
Phytic acid derivatized lignin as a thermally stable and flame retardant material†
Saba
Khodavandegar
a and
Pedram
Fatehi
 *ab
*ab
aBiorefining Research Institute, Lakehead University, 955 Oliver Road, Thunder Bay, Ontario P7B 5E1, Canada. E-mail: pfatehi@lakeheadu.ca
bLaboratory of Natural Materials Technology, Åbo Akademi University, Henrikinkatu 2, Turku FI-20500, Finland
First published on 29th July 2024
Abstract
Phosphorus-containing flame retardants have attracted attention due to their outstanding flame retardancy, enhanced thermal stability, and limited toxic smoke emission. Bio-based phosphorus-containing flame retardants could be excellent options to impart environmental benefits, renewability, and sustainability to these materials. Lignin is an underutilized but abundant and sustainable material that can be used to serve this purpose. In the present work, a lignin-derived flame retardant was produced following the facile solvent-free polycondensation reaction of kraft lignin (KL) and phytic acid (PHA) at a low temperature in an aqueous system. The optimized conditions for this reaction were 1/0.4 mol/mol KL/PHA, pH 11, 20 °C, and 20 min. By utilizing advanced NMR (H, P, and HSQC), XPS, and FTIR techniques, the covalent bonding of the phosphorus of PHA with the oxygen of aliphatic and aromatic hydroxyl groups of KL was confirmed. C–P–O and P–O–P bonds provided high decomposition temperature (Tmax), high glass transition temperature (Tg), and char formation in the product. The presence of phosphorus atoms was observed on the combusted material by EDS mapping and EDX, illustrating the increase in the intensity of this element after combustion at 800 °C. The results of this work provided a new approach for preparing a fully bio-based flame-retardant with limited smoke density (i.e., a decrease from 34% for KL to 17.7% for modified KL) and a higher limiting oxygen index (i.e., an increase from 21.8% for KL to 26% for modified KL) following a green chemistry concept.
Introduction
Flame-retarding materials have extensive use in industry to delay or prevent the spread of fire.1,2 They are necessary for sectors where the fire risk is high and safety is paramount, such as construction, textile, painting, etc., specifically where wood structures are available. Wood is a renewable and eco-friendly material with aesthetics, strength, and cost advantages, and it has been used extensively in various industries, such as interior design, construction, and furniture.3 However, its flammability restricts its use in many areas.4 Applying a flame retardant via coating the wood surface is recognized as an efficient and environmentally friendly method to shield wood products from fire.5Chemicals, such as halogenated, nitrogen-containing, silicon-containing, and nano-metric compounds, are the major types of fire retardants.6 However, these flame retardants have been proved to possess detrimental environmental and health effects, such as their persistence in nature for a long time and potential carcinogenicity.7 Earlier, halogen-based flame retardants, polychlorinated biphenyls (PCBs), and brominated flame retardants (BFRs), such as penta- and octa-bromodiphenyl ether and hexabromocyclododecane, were used due to their efficiency in interacting with fabrics and biopolymers.8 However, their use was restricted due to generating toxic and corrosive gases when burned, which is dangerous for humans, animals, and the environment.9 Mineral flame retardants, such as aluminum trihydroxide, are particularly effective in reducing fire risks and, hence, are frequently utilized. Nevertheless, their low efficiency requires their significant quantity to be used to attain the desired increased fire retardancy in materials.10 Therefore, there is a need to produce safer and more effective flame retardants that fulfill environmental and health requirements. Considering environmental protection and human safety, halogenated flame retardants have been suggested to be replaced by bio-based phosphorus materials.11 In this context, phosphorus-containing biomacromolecules have been assessed as sustainable flame retardants.12 For example, phosphorus-containing flame retardants were reported to promote carbonization, dehydrogenation, and the creation of physical protective layers via radical-capturing char formation.13
Phosphorylated flame retardants are known for their lower toxicity and reduced environmental persistence compared to their traditional counterparts. Additionally, they exhibit efficient flame-retardant properties without releasing harmful byproducts during combustion.9 In contrast, halogenated flame retardants, while effective, are often associated with higher levels of corrosivity and toxicity with environmental concerns. Moreover, they are mostly derived from non-renewable resources and may not leave significant carbon residue due to the presence of less carbon in their structure to make a char layer.2 These facts underscore the growing preference for phosphorylated flame retardants in applications where health and environmental safety are paramount.
Lignin, among other biopolymers, has received significant attention due to its three-dimensional structure, vast availability,14 and biodegradability.15 Lignin is an amorphous biopolymer of polyphenolic molecules with many functional groups, including hydroxyl, phenoxyl, carbonyl, methoxyl, and carboxyl.16 In the past, lignin was modified to be used in drug delivery,17 antimicrobial,18 thermal insulating,19 flocculant, dispersant,20 and biosensor applications.21 Lignin can also be used as a flame retardant because of its active functional groups, aromatic ring, and ability to form char.22 However, it cannot be used in pristine form as a flame retardant because of its low fire resistance.23 Interestingly, lignin can be modified to improve its flame retardancy and fire resistance. Generally, chemical reagents containing halogens, nitrogen, silane, and phosphorus elements are used to improve the flame retardancy of materials.24–27 Among these elements, phosphorus with different oxidation states from 0 to +5, which reacts differently with organic and inorganic materials, is a suitable alternative for mimicking lignin flame retardants and fire resistance.28
Phytic acid (PHA) is an eco-friendly organic acid and biocompatible material widely available in plants.29 PHA is considered an excellent flame retardant because of the six phosphate groups present in its structure, which produces a char layer when burned to protect the material underneath it.30 Interestingly, the phosphorus element in the phytic acid structure is active and covalently bound to the active functional groups, resulting in excellent flame-retardant efficiency.31,32 In the past, phytic acid was used along with chitosan, silane sol, and collagen to produce flame-retardant materials. However, phytic acid was primarily blended physically with these materials.33–35 Consequently, it tended to leach out from the blends due to a lack of chemical bonds, which could affect the mechanical properties and processability of the products.36 Also, PHA was used as a flame retardant to coat textile materials. Still, as it is soluble in water, the flame-retardant properties of the textile were reduced by washing the textile materials.37 In this regard, PHA would be more effective if it is covalently bound to the material, as it can have a permanent flame retarding effect.
In previous studies, the modification of lignin was carried out mainly using different phosphorus reagents, for example, ammonium dihydrogen phosphate (NH4H2PO4), phosphorus pentoxide (P2O5), phosphoric acid (H3PO4), 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO), O,O-dialkylthiophosphoric acids, and sodium 3-chloro-2-hydroxypropyl phosphate.38–45 However, phosphorus reagents, such as DOPO, need a high temperature to be activated for reaction.38 Generally, phosphorylation is conducted in the presence of urea and tetrahydrofuran (THF) to prevent its degradation at high reaction temperatures.46 Interestingly, it was observed that the number of phosphorus atoms in a biomaterial is directly correlated to its flame retardancy.47 Compared with the above-mentioned reagents, not only is the large number of phosphorus atoms with OH active sites in the PHA structure considerable, but also its eco-friendliness makes PHA a better option to achieve phosphorylated kraft lignin.48 Interestingly, this modified lignin can be used for different applications, such as coating wood.
This work reported the modification of kraft lignin with C6H18O24P6, PHA, a bio-based reagent, in a facile solvent-free reaction at a low temperature to produce a novel reactive bio-based flame retardant. This work aimed to optimize the reaction conditions to generate phosphorylated lignin with excellent flame retardancy. Moreover, the chemical properties of the fabricated lignin derivative were comprehensively analyzed by advanced techniques, such as 1H NMR, 31P NMR, HSQC-NMR, XPS, ICP-AES, SEM, TGA, and DSC. Also, smoke density and limiting oxygen index analyzers were used to investigate the flame retardant behavior of modified lignin for wood and paper samples.
Experimental
Modification of lignin via phosphorylation
The modification reaction used phytic acid sodium salt hydrate (PHA) as the phosphorylating reagent. In each reaction setup, KL with a molecular weight of 180 g mol−1 (1.5 g) was dispersed in deionized (DI) water (25 g L−1), and the pH of the mixture was adjusted to 12 using 1 M NaOH. The mixture was stirred for 24 hours at room temperature to complete the deprotonation. Different concentrations of the reagent (PHA) were dispersed in 30 mL of deionized water. Then, the mixtures were added to the KL solution to make different molar ratios of KL/PHA solutions. Afterward, the mixtures were transferred to three-neck round-bottom flasks in a water bath with a reflux condenser. Upon completion, the reaction mixtures were cooled to room temperature, neutralized with 1 M H2SO4, and purified for 48 hours via a dialysis membrane in DI water to remove unreacted reagents and salts. The product was then dried at 60 °C in an oven. The product of this reaction, phosphorylated lignin, was denoted as PK. This experiment was repeated under different conditions of KL/PHA molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.02, 0.06, 0.16, 0.2, 0.3, and 0.4, pH values, solvents (DI water and urea), time (20–240 minutes), and temperature (20–80 °C) to optimize the modification reaction. The pH was adjusted using 1 M NaOH solution and 1 M HCl. In strategy one, the pH dropped to 3 after adding the reagent, the reaction was exploited, and the samples were neutralized and dialyzed after the reaction. In strategy two, the pH of the reaction was adjusted to 11 after adding the reagent, and the reaction ran at pH 11. Afterward, the reaction medium was neutralized and dialyzed. A control sample (CK) of KL was produced following all steps outlined in the reaction, pH adjustment, purification, and drying procedures stated above in the absence of PHA.
:0.02, 0.06, 0.16, 0.2, 0.3, and 0.4, pH values, solvents (DI water and urea), time (20–240 minutes), and temperature (20–80 °C) to optimize the modification reaction. The pH was adjusted using 1 M NaOH solution and 1 M HCl. In strategy one, the pH dropped to 3 after adding the reagent, the reaction was exploited, and the samples were neutralized and dialyzed after the reaction. In strategy two, the pH of the reaction was adjusted to 11 after adding the reagent, and the reaction ran at pH 11. Afterward, the reaction medium was neutralized and dialyzed. A control sample (CK) of KL was produced following all steps outlined in the reaction, pH adjustment, purification, and drying procedures stated above in the absence of PHA.
