ni2+ + O3 + 1.5H2O = NiOOH + 2H+ + 1.2O2 | Oxidative | −175.159 |
In conventional AMD treatment, the removal of metals like Mn through precipitation is typically achieved by raising the pH to above 9, with agents such as lime (Ca(OH)2), sodium hydroxide (NaOH), or ammonium hydroxide (NH4OH). As such basic conditions, Mn, Co and Ni precipitate as Co(OH)2, Mn(OH)2, and Ni(OH)2 (following paths A1 in Fig. 3a, B1 in Fig. 3b, and C1 in Fig. 3c).
Mn–Co–Ni ions in solution can be oxidized by strong oxidants, such as ozone, to form unstable species that subsequently react with hydroxyl ions (OH−) to form stable oxides and hydroxides, as summarized in Table 2. The Gibbs free energy calculations support the feasibility of oxidizing and precipitating these elements with ozone, as the negative ΔG values indicate that these reactions are thermodynamically favorable and can occur spontaneously.34 The oxidation of Mn(II)–Co(II)–Ni(II) in AMD to Mn(III), Mn(IV), Co(III), and Ni(III) can be achieved through oxidative precipitation across a pH range from highly acidic (e.g., MnO2 formation via path A4 in Fig. 3a) to circumneutral pH (following paths A3 and A2 (Fig. 3a), B2 (Fig. 3b), and C2 (Fig. 3c) to form Mn2O3, Mn3O4, Co3O4, and NiOOH, correspondingly) with high Eh values above 700 mV.
This study followed the pathways A4 (Fig. 3a), B2 (Fig. 3b), and C2 (Fig. 3c) to produce high-grade precipitates at circumneutral pH, providing a chemical-less precipitation process for the Mn–Co–Ni recovery from AMD. In the oxidation–precipitation process, the production of H+ ions can lower the solution's pH. To manage the system's potential and maintain the pH, NaOH was added to neutralize the generated H+ ions.
3.2. Effect of process parameters
The effect of individual parameters, including flow rate, stirring rate, and temperature, on the ozone oxidative precipitation of Mn–Co–Ni was investigated. | ||
| Fig. 4 Mn–Co–Ni recovery and ORP values as a function of time at different gas flow rates. | ||
 | ||
| Fig. 5 Mn–Co–Ni recovery and ORP values as a function of time at different stirring rates. | ||
 | ||
| Fig. 6 Mn–Co–Ni recovery and ORP values as a function of time at different temperatures. | ||