Charge density, solubility, elemental analysis, and molecular weight measurements
The charge density of PK samples was determined using a particle charge detector (PCD 04, BTG Mütek GmbH, Germany) using a 0.005 mol L−1 PDADMAC solution as the titrant. The solubility of the samples was measured according to the process described previously.49 The organic elements of the samples were analyzed using an organic elemental analyzer, Vario EL, following the combustion method.50 The molecular weight of the samples was investigated via gel permeation chromatography (GPC, Malvern GPCmax VE2001 Module + Viscotek TDA305 with multi-detectors) with the columns of PolyAnalytic 206 and PAA203.1H, HSQC, HMBC, and 31P NMR analyses
The chemical structure of lignin samples was characterized by proton nuclear magnetic resonance (1H NMR) and two-dimensional heteronuclear single-quantum coherence NMR (HSQC), and for phosphorus element analysis, 1H–31P HMBC measurements were performed using a Bruker Avance spectrometer at room temperature.31P NMR spectroscopy was carried out quantitatively and qualitatively. Quantitative 31P NMR analysis assessed the content of phenolic hydroxyl, aliphatic hydroxyl, and carboxyl groups of KL and PK17.51
FTIR analysis
The FTIR analysis of KL and PK samples was carried out to monitor the chemical structures of the samples using a Bruker Tensor 37 instrument (Germany, Germany, ATR accessory).XPS and ICP-AES analyses
The surface chemical composition was determined by X-ray photoelectron spectroscopy using a photoelectron spectrometer (XPS, Escalab 250XL+, Thermo Fisher Scientific, USA). The phosphorus content of the samples was determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES).Thermal analysis
Thermogravimetric analysis (TGA) was performed to investigate the samples’ thermal response in a thermal analyzer (TGA i1000, Instrument Specialists Inc.).Differential scanning calorimetry (DSC) was performed to estimate the glass transition temperature (Tg) of KL and PK in a differential scanning calorimeter (DSC Q2000, TA Instruments, DE, USA).
Flame retardant behavior analysis
For flame retardant behavior analysis, wood and filter paper samples were first coated with KL and PK17 solutions. Before coating, the wood samples were washed with ethanol, rinsed with DI water, and dried at 60 °C for 2 hours. Three different concentrations (1, 2, and 3 wt%) of KL and PK17 were diluted in deionized water and labeled KC1, KC2, and KC3 for KL-coated wood and PC1, PC2, and PC3 for PK17-coated wood and UW for uncoated wood, respectively. The wood samples were immersed and dip-coated in solution at 60 °C for 4 hours. After coating, each sample was dried at 60 °C until reaching a stable weight. To further analyze the flame retardant behavior of PK17, the filter paper was immersed and coated with 3 wt% PK17 and dried at 60 °C.The fire resistance behavior of the samples was measured using a limiting oxygen index (LOI) analyzer (Netzsch Taurus Instrument, Germany) according to the ASTM D 2863 standard. The static smoke release of coated wood was tested using a smoke detector instrument (Smoke Density Advanced Instruments Co., Ltd, AIC-2843) according to the ASTM D2843 standard.
Morphological study of burned wood
Scanning electron microscopy (SEM) was used to analyze the surface morphology of the wood samples after the flame retardancy test. Specifically, after burning the wood samples in the smoke density analyzer, the burned samples were collected, and the morphologies of char formed on uncoated wood (UW), coated wood with KL (KC), and coated wood with PK17 (PC) were analyzed using SEM. Also, PK17 and KL before and after combustion at 800 °C by TGA were collected and analyzed by FE-SEM; Hitachi Su-70 with a voltage of 5 kV. Also, the surface element mapping and elemental analysis of the samples were carried out by energy dispersive spectroscopy (EDS) and energy-dispersive X-ray spectroscopy (EDX) at a voltage of 200 kV, where the samples were coated with gold and carbon glue. The details of characterization are available in the ESI.†Results and discussion
Solubility, charge density, and molecular weight
Solubility, charge density, and molecular weight measurements were carried out for reaction optimization to obtain the modified lignin with the highest phosphorylated group, and the results are reported in Table S1 in the ESI.† The effect of different molar ratios of the reagent is illustrated in this table and Fig. 1a–d. When the molar ratio of PHA increased from 0.02 to 0.4, the phosphorus (P) content, charge density, and molecular weight of phosphorylated lignin were increased from 0.8% to 4.4%, −1.8 mmol g−1 to −4.3 mmol g−1, and 3900 g mol−1 to 4560 g mol−1, respectively. Because of the presence of hydroxyl groups in KL and PHA, a further increment in the molar ratio would cause crosslinking of the P–OH groups of PHA and the OH of KL to make P–O–P and P–O–C bonds during the reaction.52 This crosslinking will cause a decrease in solubility (occurring when the molar ratio > 0.3). The elemental analysis results in Table S2 (ESI†) show that by increasing the molar ratio of KL/PHL, the carbon and hydrogen contents decreased from 62.7 wt% for KL to 42.44 wt% for PK6 and from 6.9 wt% for KL to 5.37 wt% for PK6, respectively, which is attributed to the attachment of phosphorus to the KL backbone. The ICP-AES and XPS analyses confirmed the increment of the P element in the PK by increasing the PHA molar ratio in the reaction medium (Table S2†). The sulfur content of KL was less than 1.2 wt%, which can originate from its introduction to lignin during kraft pulping and the acid-washing treatment in the LignoForce process for producing KL in industry.53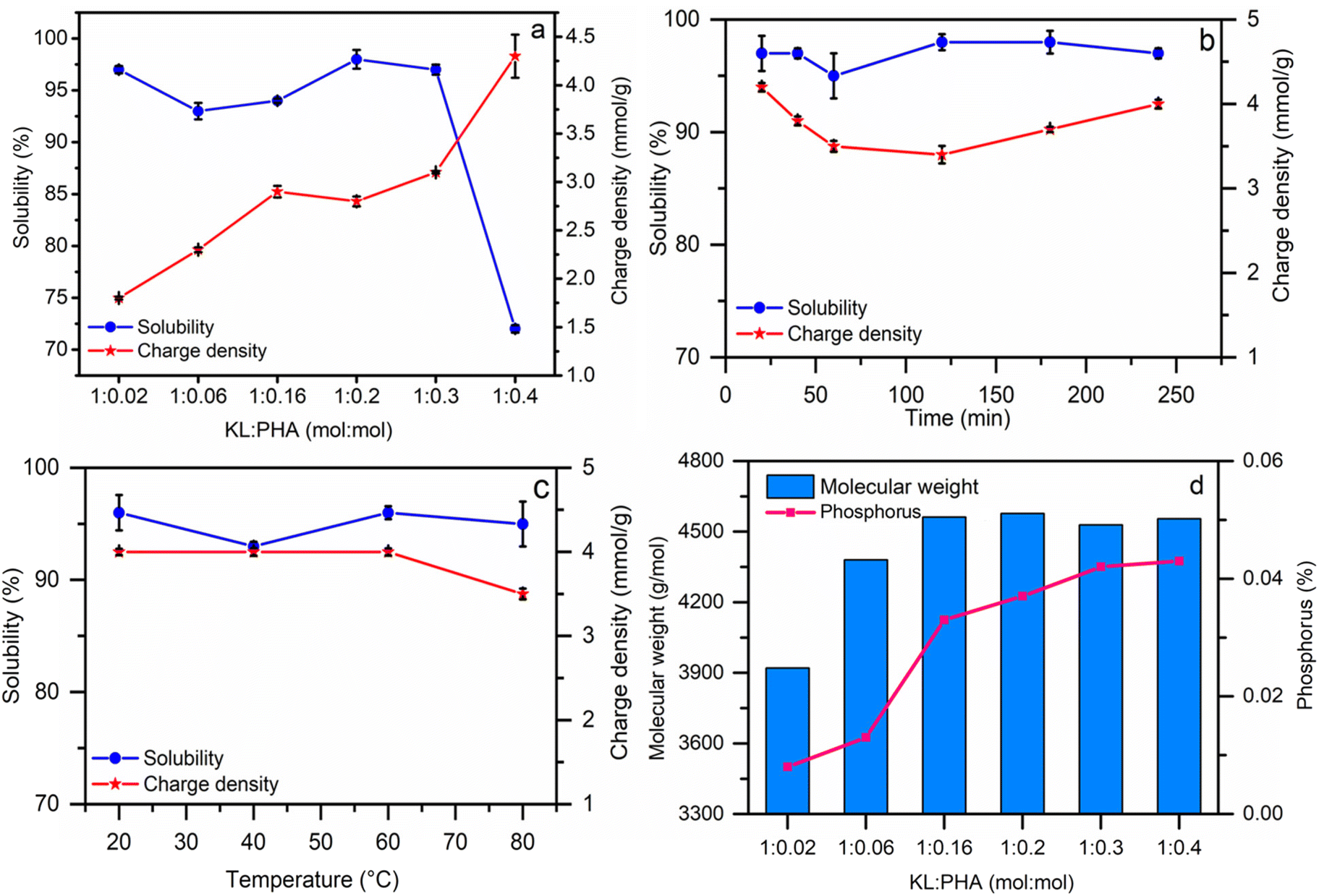 | ||
| Fig. 1 The effect of the molar ratio (a, PK1–PK6), time (b, PK11–PK16), and temperature (c, PK11, PK17–PK19) on the reaction. The effect of the molar ratio on molecular weight (Mw) and phosphorus content (d, PK1–PK6). | ||
The results show that time and temperature did not have a significant influence on the charge density and solubility of the resulting PK (Fig. 1b and c). The effect of the molar ratio of KL/PHA was investigated, and the results are available in Fig. 1a, d and Table S1.† Increasing the molar ratio decreases the pH of the reaction medium as PHA is acidic, promoting the aggregation and protonation of KL, resulting in a reduction in solubility. In addition, the activities of the hydroxyl and phosphate ions are pH dependent.54,55 Higher PHA concentrations facilitate a pH drop and suggest that the phosphate groups start to crosslink among themselves instead of reacting with KL because PHA is more active under acidic conditions, which would increase the average molecular weight of PK (Fig. 1d).56 Also, the aggregation of KL would decrease the reactivity of OH groups and its solubility due to its protonation under acidic conditions. This phenomenon will transform fractal-like KL into closed-packed aggregates, which would cause less solubility and phosphorylation.57 The effect of pH on the reaction efficiency was also investigated at the KL/PHA ratio of 1:0.4 mol:mol (PK6 and PK7 in Table S1†). The results confirmed that the reaction under alkaline conditions was preferred as the PK was produced with a higher solubility. In the pH range of 9–11, hydroxyl and phosphate groups would actively compete to interact with KL and promote different crosslinking scenarios, such as monophosphate, phosphodiester, and orthophosphate.58
Also, the effect of urea (as a solvent of the reaction) was studied at three reaction temperatures, and the results are reported in Table S1.† In earlier research, urea was utilized not only as a solvent but also as a degradation inhibitor of the lignin structure at high reaction temperatures (>90 °C), as the phosphorylation was primarily studied in the high-temperature range of 90–120 °C.59 However, our results confirmed that urea did not significantly affect the phosphorus group, charge density, and solubility of the induced phosphorylated samples (PK7 and PK8).
The reaction experiments confirmed that the optimized conditions for generating phosphorylated samples, PK17, with the highest molecular weight (Mw) of 4800 g mol−1, solubility of 97%, and charge density of −4.2 mmol g−1 were the KL/PHA molar ratio of 1:0.4 (mol:mol), water as the solvent, 20 min reaction time at 20 °C and pH 11 (i.e., strategy 2), and this sample was selected for further analysis.