During the oxidative precipitation of manganese using ozone, other ions, such as Ni and Co, can be incorporated into the manganese precipitate through adsorption and co-precipitation processes as manganese dioxide is an effective adsorption material.34,50 During precipitation, other ions present in the solution can be adsorbed onto the surface of the growing MnO2 particles. The extent of cobalt co-precipitation is influenced by temperature as increasing the temperature enhances mass transfer within the system, leading to more contact between Co–Ni ions and MnO2, and increasing the adsorption of Co–Ni lost in the manganese precipitate.
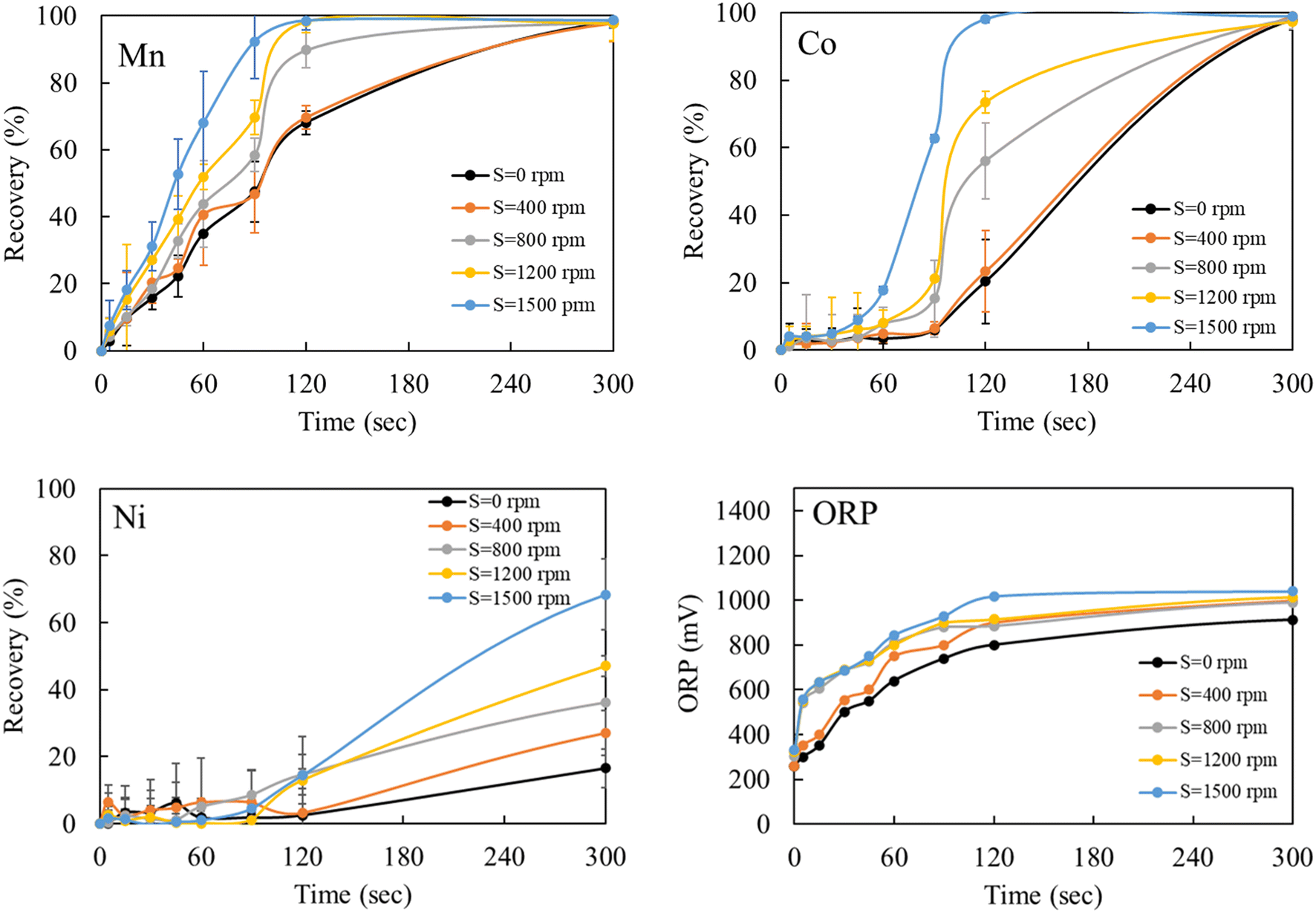
The observed rapid precipitation reactions indicate that the Mn–Co–Ni ions quickly interact with ozone, reflecting the kinetic favorability of the reaction. This rapid interaction is attributed to the strong oxidizing property of ozone, allowing it to readily react with Mn–Co–Ni ions. As the temperature increases, the kinetic energy of the molecules also rises, leading to an accelerated reaction rate (Fig. 7), as predicted by the Arrhenius equation. The data revealed a substantial precipitation of these elements within the first 120 seconds following ozone introduction, while maintaining a pH of 7. Increasing the concentration of a precipitant or oxidizer in a supersaturated solution enhances the precipitation rate, shifts the equilibrium, increases supersaturation levels, and thereby promotes a high rate of nucleation for Mn–Co–Ni. These findings are consistent with previous studies showing that elements tend to precipitate more rapidly under higher supersaturation levels.22,24
 | ||
| Fig. 7 Rate constant of Mn–Co–Ni precipitation at different temperatures (20, 40, 60, and 80 °C). | ||
After the initial seconds of reaction, a slight decrease in the precipitation of Mn, Co, and Ni was observed, followed by a gradual increase as the experiments progressed (Fig. 7). This pattern suggests the occurrence of Ostwald ripening, where smaller particles dissolve due to their higher chemical potential compared to larger particles.22,24,51 This dissolution of smaller particles continues throughout the oxidative precipitation. Notably, significant precipitation occurred within the first five minutes but continued gradually until the experiment concluded at 30 minutes. The variation in precipitation rates of Mn–Co–Ni with temperature changes can be attributed to several factors, including differences in their solubility, the role of ozone in facilitating precipitation, the required ORP levels in the solution, metal ion concentrations, and the presence of other compounds (Fig. 7).15,52
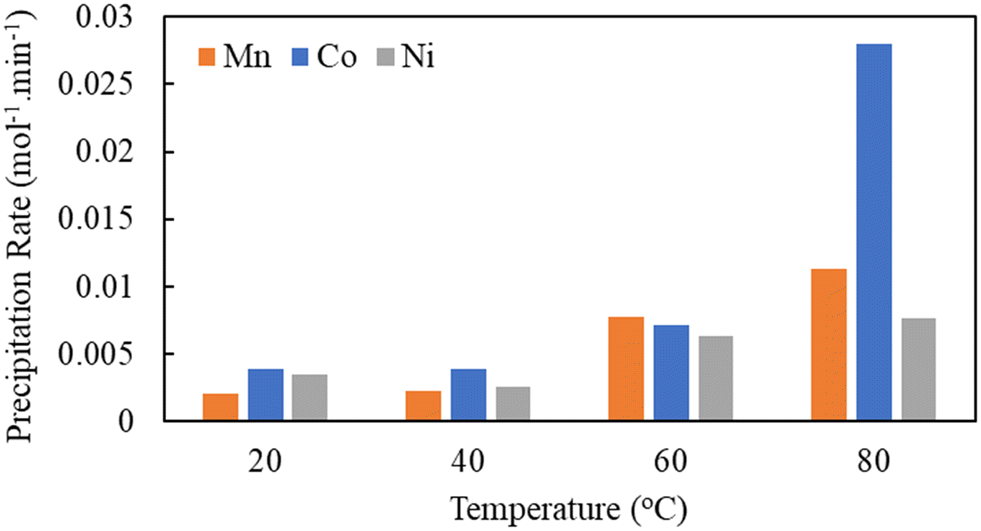
The activation energy values were determined to explore the mechanisms driving the ozone oxidative precipitation of Mn–Co–Ni within the initial 30 seconds, using three different models. The analysis demonstrated that the pseudo-homogeneous model aligned most accurately with the precipitation data, as shown in Fig. 8.
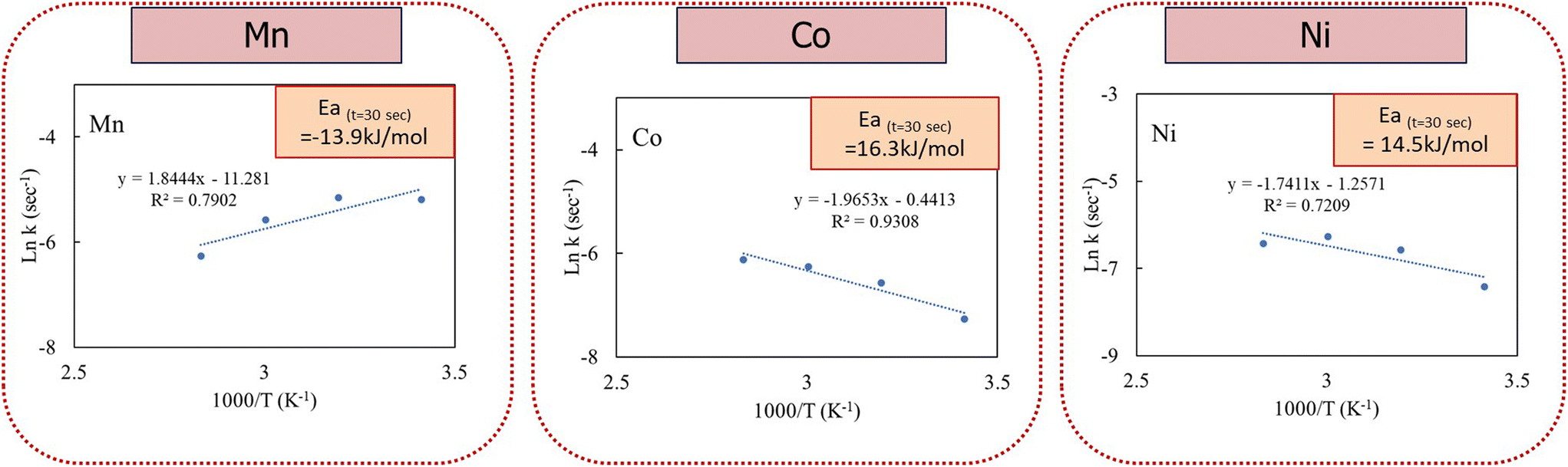 | ||
| Fig. 8 Activation energy for Mn, Co, and Ni precipitation during the first 30 seconds of the reaction. | ||
Providing a specific mechanism for Mn–Co–Ni precipitation using ozone is challenging as it involves several concurrent processes (i.e., mass transfer of ozone, induction period for precipitation, ozone conversion to oxygen, co-precipitation and adsorption effects on precipitates of elements, and competing reactions), making it difficult to deconvolute these processes. Mn–Co–Ni–(OOH) precipitation occurs through a series of sub-processes. For example, Co oxidation and subsequent precipitation can be represented by the following reactions (reaction (10)–(12)), which highlight the induction period and interdependencies within these processes:
| 2Co2+ + O3 + 2H+ = 2Co3+ + O2 + H2O | (10) |
| 2Co3+ + 4H2O = 2CoOOH + 6H+ | (11) |
| 2Co2+ + O3 + 3H2O = 2CoOOH + 4H+ + O2 | (12) |
Furthermore, the transfer of ozone across the gas–liquid interface significantly influences the reaction, as it is continuously injected throughout the process. At the interface, ozone must dissolve into the aqueous phase, with its dissolution rate affected by factors such as temperature, pressure, and ozone's solubility in the liquid, along with the convective and diffusive transport of gaseous species from the bubble surface. Once dissolved, ozone engages in various chemical reactions depending on the aqueous solution's composition.