NMR characterization
1H NMR was used to characterize the chemical structures of PK17 and KL. The spectra in Fig. 2 show the structures of KL and PK17. In the KL spectrum, the protons of phenolic and aliphatic hydroxyl groups are at chemical shifts of around 8.7 (a) and 5.5 (g and g′) ppm, respectively. After the modification, both hydroxyl groups participated in the reaction and disappeared from the spectrum of KL. The aromatic protons of the guaiacyl unit show a broad peak in the 7.5–6.1 ppm range (b and b′). The intense peaks at 4.1–3.1 ppm are associated with the protons of methoxyl groups on the lignin structure (c–e). Three strong peaks at 4.70, 2.5, and 0 ppm belong to D2O, DMSO, and TMSP, respectively, which were used as solvents for sample preparation and the internal standard. Also, two characteristic peaks in the PK17 spectrum at 4.5 and 4.2 ppm were ascribed to the PHA protons, confirming the success of phosphorylation.60 Fig. S1a in the ESI† shows the 1H NMR spectra of the samples at different molar ratios of KL/PHA in the reaction. By increasing the molar ratio of KL/PHA, both aromatic and aliphatic hydrogens decreased due to their participation in the reaction, and more phosphorus atoms were attached to the KL backbone. In addition, the intensity of the peaks in the range of 4–4.7 ppm was increased, which showed the presence of protons on phosphorus groups. By increasing the molar ratio of KL/PHA (PK1 to PK6 in Fig. S1a†), two peaks in the 1–4 ppm region had a slight decrease due to the crosslinking of the hydroxyl group of phosphorus groups.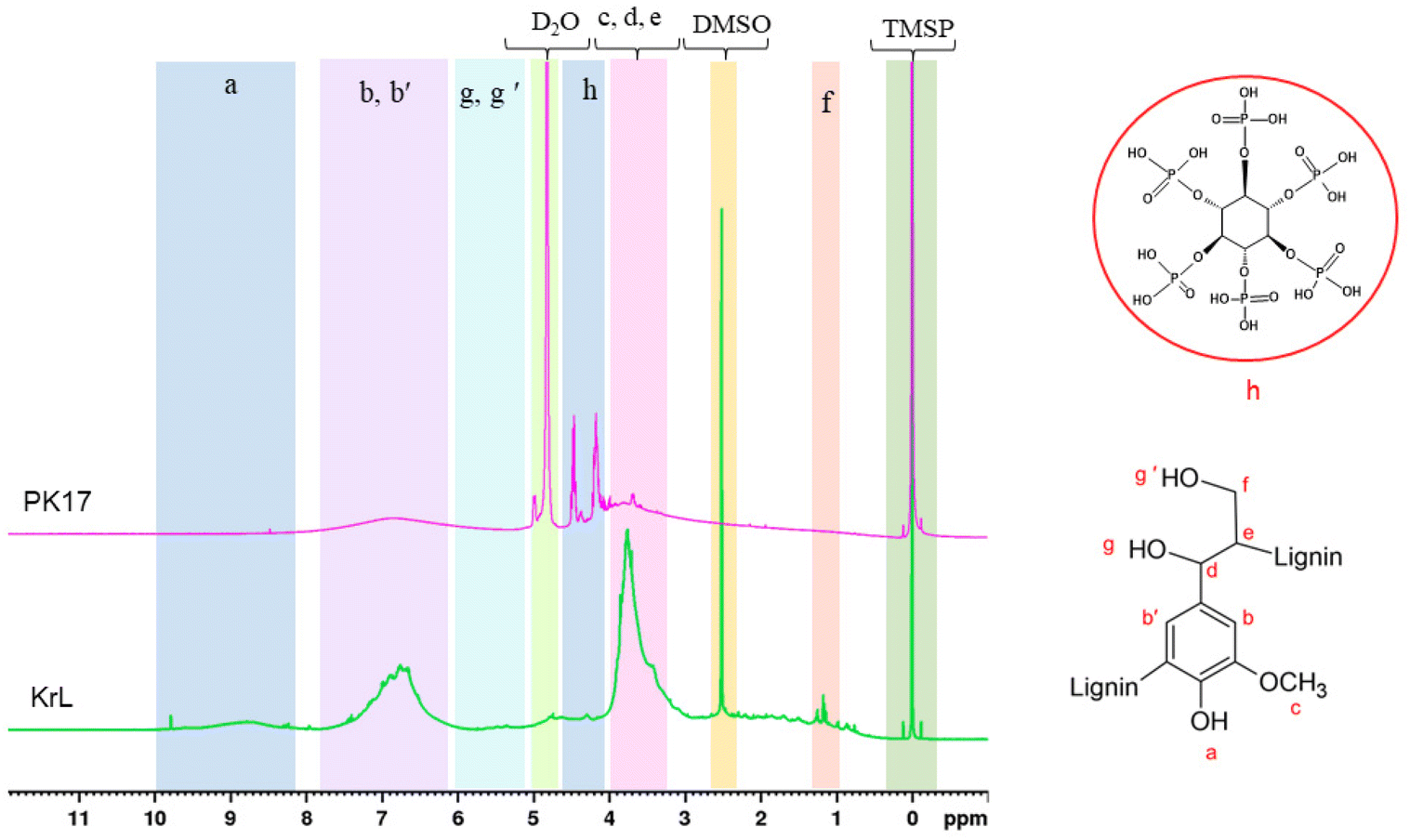 | ||
| Fig. 2 1H NMR spectra of KL and PK17. Shaded peaks are identified as described in the text. | ||
Quantitative 31P NMR spectroscopy was carried out to confirm the chemical structures of KL and PK17 samples, and the results are shown in Fig. 3 and Table 1. This analysis provided quantitative information on the concentration of each hydroxyl group of lignin participating in the reaction. The cyclohexanol peak (internal standard) appears at 144.8 ppm. The decrease in the aliphatic, phenolic, and carboxylic hydroxyl groups proved that these groups participated in the modification reaction. A significant drop in the amount of phenolic hydroxyl groups from 3.33 mmol g−1 to 0.63 mmol g−1 confirmed the high activity of phenolic hydroxyl groups in KL and phosphorylation at this site by polycondensation.61 After the phosphorylation of KL, the aliphatic hydroxyl group decreased from 1.72 mmol g−1 for KL to 0.63 mmol g−1 for PK17. In addition, there is a significant drop from 0.41 mmol g−1 to 0.06 mmol g−1 for carboxylic hydroxyl content, inferring that the reaction happened on the aliphatic, aromatic, and carboxyl hydroxyl groups. The sharp peak in the phenolic hydroxyl region is for PHA's hydroxyl groups, confirming the successful phosphorylation reaction.
 | ||
| Fig. 3 Quantitative 31P NMR of KL and PK17. Shaded peaks indicate the type of OH functional group. | ||
| Sample name | Aliphatic OH (mmol g−1) | Phenolic OH (mmol g−1) | C5-substituted (mmol g−1) | Guaiacyl OH (mmol g−1) | p-Hydroxyphenyl OH (mmol g−1) | Carboxylic acid OH (mmol g−1) |
|---|---|---|---|---|---|---|
| KL | 1.72 | 3.33 | 1.5 | 1.6 | 0.11 | 0.41 |
| PK17 | 0.63 | 0.71 | 0.29 | 0.39 | 0.02 | 0.06 |
The qualitative 31P NMR spectra of KL and PK17 are shown in Fig. 4. No peak can be characterized for KL, inferring that KL did not have any phosphorus-containing group. Based on previous studies, 31P NMR of PHA was assigned to four different peaks at 1, 0.5, 0, and −0.8 ppm.62 As explained earlier, the phosphorylation of lignin could happen on aliphatic, phenolic, and carboxylic hydroxyl groups. In addition, an esterification reaction and crosslinking could occur between phosphate and hydroxyl groups, resulting in a chemical shift from 5 ppm.38,63 Fig. S1b in the ESI† shows the 31P NMR spectra of PK generated at different molar ratios, and by increasing the molar ratio of KL/PHA, the intensity of 31P NMR peaks increased. At a higher KL/PHA molar ratio than 1:0.16 (mol:mol), PK started to exhibit different crosslinking types (phosphate (a), orthophosphate (b), and phosphodiester (c)), especially type c, that had lower solubility (Table S1†).