In this study, the activation energy for ozone oxidative precipitation of Mn–Co–Ni was assessed during the initial 30 seconds, a period in which nucleation occurs at a high rate. This is followed by the growth phase, overtaking the rate of nucleation. The influence of process parameters and the activation energy values for Mn–Co–Ni, listed in Fig. 8, suggest that Mn, Co, and Ni reactions are predominantly diffusion-controlled. This implies that the reaction rate is more influenced by the diffusion coefficient, which governs the rate at which reactants mix, than by temperature variations. Consequently, the diffusion of ozone molecules to the surface of the Co and Mn nuclei is a critical determinant of the reaction rate, underscoring the importance of ozone mass transfer in the oxidative precipitation of Mn–Co–Ni. Although negative activation energy is not feasible for an individual elementary reaction, the precipitation process involves multiple sub-processes. These include the mass transfer of ozone, which limits the reaction rate as ozone diffuses into the solution, and ozone decomposition, especially at higher gas flow rates, where the rapid breakdown of ozone reduces its availability for oxidation, thereby impacting the recovery and ORP. Additionally, co-precipitation and adsorption play a significant role, as MnO2, a key product of Mn oxidation, acts as an adsorbent for Co and Ni, facilitating their incorporation into the precipitate. Furthermore, the Ostwald ripening phenomenon, where smaller particles dissolve and larger ones grow, further complicates the kinetic profile by reducing precipitation initially, followed by a gradual increase. This complexity allows for negative apparent overall activation energy in the complete precipitation process, as documented in studies by Vaziri Hassas et al.,51 Meshram et al.,53 Vyazovkin,54 and Wang et al.55
These findings indicate that while elevated temperatures improve the kinetics of oxidative precipitation, industrial applications may benefit from operating at ambient temperature due to cost constraints. Notably, as shown in Fig. 6, high recovery of Mn, Co, and Ni was achieved within three minutes even at ∼20 °C, demonstrating that efficient recovery is feasible without heating. In this context, the slightly slower kinetics observed at lower temperatures may be an acceptable or even advantageous trade-off, particularly when longer residence times are operationally manageable. Overall, the ability to maintain high recovery rates under ambient conditions shows the practicality and scalability of the process for real-world AMD treatment scenarios. To further assess the formation and morphology of the precipitates formed, the products of Mn–Co–Ni precipitation via ozone treatment at two distinct temperatures (20 °C and 80 °C) were analyzed using SEM–EDS, as shown in Fig. 9. SEM–EDS images revealed that the precipitated Mn–Co–Ni particles predominantly ranged from micron to submicron sizes, exhibiting irregular shapes and rough surfaces. No clear differences in crystallization were observed in the SEM micrographs. The presence of a significant amount of Mn–oxygen detected in the EDS analysis suggests the formation of Mn oxide, corroborating the solution chemistry results and XRD findings reported in previous research by the authors.9 The processes of co-precipitation and adsorption play significant roles in the oxidative precipitation of metals like Mn, Co, and Ni when using ozone. However, these processes complicate the characterization of precipitation products due to their intertwined nature.
 | ||
| Fig. 9 SEM micrographs of precipitated solids from ozone oxidative precipitation and EDS mapping of precipitates at (a) 20 °C and (b) 80 °C temperatures. | ||
The analysis of the oxidative precipitation of Mn–Co–Ni using ozone highlights the significant roles of solution chemistry and process parameters, such as stirring rate, gas flow rate, and temperature. These parameters crucially affect the efficiency of metal recovery from AMD. This study demonstrates that adjustments to these parameters can significantly enhance the precipitation process, as evidenced by the rapid and high recovery of metals within the first few minutes of reaction initiation. Moreover, the findings reveal the complexity of the precipitation process, which involves not just simple chemical reactions but a series of interconnected sub-processes, including mass transfer, nucleation, and growth of precipitates. Given these complexities, a fundamental study focusing on the effect of process parameters in pure solutions of Co, Ni, and Mn is recommended. Such studies could offer a clearer understanding of the specific roles and interactions of each metal during the precipitation process. This approach could provide valuable insights into how each metal contributes to the co-precipitation and adsorption processes and would be instrumental in optimizing conditions for selective recovery and purification of each metal from mixed metal solutions. For the process scale-up, various factors, including reactor design, gas flow rate, stirring time, and other process parameters should be identified to enhance the mass transfer and reaction kinetics for the recovery of these elements. Therefore, further studies on the interactive effects of process parameters are recommended for future research.
3.3. Validation of the integrated staged precipitation process
To validate the results for application in AMD treatment, the AMD was continuously fed at a rate of 150 mL min−1 to the combined staged carbonate precipitation and ozone oxidative precipitation process.25,43 The ozone oxidative precipitation was conducted at room temperature, with a stirring rate of 400 rpm, and an air flow rate of 800 cc min−1 supplied to the ozone generator. A schematic of the process, along with the grade of the main product in each stream and the corresponding elemental recovery values, is presented in Fig. 10. The recovery values and elemental content of the treated AMD confirm the technical feasibility and effectiveness of the process in recovering multiple critical minerals while achieving environmental compliance. The product streams can be further processed through our previously developed purification process to achieve high-purity products.51 Further techno-economic assessment and life cycle analysis comparing this method with conventional treatment approaches will elucidate its economic and environmental advantages. | ||
| Fig. 10 Result of the integrated staged precipitation process developed for continuous AMD treatment at 150 mL min−1 flow rate for Fe removal and selective recovery of Al, REEs, and Mn–Co–Ni. | ||
4. Conclusions
This research investigated the effect of gas flow rate, stirring rate, and temperature on the oxidative precipitation of Mn–Co–Ni from AMD using ozone, as these parameters significantly affect the mass transfer of ozone and resulting reaction kinetics. By reducing mass transfer resistance through improved mixing and higher ozone availability, higher recovery rates of Mn–Co–Ni were expected. Saturation index calculations and Pourbaix diagram analyses demonstrated that the recovery of Mn–Co–Ni using ozone is feasible across a wide pH range (pH 2–8 for Mn and Co, and pH > 6 for Ni). Detailed experimental analyses and solution chemistry revealed that increased gas flow rate was not significant for Mn recovery as the ORP was within the range of rapid oxidation of Mn even at low flow rate. However, increased flow rate till 1400 cc min−1, beyond which ozone started decomposing, enhanced Co and Ni recovery due to the increased volumetric transfer of ozone into the solution, thereby elevating the concentration of dissolved ozone available for reaction with metal ions. Temperature and stirring rates significantly improved Mn–Co–Ni recovery in general due to enhanced mass transfer of ozone, thereby improving the Mn–Co–Ni oxidation and precipitation. However, these increases were not statistically significant at a 95% confidence level within the lower range of these parameters (i.e., a gas flow rate of 800 cc min−1, a stirring rate up to 400, and a temperature up to 40 °C). Activation energy and kinetic rate measurements, conducted during the initial 30 seconds of the reaction, showed that the reaction kinetics, with activation energies of 16.3 kJ mol−1 for Co, 14.5 kJ mol−1 for Ni, and −13.9 kJ mol−1 for Mn, are best described by the pseudo-homogeneous model. These findings suggest that the oxidative precipitation of Mn–Co–Ni predominantly follows a diffusion-controlled reaction mechanism. Despite these findings, the specific mechanisms underlying the oxidative precipitation of Mn–Co–Ni using ozone were not definitively identified due to the complexity of the process and the interaction of various phenomena, including the mass transfer of ozone, co-precipitation, adsorption effects on precipitates, and competing reactions. This study provides a foundation for further research to unravel the interaction effects among process parameters and metal ions.Data availability
All data supporting the findings of this study are presented within the article as figures and tables. No additional datasets were generated or deposited in external repositories.Author contributions
Younes Shekarian: conceptualization, methodology, software, data curation, validation, formal analysis, investigation, visualization, writing – original draft. Mohammad Rezaee: supervision, project administration, funding acquisition, conceptualization, methodology, formal analysis, validation, data curation, resources, writing – review & editing. Sarma Pisupati: conceptualization, methodology, validation, funding acquisition, resources, writing – review & editing.Conflicts of interest
Younes Shekarian, Dr. Mohammad Rezaee, and Dr. Sarma Pisupati have patent #63/392 2022 330 pending to The Pennsylvania State University. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are grateful to Penn State Center for critical Minerals (C2M), EMS Energy Institute, Material Research Institute (MRI), and Laboratory for Isotopes and Metals in the Environment (LIME) for providing technical facilities. Partial funding for this study was provided by Leonardo Technologies, Inc., USA (under USDOE agreement #DE-FE0022594). Appreciation is also extended to the Pennsylvania Department of Environmental Protection (PADEP) for the valuable contributions to this research project through providing samples, process data, and access to the treatment facilities. Special thanks are extended to Hari Jammulamadaka, and Mamoru Fujii for the fabrication and testing of the continuous process, and Chandima Subasinghe for his assistance in performing the kinetic experiments.Notes and references
- U.S. Geological Survey, Mineral Commodity Summaries, 2022, pp. 52–104 Search PubMed.