 | ||
| Fig. 4 Qualitative 31P NMR of KL and PK17. | ||
HSQC NMR was used to understand the changes in the structure of lignin, and the 1H–13C HSQC spectra of KL and PK17 are displayed in Fig. 5a–d. The detailed structures of signal assignment for each bond of aromatic (δC/δH 100–140/6.1–7.9), aliphatic (δC/δH 45–100/3–5.6), and side chain (δC/δH 10–60/0.2–2.8) groups are shown in Table S3.†64,65 In Fig. 5a and b, the aromatic region, G-type structure, appears in both KL and PK17, implying that no degradation of KL happened after modification.66 In addition, Fig. 5c shows the aliphatic region in both KL and PK17.67 The large area in Fig. 5d and e belongs to C–H in –OCH3 (methoxy) in the aliphatic region (δC/δH 57.3/3.7). Two linkages of Bγ (δC/δH 72.7/3.7–4.1) and Bβ (δC/δH 55/3.2) disappeared and cleaved after modification (PK17 compared with KL), which can be attributed to the crosslinking or polycondensation of the hydrogen element of these linkages during modification.68
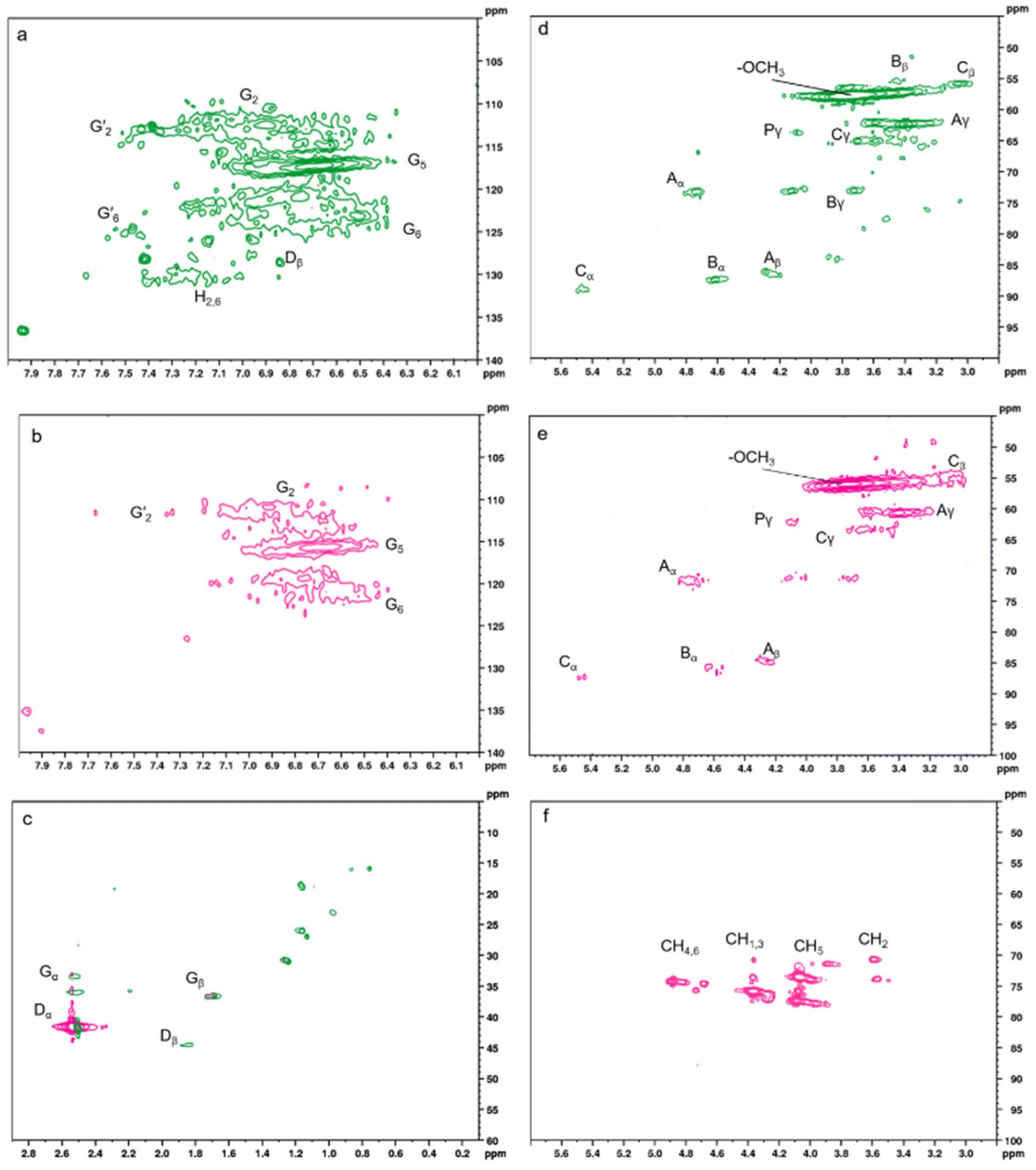 | ||
| Fig. 5 HSQC NMR spectra (a and d) of KL, (b and e) PK17 in DMSO, (c) aliphatic region of both KL and PK17 in DMSO, and (f) PK17 in D2O. | ||
Due to the proton transferring effect of aromatic groups on the lignin backbone, the aromatic region does not correlate in the HSQC NMR spectra. This behavior was more vital when the sample was prepared in D2O, as exchangeable protons such as hydroxyl groups increased the typical widening in the resonances.69 A detailed study was conducted in D2O because phosphorus groups were not detectable in DMSO due to the high polarity and less solubility of PK17.65Fig. 5f shows the presence of a new linkage of lignin with PHA based on the four different protons and their correlation with carbons on the PHA structure.70 The HSQC NMR spectrum of PHA is shown in Fig. S2.† It is observable that the linkages of PHA mostly appear in the aliphatic region of KL (δC/δH 75–85/3.5–5). These linkages were not detectable for the sample prepared in DMSO due to the high negative charge density around the nucleus and the high opposite magnetic field around the copolymer, which will provide a great shield covering the linkages in this region. Due to this shielding, 1H–31P HMBC NMR was used (Fig. S3†). The P and H linked to the same C on the 6-membered ring were detected by the cross-peaks on the HMBC spectrum.71 These findings show that the skeletal structure of lignin was preserved during phosphorylation, which might be attributed to the moderate reaction conditions.72
FTIR analysis
The FTIR spectra of KL and PK17 are shown in Fig. 6. The characteristic bands in KL were comprehensively determined in previous studies.73 The O–H aliphatic and aromatic stretching vibration groups show a broad transmittance band at 3400 cm−1 (region a). After phosphorylation, the band intensity decreased, which could prove the participation of these O–H functional groups in the phosphorylation reaction. The aliphatic C–H stretching vibration of methyl and methylene groups showed transmittance bands at around 2930 cm−1 and 2830 cm−1 (region b). The phenylpropane monomer bands in KL were established at 1591 cm−1, 1510 cm−1, and 1450 cm−1, which are assigned to the stretching vibrations of the C–C bonds. However, the intensity of these peaks after modification decreased (region c). Both ether bands of C–O–C and the stretching vibration of C–O were assigned to the bands at 1280 cm−1, 1200 cm−1, 1120 cm−1, and 1030 cm−1 (region d). IR spectroscopy does not show intense bands of phosphate moieties.74 However, minor alterations in the 1250 cm−1 and 1300 cm−1 bands belong to P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. After phosphorylation, the appearance of three new bands proved that KL was modified successfully with PHA. These bands are PO43−, P–O–C aliphatic, and the P–O stretching band (region f), which are assigned to 990 cm−1, 960 cm−1, and 850 cm−1, respectively.75–77 In region f, two bands of bending and stretching are assigned to P–O–P, which can be determined at 760 cm−1 and 810 cm−1, respectively.78 The P–O bond in PK17 overlapped with the aromatic C–O–C structure, which shifted the band from 1120 cm−1 to 1020 cm−1 in the C–O stretching of the guaiacyl group of KL (region e).79 All these changes in the FTIR bands confirmed that KL was modified with phosphorus groups on phenolic, aromatic, and carboxylic hydroxyl groups for generating PK17.
O. After phosphorylation, the appearance of three new bands proved that KL was modified successfully with PHA. These bands are PO43−, P–O–C aliphatic, and the P–O stretching band (region f), which are assigned to 990 cm−1, 960 cm−1, and 850 cm−1, respectively.75–77 In region f, two bands of bending and stretching are assigned to P–O–P, which can be determined at 760 cm−1 and 810 cm−1, respectively.78 The P–O bond in PK17 overlapped with the aromatic C–O–C structure, which shifted the band from 1120 cm−1 to 1020 cm−1 in the C–O stretching of the guaiacyl group of KL (region e).79 All these changes in the FTIR bands confirmed that KL was modified with phosphorus groups on phenolic, aromatic, and carboxylic hydroxyl groups for generating PK17.
 | ||
| Fig. 6 FTIR spectra of KL and PK17. Shaded peaks are identified as described in the text. | ||
XPS and ICP-AES analyses
The chemical compositions of the samples were analyzed by X-ray photoelectron spectroscopy. The broad XPS spectra of KL and PK17 are shown in Fig. 7a. These graphs showed two major peaks of C 1s and O 1s. In the broad spectrum of KL, two other signals are assigned to S 2p and Si 2p that remained from the pulping process of lignin extraction.80 The presence of two new peaks in the spectrum of PK17 was attributed to the phosphorus element after modification with PHA.81 These peaks were found at 134.5 eV and 199 eV, respectively, assigned to P 2p and P 1s. The fitted C 1s, O 1s, and P 2p spectra for KL and PK17 are shown in Fig. 7b–f and Tables S4–S7.† According to the C 1s spectra and the result of mass concentration, three significant peaks are shown in KL at 281.1 eV for C–C, 284.2 eV for C–OH, and 285.7 eV for OC–OH with a mass concentration of 42.44%, 55.2%, and 2.35%, respectively. After the modification, PHA covalently bonded to KL, and by adding PHA to the KL backbone, the C–C bond increased to 48.41%.
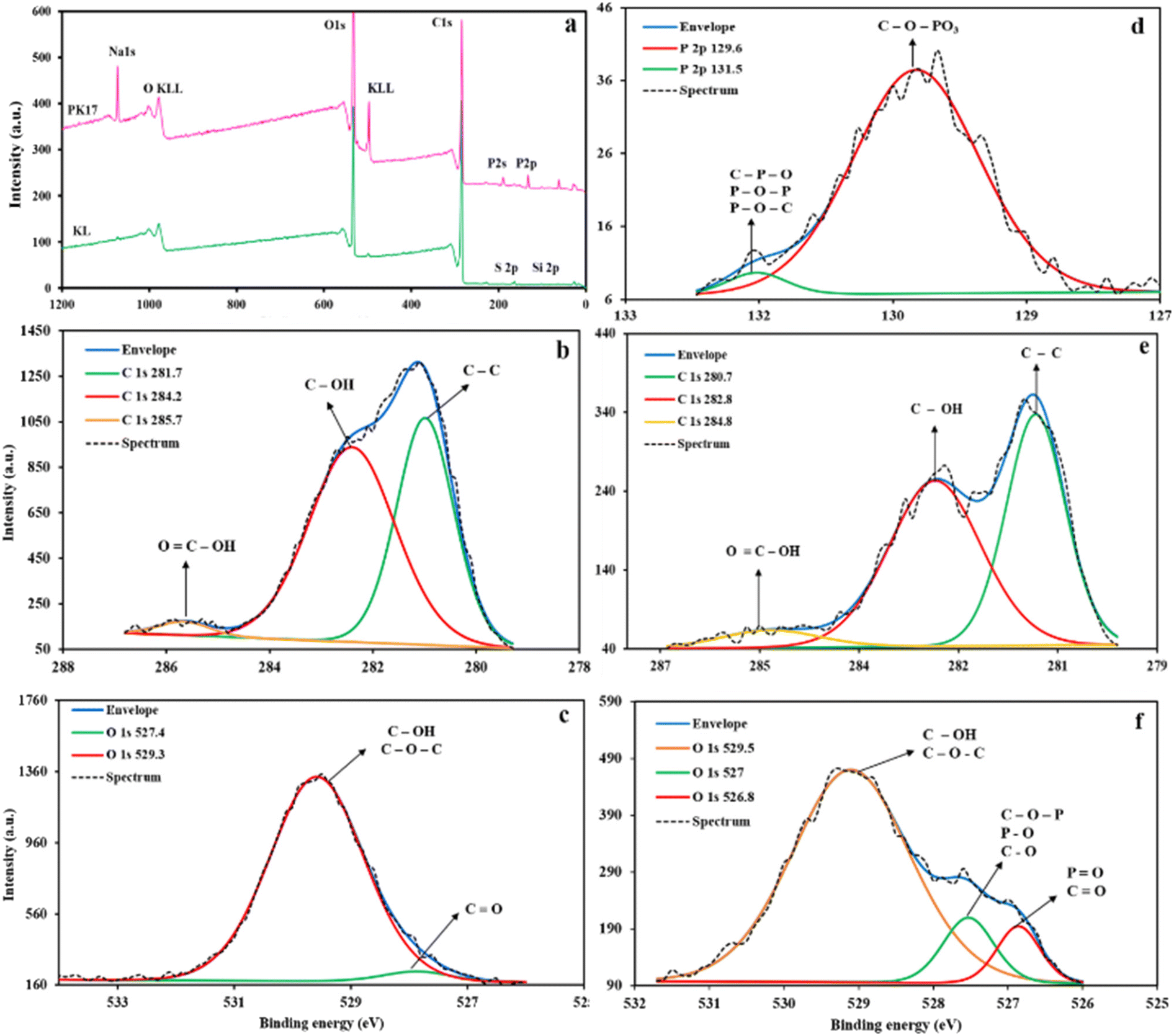 | ||
| Fig. 7 XPS broad spectra (a), C 1s and O 1s spectra of KL (b and c), C 1s and O 1s, and P 2p spectra of PK17 (d–f). | ||
In Fig. 7d, two new bonds in PK17 (C–O–PO3 and C–P–O) proved that the reaction happened via polycondensation with the hydroxyl groups on KL, confirming the results verified by 31P NMR analysis. The phosphorus content of the samples was also determined from XPS and ICP-AES data, and the results are reported in Table S2.† The phosphorus group content increased from 0 wt% to 7 wt% after the modification of KL with PHA. As noted, the amount of P determined in both ICP-AES and XPS results is similar.