- International Energy Agency (IEA), Critical Minerals Market Review, 2023 Search PubMed.
- P. L. Rozelle, N. Mamula, B. J. Arnold, T. O'Brien, M. Rezaee and S. V. Pisupati, Secondary Cobalt and Manganese Resources in Pennsylvania: Quantities, Linkage with Mine Reclamation, and Preliminary Flowsheet Evaluation for the US Domestic Lithium-Ion Battery Supply Chain, The Pennsylvania State University, Center for Critical Minerals, prepared for Leonardo Technologies, Inc. (LTI) of Bannock, OH, under the US Department of Energy's Agreement Number DE-FE0022594, 2021, pp. 1–55 Search PubMed.
- D. B. Johnson and K. B. Hallberg, Acid mine drainage remediation options: a review, Sci. Total Environ., 2005, 338(1–2), 3–14 CrossRef CAS PubMed.
- J. G. Skousen, P. F. Ziemkiewicz and L. M. McDonald, Acid mine drainage formation, control and treatment: Approaches and strategies, Extr. Ind. Soc., 2019, 6(1), 241–249 Search PubMed.
- B. V. Hassas, M. Rezaee and S. V. Pisupati, Precipitation of rare earth elements from acid mine drainage by CO2 mineralization process, Chem. Eng. J., 2020, 399, 125716 CrossRef.
- A. R. Chowdhury, D. Sarkar and R. Datta, Remediation of Acid Mine Drainage-Impacted Water, Curr. Pollut. Rep., 2015, 1(3), 131–141 CrossRef.
- M. Rezaee and R. Q. Honaker, Long-term leaching characteristic study of coal processing waste streams, Chemosphere, 2020, 249, 126081 CrossRef CAS PubMed.
- Y. Shekarian, B. V. Hassas, M. Rezaee and S. V. Pisupati, Development of a chemical-free process utilizing ozone oxidative precipitation for the recovery of cobalt and manganese from acid mine drainage, J. Environ. Chem. Eng., 2022, 10(5), 108533 CrossRef CAS.
- M. Rabbani, J. Werner, A. Fahimi and E. Vahidi, Innovative pilot-scale process for sustainable rare earth oxide production from coal byproducts: A comprehensive environmental impact assessment, J. Rare Earths, 2025, 43(2), 397–404 CrossRef.
- L. E. Calzado, C. O. Gomez and J. A. Finch, Nickel recovered from solution by oxidation using ozone: some physical properties, Miner. Eng., 2005, 18(5), 537–543 CrossRef CAS.
- J. Van Rooyen, S. Archer and M. Fox, Manganese removal from cobalt solutions with dilute sulphur dioxide gas mixtures, in The Fourth Southern African Conference on Base Metals, The Southern Africa Institute of Mining and Metallurgy, 2007, pp. 365–376 Search PubMed.
- W. Zhang and C. Y. Cheng, Manganese metallurgy review. Part II: Manganese separation and recovery from solution, Hydrometallurgy, 2007, 89(3–4), 160–177 CrossRef CAS.
- S. H. Joo, J. Kang, K. Woong and S. M. Shin, Production of Chemical Manganese Dioxide from Lithium Ion Battery Ternary Cathodic Material by Selective Oxidative Precipitation of Manganese, Mater. Trans., 2013, 54(5), 844–849 CrossRef CAS.
- Z. T. Ichlas, M. Z. Mubarok, A. Magnalita, J. Vaughan and A. T. Sugiarto, Processing mixed nickel–cobalt hydroxide precipitate by sulfuric acid leaching followed by selective oxidative precipitation of cobalt and manganese, Hydrometallurgy, 2020, 191, 105185 CrossRef CAS.
- I. V. Chernyshova, M. Suup, C. Kihlblom, H. R. Kota and S. Ponnurangam, Green mining of mining water using surface e-precipitation, Sep. Purif. Technol., 2023, 327, 125001 CrossRef CAS.
- G. L. Bolton, V. B. Sefton and N. Zubryckyj, Removal of manganese and chloride ions from aqueous acidic zinc sulphate solutions, US Pat., US4379037, 1983 Search PubMed.
- A. R. Weshahy, A. K. Sakr, A. A. Gouda, B. M. Atia, H. H. Somaily and M. Y. Hanfi, et al., Selective Recovery of Cadmium, Cobalt, and Nickel from Spent Ni–Cd Batteries Using Adogen® 464 and Mesoporous Silica Derivatives, Int. J. Mol. Sci., 2022, 23(15), 8677 CrossRef CAS PubMed.
- C. Wang, H. Xia, Y. Xu, Z. Lu, Q. Pei and L. Dai, et al., Efficient recovery of valuable metals from low-grade zinc residue by ultrasonic strengthening, Chem. Eng. Process., 2025, 211, 110240 CrossRef CAS.
- B. Xiong, S. Han, S. Yang, K. Xie, K. Wei and W. Ma, Facilitated removal of metal from diamond wire saw silicon powder waste via surfactant-assisted HCl + L-tartaric acid mixed leaching, Chem. Eng. J., 2025, 505, 159632 CrossRef CAS.
- C. Peng, X. Fu, Z. Niu, W. Sun and T. Yue, Protonation behavior study of the active sites on typical sulfide minerals surface using surface complexation model, Colloids Surf., A, 2025, 710, 136307 CrossRef CAS.
- A. Myerson, Handbook of IndustriaI, CrystalIization Second Edition, 2002 Search PubMed.
- M. Free, Hydrometallurgy: Fundamentals and Applications, Wiley, 2013, pp. 212–214 Search PubMed.
- B. V. Hassas, Y. Shekarian and M. Rezaee, Selective precipitation of rare earth and critical elements from acid mine drainage – Part I: Kinetics and thermodynamics of staged precipitation process, Resour., Conserv. Recycl., 2023, 188, 106654 CrossRef.
- M. Rezaee, B. V. Hassas and S. V. Pisupati, Recovery of rare earth elements from acidic solutions, WO2021155224A1, World Intellectual Property Organization, 2021.
- M. Balintova and A. Petrilakova, Study of pH Influence on the Selective Precipitation of Heavy Metals from Acid Mine Drainage, Chem. Eng. Trans., 2021, 25, 345–350 Search PubMed.
- R. M. Freitas, T. A. G. Perilli and A. C. Q. Ladeira, Oxidative Precipitation of Manganese from Acid Mine Drainage by Potassium Permanganate, J. Chem., 2013, 2013(1), 287257 CrossRef.
- E. E. Coyle, L. E. Ormsbee and G. M. Brion, Peracetic Acid as an Alternative Disinfection Technology for Wet Weather Flows, Water Environ. Res., 2014, 86(8), 687–697 CrossRef CAS PubMed.
- Z. B. Guzel-Seydim, A. K. Greene and A. C. Seydim, Use of ozone in the food industry, Lebensm.-Wiss. Technol., 2004, 37(4), 453–460 CrossRef CAS.
- E. I. Epelle, A. Macfarlane, M. Cusack, A. Burns, J. A. Okolie and W. Mackay, et al., Ozone application in different industries: A review of recent developments, Chem. Eng. J., 2023, 454, 140188 CrossRef CAS PubMed.
- M. Roustan, J. Mallevialle, H. Roques and J. P. Jones, Mass Transfer of Ozone to Water: A Fundamental Study, Ozone: Sci. Eng., 1980, 2(4), 337–344 CrossRef CAS.
- B. Qu, T. Li, Z. Yang, L. Ren, C. Long and H. Deng, Kinetics Analysis of Ozonation Process of Recovering Manganese Dioxide from Aqueous Phase in the Presence of Manganese Dithionate, Research Square, 2022, preprint, DOI:10.21203/rs.3.rs-2265190/v1.
- J. Ren, S. He, C. Ye, G. Chen and C. Sun, The ozone mass transfer characteristics and ozonation of pentachlorophenol in a novel microchannel reactor, Chem. Eng. J., 2012, 210, 374–384 CrossRef CAS.
- Q. Tian, Y. Xin, H. Wang and X. Guo, Potential-controlled selective recovery of manganese and cobalt from cobalt slag leaching solution, Hydrometallurgy, 2017, 169, 201–206 CrossRef CAS.
- Q. H. Tian, X. Y. Guo, Y. I. Yu and Z. H. Li, Kinetics of Oxidation-Precipitation of Cobalt (II) from Solution by Ozone, Trans. Nonferrous Met. Soc. China, 2010, 20, s42–s45 CrossRef CAS.