One strong peak at 527.4 eV was observed in the fitted O 1s spectra of KL and PK17 (Fig. 7), corresponding to C–OH and C–O–C with 96.49% mass concentration dropping to 81.31% after phosphorylation. The peak intensity at 526.8 eV increased due to the overlap between the new PO bond and the existing CO bond in this region. Previous studies suggest that the CO and PO groups in the phosphate group exhibited the same binding energy in the spectrum.82,83 The increase in these bands from 3.51% to 7.93% is the reason for this overlap. The appearance of the C–O–P bond in the oxygen graph after modification at 527 eV is evidence of polycondensation in the route of phosphorylation.
The fitted P 2p spectrum, Fig. 7d, shows one prominent intense peak at 129.6 eV for C–O–PO3, which was characteristic of phosphorus in both polyphosphates and phosphates, and one small shoulder peak at 131.5 eV for both C–P–O and P–O–P.84 Such evidence confirmed that the –PO3 group replaced a part of the aliphatic, phenolic, and carboxylic hydroxyl groups on the KL structure through the nucleophilic reaction.85
Mechanism of reaction
The mechanism of reaction is proposed in Fig. 8a and b. The reaction occurs in two repetitive steps: first, phosphoric anhydride is formed with the elimination of water by a polycondensation reaction, and second, the phosphate atom leads to electron transfer from the oxygen in PA to the hydrogen of hydroxyl in the KL to form a new ester bond on the KL (Fig. 8a).43 The appearance of new peaks related to phosphorus groups in 1H NMR in the range of 4–4.7 ppm, C–O–PO3 and C–P–O bonds in XPS at 130 eV and 132 eV, and PO43−, P–O–C aliphatic, and P–O stretching bands at 990 cm−1, 960 cm−1, and 850 cm−1, in FTIR, respectively, confirms the reaction between KL and PHA. Moreover, quantitative 31P NMR results approved the reduction of the hydroxyl groups of KL after modification. Phosphate groups on PHA are covalently bonded with different crosslinking types (a–c). This reaction can occur through both intra-chain and inter-chain routes, as shown in Fig. 8b.86,87 According to the results of qualitative 31P NMR (Fig. 4), the intensities of two peaks, a and b, are high, confirming that the modification mainly occurred via the intra-chain route.55 It can be concluded that PHA reacted with the hydroxyl groups of KL and exhibited two types of a and b crosslinks that were stronger than the c type (Fig. 4). | ||
| Fig. 8 (a) The reaction scheme of phosphorylation and (b) scheme of all possibilities of crosslinking (a–c). | ||
Thermal properties
The thermal behavior of KL and PK17 was evaluated by thermogravimetric analysis (TGA) and is reported in Fig. 9a. The decomposition of lignin is divided into three main stages: the initial slight weight loss in the range of 25–100 °C is because of the evaporation of moisture, CO and CO2. The weight loss between 175 °C and 550 °C is attributed to the degradation of aromatic rings and C–C linkages, such as C–C and β–β in KL.88 The final weight loss at >550 °C was less and related to the decomposition and degradation of organic materials and carbonization.89,90 After the moisture removal from KL, the temperature corresponding to 5 wt% weight loss (Td) increased from 210 ± 2 °C to 262 ± 2 °C after modification. Tonset is the temperature at which the major decomposition starts to occur. This temperature for KL and PK17 is 260 ± 2 °C and 300 ± 2 °C, respectively, implying that the modification improved the Tonset temperature by about 40 °C. Introducing the phosphorus group to KL increased the maximum decomposition temperatures (Tmax) from 545 °C to 650 °C, and the weight loss rate decreased from 0.064 wt% °C−1 to 0.026 wt% °C−1.86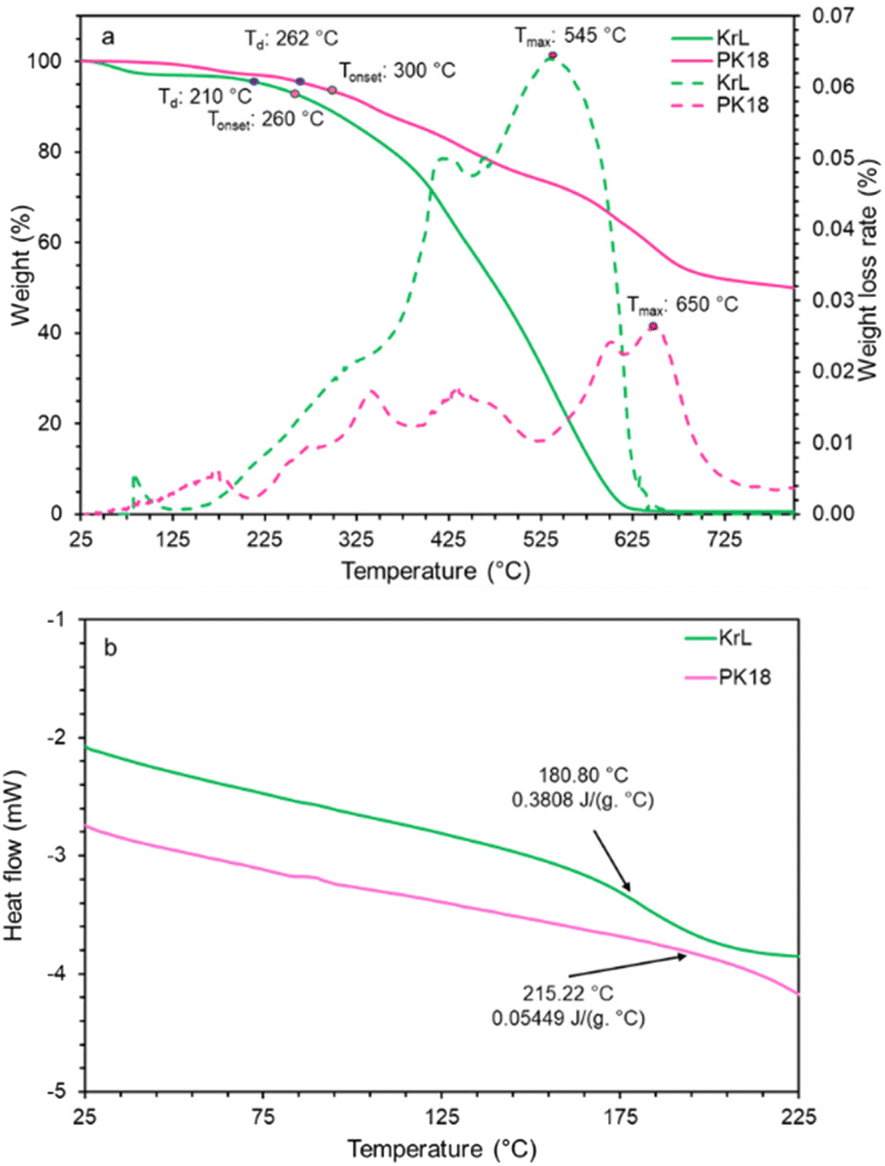 | ||
| Fig. 9 Thermal analysis of KL and PK17. TGA curves with an arrow indicating Tmax (a). DSC curves showing the Tg and Cp (b). | ||
It is also seen that KL did not remain at >620 °C, while 65 wt% of PK17 remained. At 800 °C, the char mass of PK17 was about 50 wt%, implying the improved char formation via phosphorylation.
The differential scanning calorimetry (DSC) analysis of the samples is shown in Fig. 9b. The molecular weight, crosslinking degree, branching degree, and chain length can affect the Tg of the material due to their impacts on segment mobility when heated/cooled.91,92 As reported in Fig. 9b, the Tg for KL was found at 180.8 °C, which increased to 215.2 °C after modification. The presence of phosphorus atoms after the modification and crosslinking of hydroxyl groups on the KL in the presence of the active sites of PHA contributed to an increased Tg.93 In addition, PK17 had a lower heat capacity (Cp) than KL, 0.0544 J (g °C)−1 and 0.3808 J (g °C)−1, respectively. Less mobility, resistance to breakdown, and crosslinking would be the causes of this decreased heat capacity.94,95 In addition, inorganic compounds, such as phosphor, contributed to the increased Tg and decreased Cp.96
Flame retardant performance
The flame retardancy of materials is commonly determined by the limiting oxygen index (LOI), and the results are listed in Fig. 10. Generally, with the increase in LOI values, the flame retardancy of the sample increased. As shown in Fig. 10, the LOI value of uncoated wood (UW) was 21.8%, implying that it could burn quickly with a small ignition. Different concentrations (1, 2, and 3 wt%) of KL (KC1, KC2, and KC3) were used for coating the wood species, respectively. Upon increasing the concentration of PK17 to 1, 2, and 3 wt%, the LOI value increased to 23.8%, 24.6%, and 26.0% for the samples of PC1, PC2, and PC3, respectively. Moreover, filter paper coated with 3 wt% PK17 demonstrated an increment in the LOI from 18% to 21%. Jiang et al. found that 10 wt% of N,N-biguanide-diethyl phosphonic acid can increase the LOI value of filter paper by >10%.97 | ||
| Fig. 10 LOI values of the UW, KL, and PC (PK17 coated) samples. | ||
As shown in Fig. 11a and b, the smoke density rate (SDR) in 240 s and light absorption of the uncoated wood (UW) were 34% and 50%, respectively. Generally, introducing KL to wood will affect wood species’ flame retardancy. Coating wood with 1, 2, and 3% KL decreased the SDR value to 23.8%, 24.7%, and 27.8%, respectively. The introduction of 1, 2, and 3 wt% of PK17 reduced the light absorption to about 30%. The PK17-coated woods indicated a decrease in smoke production after 200 seconds, mainly because of the protective char layer of phosphorus–carbon.98 Interestingly, the light absorption value peaked at around 180 s and subsequently declined since less smoke was released due to the char formation acting as a protective layer to lower the smoke density. PC3 (coated wood with 3 wt% of PK17) showed the lowest light absorption of 17.7% (i.e., 16% lower than UW).
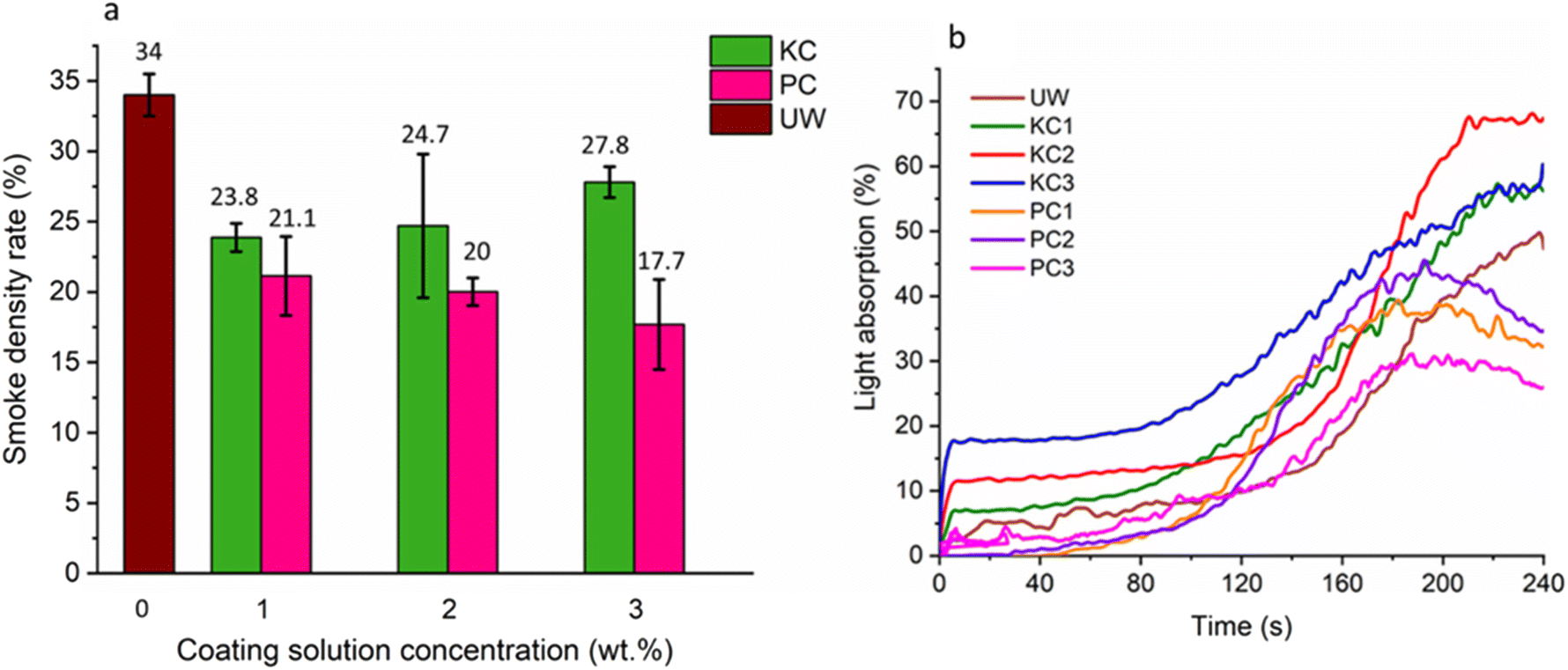 | ||
| Fig. 11 Smoke density rating values (a) and light absorption curves (b) of the UW, KL, and PC (PK17 coated) samples. | ||
Morphological study of burned wood
The influence of phosphorylated lignin on the surface morphology of wood is generally determined by investigating char residues with SEM after smoke detector analysis.88 Fig. S4 (in the ESI†) and Fig. 12 show char morphology after completing the combustion of UW and PC3, respectively. The spherical pores of some broken structures (Fig. S4a–d†) show the released gas caused by the combustion of UW, while these pores are compact in PC3.99Fig. 12a–d show the blocked pores because of phosphoric acid production during thermal degradation. This would lead to increased dehydration and carbonization of PK17 to form char and phosphorus crystals to penetrate the pores and block them.100,101Fig. 12d shows the phosphorus crystals after combustion. A char layer covered the PC3 after burning due to phosphorus groups’ dehydration of PK17 and wood.102 In addition, EDX observation indicated the presence of phosphorus atoms after burning PC3 (Fig. 12f). | ||
| Fig. 12 SEM images (a–d), digital picture (e), and EDX graph (f) of char residue after burning PC3 in smoke density analysis. | ||
The mechanism of flame retardancy
FRs are employed to interrupt the combustion process in two ways: they create a dense char that shields the substrate from combustible gases (i.e., oxygen) and heat (condensed phase mechanism), and they generate radical scavengers that can halt the chain propagation of the combustion reaction (vapor phase mechanism).103 The flame retardancy of PK17 can happen in both gas and condensed phases, chemically and physically. Phosphorus is an inorganic element that will produce radicals after decomposition to accelerate char formation as a protective layer from heat degradation.104 During combustion, triphenyl phosphate and triphenylphosphine oxide can break down into small radicals, such as HPO˙, PO2˙, PO˙, and P2˙.105 Because of ignition, these phosphorus radicals can function as an adsorbent for H˙ and OH˙ radicals released in the gas phase needed for ignition. A decrease in these radicals will reduce the flame.106In the condensed phase, phosphorus compounds start to decompose, and the protective char layer of phosphorus–carbon begins to form.92 This protective layer could hold the flammable gases released from inner layers and keep away the heat from unburned wood layers.107,108
For a better understanding of how PK17 works as a flame retardant fundamentally, XPS analysis was carried out on the samples collected from TGA after treatment at four different temperatures (220, 320, 600, and 800 °C). Fig. S5–S8† show how the intensity of each peak changed, and the mass concentrations of each element and bond are reported in Tables S4–S7† in detail. The mass concentration of P 2p increased from 7% at 25 °C to 26.9% at 800 °C (Table S4†). Also, the C–C bond concentration remained almost the same (about 42%) (Table S5†), but there was a decrease in the mass concentration of C–OH and C–O–C bonds from 81.31 to 15.22%. However, there was an increment in P–O, CO, and PO bonds from 3.51% to 49.49% (Table S6†). These results confirmed that P and C remained as by-products of burning in the form of –PO, CO, C–O–P, P–O, P–O–P, and C–P–O, while oxygen was released. Also, the mass concentration of C–P–O and P–O–P increased from 4.36% to 70.33% (Table S7†).109 Wang et al. argued that thermal oxidation resistance can be confirmed by an increment in CO, which exhibits a thermal flame retardancy mechanism.110
A deep analysis was carried out on a microscale using SEM, EDS, and EDX for PK17 before and after combustion at 800 °C. It was found (Fig. 13a and c) that the morphology of PK17 changed from a regular structure to irregular with some crystal structures,111 which was confirmed by SEM in coated wood after burning (Fig. 12d). In addition, the homogeneous distribution of C, P, and N elements in the PK17 and KL samples before and after combustion (Fig. 13 and S9b, d†) was confirmed in the EDS images. Still, the intensity of the phosphorus element increased after combustion for PK17. Flame retardancy is mainly related to the concentration of phosphorus that can delay the transfer of heat, block the diffusion of flame efficiently, and increase oxygen demand for burning, inducing a more efficient flame retardancy (Fig. 10 and 11b).112
 | ||
| Fig. 13 The SEM images (a and c), EDS elemental mapping, and EDX (b and d) of the surface of PK17 before and after combustion. | ||
To understand how PK17 would function as a flame retardant, the SEM and EDX images of PC3 samples after combustion were analyzed in detail. The results (Fig. S7† and 12) provide evidence for the elemental composition of phosphorus crystals, char residue formation, filled porous structures, gaps, and cracks in wood after burning. These results imply that PC3 acted as a protection shield to disrupt the penetration of gas (oxygen) and heat flow during combustion.107 In addition, the char formation would work as a protective layer that would decrease smoke release, leading to a lower smoke density rate and light adsorption (Fig. 11).
The performance and reaction route of phosphorylated lignin products were compared with other phosphorylated lignin products in Table 2. Our sample, a pioneering biomaterial, sets itself apart by being free from solvent (water base) and bio-phosphorus reagent (PHA) while exhibiting outstanding high heat resistance and flame retardancy. Unlike conventional materials that are listed in Table 2, made of oil-derived chemical reagents and under harsh and environmentally unfriendly conditions, our biomaterial was produced using environmentally friendly reagents, a water-based reaction at room temperature, ensuring a safer and more sustainable approach. The elimination of solvents not only reduces the environmental impact but also minimizes potential health hazards associated with their use. Additionally, our biomaterial's heat resistance is outstanding, offering enhanced heat resistance. This combination of solvent-free processing and excellent heat resistance with high thermal performance underscores the material's potential in advancing eco-friendly and high-performance applications. Theoretically, other types of lignin, e.g., alkali, lignosulfonate, and organosolv, may be used for reacting with PHA in a similar manner as they all have OH functional groups. However, their efficiency may be different, which will be attested in our future work.
| Source of lignin | Reagent (reagent:lignin) |
Reaction conditions | Solvent | TGA and DTG | Char residue at 800 °C | Application | Ref. |
|---|---|---|---|---|---|---|---|
| Softwood kraft lignin | Ammonium dihydrogen phosphate 9:1 (mol:mol) |
1 h at 70 °C | Urea | T max from 541 °C to 620 °C | From 0% to 15% | Not tested | 38 |
| Softwood kraft lignin | Phosphoric acid (85%) 0.5:0.14 (g:g) |
50 °C | Acetone and urea | Not reported | Not reported | Not tested | 113 |
| Kraft lignin | Phosphorus pentoxide 2.5:1 (phr:g) |
1 to 24 h at 70 °C | Tetrahydrofuran | T max not reported | From 0% to 38% | Polypropylene-based composites | 114 |
| Alkaline lignin | Phosphorus pentoxide 5.5:1 and 2.5:1 (mol:mol) |
2 h at 5 °C; nitrogen gas | Methane sulfonic acid, purification with diethyl ether, acetone, or methanol | T max from 465 °C to 572 °C and 607 °C | From 44.4% to 13.5% and 10.3% | Epoxy resin | 115 |
| Kraft lignin | Phosphorus pentoxide; not reported | 7 to 8 h at room temperature | Tetrahydrofuran | T max from 370 °C to 409 °C | From 45% to 53% | Not tested | 116 |
| Kraft lignin | Phosphorus chloride 6.5:10 (g:g) |
17 h at 60 °C | A mixture of CHCl3 and N-methyl-2-pyrrolidone precipitated and washed with cooled isopropyl alcohol and diethyl ether | Not available | Not available | Polyester-based composites | 117 |
| Kraft lignin | Phytic acid 0.4:1 (mol:mol) |
20 min at room temperature | Water | T max from 545 °C to 640 °C | From 0% to 50% | Coating | This work |
Conclusions
A sustainable flame retardant was prepared in an aqueous medium, i.e., green solvent, from KL and PHA. The reaction conditions that yielded the lignin derivative with the highest phosphorus element were a KL:PHA molar ratio of 1:0.4 mol/mol, a reaction time of 20 min, a temperature of 20 °C, and a pH of 11. These optimized conditions resulted in PK17 with −4.2 mmol g−1 charge density, 9.7 g L−1 solubility in water, and an Mw of 4800 g mol−1. The reaction under the optimized conditions decreased the aliphatic and phenolic hydroxyl groups of KL from 1.72 to 0.63 mmol g−1 and 3.33 to 0.71 mmol g−1, respectively. 1H, HSQC, and 31P NMR confirmed that the aliphatic and aromatic OH groups of KL participated in the reaction with PHA and generated a covalent bond of P–O–C. The qualitative 31P NMR confirmed the three different crosslinking types of phosphate groups based on the reaction conditions, such as monophosphate, phosphodiester, and orthophosphate. Also, FTIR, XPS, and ICP-AES analyses confirmed the presence of phosphorus atoms and the new C–O–P bond between KL and PHA.
The TGA analysis confirmed that the phosphorylation of KL improved the thermal stability of lignin by improving its decomposition temperature from 545 °C to 650 °C with char formation at 800 °C, while KL did not produce any char at 625 °C. The DSC results confirmed the increment in Tg from 180.8 °C to 215.2 °C after phosphorylation. The combustion completion was analyzed by SEM, EDX, and EDS for PK17, and the change in sample morphology from regular to irregular and the increment in the intensity of phosphorus were confirmed after combustion. By increasing the concentration of PK17 solution from 1 wt% to 3 wt% in the wood coating process, the LOI of wood rose from 21.8% to 26.0%, and the smoke density rate decreased from 34.0% to 17.7% due to the formation of thermally stable char and the release of non-flammable gases. The promising flame retardant performance of PK17 on wood and filter paper paves the way for the production pathway of PK17 as a simple and effective aqueous-based reaction to produce phosphorylated lignin.
Author contributions
Saba Khodavandegar: methodology, investigation, experiment, data analysis, and writing – original draft. Pedram Fatehi: supervision, review & editing, conceptualization, resources, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.† No software or codes were used in this article, and extra data can be obtained upon request from the corresponding author.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank the NSERC, Canada Research Chairs, and the Canada Foundation for Innovation for their financial support.Notes and references
- E. D. Weil and S. V. Levchik, J. Fire Sci., 2008, 26, 243–281 CrossRef CAS.
- A. B. Morgan and J. W. Gilman, Fire Mater., 2013, 37, 259–279 CrossRef CAS.
- S. K. Pandit, B. K. Tudu, I. M. Mishra and A. Kumar, Prog. Org. Coat., 2020, 139, 105453 CrossRef CAS.
- M. Li, X. Hao, M. Hu, Y. Huang, C. Tang, Y. Chen and L. Li, Prog. Org. Coat., 2022, 172, 107161 CrossRef CAS.
- T. Ma, L. Li, Q. Wang and C. Guo, Composites, Part B, 2019, 177, 107357 CrossRef CAS.
- M. Dogan, S. Dilem Dogan, L. Atabek Savas, G. Ozcelik and U. Tayfun, Composites, Part B, 2021, 222, 109088 CrossRef CAS.
- J. De Boer and H. M. Stapleton, Science, 2019, 364, 231–232 CrossRef CAS PubMed.
- F. Xu, L. Zhong, C. Zhang, P. Wang, F. Zhang and G. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 13999–14008 CrossRef CAS.
- I. van der Veen and J. de Boer, Chemosphere, 2012, 88, 1119–1153 CrossRef CAS PubMed.
- L. Costes, F. Laoutid, S. Brohez and P. Dubois, Mater. Sci. Eng., R, 2017, 117, 1–25 CrossRef.
- H. S. Hendriks, M. Meijer, M. Muilwijk, M. Van Den Berg and R. H. S. Westerink, Arch. Toxicol., 2014, 3, 857–869 CrossRef PubMed.
- M. Ghanadpour, F. Carosio, P. Tomas Larsson and L. Wa, J. Biol. Macromol., 2015, 16, 3399–3410 CAS.
- M. Wang, G.-Z. Yin, Y. Yang, W. Fu, J. Luis Díaz Palencia, J. Zhao, N. Wang, Y. Jiang and D.-Y. Wang, Adv. Ind. Eng. Polym. Res., 2022, 6, 132–155 Search PubMed.
- K. Ahmad, H. R. Ghatak and S. M. Ahuja, J. Environ. Chem. Eng., 2022, 275–309 Search PubMed.
- G. Atiwesh, C. C. Parrish, J. Banoub and T. A. T. Le, Biotechnol. Prog., 2021, 38, 3226 CrossRef PubMed.
- Q. Shao, Y. Luo, M. Cao, X. Qiu and D. Zheng, Chem. Eng. J., 2023, 476, 146678 CrossRef CAS.
- Y. Li, X. Qiu, Y. Qian, W. Xiong and D. Yang, Chem. Eng. J., 2017, 327, 1176–1183 CrossRef CAS.
- Y. F. Gao, C. Y. Ma, Q. Sun, X. X. Zhang, J. Liu, J. L. Wen, R. X. Chen, H. L. Wang and T. Q. Yuan, Chem. Eng. J., 2024, 481, 148514 CrossRef CAS.
- B. Yan, M. Li, H. Lu, M. Pi, J. Mu, W. Cui and R. Ran, Chem. Eng. J., 2024, 481, 148540 CrossRef CAS.
- B. Wang, X. F. Cao, S. X. Yu, Z. H. Sun, X. J. Shen, J. L. Wen and T. Q. Yuan, J. Cleaner Prod., 2023, 387, 135801 CrossRef CAS.
- L. Meng, S. Chirtes, X. Liu, M. Eriksson and W. C. Mak, Biosens. Bioelectron., 2022, 218, 114742 CrossRef CAS PubMed.
- T. Yao, R. Yang, C. Sun, Y. Lin, R. Liu, H. Yang, J. Chen and X. Gu, Molecules, 2023, 28, 2699 CrossRef CAS PubMed.
- D. Zhang, J. Zeng, W. Liu, X. Qiu, Y. Qian, H. Zhang, Y. Yang, M. Liu and D. Yang, Green Chem., 2021, 23, 5972–5980 RSC.
- H. Yang, B. Shi, Y. Xue, Z. Ma, L. Liu, L. Liu, Y. Yu, Z. Zhang, P. K. Annamalai and P. Song, J. Compos. Sci., 2021, 22, 1432–1444 CAS.
- L. Costes, F. Laoutid, M. Aguedo, A. Richel, S. Brohez, C. Delvosalle and P. Dubois, Eur. Polym. J., 2016, 84, 652–667 CrossRef CAS.
- Y. Liu, W. Zhao, X. Yu, J. Zhang, Y. Ren, X. Liu and H. Qu, Polym. Degrad. Stab., 2022, 203, 110062 CrossRef CAS.
- Y. Guo, C. Cheng, T. Huo, Y. Ren and X. Liu, Polym. Degrad. Stab., 2020, 181, 109362 CrossRef CAS.
- T. Mariappan, Y. Zhou, J. Hao and C. A. Wilkie, Eur. Polym. J., 2013, 49, 3171–3180 CrossRef CAS.
- Z. Xiong, Y. Zhang, X. Du, P. Song and Z. Fang, ACS Sustainable Chem. Eng., 2019, 7, 8954–8963 CrossRef CAS.
- X. Cheng, L. Shi, Z. Fan, Y. Yu and R. Liu, Polym. Degrad. Stab., 2022, 199, 109898 CrossRef CAS.
- X. Qian, L. Song, S. Jiang, G. Tang, W. Xing, B. Wang, Y. Hu and R. K. K. Yuen, Ind. Eng. Chem. Res., 2013, 52, 7307–7315 CrossRef CAS.
- X. W. Cheng, Z. Y. Wang, W. J. Jin and J. P. Guan, Ind. Crops Prod., 2022, 187, 115332 CrossRef CAS.
- X. Cheng, L. Shi, Z. Fan, Y. Yu and R. Liu, Polym. Degrad. Stab., 2022, 199, 109898 CrossRef CAS.
- M. Barbalini, L. Bertolla, J. Toušek and G. Malucelli, Polym. J., 2019, 11, 1664 CAS.
- L. Li, Z. Chen, J. Lu, M. Wei, Y. Huang and P. Jiang, ACS Omega, 2021, 6, 3930 Search PubMed.
- T. C. Mokhena, E. R. Sadiku, S. S. Ray, M. J. Mochane, K. P. Matabola and M. Motloung, J. Appl. Polym. Sci., 2022, 139, 52495 CrossRef.
- L. Costes, F. Laoutid, L. Dumazert, J. M. Lopez-Cuesta, S. Brohez, C. Delvosalle and P. Dubois, Polym. Degrad. Stab., 2015, 119, 217–227 CrossRef CAS.
- C. Gao, L. Zhou, S. Yao, C. Qin and P. Fatehi, Int. J. Biol. Macromol., 2020, 162, 1642–1652 CrossRef CAS PubMed.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, Polym. Degrad. Stab., 2016, 127, 32–43 CrossRef CAS.
- G. L. Bykov and B. G. Ershov, Russ. J. Appl. Chem., 2010, 83, 316–319 CrossRef CAS.
- Y. Guo, C. Cheng, T. Huo, Y. Ren and X. Liu, Polym. Degrad. Stab., 2020, 181, 109362 CrossRef CAS.
- S. J. Marciano, F. Avelino, L. R. R. da Silva, S. E. Mazzetto and D. Lomonaco, Biomass Convers. Biorefin., 2022, 12, 619–631 CrossRef CAS.
- Y. M. Zhang, Q. Zhao, L. Li, R. Yan, J. Zhang, J. C. Duan, B. J. Liu, Z. Y. Sun, M. Y. Zhang, W. Hu and N. N. Zhang, RSC Adv., 2018, 8, 32252–32261 RSC.
- M. V. Efanov and A. I. Galochkin, Chem. Nat. Compd., 2012, 48, 457–459 CrossRef CAS.
- W. Xiong, X. Qiu, R. Zhong and D. Yang, Holzforschung, 2016, 70, 937–945 CrossRef CAS.
- V. Carretier, M. F. Pucci, C. Lacoste, A. Regazzi and J. M. Lopez-Cuesta, Appl. Surf. Sci., 2022, 579, 152159 CrossRef CAS.
- H. Qiao, Y. Liang, G. Xu, Y. Wang, J. Yang and J. Hu, Des. Monomers Polym., 2019, 22, 114–121 CrossRef CAS PubMed.
- L. Oatway, T. Vasanthan and J. H. Helm, Food Rev. Int., 2007, 17, 419–431 CrossRef.
- S. Gharehkhani, N. Ghavidel and P. Fatehi, ACS Sustainable Chem. Eng., 2019, 7, 2370–2379 CrossRef CAS.
- G. S. Dhaliwal, N. Gupta, S. S. Kukal and M. Kaur, Commun. Soil Sci. Plant Anal., 2011, 42, 971–979 CrossRef CAS.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- R. Ravichandran, V. Seitz, J. Reddy Venugopal, R. Sridhar, S. Sundarrajan, S. Mukherjee, E. Wintermantel and S. Ramakrishna, Macromol. Biosci., 2013, 13, 366–375 CrossRef CAS PubMed.
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53–58 CAS.
- D. R. Bhumkar and V. B. Pokharkar, AAPS PharmSciTech, 2006, 7, 138–143 CrossRef PubMed.
- C. Cagnin, B. M. Simões, F. Yamashita, G. M. de Carvalho and M. V. E. Grossmann, Polym. Eng. Sci., 2021, 61, 388–396 CrossRef CAS.
- L. Xia, F. Song, F. Meng, L. Wang and S. Zhao, ACS ES&T Water, 2023, 3, 1923–1934 Search PubMed.
- L. Jiang, J. Guan, L. Zhao, J. Li and W. Yang, Colloids Surf., A, 2009, 346, 216–220 CrossRef CAS.
- J. Y. Park and E. Y. Park, Int. J. Biol. Macromol., 2023, 226, 312–320 CrossRef CAS PubMed.
- H. Huo, Y. Yu, X. Zhang, M. Tang, C. Chen, S. Wang and D. Min, Ind. Crops Prod., 2022, 188, 115727 CrossRef CAS.
- D. Wang, Y. Wang, T. Li, S. Zhang, P. Ma, D. Shi, M. Chen and W. Dong, ACS Sustainable Chem. Eng., 2020, 8, 10265–10274 CrossRef CAS.
- J. Pretula, K. Kaluzynski, B. Wisniewski, R. Szymanski, T. Loontjens and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2007, 46, 830–843 CrossRef.
- S. P. J. Brooks, D. Oberleas, B. A. Dawson, B. Belonje and B. J. Lampi, J. AOAC Int., 2001, 84, 1125–1129 CrossRef CAS PubMed.
- F. Rol, C. Sillard, M. Bardet, J. R. Yarava, L. Emsley, C. Gablin, D. Léonard, N. Belgacem and J. Bras, Carbohydr. Polym., 2020, 229, 115294 CrossRef CAS PubMed.
- S. N. Sun, M. F. Li, T. Q. Yuan, F. Xu and R. C. Sun, Ind. Crops Prod., 2013, 43, 570–577 CrossRef CAS.
- N. M. Stark, D. J. Yelle and U. P. Agarwal, Cellulose, 2016, 4, 6811–6831 Search PubMed.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Curr. Green Chem., 2017, 19, 4104 RSC.
- Z. Shomali and P. Fatehi, ACS Sustainable Chem. Eng., 2022, 10, 16563–16577 CrossRef CAS.
- A. Hoffmann, J. P. Nong, A. Porzel, M. Bremer and S. Fischer, React. Funct. Polym., 2019, 142, 112–118 CrossRef CAS.
- C. Heitner, D. Dimmel and J. A. Schmidt, Lignin and Lignans, Advances in Chemistry, CRC Press, 2010 Search PubMed.
- A. T. Bauman, G. M. Chateauneuf, B. R. Boyd, R. E. Brown and P. P. N. Murthy, Tetrahedron Lett., 1999, 40, 4489–4492 CrossRef CAS.
- A. Chen, L. Zhu and Y. Arai, Sci. Total Environ., 2022, 840, 156700 CrossRef CAS PubMed.
- G. J. Jiao, J. Ma, J. Hu, X. Wang and R. Sun, J. Hazard. Mater., 2023, 448, 130988 CrossRef CAS PubMed.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, Polym. Degrad. Stab., 2016, 127, 32–43 CrossRef CAS.
- G. Nourry, D. Belosinschi, M. P. Boutin, F. Brouillette and R. Zerrouki, Cellulose, 2016, 23, 3511–3520 CrossRef CAS.
- F. Z. Semlali, A. Ait Benhamou, K. El Bourakadi, A. E. K. Qaiss, R. Bouhfid, J. Jacquemin and M. El Achaby, Chem. Eng. J., 2023, 473, 145268 CrossRef CAS.
- R. J. Coleman, G. Lawrie, L. K. Lambert, M. Whittaker, K. S. Jack and L. Grøndahl, Biomacromolecules, 2011, 12, 889–897 CrossRef CAS PubMed.
- F. Zhang, W. Liu, C. Liu, S. Wang, H. Shi, L. Liang and K. Pi, Colloids Surf., A, 2021, 617, 126390 CrossRef CAS.
- H. Liu, J. Ma, J. Gong and J. Xu, J. Non-Cryst. Solids, 2015, 419, 92–96 CrossRef CAS.
- Y. Yu, S. Fu, P. Song, X. Luo, Y. Jin, F. Lu, Q. Wu and J. Ye, Polym. Degrad. Stab., 2012, 97, 541–546 CrossRef CAS.
- J. Yang, Z. Feng, Q. Gao, L. Ni, Y. Hou, Y. He and Z. Liu, Renewable Energy, 2021, 174, 178–187 CrossRef CAS.
- X. Gao, Z. Cao, C. Li, J. Liu, X. Liu and L. Guo, New J. Chem., 2022, 46, 18952–18960 RSC.
- B. Tian, B. Tian, B. Smith, M. C. Scott, Q. Lei, R. Hua, Y. Tian and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4345–4350 CrossRef CAS PubMed.
- C. Viornery, Y. Chevolot, D. Léonard, B.-O. Aronsson, P. Péchy, H. J. Mathieu, P. Descouts and M. Grä, Langmuir, 2002, 18, 2582–2589 CrossRef CAS.
- M. Shimizu, Y. Tsushima and S. Arai, ACS Omega, 2017, 2, 4306–4315 CrossRef CAS PubMed.
- Q. Zhang, X. Wang, Z. Li, W. Wu, J. Liu, H. Wu, S. Cui and K. Guo, RSC Adv., 2014, 4, 19710–19715 RSC.
- Y. J. Lee, B. Bingöl, T. Murakhtina, D. Sebastiani, W. H. Meyer, G. Wegner and H. W. Spiess, J. Phys. Chem. B, 2007, 111, 9711–9721 CrossRef CAS PubMed.
- M. Farrokhi, M. Abdollahi and A. Alizadeh, Polymer, 2019, 169, 215–224 CrossRef CAS.
- Z. Ma, Q. Sun, J. Ye, Q. Yao and C. Zhao, J. Anal. Appl. Pyrolysis, 2016, 117, 116–124 CrossRef CAS.
- C. Gao, L. Zhou, S. Yao, C. Qin and P. Fatehi, Int. J. Biol. Macromol., 2020, 162, 1642–1652 CrossRef CAS PubMed.
- M. Wądrzyk, R. Janus, M. Lewandowski and A. Magdziarz, J. Renewable Energy, 2021, 177, 942–952 CrossRef.
- I. E. Raschip, G. E. Hitruc, C. Vasile and M. C. Popescu, Int. J. Biol. Macromol., 2013, 54, 230–237 CrossRef CAS PubMed.
- I. Ribca, M. E. Jawerth, C. J. Brett, M. Lawoko, M. Schwartzkopf, A. Chumakov, S. V. Roth and M. Johansson, ACS Sustainable Chem. Eng., 2021, 9, 1692–1702 CrossRef CAS.
- S. Luo, J. Cao and A. G. McDonald, Ind. Crops Prod., 2018, 121, 169–179 CrossRef CAS.
- S. Montserrat, Polymer, 1995, 36, 435–436 CrossRef CAS.
- H. Li and A. G. McDonald, Ind. Crops Prod., 2014, 62, 67–76 CrossRef CAS.
- D. Tarasov, M. Leitch and P. Fatehi, Fuel Process. Technol., 2017, 158, 146–153 CrossRef CAS.
- S. Jiang, L. Meng, Y. Lou, Z. Yan, J. Xi, H. Bian, W. Wu and H. Xiao, J. Environ. Chem. Eng., 2023, 11, 111540 CrossRef CAS.
- S. Zhao, B. Xu, H. Shan, Q. Zhang and X. Wang, Polymers, 2023, 15, 1917 CrossRef CAS PubMed.
- A. Sethurajaperumal, A. Manohar, A. Banerjee, E. Varrla, H. Wang and K. Ostrikov, Nanoscale Adv., 2021, 14, 4235–4243 RSC.
- F. Wang, J. Y. Cheong, Q. He, G. Duan, S. He, L. Zhang, Y. Zhao, I. D. Kim and S. Jiang, Chem. Eng. J., 2021, 414, 128767 CrossRef CAS.
- H. Bin Yuan, R. C. Tang and C. B. Yu, Int. J. Mol. Sci., 2021, 22, 9631 CrossRef PubMed.
- J. Liu, P. Qi, D. Meng, L. Li, J. Sun, H. Li, X. Gu, S. Jiang and S. Zhang, Ind. Crops Prod., 2022, 186, 115239 CrossRef CAS.
- P. Parcheta-Szwindowska, J. Habaj, I. Krzemińska and J. Datta, Int. J. Mol. Sci., 2024, 25, 5512 CrossRef CAS PubMed.
- P. Wang and Z. Cai, Polym. Degrad. Stab., 2017, 137, 138–150 CrossRef CAS.
- D. Niu, W. Yu, W. Yang, P. Xu, T. Liu, Z. Wang, X. Yan and P. Ma, Chem. Eng. J., 2023, 474, 145753 CrossRef CAS.
- M. M. Velencoso, A. Battig, J. C. Markwart, B. Schartel and F. R. Wurm, Angew. Chem., Int. Ed., 2018, 57, 10450–10467 CrossRef CAS PubMed.
- L. Yan, Z. Xu and N. Deng, Prog. Org. Coat., 2019, 135, 123–134 CrossRef CAS.
- K. Bansal, S. Swarup and M. Quadir, Prog. Org. Coat., 2022, 172, 107093 CrossRef CAS.
- H. Zheng, X. Han, Q. Wei, C. Zheng, C. Huang, Z. Jin, Y. Li and J. Zhou, Chem. Eng. J., 2022, 437, 135412 CrossRef CAS.
- K. Wang, D. Meng, S. Wang, J. Sun, H. Li, X. Gu and S. Zhang, Ind. Crops Prod., 2021, 176, 114364 CrossRef.
- H. Yu, Y. Xia, X. Xu, N. Zarshad, M. Wu and H. Ni, J. Appl. Polym. Sci., 2020, 137, 49256 CrossRef CAS.
- S. Gaan and G. Sun, J. Anal. Appl. Pyrolysis, 2007, 78, 371–377 CrossRef CAS.
- O. Karlsson, C.-F. Lin, D. Jones and D. Sandberg, in Proceedings of the Tenth European Conference on Wood Modification, 2022, pp. 25–26.
- J.-H. Lee, D. Jang, I. Yang, S. Mu Jo and S. Lee, J. Appl. Polym. Sci., 2022, 139, e52519 CrossRef CAS.
- B. Ramadhoni, A. Rifathin and F. Yuliati, J. Inst. Eng. (India): Ser. D, 2023, 97 DOI:10.1007/s40033-023-00564-w.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, RSC Adv., 2017, 7, 16866–16877 RSC.
- J. Rusmirović, V. Lukić, T. Kovačević, M. Bogosavljević, S. Brzić, A. Marinković and T. Stevanović, Sci.-Tech. Rev., 2019, 69, 16–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03169e |
| This journal is © The Royal Society of Chemistry 2024 |