- Q. K. Jing, X. Y. Liu and J. K. Wen, A Novel Iron Oxidation Process in Zinc Leaching Solution by Ozone, Adv. Mater. Res., 2014, 900, 35–38 Search PubMed.
- M. R. Cruz-Díaz, Y. Arauz-Torres, F. Caballero, G. T. Lapidus and I. González, Recovery of MnO2 from a spent alkaline battery leach solution via ozone treatment, J. Power Sources, 2015, 274, 839–845 CrossRef.
- B. P. Oruê, A. B. Botelho Jr, J. A. S. Tenório, D. C. R. Espinosa and M. d. P. G. Baltazar, Kinetic Study of Manganese Precipitation of Nickel Laterite Leach Based-solution by Ozone Oxidation, Ozone: Sci. Eng., 2021, 43(4), 324–338 CrossRef.
- U. Kogelschatz and P. Baessler, Determination of Nitrous Oxide and Dinitrogen Pentoxide Concentrations in the Output of Air-Fed Ozone Generators of High Power Density, Ozone: Sci. Eng., 1987, 9(3), 195–206 CrossRef CAS.
- M. A. T. Alsheyab and A. H. Muñoz, Optimisation of ozone production for water and wastewater treatment, Desalination, 2007, 217(1–3), 1–7 CrossRef CAS.
- C. von Sonntag and U. von Gunten, Chemistry of ozone in water and wastewater treatment: from basic principles to applications, IWA Publishing, 2012 Search PubMed.
- U. Kogelschatz, Dielectric-barrier discharges: their history, discharge physics, and industrial applications, Plasma Chem. Plasma Process., 2003, 23(1), 1–46 CrossRef CAS.
- M. Rezaee, Y. Shekarian and S. V. Pisupati, Ozone Oxidative Precipitation of Elements from Aqueous Streams, WO2025048766A2, World Intellectual Property Organization, 2022.
- Y. Shekarian, M. Rezaee and S. V. Pisupati, Kinetics of Cobalt and Manganese Precipitation from Acid Mine Drainage Using Ozone, in XXXI International Mineral Processing Congress, Society for Mining, Metallurgy, and Exploration (SME), Washington DC, 2024, pp. 1722–1732 Search PubMed.
- E. A. Vieira, J. R. de Oliveira, G. F. Alves, D. C. R. Espinosa and J. A. S. Tenório, Use of Chlorine to Remove Magnesium from Molten Aluminum, Mater. Trans., 2012, 53(3), 477–482 CrossRef CAS.
- A. Lewis, M. Seckler, H. Kramer and G. V. Rosmalen, Industrial Crystallization: Fundamentals and Applications, 2015 Search PubMed.
- B. V. Hassas, The Graduate School Process Development for Selective Separation of Critical Elements From Secondary Resources, 2022 Search PubMed.
- S. T. Martin, Precipitation and dissolution of iron and manganese oxides, Environ. Catal., 2005, 1, 61–68 Search PubMed.
- M. K. Sinha and W. Purcell, Reducing agents in the leaching of manganese ores: A comprehensive review, Hydrometallurgy, 2019, 187, 168–186 CrossRef CAS.
- S. Q. Kong, Y. X. Wang, X. W. Huang and C. Wang, As(III) and As(V) Removal on Manganese Dioxide, Adv. Mater. Res., 2012, 573–574, 39–42 CAS.
- B. V. Hassas, Y. Shekarian, M. Rezaee and S. V. Pisupati, Selective recovery of high-grade rare earth, Al, and Co-Mn from acid mine drainage treatment sludge material, Miner. Eng., 2022, 187, 107813 CrossRef.
- Y. W. Kim and J. K. Baird, Chemical equilibrium and critical phenomena: The solubilities of manganese dioxide and aluminum oxide in isobutyric acid + water near its consolute point, J. Phys. Chem. B, 2005, 109(36), 17262–17266 CrossRef CAS PubMed.
- P. Meshram, B. D. Pandey and T. R. Mankhand, Hydrometallurgical processing of spent lithium ion batteries (LIBs) in the presence of a reducing agent with emphasis on kinetics of leaching, Chem. Eng. J., 2015, 281, 418–427 CrossRef CAS.
- S. Vyazovkin, A time to search: finding the meaning of variable activation energy, Phys. Chem. Chem. Phys., 2016, 18(28), 18643–18656 RSC.
- Y. Wang, H. Liu, Q. Duan and Z. Li, Understanding the Negative Apparent Activation Energy for Cu2O and CoO Oxidation Kinetics at High Temperature near Equilibrium, Catalysts, 2024, 14(11), 832 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |