
Rhenium-based catalysts for biomass conversion
Julian Skagfjörd
Reinhold
ab,
Jifeng
Pang
*a,
Bo
Zhang
 *a,
Fritz E.
Kühn
*b and
Tao
Zhang
a
*a,
Fritz E.
Kühn
*b and
Tao
Zhang
a
aCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: jfpang@dicp.ac.cn; bo.zhang@dicp.ac.cn
bMolecular Catalysis, Department of Chemistry and Catalysis Research Center, TUM School of Natural Sciences, Technical University of Munich, Lichtenbergstr. 4, D-85748, Garching bei München, Bavaria, Germany. E-mail: fritz.kuehn@ch.tum.de
First published on 14th September 2024
Abstract
Biomass is the most abundant and renewable resource in the pursuit of bio-value added chemicals. Platinum-group metals and typical nonprecious transition metals are considered as the active metals for catalytic conversion of biomass into value added chemicals and fuels. Rhenium (Re) in particular is a versatile, oxophilic element with a broad variety of easily accessible oxidation states ideally suited for catalytic applications. Taking advantage of Re's unique properties and the green concept of biomass, the catalytic transformation of lignocellulose with Re-based catalysts has gained considerable attention, leading to the preparation of value-added chemicals including olefins through deoxydehydration from the conversion of polyols, aromatics through depolymerization of lignin, and alkane fuels through a hydrodeoxygenation reaction. This review focuses on the catalytic conversion of lignocellulosic main components (cellulose, hemicellulose, and lignin) and their derived platform chemicals into value-added chemicals and fuels over homogeneous and heterogeneous Re-based catalysts. The reaction pathways for biomass conversion over Re-based catalysts are introduced based on different feedstocks or chemical bonds. The unique role of Re species in tailoring the active sites of catalysts for these reactions is summarized and possible reaction mechanisms are discussed. Finally, an outlook is provided to underscore both the challenges and opportunities associated with this interesting and important field, offering a comprehensive overview on the current state of Re catalysis with a low carbon footprint.
1. Introduction
The excessive consumption of fossil resources increases CO2 emissions, inducing global climate change and several other serious environmental issues. Since the Paris Agreement was signed in 2015, an increasing number of countries have pledged to limit CO2 emissions and now regard carbon neutrality as a national strategy.1 To achieve this ambitious goal, renewable energy resources such as solar, wind, hydro, biomass, tidal, geothermal, and ocean thermal, are gradually incorporated into energy systems to approach a future without further consumption of fossil feedstock.2,3 As the only carbon-containing renewable energy resource, biomass is an ideal resource for upgrading into a variety of valuable chemicals, fuels and materials, gradually replacing the current fossil sources also as a basis for the chemical industry.4–7Biomass, mainly lignocellulosic biomass, is composed of cellulose (30–50%), hemicellulose (20–40%), and lignin (10–30%).8,9 Cellulose and hemicellulose are polysaccharides with basic units of pentose and hexose, respectively, linked by glycosidic bonds.9 Lignin is a polymer of aromatic alcohols that is branched and has different C–O and C–C bond combinations.10 In plant cells, these three components interact strongly to form a rigid structure via hydrogen and covalent bonds and are hard to degrade under chemical and biological conditions. In recent decades, significant progress has been made in the development of novel catalysts and the study of reaction mechanisms, aimed at efficiently and selectively converting biomass into value-added products.5,9,11,12 Considering the different structures of (hemi)cellulose and lignin, most works first separate these components from lignocellulosic biomass and afterwards transform them into platform chemicals via chemical or biological methods, followed by further upgrading into key chemicals and fuels. Specifically, the oxygen-containing functional groups in biomass or platform chemicals are treated with metal and/or oxide catalysts via hydrodeoxygenation, hydrogenolysis, decarbonylation, decarboxylation, oxidation, esterification, isomerization, dehydration and hydrolysis reactions to meet the current requirements for achieving industrially useful chemical compounds.13 For instance, (hemi)cellulose or lignin is catalytically converted into glycols, furfural, 5-hydroxymethylfurfural or aromatics, and then upgraded into fine chemicals and advanced fuels, or the carbohydrates in biomass are fermented into alcohols and carboxylic acids for further catalytic conversions.14–16
During these catalytic processes, transition metal catalysts have gained great attention due to their specific electronic structure and ability to easily give or take electrons from other molecules. Among them, rhenium (Re) is a versatile element and has widely been applied in catalyzing oxygen-containing functional groups in biomass conversions.17,18 Because Re has a variety of easily accessible oxidation states, it provides a number of possibilities to adjust the interaction between catalysts and biomass-derived substrates.19 Typically, organometallic Re species like organo(trioxo-Re) complexes demonstrate soft “enzyme-like” structures for deoxydehydration reactions.20–25 Furthermore, ReOx have acidic, oxophilic and metallic properties and demonstrate great potential in dissociating H2 molecules or suitable interactions for –OH containing compounds’ conversion. Thus, Re species are used directly as a notable promoter to modify the coordination environment, electronic structure and acid–base properties of other metal catalysts in typical redox reactions, including dehydration,26 hydrogenation,27–29 hydrogenolysis,30,31 hydrodeoxygenation32,33 and deoxydehydration reactions.24,29,34
Because of the versatile application of Re-based catalysts, several reviews have been dedicated to this topic. For instance, Dilworth provided a detailed review of recent developments in Re chemistry from 2015 to 2020 concerning properties and applications.19 Raju et al. reviewed recent progress in dehydration and deoxydehydration reactions of biomass-derived alcohols and polyols into olefins over Re-based catalysts.26 DeNike and Kilyanek surveyed the mechanism of the deoxydehydration reaction over homogeneous early metal-oxo catalysts.35 Recently, the achievements in the deoxydehydration reaction were revisited over transition metal-based catalysts for upgrading bio-based polyols into value-added chemicals and fuels.22,36,37 However, most of the reviews focus on some specific reactions. Highlighting the achievements in the catalytic conversion of biomass over both homogeneous and heterogeneous Re-based catalysts has not yet been covered, especially with the newly emerging lignin depolymerization reactions38–44 and raw lignocellulosic biomass upgrading.45
In this review, the application of Re-based catalysts for biomass conversion including cellulosic biomass, lignin, and platform chemicals catalyzed by homogeneous and heterogeneous Re-based catalysts is comprehensively reviewed and discussed (Fig. 1). In detail, catalytic conversion of sugar derivatives and lignin is first introduced, focusing on homogeneous organic Re complexes and the discussion of mechanistic studies. Then, works on the conversion of hemicellulose, lignin and platform chemicals over heterogeneous Re catalysts are discussed from the viewpoint of the key role of Re species in tailoring different reaction pathways. Moreover, challenges and opportunities for biomass conversions over Re-based catalysts are addressed. This review offers a useful resource to explore alternative Re-based catalysts for biomass conversion in industrial applications and to encourage researchers to develop feasible, sustainable and economical technologies to maximize the chemical, fuel and material production from biomass feedstock.
 | ||
| Fig. 1 Overview of biomass conversion over homogeneous and heterogeneous Re-based catalysts. | ||
2. Homogeneous Re-based catalysts for biomass conversion
Subdividing catalysis into homogeneous and heterogeneous reactions is a traditional approach, relating to the thermodynamic phases of catalyst and reactants. The characteristic feature of homogeneous catalysis is the coexistence of the catalyst and reactants in the same phase, which leads to high catalytic efficiency owing to sufficient contact between the active center and reactants and only a small influence of diffusion, as long as enough substrate is available in the proximity of the catalyst.46 Additionally, homogeneous catalysis allows for the catalytic selectivity to be rationally tuned by adjusting the ligand associated with the active metal. However, separating homogeneous catalysts from the reaction can be challenging and/or associated with considerable energy consumption, which limits industrial applications. Another issue is that homogeneous catalysis is usually not well compatible for continuous processes.Notwithstanding these limitations, homogeneous Re-based compounds and their applications have gained quite some interest during recent decades.19,47–49 In particular, organorhenium oxides are in rapid progress in both catalysis and materials science,47,49–52 producing a large number of useful active and selective Re-complex candidates for catalytic reactions. For instance, methyltrioxorhenium(VII) is utilized as an oxidation catalyst with a wide variety of applications, displaying robust activity and product selectivity.53–55 Building upon this highly promising foundation, there has been a rapid increase in studies focusing on organometallic derivatives and their catalytic reactions. While extensive reviews have covered aspects such as synthesis, structure, spectroscopy, and chemical behavior, recent attention has been directed towards exploring new applications, particularly in the realm of biomass conversion. In this section, particular emphasis is given to biomass conversion catalyzed by homogeneous Re-based catalysts.
2.1 Conversion of (hemi)cellulose
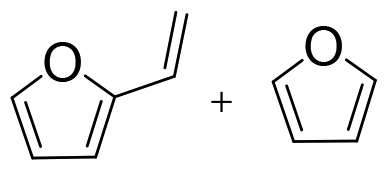
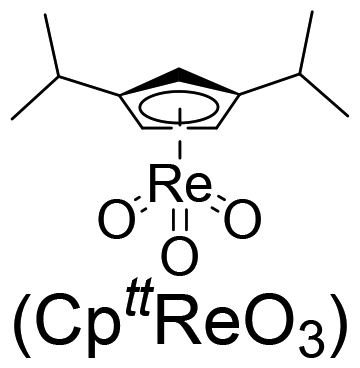
Cellulose is a polymer that is both chemically stable and insoluble in the majority of solvents, since glucose is joined by β-1,4-glycosidic linkages to form a strong crystal structure with a network of intra- and intermolecular H-bonds.56,57 Thus, it is still a challenge to degrade cellulose into target products with high efficiency.58 In contrast, monosaccharides derived from carbohydrates are preferably selected as model substrates for catalytic conversions because of their good solubility and enhanced reactivity. In addition, previous works show that homogeneous organometallic Re compounds have outstanding performance in removing hydroxy groups, especially in the deoxydehydration reaction, which allows the removal of two adjacent hydroxy groups from diols, leading to an olefin in the presence of a reducing agent.17,24 Since deoxydehydration reactions of polyols (<C4) catalyzed by homogeneous Re-based catalysts have already been reviewed,22,24 the focus of this part is on the summary of deoxydehydration reactions, categorized by sugar, sugar alcohols and sugar acids (>C3) along with the typical ranges of reaction conditions.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8 (Table 1, entry 1).59 Substituted homogeneous Re-based catalysts also enable deoxydehydration reactions towards olefins. For instance, cyclopentadiene substituted homogeneous Re (CpttReO3) (Cptt = 1,3-di-tert-butylcyclopentadienyl) and L4Re(CO)3 (L4 = 2,4-di-tert-butyl-6-(tetramethylethane-1,2-diamine)phenol) can be applied for the conversion of D-glucose, yielding 8% and 6% 2-vinylfuran and furan at a ratio of 1.7:1 and 1.8:1, respectively (Table 1, entry 1).60,61 The low yield of product using sugar as the feedstock might be attributed to difficulties in coordination of the catalyst and substrate due to the multitude of isomeric states and the thermal instability of substrates.59 Besides D-glucose, other monosaccharides like D-galactose, D-mannose and D-allose can also be converted to 2-vinylfuran and furan applying these Re-based catalysts. Up to 40% yield of products in the deoxydehydration reaction were achieved (Table 1, entries 2–4).59,60 In addition, D-xylose and L-arabinose catalyzed by methyltrioxorhenium leads to the ether 2-((octan-3-yloxy)mehtyl)furan as the main product, however in rather low yields (<7%), presumably owing to the reactivity of furfuryl alcohol (Table 1, entries 5 and 6). On the other hand, the deoxydehydration reaction of D-erythrose and D-threose catalyzed by methyltrioxorhenium results in 60% and 47% furan yields, respectively (Table 1, entries 7 and 8). This comparatively high yield is attributed to two reactions occurring within the system, including a deoxydehydration reaction of two carbon, C1 and C2, with hydroxy groups, and the epimerization of the C2 hydroxy group via erythrulose.59 Based on the aforementioned findings, achieving a high yield of olefins from the deoxydehydration reaction of sugars remains a significant challenge. This is primarily attributed to the complex structure of sugars, which leads to an equilibrium between multiple isomeric forms, coupled with their thermal instability under hydrothermal conditions.
:1.8 (Table 1, entry 1).59 Substituted homogeneous Re-based catalysts also enable deoxydehydration reactions towards olefins. For instance, cyclopentadiene substituted homogeneous Re (CpttReO3) (Cptt = 1,3-di-tert-butylcyclopentadienyl) and L4Re(CO)3 (L4 = 2,4-di-tert-butyl-6-(tetramethylethane-1,2-diamine)phenol) can be applied for the conversion of D-glucose, yielding 8% and 6% 2-vinylfuran and furan at a ratio of 1.7:1 and 1.8:1, respectively (Table 1, entry 1).60,61 The low yield of product using sugar as the feedstock might be attributed to difficulties in coordination of the catalyst and substrate due to the multitude of isomeric states and the thermal instability of substrates.59 Besides D-glucose, other monosaccharides like D-galactose, D-mannose and D-allose can also be converted to 2-vinylfuran and furan applying these Re-based catalysts. Up to 40% yield of products in the deoxydehydration reaction were achieved (Table 1, entries 2–4).59,60 In addition, D-xylose and L-arabinose catalyzed by methyltrioxorhenium leads to the ether 2-((octan-3-yloxy)mehtyl)furan as the main product, however in rather low yields (<7%), presumably owing to the reactivity of furfuryl alcohol (Table 1, entries 5 and 6). On the other hand, the deoxydehydration reaction of D-erythrose and D-threose catalyzed by methyltrioxorhenium results in 60% and 47% furan yields, respectively (Table 1, entries 7 and 8). This comparatively high yield is attributed to two reactions occurring within the system, including a deoxydehydration reaction of two carbon, C1 and C2, with hydroxy groups, and the epimerization of the C2 hydroxy group via erythrulose.59 Based on the aforementioned findings, achieving a high yield of olefins from the deoxydehydration reaction of sugars remains a significant challenge. This is primarily attributed to the complex structure of sugars, which leads to an equilibrium between multiple isomeric forms, coupled with their thermal instability under hydrothermal conditions.
| Entry | Substrate | Catalyst | Product | Yield | Reducing agent | Reaction conditions | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | D-Glucose |

|
|
25% (1:1.8) |
3-Pentanol | 155 °C, 3 h, N2 | 59 |
|
8% (1.7:1) |
3-Octanol | 135 °C, 15 h, N2 | 61 | |||

|
6% (1.8:1) |
3-Pentanol | 155 °C, 3 h, air | 60 | |||
| 2 | D-Galactose | MTO |

|
32% (1:3.0) |
3-Pentanol | 155 °C, 3 h, N2 | 59 |
| CpttReO3 | 22% (1.2:1) |
3-Octanol | 135 °C,15 h, N2 | 61 | |||
| L4Re(CO)3 | 11% (2.0:1) |
3-Pentanol | 155 °C, 3 h, air | 60 | |||
| 3 | D-Mannose | MTO |

|
30% (1:2.3) |
3-Pentanol | 155 °C, 3 h, N2 | 59 |
| CpttReO3 | 39% (2.3:1) |
3-Pentanol | 155 °C, 12 h, N2 | 61 | |||
| L4Re(CO)3 | 32% (1.5:1) |
3-Pentanol | 155 °C, 3 h, air | 60 | |||
| 4 | D-Allose | MTO |

|
40% (1:2.1) |
3-Pentanol | 155 °C, 3 h, N2 | 59 |
| 5 | D-Xylose | MTO |

|
Trace | 3-Octanol | 155 °C, 2 h, N2 | 59 |
| 6 | L-Arabinose | MTO |
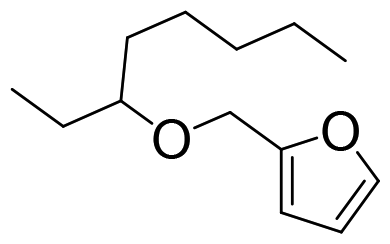
|
7% | 3-Octanol | 155 °C, 2 h, N2 | 59 |
| 7 | D-Erythrose | MTO |
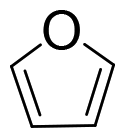
|
60% | 3-Octanol | 155 °C, 1.25 h, N2 | 59 |
| 8 | L-Threose | MTO |

|
47% | 3-Octanol | 155 °C, 1.25 h, N2 | 59 |
 | ||
| Fig. 2 Deoxydehydration reactions of typical C6 and C5 sugar alcohols59 (MTO: methyltrioxorhenium). | ||
Both glucose fermentation and pentose decarbonylation produces erythritol alcohol,62 which is therefore regarded as a platform chemical in a biomass refinery. A variety of C4 chemicals can be obtained from erythritol, of which the deoxydehydration reaction of erythritol to alkenes has caught rapidly growing attention. Homogeneous Re-based catalysts with a reducing agent, such as methyltrioxorhenium with 3-octanol, 1-heptanol or indoline, (NBu4)ReO4 with Na2SO3, or L4Re(CO)3 with 3-octanol can be employed for the deoxydehydration reaction of erythritol. Typical product distributions and reaction conditions are summarized in Fig. 3. With Re-based catalysts, many olefinic products can be obtained in good yields. In detail, methyltrioxorhenium enables the double deoxydehydration of erythritol to 1,3-butadiene in 89% yield within 1.5 h at 167 °C. The reaction, maintained by the reducing agent 3-octanol, also yields 11% 2,5-dihydrofuran as the deoxydehydration coupled dehydration (DH) product.59 Additionally, reducing agents also greatly affect the deoxydehydration reaction of polyols when utilizing the same Re catalysts. For instance, (NBu4)ReO4 catalyzes the transformation of tetrol erythritol to olefins with Na2SO3 as a reductant at 150 °C within 100 h. This process leads to a liquid biphasic mixture, ultimately resulting in a 27% yield of 1,3-butadiene.63 Utilizing 3-octanol as the reductant, Li et al. also have been able to show that L4Re(CO)3 is efficient in converting erythritol to butadiene with a yield up to 78% at 180 °C within 3 h under air.60 Similarly, with 1-heptanol as the reductant, meso-erythritol can be converted into 2,5-dihydrofuran and allyl alcohol at a total yield of 58% in a 1:0.28 ratio using methyltrioxorhenium as the catalyst. However, keeping the reaction for 1 h at 165 °C does not lead to higher product yields.18 When indoline is used as the reducing agent, erythritol is also converted into 1,3-butadiene in 43% yield with methyltrioxorhenium as the catalyst. After 4 h of reaction at 170 °C, 61% of the oxidized indole compounds are obtained, when using 1-butanol as the solvent.64 1,4-Anhydroerythritol, obtained through the dehydration of erythritol, is efficiently converted to 2,5-dihydrofuran in more than 80% yield with catalytic amounts of organorhenium compounds including methyltrioxorhenium, CpttReO3 and L4Re(CO)3.
 | ||
| Fig. 3 Erythritol conversion catalyzed by homogeneous Re compounds (MTO: methyltrioxorhenium). | ||
When vegetable oils are transesterified to produce biodiesel fuel on a big scale, the primary waste is glycerol. Because of its distinct structural characteristics, chemoselective transformation will produce a variety of platform molecules, including acrolein,65 hydroxyacetone,66 propanediols,67 propene,68 and propane.69 Besides these products, allyl alcohol, obtained through the deoxydehydration reaction of glycerol has emerged as a sustainable and economically viable alternative. Most works focus on Re-based catalysts including NH4ReO4, HReO3, methyltrioxorhenium and L4Re(CO)3 for the deoxydehydration reaction of glycerol. The experimental data and reaction conditions are summarized in Table 2. In detail, methyltrioxorhenium is able to catalytically transform glycerol into the corresponding allyl alcohol in yields ranging from 66% to 90% in the presence of a reducing agent (Table 2, entries 1–5). The N,N,O coordinated Re complex (Table 2, entry 6) shows outstanding performance for the deoxydehydration reaction, leading to 97% yield of allyl alcohol at 180 °C within 3 h.60 Likewise, inorganic salts including KReO4, NaReO4, and NH4ReO4 and ReO3 also enable the catalytic transformation of glycerol into allyl alcohol in acceptable yields (Table 2, entries 7–13).18,64,70–72
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Reductant | Yield | Condition | Ref. |
| a NH4Cl as additive. b KCl as additive; L4 = 2,4-di-tert-butyl-6-(tetramethylethane-1,2-diamine)phenol; DMP = 2,4-dimethyl-3-pentanol. MTO = methyltrioxorhenium. | |||||
| 1 | MTO | Glycerol | 54% | 165 °C, 1 h air | 18 |
| 2 | MTO | 3-Octanol | 90% | 170 °C, 2.5 h air | 59 |
| 3 | MTO | 3-Octanol | 70% | 170 °C, 2.5 h air | 71 |
| 4 | MTO | DMP | 87% | 140 °C, 16.5 h, H2 | 70 |
| 5 | MTO | Indoline | 66% | 150 °C, 24 h, N2 | 64 |
| 6 | L4Re(CO)3 | 3-Octanol | 97% | 180 °C, 3 h, air | 60 |
| 7 | KReO4 | Glycerol | 17% | 165 °C, 10 h air | 18 |
| 8a | NaReO4 | Glycerol | 76% | 165 °C, 1 h air | 18 |
| 9b | NH4ReO4 | Glycerol | 75% | 165 °C, 1.5 h air | 18 |
| 10 | NH4ReO4 | 3-Octanol | 59% | 170 °C, 2.5 h air | 71 |
| 11 | NH4ReO4 | Indoline | 80% | 150 °C, 24 h, N2 | 64 |
| 12 | ReO3 | DMP | 91% | 140 °C, 11.5 h, H2 | 70 |
| 13 | ReO3 | DMP | 91% | 140 °C, 13.5 h, H2 | 72 |

![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to obtain the aliphatic diacid is very attractive owing to the wide application of aliphatic diacids in biodegradable polymer sectors. Coupling these cascade steps provides a sustainable and promising strategy for the production of bio-acids.
C bond to obtain the aliphatic diacid is very attractive owing to the wide application of aliphatic diacids in biodegradable polymer sectors. Coupling these cascade steps provides a sustainable and promising strategy for the production of bio-acids.
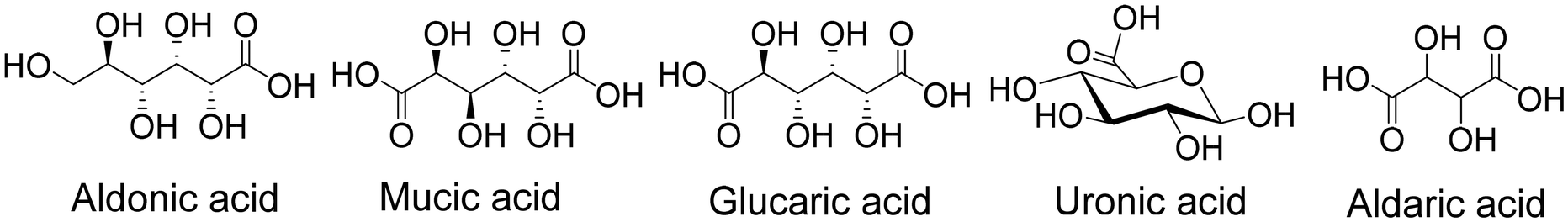 | ||
| Fig. 4 The structure of typical sugar acids. | ||
Glucaric and mucic acids are C6 sugar acids or aldaric acids, which are produced by the oxidation of glucose and galactose.36 Specifically, mucic acid is a dicarboxylic acid of considerable interest. Deoxydehydration reactions of such acids yield muconic acid or muconic acid diester (esterification product in acidic conditions), which are potential feedstock for the production of adipic acid, which is the basic monomer for the production of nylon 66. To bridge the gap between abundant sugars and value-added adipic acids, developing an efficient synthesis system is therefore in high demand.
Re-based catalysts show high potential for the conversion of sugar acids to adipic acid. The performance of homogeneous Re-based catalysts for mucic acid conversion is summarized in Table 3. Re-based catalysts with secondary and primary alcohols including 3-pentanol, 3-octanol and n-butanol are powerful systems to deoxygenate sugar acids. Owing to the excess use of alcohols, mixtures of carboxylic acids and esters are commonly obtained.73 In detail, methyltrioxorhenium catalyzes mucic acid to muconic acid in 43–99% yields (Table 3, entries 1–3).73–75 It has been found that Brønsted acids such as para-toluene sulfonic acid (TsOH) as a co-catalyst lead to a considerable reactivity increase for the deoxydehydration of mucic acid, and selectivity increases towards esterified products,73 allowing for methyltrioxorhenium loadings as low as 0.5 mol%. This is mainly attributed to the assistance of the acid additive in olefin extrusion via Re diolate intermediate protonation.76,77 Similarly, other Re compounds including CpttReO3 and L4Re(CO)3 also have high catalytic activity for this reaction, leading to 75% and 46% yield of muconates (Table 3, entries 4 and 5). The experiment in Table 3, entry 6 shows that Re2O7 catalyzes deoxydehydration of mucic acid in 3-pentanol with high reaction rates. In moist environments, Re2O7 is transformed to HReO4, which promotes the esterification and olefin extrusion steps in the deoxydehydration reaction. Thus, employing HReO4 as a sole catalyst gives 62% and 71% yield of olefin with n-butanol as the solvent (Table 3, entries 8 and 9). Furthermore, Zhang et al. found that tuning the Lewis or Brønsted acidity of the reaction system is an efficient method to obtain high yields of carboxylic acids. For example, the methyltrioxorhenium system modified by pyridine can influence the selectivity of the reaction towards muconic acid with a yield of 74% by reducing the acidity of methyltrioxorhenium (Table 3, entry 3). It is noteworthy that coupling deoxydehydration reaction and heterogeneous hydrogenation catalysts such as Pt/C and Pd/C results in good yields of dibutyl adipate (Table 3, entries 2 and 9).73,74
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. 1 | Reductant | R1 | R2 | Yield/% Pro. 1 | Condition | Cat. 2 | Yield/% Pro. 2 | Ref. |
| L4 = 2,4-di-tert-butyl-6-(tetramethylethane-1,2-diamine)phenol; Cptt = 2,5-di-isopropyl-cyclopentadienyl; nBuO = n-butanol. MTO = methyltrioxorhenium. | |||||||||
| 1 | MTO | 3-Pentanol | HO | HO | 43 | 155 °C, 15 h, air | — | — | 74 |
| 2 | MTO | 3-Pentanol | HO/OCH(C2H5)2 | OCH(C2H5)2 | 99 | 120/160 °C, 12/12 h, air | Pt/C | 99 | 73 |
| 3 | MTO | 3-Pentanol | HO | HO | 74 | 120 °C, 24 h, N2 | — | — | 75 |
| 4 | CpttReO3 | 3-Pentanol | HO/OCH(C2H5)2 | OCH(C2H5)2 | 75 | 120 °C, 12 h, N2 | — | — | 61 |
| 5 | L4Re(CO)3 | 3-Octanol | HO/OC8H17 | OC8H17 | 46 | 180 °C, 6 h, air | — | — | 60 |
| 6 | Re2O7 | nBuOH | nBuO | nBuO | 99 | 120 °C, 12 h, N2 | — | — | 73 |
| 7 | NH4ReO4 | Indoline | nBuO | nBuO | 57 | 150 °C, 24 h, N2 | — | — | 64 |
| 8 | HReO4 | nBuOH | nBuO | nBuO | 71 | 170 °C, 7.5 h, N2 | — | — | 74 |
| 9 | HReO4 | nBuOH | nBuO | nBuO | 62 | 170 °C/RT, 15/4 h, air/H2 | Pd/C | 62 | 74 |

According to the results shown above, the deoxydehydration reaction catalyzed by Re-based catalysts along with an alcohol reducing reagent can be regarded as the most efficient method for the removal of adjacent hydroxyl groups to form olefins. The advantages of employing an alcohol as both a reductant and solvent is that polyols or sugar acids have good solubility in alcohol. Additionally, the alcoholic reductants can be obtained via hydrogenation of alcohol oxidation products (ketones or aldehydes).
With the aim to provide insights into the mechanism of the reaction (Fig. 5), the three main pathways are discussed. Toste et al. reasoned the reduction of the ReVII center of methyltrioxorhenium to a ReV specie, then followed by a condensation of a vicinal diol with the ReV specie. The reaction, it has been assumed, proceeds via extrusion of olefin and catalyst regeneration (A in Fig. 5).59 Abu-Omar et al. postulated route B in Fig. 5.18 However, theoretical calculations suggested that their overall barriers are high. Therefore, Qu et al. provided route C in Fig. 5 as the most likely pathway of the reaction. According to density functional theory, the reduction of Re catalyst in a first step is more likely than the direct substrate condensation. Moreover, reduction with alcohol assisted by hydrogen transfer via the rhenium dihydroxy intermediate MeRe(OH)2O is more stable than the dioxorhenium compound (MeReO2). The olefin extrusion process is achieved by a concerted process of diolate cleavage to form the alkene.78 Thus, alcohols apparently act as a shuttle to promote these hydrogen-transfer steps.
 | ||
| Fig. 5 Proposed mechanism C and alternative mechanisms A and B for deoxydehydration reaction of diols over MTO catalysts with alcohol reductants78 (MTO: methyltrioxorhenium). | ||
Tartaric acid, being a C4 dicarboxylic acid (C4H6O6), can be obtained from wine fermentation and oxidation of glucose,79 which is extensively utilized in the pharmaceutical and cosmetic industries. Similar with deoxydehydration of mucic acid to adipic acid, deoxydehydration of tartaric acid to maleic acid is regarded as the most efficient and promising method for the production of biopolymer precursors. Homogeneous Re-based catalysts such as methyltrioxorhenium, NBu4ReO4, NH4ReO4, and L4Re(CO)3 with a reducing agent enable transformation of tartaric acid or ester to maleic acid (Table 4). Specifically, the substrate (+)-diethyl L-tartrate can be converted to the trans-alkene, leading to 35% yield of diethyl fumarate with no cis products catalyzed by methyltrioxorhenium (Table 4, entry 1). However, the organometallic Re-based complexes L4Re(CO)3 vastly improve the yield reaching up to 81% with 3-octanol as the reductant at 180 °C (Table 4, entry 2).60 NH4ReO4 has been applied with various reducing agents including indoline, benzyl alcohol, and 3-pentanol, to catalyze deoxydehydration reactions and yields >90% target products (Table 4, entries 3–5),64,71 in contrast to the 10% diethyl fumarate yield for the NBu4ReO4 catalyst (Table 4, entry 6). Due to the versatile application of succinic acid in the synthesis of valuable C4 commodity chemicals and biodegradable polymers, it has been produced using tartaric acid as substrate via a two-step method. In detail, tartaric acid undergoes conversion to maleic acid with a notable yield of 91%, utilizing either methyltrioxorhenium-modified pyrimidine or NH4ReO4 catalysts (Table 4, entry 5), following further hydrogenation to succinic acid in 95% yield over Pt/C catalysts.75
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | R | Reductant | Yield | Condition | Ref. |
| L4 = 2,4-di-tert-butyl-6-(tetramethylethane-1,2-diamine)phenol. | ||||||
| 1 | MTO | C2H5 | Na2SO3 | 35% | 150 °C, 84 h | 63 |
| 2 | L4Re(CO)3 | H/C8H17 | 3-Octanol | 81% | 180 °C, 3 h | 60 |
| 3 | NH4ReO4 | n-Bu | Indoline | 78% | 150 °C, 24 h | 64 |
| 4 | NH4ReO4 | C2H5 | Benzyl alcohol | 95% | 145 °C, 24 h | 71 |
| 5 | NH4ReO4 | H | 3-Pentanol | 91% | 120 °C, 24 h | 75 |
| 6 | (Bu4N)ReO4 | C2H5 | Na2SO3 | 10% | 150 °C, 84 h | 63 |
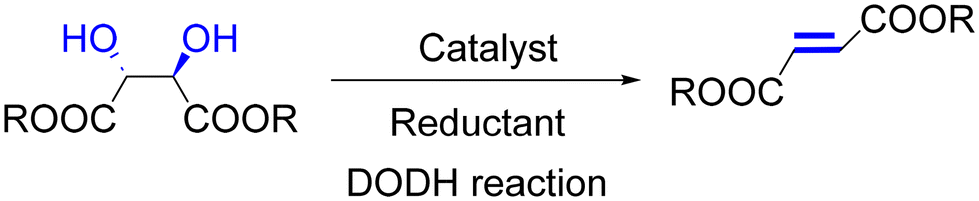
Since γ-butyrolactone is a useful precursor for a multitude of polymers, the conversion of different lactone derivatives to γ-butyrolactone is an attractive reaction. With methyltrioxorhenium as the catalyst, conversion of D-erythronolactone to γ-butyrolactone is achieved at 155 °C in argon within 2.5 h and utilizes 3-pentanol as both the solvent and reductant. The subsequent hydrogenation is also achieved with the Pd/C catalyst (Fig. 6).74 It is noteworthy that the final product γ-butyrolactone is the key precursor for the production of γ-hydroxybutanoic acid and γ-aminobutanoic acid through ring-opening reactions.
 | ||
| Fig. 6 Conversion of D-erythronolactone to γ-butyrolactone via deoxydehydration and hydrogenation reactions with methyltrioxorhenium as the catalyst74 (MTO: methyltrioxorhenium). | ||
2.2 Conversion of lignin
Lignin, the second main component in lignocellulosic biomass, is considered as the only abundant source of aromatics in nature,80,81 and regarded as a potential feedstock for aromatic bio-fuels and bio-chemicals.10,81–83 However, its intrinsic recalcitrant nature and complex structure has so far made it more difficult to convert into fuels and aromatics with additional value. Model compounds of lignin, featuring typical and well-known structural bonds such as β-O-4, α-O-4, 4-O-5, β-1, β-β, and α-1, are frequently chosen as starting materials for the conversion processes. This choice mirrors the depolymerization of actual lignin, facilitating the study of its realistic breakdown.84,85The organorhenium compounds methyltrioxorhenium and Re2O7 were employed as catalysts for the deconstruction of lignin model compounds containing β-O-4, and α-1 bonds through oxidations, redox-neutral reactions, and hydrogenation reactions (Fig. 7). The oxidation of lignin model compound catalyzed by methyltrioxorhenium/H2O2 has been the first attempt in disintegrating lignin.86,87 When using the monomer vanillyl alcohol as the substrate, the oxidation products including vanillin and 4-hydroxy-3-methoxybenzoic acid are formed as major products in 3.7% and 4.9% yield (Fig. 7, Route 1). The β-O-4 patterns, being the major linkages, are further oxidized in the same catalytic system of methyltrioxorhenium/H2O2, leading to 40.5% carboxylic acid derivatives.86 Additionally, an α-1 model compound has been modified via oxidation of side functional groups, and the C–C bond was maintained due to its high bonding energy. Interestingly, real lignin including spruce kraft lignin, hydrolytic sugar cane lignin and hardwood organosolv lignin can be degraded by using methyltrioxorhenium/H2O2. 31P NMR has been employed to determine the amount of the different unstable OH groups. After methyltrioxorhenium oxidation degradation, high amounts of carboxylic acid moieties are obtained with low contents in aliphatic and condensed OH groups, indicating that methyltrioxorhenium is also highly efficient in oxidative depolymerization of real lignin.
 | ||
| Fig. 7 Lignin model compounds catalyzed by Re-based compounds (MTO: methyltrioxorhenium). | ||
Apart from the oxidation process, hydrogenolysis of such segments catalyzed by Re2O7 has been developed in the tetrahydrofuran and under 1 MPa H2, affording 71% yield of phenylethyl alcohol and 98% yield of guaiacol.42 In contrast, other metal oxide catalysts (WO3, MoO3, Fe2O3, CeO2) show negligible activity in converting the same substrate.42 When using pinewood as the feedstock, 19% yield of lignin oil is obtained at 200 °C and 3 MPa H2 pressure within 8 h. Interestingly, the distribution of monomer products is similar to that obtained from isolated lignin.42 Using pinewood lignin as the reactant, 76 wt% liquid oil with phenol compounds as the main products is obtained after 8 h of reaction at 200 °C, at 3 MPa H2.42 After characterization, the Re2O7 species in aromatic solvents shows Lewis-acidic properties for the cleavage of β-O-4 bonds in lignin.44
Since no additional H and O agents were present in the redox neutral process, it is regarded as the most promising approach for the depolymerization of lignin via C–O bond cleavage of β-O-4 groups. Notably, with methyltrioxorhenium as the catalyst, the redox neutral process of 2-(2-methoxyphenoxy)-1-phenylethan-1-ol yields 95% of phenylacetaldehyde and 98% of guaiacol.43 Although this catalytic system is homogeneous, methyltrioxorhenium can be reused at least five times without loss of activity through removing all volatiles in vacuo. The catalytic system has been extended to the aliphatic ether 2-phenoxycyclohexan-1-ol, conducted at 155 °C for 45 h in p-xylene solvent. This reaction yields 18% ketone, 26% phenol, and 15% dehydration products.
Methyltrioxorhenium is also active as a catalyst for the cleavage of β-O-4 bonds in model compounds and real lignin in the ionic liquid of 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (B[mim]NTf2).88 When the reaction medium is treated with microwave irradiation for 2 min at 240 W and 180 °C, 64% yield of guaiacol and 98% yield of phenylacetaldehyde (Fig. 7, Route 2) are obtained. Microwave irradiation shortens the reaction time to 2 min, which might be attributed to the enhancement of the catalyst's activity and the substrate's solubility under electromagnetic radiation. Notably, organosolv birch lignin catalyzed by methyltrioxorhenium in [Bmim]NTf2 for 2 min at 180 °C produces 34.2% yield of low molecular weight lignin oil, mainly consisting of aromatic monomers.88 Besides methyltrioxorhenium, Re2O7 catalyzes the deconstruction of β-O-4 model compounds through a redox neutral reaction, leading to 73% yield of phenylacetaldehyde and 82% yield of guaiacol (Fig. 7, Route 2).44 In addition, modification of an α-1 model compound catalyzed by methyltrioxorhenium with H2O2 is achieved at room temperature although C–C bonds remain in the final products (Fig. 7, Route 3).86
The major pathway for the cleavage of the C–O bond in lignin β-O-4 model compounds proceeds as an initial excitation by dehydration of the β-O-4 model compound to the alkenyl ether intermediate, then the cleavage of C–O bond with organorhenium catalysts (Fig. 8). This mechanism diverges from previous redox-neutral processes by involving the cleavage of β-O-4 bonds through α-ketone formation via dehydrogenation reactions.89 Isotope labeling experiments and density functional theory calculations indicate the reduction of ReVII to ReV by the lignin model compound. This reduction suggests that methylrhenium dioxide is responsible for the cleavage of C–O bonds. According to the control experiments and density functional theory calculations, the different reaction mechanism should be attributed to the O shift of the substrate OH group and methyltrioxorhenium, which is typical of Re catalysts in lignin depolymerization reactions.
 | ||
| Fig. 8 The mechanistic pathway of Re-catalyzed cleavage of lignin β-O-4 model compound (MTO: methyltrioxorhenium; MDO: methyldioxorhenium). | ||
3. Heterogeneous Re-based catalysts for biomass conversion
3.1 Conversion of (hemi)cellulose
Inspired by the works in the conversion of cellulose to polyols and glycols,90–92 extensive works have been conducted on the transformation of cellulosic biomass over heterogeneous Re-based catalysts (Fig. 9, Route 1). Liu et al. employed Re oxides to modify Ir catalysts for cellulose conversion. Over the binary Ir–ReOx/SiO2 and HZSM-5 catalysts, the hydrolysis and hydrogenation–hydrogenolysis reactions were coupled to yield 83% of n-hexane in a biphasic reaction system of n-dodecane and water.93 However, the main product shifted to hexanols over the same Ir–ReOx/SiO2 catalyst after modifying the reaction conditions to 140 °C for 24 h at 10 MPa H2. Under these reaction conditions, cellulose is depolymerized to glucose by the mechanocatalytic method. Next, using Ir–ReOx/SiO2 catalysts, the glucose is hydrogenated to produce sorbitol. Finally, hydrogenolysis of sorbitol into hexanols is achieved by the synergy of Ir–ReOx/SiO2 and H2SO4, and hexane is obtained as the final product (Fig. 9, Route 1).94 In subsequent work, the same group employed hemicellulose as the feedstock, and also obtained n-pentane, pentanols or xylitol by simply adjusting the reaction conditions over the same Ir–ReOx/SiO2 catalyst combined with acids. According to extensive characterization studies, hydroxorhenium sites (Re–OH) were found to be the active sites of Ir–ReOx/SiO2 for the C–O bond hydrogenolysis in carbohydrates.33 | ||
| Fig. 9 Catalytic conversion of cellulosic biomass into chemicals over Re-based catalysts. | ||
Carbohydrates such as glucose and fructose are the main components of biomass and have been selected as probe molecules for catalytic conversions. When using glucose as the feedstock, over the Ir–ReOx/SiO2 catalyst, around 95% of n-hexane is obtained after 84 h of reaction at 140 °C in the presence of 8 MPa H2, much higher than the cellulose conversion counterpart (Fig. 9, Route 1).32 During the conversion of sugars, the same group found that ReOx–Pd/CeO2 catalysts selectively transform sugars into chiral polyols with retention of the intrinsic configuration (Fig. 9, Route 2). Specifically, employing methyl glycosides as the feedstock, vicinal OH groups can be selectively removed via the deoxydehydration–hydrogenation reaction without breaking of the C–C bonds. Under typical reaction conditions of 140 °C for 24 h with a H2 pressure of 8 MPa, the methyl-D-2,3-dideoxy-mannopyranoside yield surpasses 90%, serving as an effective reaction process for producing stereostructure chemicals. Additionally, this product is an ideal feedstock for producing chiral polyol building blocks and diols via hydrolysis and/or hydrogenation reactions.95
Apart from these deoxydehydration and hydrodeoxygenation reactions, Re-based catalysts were also employed for carbohydrate dehydration due to its specific acidic properties. By adjusting reaction conditions, different carbohydrate sources can be transformed into ethyl levulinate, 5-ethoxymethylfurfural, 5-hydroxymethylfurfural and levulinic acid over a HReO4 catalyst.96 To solve the recycling issues, Re species were incorporated into metal catalysts for sugar conversions. Chia et al. synthesized RhRe/C catalysts for the dehydration of fructose to 5-hydroxymethylfurfural, and confirmed the acidic properties of Re species in liquid water.97 Recently, Avramescu et al. loaded Re onto TiO2 for carbohydrate conversion. The balanced Brønsted and Lewis acid sites catalyze the glucose isomerization and dehydration reactions, and yield 57% of levulinic acid over 10% Re–TiO2 catalyst in four catalytic cycles (Fig. 9, Route 3).98
3.2 Conversion of lignin
Due to the high activity of methyltrioxorhenium(VII) in catalyzing the oxidation of aromatic derivatives, immobilized methyltrioxorhenium was developed for the transformation of lignin utilizing polymeric support, such as poly(4-vinylpyridine and polystyrene). Employing immobilized methyltrioxorhenium as the catalyst and H2O2 as the oxidant agent, the lignin derived from sugarcane and red spruce decomposes at room temperature into soluble lignin fragments with higher amounts of carboxylic acid groups.87To make full use of the ReOx sites, they are loaded on different supports for depolymerization of lignin model compounds and real lignin to aromatics and cycloalkanes through hydrogenation and hydrodeoxygenation reactions. Currently, the hydrogenation and hydrodeoxygenation reactions of lignin concentrate on lignin monomers including guaiacol, phenol, anisole, dimers containing β-O-4, 5-5 model compounds, and real lignin. Controlled selective hydrogenation of lignin model compounds still remains a challenge due to some side reactions. Selecting guaiacol as an example, guaiacol can be converted through several pathways, as shown in Fig. 10: guaiacol can be demethoxylated to phenol, and further hydrogenolyzed to benzene. The conversion of guaiacol to anisole through hydrogenolysis is achieved by alkylation/transalkylation of anisole to methyl anisole. The final products toluene and methyl cyclohexane are produced through hydrogenation of cresol. The whole reaction system is rather complicated, bearing a huge challenge to achieve a catalyst for improving the selectivity for the target products. Thus, Re-based compounds could be ideal candidates for the controlled hydrogenation reaction owing to the multitude of their accessible valences. For instance, Re supported on active carbon (AC) has been carburized under different temperatures between 500 °C and 700 °C under H2/C2H4 with the ratio of 75:25, forming ReC at 700 °C, RexC at 650 °C, and a mixture of Re metal, oxide, oxycarbide and carbide between 500 °C and 600 °C. Phenol as the main product is obtained for all the catalysts, while at high conversion (>80%) phenol starts to further convert into benzene. Utilizing only RexC as the predominant active phase, a benzene yield of 51% is achieved, attributed to the suppression of phenol hydrogenolysis into benzene.99 Later, Re-based bimetallic catalysts were developed for improving the catalytic activity. For instance, FeReOx/ZrO2 has been prepared by the incipient wetness co-impregnation method, and used for hydrodeoxygenation of phenolics such as guaiacol, m-cresol and anisole.100 Compared to Fe/H-Beta(38), FeReOx/ZrO2 shows clear enhancement in terms of performance for hydrodeoxygenation of phenolics. The yield of BTX mixture products (BTX = benzene, toluene and xylenes) is almost two times higher than in the case of Fe/H-Beta(38) at 350 °C and 0.1 MPa H2. The variations in performance can be attributed to the finely balanced acidity resulting from both the presence of Re oxide and the Zr support, leading to enhanced dehydration efficiency. A range of catalysts, comprising a base-metal (Re, Ga, Ni, and Co) combined with reducible metal oxides (Mo and V), were developed for the hydrodeoxygenation of anisole into benzene/toluene products.101 Among these catalysts, Re–MoOx/TiO2 and Re–VOx/TiO2 exhibit notably high catalytic activity in breaking C–O bonds to produce benzene and toluene products at 300 °C and 3 MPa H2. The catalytic performance of Re–MoOx/TiO2 primarily stems from exposed Mo5+ sites, whereas that of Re–VOx/TiO2 is governed by Re4+ sites. Control experiments and characterization studies have led to the conclusion that despite the Mo5+ sites having the highest intrinsic activity, the presence of Re as a specific type of active site in an appropriate amount is required to enhance benzene/toluene production. Besides the abovementioned study, hydrodeoxygenation of monomers to cycloalkanes is regarded as a direct way to obtain alkanes. When using cresol, anisole, and phenolic compounds as substrates, the inclusion of Re causes the bulk surface of Ni/Cu/Fe to fragment into smaller ensembles, leading to a reduction in the electron density of the metals’ d-band through both geometric and electronic influence, resulting in a high activity in the hydrodeoxygenation of lignin monomers without the hydrogenolysis of C–C bonds of the products, affording high alkane yields.100,102–104
 | ||
| Fig. 10 The possible pathways for guaiacol, phenol and cresol conversion. | ||
Re-based catalysts also have outstanding performance for the selective cleavage of C–O bonds in lignin β-O-4, α-O-4, β-1, α-5, and 5-5 model compounds and real lignin through hydrogen and a hydrogen donating solvent, as shown in Fig. 11. According to the results of model compounds’ hydrogenation, different phenols and alkanes can be synthesized selectively. Zhang et al. found ReOx/AC catalyzes the selective cleavage of C–O bonds of lignin β-O-4 model compounds delivering ethylbenzene and guaiacol as major products in 81.6% and 83.0% yield through a H-transfer reaction at 200 °C and 0.7 MPa N2 (Fig. 11a).41iPrOH has been utilized as both the solvent and hydrogen donor for reductive cleavage of the ether bonds in this case. The reaction involves dehydration, dehydrogenation, and direct hydrogenolysis, with dehydration to vinyl ether intermediates being the major route.41 Furthermore, the ReOx/AC catalyst can also be utilized for the deconstruction α-O-4 model compounds, leading to 80.3% yield of toluene and 62.5% yield of phenol at 200 °C and 3 MPa H2.39 Re 4f XPS showed the conversion of ReOx species into low valence states during catalysis. However, the species is easily oxidized with air. Lignin linkages involving C–C bonds are regarded as the most stable bonds due to their high bond dissociation energies.105 In further research progress, a RuRe alloy was developed for the deconstruction of β-1, α-5, and 5-5 model compounds, affording a mixture of alkanes and aromatics in 36.9–86.6% carbon yields at 300 °C and 2 MPa H2.40 Furthermore, both kraft lignin and alkaline lignin underwent degradation, involving the breaking of C–C and C–O bonds, ultimately yielding monocyclic products at the maximum theoretical yield based on the cleavage of C–O bonds in real lignin.
 | ||
| Fig. 11 Cleavage of C–O/C–C bonds in lignin model compounds over Re-based catalysts. | ||
The hydrodeoxygenation degradation of lignin is another effective way for the production of alkanes, which is the most direct protocol for the production of fuels. For instance, an Ir–ReOx/SiO2 catalyst was prepared by the wetness impregnation method, and used for the one-pot conversion of various compounds modeling lignin structures and three kinds of lignin (enzymolysis lignin, organosolv poplar lignin and commercial alkaline lignin) into alkanes.38 Various lignin models including β-O-4 and 5-5 were converted to cyclohexane in 95% and 63% carbon yield as well (Fig. 11c).
Compared to model compounds, lignin causes more problems due to lower solubility and more complicated structure, when degradation to phenolics is to be achieved (Fig. 12). Depolymerization of lignin is divided into two processes: oxidation and hydrogenolysis reactions. In the oxidative depolymerization process, the challenge lies in controlling bond cleavage and catalyst inefficiencies. NiO–Re2O7/GO has been developed to catalyze fractionated bagasse lignin into 28.4% yield of phenolics, producing guaiacol as the main product, which is higher compared to the conversion without incorporating Re into NiO/GO (23.7%).106 This catalyst can be reused five times. The significant improvement observed can be attributed to the capability of Re to enhance both metal dispersion properties and selectivity in ether bond cleavage within lignin.
 | ||
| Fig. 12 Depolymerization of lignin to phenolics over Re-based catalysts. | ||
Hydrogenolysis stands as another highly effective method for lignin depolymerization, involving either the addition of H2 or hydrogen-transfer reactions. Using ReOx/AC with iPrOH as both the solvent and H-source, catalysis of organosolv poplar lignin and organosolv wheat lignin yields monophenolics at 11.4 wt% and 5.3 wt%, respectively, at 200 °C and 100 psi N2. Incorporating noble metals significantly enhances selectivity toward aromatics under H2 atmosphere. For example, PtRe/TiO2 catalysts show activity in β-O-4 bond cracking in extracted birch lignin. After 12 h of reaction at 240 °C, a notable 18.7 wt% monophenol yield, including 7.5 wt% of 4-propylsyringol, is achieved using isopropanol as an in situ hydrogen donor.107
Apart from H-transfer reactions, depolymerization by H2 addition could favor the degradation of lignin into small molecules. ReS2/Al2O3 catalysts were obtained after sulfidation with dimethyl disulfide in the presence of H2, and they convert lignin into 21.5 wt% monocyclic product with 72.4% lower char yield due to the high oxophilicity. The metal-like behavior of Re sulfide stabilizes the depolymerized lignin fragments.108 By means of the precipitation–deposition and impregnation procedures, magnetic nanoparticles of Fe3O4@SiO2@Re can be synthesized for the fragmentation of lignin into aromatics. Over the recyclable catalyst, the Re species were found to catalyze both C–C and C–O bonds in the presence of H2 at 180 °C.109 Different characterization studies of this catalyst showed that Re was not reduced to the metallic state and weakly acidic Brønsted-type centers were created. Furthermore, Fe3O4@Nb2O5@Co@Re was developed via a multistep process and catalyzed lignin to lignin fragments. Compared to a maximal 40% yield in water-soluble fragments over monometallic Fe3O4@Nb2O5@Co, the addition of Re improved the catalytic activity and up to 85% yield of light fragments was achieved due to the synergetic effect between Co and Re metals.110 In subsequent advancements, bimetallic ReMo@zeolitic imidazolate framework (ZIF) catalysts have been able to crack kraft lignin, yielding 92% of monophenols in the liquid products. This outcome aligns with the observed synergistic effect between ReOx and MoOx in promoting phenol conversions.111,112 Similarly, in the presence of Ni over NbOx, kraft lignin is catalytically transformed with 96.7 wt% yield of oil after 3 h of reaction at 330 °C over 5Ni–5Re/Nb2O5 catalysts.113
In reductive approaches, an Ir–ReOx/SiO2 catalyst with both acidic and basic sites was used for one-pot conversion of lignin, resulting in 44.3 wt% yield of naphthenes from organosolv lignin at 260 °C at 4 MPa H2. This high performance is attributed to a synergistic effect between Ir and ReOx in Ir–ReOx/SiO2. During the mechanistic study, ReOx and Ir species were proved to be responsible for C–O cleavage and hydrogenation of aromatic rings, respectively.38
Converting woody biomass, composed of cellulose, hemicellulose, and lignin, into value-added chemicals remains a considerable challenge, primarily due to the intricate structure and inherently resistant chemical composition of raw lignocellulosic biomass. Currently, pyrolysis and pretreatment processes are utilized to extract individual components from woody biomass for utilization. However, these methods require extensive energy input and incur high costs. The catalytic utilization of all three components simultaneously towards different chemicals offers the potential for maximum conversion efficiency. Designing a robust and multifunctional catalyst is crucial for this purpose. Li et al. have developed Ni–ReOx/CeO2, which catalyzes the transformation of woody biomass to both aromatics and alkanes, as shown in Fig. 13. Under mild reaction conditions, Ni–ReOx/CeO2 achieves an 88.4% yield of lignin oil from woody biomass. Conversely, under harsh conditions, it converts the same substrate into alkanes. These results demonstrate that the catalyst can operate in two modes: first, preserving cellulose in a lignin-first reaction protocol, and second, converting the entire substrate into alkanes with 43.8% carbon yield.45 Additionally, the catalyst possesses magnetic properties, facilitating easy separation and enabling its reuse for up to seven cycles. This protocol presents a clear advantage for cost-efficient production of aromatics and fuels, while bypassing the need for pretreatment processes.45
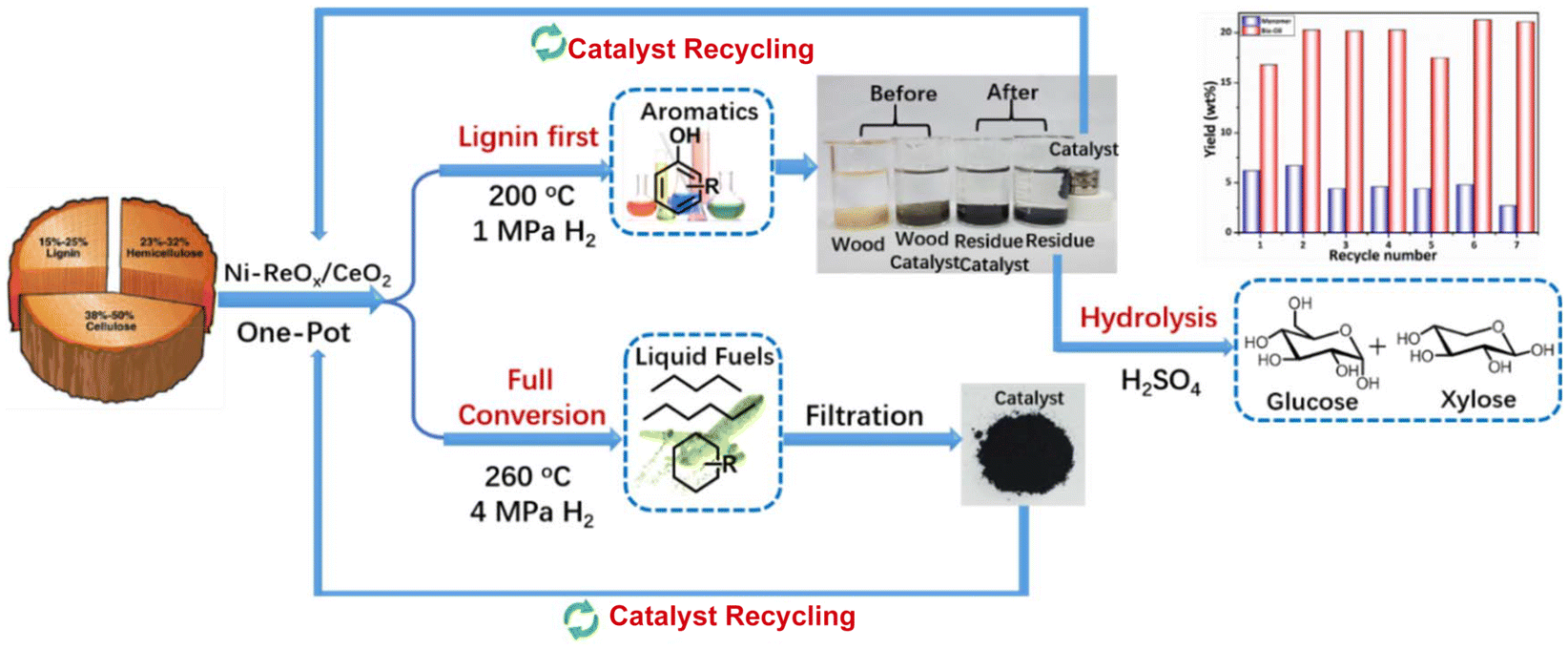 | ||
| Fig. 13 Conversion of woody biomass to aromatics and alkanes over Ni–ReOx/CeO2.45 | ||
3.3 Conversion of platform chemicals
Besides applications in the upgrading of cellulosic biomass and lignin, Re-based catalysts have also been widely used in upgrading platform chemicals via hydrogenation, hydrogenolysis, hydrodeoxygenation and deoxydehydration reactions.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C bonds.
Similar to some precious metal catalysts, supported Re catalysts show high activity in CC bond hydrogenation without affecting the aromatic rings or carboxylic groups. Over the ReOx/TiO2 catalyst with weak binding between metal oxide and support (Fig. 14), trans, trans-muconic acid can be selectively hydrogenated into adipic acid with >88% selectivity to dimethyl adipate after 5 h of reaction at 210 °C and a hydrogen pressure of 6.89 MPa.114 By the atomic layer deposition method, methyltrioxorhenium can be introduced into the metal–organic framework of NU-1000. This material can be applied in ethene hydrogenation for at least 1 day.115 Under these reaction conditions, a large number of reduced Re sites are formed, attributed to the improved hydrogenation activity in CC bond conversions.
C bonds.
Similar to some precious metal catalysts, supported Re catalysts show high activity in CC bond hydrogenation without affecting the aromatic rings or carboxylic groups. Over the ReOx/TiO2 catalyst with weak binding between metal oxide and support (Fig. 14), trans, trans-muconic acid can be selectively hydrogenated into adipic acid with >88% selectivity to dimethyl adipate after 5 h of reaction at 210 °C and a hydrogen pressure of 6.89 MPa.114 By the atomic layer deposition method, methyltrioxorhenium can be introduced into the metal–organic framework of NU-1000. This material can be applied in ethene hydrogenation for at least 1 day.115 Under these reaction conditions, a large number of reduced Re sites are formed, attributed to the improved hydrogenation activity in CC bond conversions.
 | ||
| Fig. 14 Hydrogenation of muconic acid into different adipic acid esters in methanol over ReOx/TiO2 catalysts.114 | ||
O bonds.
In most cases, carboxylic acids or esters with CO bonds are difficult to be hydrogenated due to the low electrophilicity of carbonyl carbon atoms.116,117 Interestingly, Re based catalysts demonstrate high activity for CO bond hydrogenation, especially in the presence of metal catalysts (Table 5).
O bond hydrogenation over Re based catalysts
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalysts | Reaction type | Solvent | T/°C | P/MPa | Time/h | Con./% | Sel./% | Ref. |
| 1 | Reduced Re2O7 | Batch | Water–dioxane | 265 | 24.5 | 23.5 | 71 | 60.5 | 118 |
| 2 | ReOx/TiO2 | Batch | Dodecane | 220 | 2.0 | — | 80 | 88 | 119 |
| 3 | Re–Pd/SiO2 | Batch | 1,4-Dioxane | 140 | 8.0 | 4 | 18 | 94 | 121 |
| 4 | Ir–ReOx/SiO2 | Batch | Cyclohexane | 180 | 2.0 | 1 | 100 | 94 | 122 |
| 5 | Pt–Re/TiO2 | Batch | Dodecane | 130 | 2.0 | 5 | 62 | 83 | 123 |
| 6 | Ni1Re1 | Batch | Cyclohexane | 150 | 4.0 | 5 | 99 | 94.5 | 124 |

|
|||||||||
| 7 | Pd–Re/TiO2 | Batch | Water | 160 | 15.0 | 48 | 100 | 83 | 129 |
| 8 | Re–Ru/MC | Batch | 1,4-Dioxane | 200 | 8.0 | 7 | 100 | 71.2 | 130 |
| 9 | Re–Pd | Batch | 1,4-Dioxane | 140 | 8.0 | 96 | 100 | 89 | 131 |
| 10 | Re–Pt/C | Batch | Water | 160 | 8.0 | — | >99 | 80 | 132 |

|
|||||||||
| 11 | ReOx/ZrO2 (SiO2) | Batch | 1,4-Dioxane | 200 | 5.0 | 3 | 100 | >90 (GVL) | 138 |
| 12 | Re–Ru–O/HZSM-5 | Batch | — | 230 | 4.0 | 4 | — | 65% yield valeric acid/esters | 140 |

|
|||||||||
| 13 | PdRe/Al2O3 | Fixed-bed reactor | — | 150 | 0.1 | — | 10 | 66.7 | 142 |
| 14 | ReOx/SiO2 | Batch | Dodecane | 200 | 4.0 | 4 | 10 | 70 | 143 |

During the transformation of C6–C18 fatty acids to alcohols, ReOx shows high selectivity for alcohols only under harsh reaction conditions.118,119 In the presence of metals, the activity and product selectivity were greatly improved due to the synergistic effect between metal and ReOx species.120–123 In Fig. 15, among both monometallic and bimetallic catalysts, the Ni1Re1 catalyst demonstrates the highest TOF (turnover frequency), underscoring the pivotal contribution of Re species in bolstering catalytic activity. The H2-TPD findings suggest that the introduction of Re species amplifies the catalyst's capability for hydrogen activation and dissociation, thereby generating a greater abundance of active hydrogen atoms crucial for the hydrogenation reaction. Moreover, the NiRe alloys promote fatty acid adsorption on the catalyst surface, favouring the conversion of fatty acids to alcohols. As a result, over the Ni1Re1 catalyst, fatty acid conversion surpasses 95% with >90% selectivity for fatty alcohols under mild conditions (150 °C and 4.0 MPa hydrogen).124 Additionally, the presence of Re species enhances the rate of hydrodeoxygenation and shifts the product from n-heptadecane to n-octadecane for ethyl stearate conversions. Over the Ru–Re/TiO2 catalyst, the ratio of n-octadecane/n-heptadecane reached 3.2 due to the newly formed weak acid sites of the catalyst.125
 | ||
| Fig. 15 (a) Comparison of TOF values over different reduced catalysts (0.1 g of fatty acid, 0.02 g of activated catalyst in 10 mL of cyclohexane solvent at 150 °C and 4.0 MPa hydrogen); (b) H2-TPD profiles of the reduced Ni1 and Ni1Re1 catalysts.124 | ||
Hydrogenation of succinic acid into value-added chemicals is highly attractive but challenging over most metal catalysts.126 In contrast, over ReOx/C catalysts, γ-butyrolactone, the key intermediate for 1,4-butanediol production, is preferably formed, due to the weak activity of Re species for the ring-opening of γ-butyrolactone.127,128 After introducing metal sites, the main product shifts to 1,4-butanediol due to the synergistic effect between metal and Re species.129 Over Pd(Ru, Pt)–ReOx catalysts, the succinic acid conversion reaches 100% with >70% 1,4-butanediol selectivity.129–132 According to in situ characterization, metallic Re and Re3+ species are present, which interact with high coordination metal species to bifunctional sites for the selective hydrogenolysis of the intermediate of γ-butyrolactone to 1,4-butanediol.133–135 In detail, over a Pd–ReOx/TiO2 catalyst with active Pd–ReOx interfaces, the activation of γ-butyrolactone occurs on protonated Re species bound to Pd atoms, forming an intermediate with both the carbonyl group and the oxygen in the cycle, which are then transformed into an ester intermediate and further hydrogenated over Pd0 sites to yield 1,4-butanediol (Fig. 16).133
 | ||
| Fig. 16 Schematic reaction pathway for the conversion of γ-butyrolactone to 1,4-butanediol.133 | ||
As one of the ten high-value biomass-derived compounds from lignocellulosic biomass, levulinic acid is employed for producing a large number of commodity chemicals.136,137 Over Re oxides supported on ZrO2 and SiO2, the γ-valerolactone selectivity exceeds 90% regardless of the support used, demonstrating the bifunctional properties of ReOx species under reaction conditions.138 Over a RuRe/C catalyst, 1,4-pentanediol is preferably produced with selectivity up to 75% at 130 °C and 50 bar H2. According to the in situ FT-IR spectroscopy results, the presence of Re species promoted the adsorption of the reactant and intermediates, and then facilitated the ring-opening hydrogenation of γ-valerolactone to 1,4-pentanediol.139 The product changes to methyl valerate and valeric acid when using neat methyl levulinate as the feedstock over Re–Ru–O/HZSM-5 catalysts. The authors propose that the ReOx species catalyzes the conversion of methyl levulinate to γ-valerolactone, the acidic HZSM-5 promots the ring-opening of γ-valerolactone, and the ReOx modified Ru hydrogenates the intermediate to valeric compounds.140 Similarly, ethyl levulinate is transformed into ethyl-4-ethoxy pentanoate with 83% yield at 160 °C for 2 h under a pressure of 1 MPa H2 over NiReOx/Beta catalysts. The presence of ReOx increases the dispersion and reducibility of NiO over the support of Beta zeolite, affording large amounts of Lewis acid sites and Brønsted acid sites for ethyl levulinate conversions.141
Re based catalysts have also been used for other CO bond hydrogenations in biomass conversions. For instance, PdRe/Al2O3 catalysts show greater furfuryl alcohol selectivity and activity than Pd/Al2O3 catalysts for furfural conversions due to the intimate contact of Re clusters with the surface Pd particles.142 At a higher reaction temperature of 200 °C, furfuryl alcohol is produced over ReOx/SiO2 catalysts, and then shifted to a mixture of furfuryl alcohol and 2-methyl furan, when the reaction time is extended, confirming the high hydrogenation activity of partial reduced ReOx species.143
 | ||
| Fig. 17 Potential reaction pathway for glycerol hydrogenolysis over Ir–ReOx/SiO2 (left) and model structure of Ir–ReOx/SiO2 (20 wt%-Ir, Re/Ir = 0.34, actual) during the reaction (right).149,150 | ||
Besides the structure of Re species, the type of metal also greatly affects the product selectivity. For instance, 1,2-propanediol is preferably produced over Pt–ReOx based catalysts. Under the reaction conditions, strong Brønsted acid sites are formed over the bimetallic catalyst, which catalyzes the dehydration of glycerol to aldehyde, followed by hydrogenation reactions over Pt sites.151–153 Similarly, the addition of Pt on Ir–ReOx/SiO2 catalysts shifts the main product from 1,3-propanediol to 1,2-propanediol for glycerol conversions without hydrogen being present.154 Over non-modified Ce-supported Re oxide catalysts (10 wt% ReOx/CeO2 + SiO2), the deoxydehydration of glycerol to allyl alcohol is achieved with a yield up to 86%, using 2-hexanol and 4-methyl-2-pentanol as both a H-donor and a solvent at 175 °C. Based on the experimental results, the redox couples of ReVII/ReV and ReVI/ReIV, should be the active sites for this reaction.155 Additionally, Re based catalysts have been applied in dehydration reactions. For instance, over Re2O7 catalysts, benzylic alcohols were dehydrated into styrenes in the presence of toluene, at 100 °C and under ambient atmosphere.156 In a later work, the reactants were extended to a wide range of allylic, aliphatic, and homoallylic alcohols. Interestingly, the heterogeneous Re2O7 catalyst demonstrates superior activity and selectivity when compared to sulfuric acid and most common solid acids.157
Another attractive deoxygenation reaction is the catalytic deoxydehydration reaction, which removes vicinal hydroxyl groups and forms CC double bonds with the help of a sacrificial reductant. Learning from the robust activity of homogeneous Re species in deoxydehydration reactions, Re-based heterogeneous catalysts were also synthesized for this reaction and showed excellent performances. In 2013, Denning et al. utilized ReOx on carbon for the deoxydehydration of glycols into olefins, but these catalysts suffered from leaching issues.158 Afterwards, more stable ReOx nanoparticles and supported ReOx were synthesized for deoxydehydration reactions. Under reaction conditions, a mixture of ReVII, ReIV, and Re0 species is formed in the presence of alcohols, providing a redox pool for deoxydehydration reactions with improved catalyst stability.159–161
To further improve the catalyst stability and activity, metals have been introduced into the ReOx system. For instance, over ReOx–Pd/CeO2 catalysts, tetrahydrofuran is obtained with a selectivity >99% using 1,4-anhydroerythritol as the reactant, as reported by Tomishige's group. With the aid of metallic Pd sites, the high-valent Re species was reduced to ReIV by H2, which was coordinated with vicinal OH groups of 1,4-anhydroerythritol to diolate. After removing the –OH groups, the alkene can be hydrogenated to tetrahydrofuran over metallic Pd sites in the presence of hydrogen (Fig. 18).21,162 In follow-up studies, additional metals including Au, Ag, Ni were introduced onto ReOx/CeO2 catalysts for the deoxydehydration of different vicinal diols, and also described achieving >90% yield of deoxydehydration products.25,163–166
 | ||
| Fig. 18 Proposed reaction mechanism for the deoxydehydration reaction over ReOx catalysts.162 | ||
Due to the versatile applications of 1,4-butanediol in the polyester sectors, the deoxydehydration reaction was applied in the conversion of erythritol or 1,4-anhydroerythritol to 1,4-butanediol. Over the physical mixture of ReOx(Au)/CeO2 + ReOx/C, the 1,4-butanediol yield reaches 90% at 140 °C with H2 as the reductant. Over ReOx(Au)/CeO2 as the catalyst, the rate-determining step of the deoxydehydration of 1,4-anhydroerythritol to 2,5-dihydrofuran has been described as being the activation of H2 by ReOx/C. Afterwards, the 2,5-dihydrofuran is isomerized to 2,3-dihydrofuran over ReOx/C, which is then further hydrated to hydroxytetrahydrofuran and at last hydrogenated forming 1,4-butanediol.167–169 Using more abundant erythritol as feedstock, the deoxydehydration reactions, dehydration and hydrogenation reactions were coupled, obtaining a selectivity of 33% to 1,4-butanediol with a conversion of 74.2% cover Ir–ReOx/SiO2 as the catalyst.77 When changing the support to rutile-TiO2, the yield of 1,4-butanediol slightly decreases but with a high selectivity, especially at low conversion levels.170 According to kinetic analysis results, C–O bond cleavage shows lower activation energy than C–C bond cleavage, in the presence of ReOx species, resulting in a high selectivity for erythritol to 1,4-butanediol conversion.171,172 During the reaction kinetics study of deoxydehydration and hydrogenation of erythritol to butanediols and n-butane over the ReOx–Pd/CeO2 catalyst, Cao et al. proposed that the deactivation of catalyst should not be attributed to the deposition of organic species, but the aggregation of ReOx species, as evidenced by Raman spectroscopy and supported by density functional theory calculations.173
Apart from these applications, Re-based catalysts have also been used in deoxydehydration of multiple –OH chemicals. For example, ReOx/ZrO2 catalysts effectively facilitate the deoxydehydration of D-glucaric acid into valuable adipic acid ester, achieving a high yield of up to 82% when combined with subsequent hydrogenation over Pd/C catalysts.174 Additionally, a two-step pathway has been devised for converting glucose into adipic acid. In the first step, glucose is oxidized into glucaric acid over Pt/CNT catalysts, obtaining 82% product yield after 4 h at 60 °C and 1 MPa O2 pressure. In the second step, the four –OH groups are removed from adipic acid via the deoxydehydration reaction over ReOx/AC catalysts with yields up to 99%, and then the CC bonds were hydrogenated to adipic acid over Pd sites. According to conditional experiments and density functional theory calculations, the authors propose a possible reaction mechanism for the second step (see Fig. 19). Initially, potassium glucarate dissolved in methanol undergoes esterification with methanol to form lactone and linear esters, facilitated by a solid acid catalyst. Subsequently, the ReOx species, featuring the binuclear Re–O–Re site on ReO3 surfaces, catalyzes the initial deoxydehydration reaction of methyl glucarate into corresponding olefins, concurrently oxidizing the Re–O–Re site. Following this, Pd nanoparticles facilitate the hydrogenation of the olefin by supplying active hydrogen through H2 dissociation. Pd also facilitates the reduction of the oxidized rhenium sites with H2. Finally, the saturated intermediates such as 1 and 2′ are transformed further into 2 with subsequent deoxydehydration and hydrogenation over ReOx species and Pd nanoparticles to yield adipic acid.175 With pure isopropanol as the solvent, a key hydrogen transfer agent, glucaric acid is efficiently converted into adipic acid over the same Pt–ReOx/C catalyst via balancing the deoxydehydration and hydrogenation reactions.176
 | ||
| Fig. 19 Proposed reaction mechanism for the conversion of glucaric acid (potassium glucarate) to adipic acid (methyl adipate) catalyzed by Pd–ReOx/AC175 (DODH: deoxydehydration). | ||
C, C–O and CO bonds (Fig. 20). The Ir metal heterolytically dissociates H2 for the hydrogenation of the furan rings, and then the strong synergy between Ir and ReOx activates furan rings and alcoholic C–O bonds for hydrogenation to alkanes.180
 | ||
| Fig. 20 Intermediate oxygen-containing compounds generated during the trifuran conversion process over an Ir–ReOx/SiO2 catalyst were discerned via GC-MS analysis of the resultant product solutions.180 | ||
5-Hydroxymethylfurfural can be hydrogenated to tetrahydropyran-2-methanol, which is further converted into 1,6-hexanediol at 84% yield over Rh–ReOx/C as the catalyst. The ReOx and Rh species show a synergistic effect between the Rh metal surface and attached ReOx species for hydrogenolysis of tetrahydropyran-2-methanol to 1,6-hexanediol.181 In another attempt, 5-hydroxymethylfurfural has been selectively converted into 1,6-hexanediol (1,6-hydrodeoxygenation) over the double-layered catalysts of Pd/SiO2 + Ir–ReOx/SiO2 in a fixed-bed reactor, obtaining yields as high of 57.8% at 100 °C and 7 MPa H2. Remarkably, the double-layered catalysts exhibit superior performance when contrasted with single-layer catalysts such as Pd–Ir–ReOx/SiO2, despite containing an equivalent amount of active components. As shown in Fig. 21, the upper layer Pd/SiO2 catalyzes the direct hydrogenation of 5-hydroxymethylfurfural to 2,5-bis(hydroxymethyl)-tetrahydrofuran, which are further hydrogenated into 1,6-hexandiol via the intermediate 1,2,6-hexanetriol over Ir–ReOx/SiO2 catalysts. Simultaneously, side reactions including the hydrodeoxygenation of products, reactants and intermediates take place, lowering the final product yield of 1,6-hexandiol.182
 | ||
| Fig. 21 Reaction routes of hydroxymethylfurfural conversion to 1,6-hydrodeoxygenation over Pd/SiO2 + Ir–ReOx/SiO2 catalysts.182 | ||
4. Conclusions and outlook
Re-based catalytic compounds have emerged as highly useful materials for various catalytic reactions, encompassing olefin metathesis, aldehyde olefination, oxidation, and dehydrogenation reactions. Biomass, recognized as a sustainable and renewable resource, serves as an ideal starting material for the production of value-added chemicals through catalytic processes. The integration of Re-based compounds with biomass conversion has been elegantly demonstrated in previous studies.Due to rapid progress in this field, this work focuses on the catalysis of the three main components of lignocellulose (cellulose, hemicellulose, and lignin) and their derivatives through C–O bond cleavage, oxidation, hydrogenation, deoxydehydration, and hydrodeoxygenation reactions using Re-based compounds. The reaction pathways, catalytic systems, the key role of Re species in controlling product selectivity and the reaction mechanisms are listed and discussed. This provides insights into the delivery of value-added chemicals from biomass over Re catalysts. This review serves as a comprehensive approach for summarizing and capturing the key highlights, identifying knowledge gaps, and suggesting potential research directions for the advancing Re-catalyzed biomass conversion.
Although significant progress has been made, there are still some challenges in the application of Re-based catalysts in biomass conversions:
(1) Stability: different from other transition metals, Re-based catalysts demonstrate unique activity and selectivity in oxygen-containing feedstock conversions. However, leaching issues of Re-catalytic systems in both homogeneous and heterogeneous phases often occur. To date, keeping both high catalytic activity and stability still remains a huge challenge for Re-based catalysts. Thus, it is crucial to design supports with a uniform and well-defined surface structure so as to increase the interaction between Re sites. Thanks to the rapid development of novel porous materials such as metal organic frameworks (MOFs), covalent organic frameworks (COFs) and polymers, there are a number of options for highly dispersed Re catalysts synthesis with definite structures. Alternatively, synthesis of high stability Re single atom catalysts would be an appropriate strategy to maximize the Re activity per atom with every site featuring unique electronic structures, while preserving distinct metal support interactions. Here, single atom catalysis bridges the gap between homogeneous and heterogeneous catalysts and provides a unique viewpoint on Re catalysis.183,184 For example, the Re–Ni–Fe alloy nanoparticles over lanthanum ferrite (LaFeO3) showed long-term resistance to coking and sintering for methane dry reforming due to the inhibition of Re evaporation issues.185 This alloy strategy provides valuable information for metallic Re catalysis and high- stability catalyst design.
(2) Mechanistic understanding: the incomplete understanding of catalytic behavior of Re-based catalysts hampers the discovery of real active sites for the catalytic reactions due to the polyvalent behavior of Re compounds. To elucidate the catalytic mechanisms, advancement of in situ and operando characterization techniques would be helpful for in-depth analysis. Biomass conversion reactions often take place in organic solvents, but their use poses challenges for operando characterization. The presence of liquid organic solvents can lead to interference due to their significant absorptions in infrared (IR) or ultraviolet-visible (UV/Vis) spectral ranges. Additionally, the high viscosity of organic solvents may have a strong impact on the mass transfer, reactant concentration and diffusion rates of the reactants. Therefore, the development of appropriate operando cells and techniques for the conversion in organic solvents remains an active area of research.
(3) New applications: the high electronic richness combined with the tunable stereochemical bulk of Re-based catalysts provides numerous opportunities in biomass reactions. The renewed interest in lignocellulose-based reactions catalyzed by Re-based compounds has led to the production of other value-added biochemicals. For instance, heteroatoms (X) participate in biomass conversion through the cleavage of C–O/C–C bonds and the construction of C–X bonds catalyzed by Re-based compounds, resulting in monomers containing heteroatoms. This expands the applications and functionalization of lignocellulosic production. Additionally, single Re atom catalysis is also an attractive option for biomass conversion due to its definite structure and active sites. The transformation of biomass-derived platform chemicals into high yields of a single monomer, rather than a mixture of products, should receive significant attention. Therefore, further advancements in designing Re-based catalysts for suitable reactions to access value-added monomers are crucial requirements for cost-competitive and sustainable large-scale production in industry.
(4) Economic feasibility: Re-catalyzed deoxydehydration has undergone significant advancements, particularly in the presence of a reductant, resulting in the production of olefin products. Assessments such as life cycle analysis, economic evaluation, and reaction efficiency are essential in determining the most suitable processes for industrial application. Given the high cost associated with Re metal, more efforts should be devoted on the following aspects: (1) developing inexpensive and abundantly available metals like molybdenum, vanadium, and tungsten holds promise for large-scale and economically viable deoxydehydration reactions. (2) Re based catalysts should be efficient and recyclable to ensure sustainability and economic viability. (3) The Re sites should be 100% utilized, i.e. single Re catalysts for biomass conversions. These methodologies hold great potential for overcoming the current limitations and unlocking new opportunities for advancement in the field.
(5) Carbon neutrality: the recent achievements of Re-based catalysts for biomass conversion are beneficial for reaching the goal of carbon neutrality. First, the conversion of biomass to chemicals, fuels and materials reduces CO2 emissions since biomass is the fixed CO2 in the atmosphere via the photosynthesis process. Then, the Re-based catalysts demonstrate robust activity and efficiency in tailoring the –OH groups in biomass conversion. For instance, over Re-based catalysts, high olefin product yields were obtained through the deoxydehydration reaction, thus being a green and atom economic reaction for biomass conversion. Thereafter, the product separation energy and the treatment of waste water should be largely reduced. Finally, the rapid developments of Re catalysis also inspire the application of Re-based catalysts in other energy sectors, which will upgrade some energy systems and promote the construction of a low carbon-footprint society. For instance, atomically dispersed ReO4 exhibited >93% selectivity to acetic acid for halide-free, gas phase carbonylation of methanol, representing a new class of promising catalysts for alcohol carbonylation.186
To tackle these persistent challenges, it is essential to foster close collaboration across various disciplines and facilitate dialogue between academia and industry. This review aims to spark significant interest among researchers in both Re chemistry and biomass conversion. It will encourage further exploration of Re-based compounds as promising alternative catalysts, thereby meeting the current and future needs of biomass conversion.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Natural Science Foundation of China (22078317, 22378383) is acknowledged.References
- D. Welsby, J. Price, S. Pye and P. Ekins, Nature, 2021, 597, 230–234 CrossRef CAS
.
- M. S. Nazir, Z. M. Ali, M. Bilal, H. M. Sohail and H. M. N. Iqbal, Environ. Sci. Pollut. Res., 2020, 27, 33516–33526 CrossRef
- K. M. Yavor, V. Bach and M. Finkbeiner, Sustainability, 2021, 13, 6107 CrossRef CAS
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS
- B. Zhang, B. K. Biswal, J. Zhang and R. Balasubramanian, Chem. Rev., 2023, 123, 7193–7294 CrossRef CAS
- Y. Feng, S. Long, X. Tang, Y. Sun, R. Luque, X. Zeng and L. Lin, Chem. Soc. Rev., 2021, 50, 6042–6093 RSC
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef PubMed
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 Search PubMed
- G. Li, R. Wang, J. Pang, A. Wang, N. Li and T. Zhang, Chem. Rev., 2024, 124, 2889–2954 CrossRef PubMed
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef
- A. A. Koutinas, A. Vlysidis, D. Pleissner, N. Kopsahelis, I. Lopez Garcia, I. K. Kookos, S. Papanikolaou, T. H. Kwan and C. S. K. Lin, Chem. Soc. Rev., 2014, 43, 2587–2627 RSC
- T. A. Ewing, N. Nouse, M. van Lint, J. van Haveren, J. Hugenholtz and D. S. van Es, Green Chem., 2022, 24, 6373–6405 RSC
- A. J. J. Straathof, Chem. Rev., 2014, 114, 1871–1908 CrossRef CAS PubMed
- S. Dutta, ChemSusChem, 2012, 5, 2125–2127 CrossRef CAS PubMed
- J. Yi, S. Liu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 1401–1404 CrossRef CAS
- J. R. Dilworth, Coord. Chem. Rev., 2021, 436, 213822 CrossRef CAS
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 1897–1900 CrossRef CAS PubMed
- N. N. Tshibalonza and J.-C. M. Monbaliu, Green Chem., 2020, 22, 4801–4848 RSC
- L. J. Donnelly, S. P. Thomas and J. B. Love, Chem. – Asian J., 2019, 14, 3782–3790 CrossRef CAS PubMed
- J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 CrossRef CAS PubMed
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 Search PubMed
- S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS
- X. Huang, K. Liu, W. L. Vrijburg, X. Ouyang, A. Iulian Dugulan, Y. Liu, M. W. G. M. Tiny Verhoeven, N. A. Kosinov, E. A. Pidko and E. J. M. Hensen, Appl. Catal., B, 2020, 278, 119314 CrossRef CAS
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, ACS Catal., 2016, 6, 3600–3609 CrossRef CAS
- M. L. Gothe, K. L. C. Silva, A. L. Figueredo, J. L. Fiorio, J. Rozendo, B. Manduca, V. Simizu, R. S. Freire, M. A. S. Garcia and P. Vidinha, Eur. J. Inorg. Chem., 2021, 4043–4065 CrossRef CAS
- Y. Nakagawa, K. Mori, K. Chen, Y. Amada, M. Tamura and K. Tomishige, Appl. Catal., A, 2013, 468, 418–425 CrossRef CAS
- Y. Nakagawa, X. Ning, Y. Amada and K. Tomishige, Appl. Catal., A, 2012, 433–434, 128–134 CrossRef CAS
- K. Chen, M. Tamura, Z. Yuan, Y. Nakagawa and K. Tomishige, ChemSusChem, 2013, 6, 613–621 CrossRef CAS PubMed
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2016, 18, 165–175 RSC
- J. Cao, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Catal., 2019, 9, 3725–3729 CrossRef CAS
- K. A. DeNike and S. M. Kilyanek, R. Soc. Open Sci., 2019, 6, 191165 CrossRef PubMed
- K. Tomishige, M. Yabushita, J. Cao and Y. Nakagawa, Green Chem., 2022, 24, 5652–5690 RSC
-
C. Boucher-Jacobs and K. M. Nicholas, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, 2014, vol. 353, pp. 163–184 Search PubMed
- X. Li, B. Zhang, X. Pan, J. Ji, Y. Ren, H. Wang, N. Ji, Q. Liu and C. Li, ChemSusChem, 2020, 13, 4409–4419 CrossRef
- B. Zhang, Z. Qi, X. Li, J. Ji, W. Luo, C. Li, A. Wang and T. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 208–215 CrossRef
- X. Li, Y. Ding, X. Pan, Y. Xing, B. Zhang, X. Liu, Y. Tan, H. Wang and C. Li, J. Energy Chem., 2022, 67, 492–499 CrossRef
- B. Zhang, Z. Qi, X. Li, J. Ji, L. Zhang, H. Wang, X. Liu and C. Li, Green Chem., 2019, 21, 5556–5564 RSC
- Z. J. Qi, B. Zhang, J. W. Ji, X. X. Li, T. Dai, H. W. Guo, A. Q. Wang, L. C. Lu and C. Z. Li, ChemPlusChem, 2018, 83, 500–505 CrossRef
- R. G. Harms, I. I. E. Markovits, M. Drees, W. A. Herrmann, M. Cokoja and F. E. Kühn, ChemSusChem, 2014, 7, 429–434 Search PubMed
- B. J. Hofmann, R. G. Harms, S. P. Schwaminger, R. M. Reich and F. E. Kuhn, J. Catal., 2019, 373, 190–200 CrossRef CAS
- X. Li, Q. Qiang, Y. Ding, Y. Xing, J. Ji, H. Wang, X. Pan, B. Zhang and C. Li, Chem. Eng. Sci., 2024, 285, 119535 Search PubMed
- F. Chen, X. Jiang, L. Zhang, R. Lang and B. Qiao, Chin. J. Catal., 2018, 39, 893–898 CrossRef CAS
- J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43–55 RSC
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS
- W. A. Herrmann and F. E. Kühn, Acc. Chem. Res., 1997, 30, 169–180 CrossRef
- C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef
- J. E. Fergusson, Coord. Chem. Rev., 1966, 1, 459–503 CrossRef
- S. Liu, A. Senocak, J. L. Smeltz, L. Yang, B. Wegenhart, J. Yi, H. I. Kenttämaa, E. A. Ison and M. M. Abu-Omar, Organometallics, 2013, 32, 3210–3219 CrossRef
- T. J. Korstanje, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, Chem. – Eur. J., 2013, 19, 13224–13234 CrossRef
- Y. Cai and J. H. Espenson, Inorg. Chem., 2005, 44, 489–495 CrossRef CAS PubMed
- H. Kobayashi, T. Komanoya, S. K. Guha, K. Hara and A. Fukuoka, Appl. Catal., A, 2011, 409–410, 13–20 CrossRef CAS
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef
- J. Li, M. Lutz and R. J. M. Klein Gebbink, Catal. Sci. Technol., 2020, 10, 3782–3788 RSC
- J. Li, M. Lutz, M. Otte and R. J. M. Klein Gebbink, ChemCatChem, 2018, 10, 4755–4760 CrossRef
- E. S. Koh, T. H. Lee, D. Y. Lee, H. J. Kim, Y. W. Ryu and J. H. Seo, Biotechnol. Lett., 2003, 25, 2103–2105 CrossRef
- I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810–2818 CrossRef
- C. Boucher-Jacobs and K. M. Nicholas, Organometallics, 2015, 34, 1985–1990 CrossRef CAS
- L. G. Possato, T. F. Chaves, W. H. Cassinelli, S. H. Pulcinelli, C. V. Santilli and L. Martins, Catal. Today, 2017, 289, 20–28 CrossRef CAS
- J. Mazarío, P. Concepción, M. Ventura and M. E. Domine, J. Catal., 2020, 385, 160–175 CrossRef
- M. El Doukkali, A. Iriondo and I. Gandarias, Mol. Catal., 2020, 490, 110928 CrossRef CAS
- G. Scioli, L. Tonucci, P. Di Profio, A. Proto, R. Cucciniello and N. d'Alessandro, Sustainable Chem. Pharm., 2020, 17, 100273 CrossRef
- D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schlaf, Chem. – Eur. J., 2009, 15, 10132–10143 CrossRef CAS
- V. Canale, L. Tonucci, M. Bressan and N. d'Alessandro, Catal. Sci. Technol., 2014, 4, 3697–3704 RSC
- C. Boucher-Jacobs and K. M. Nicholas, ChemSusChem, 2013, 6, 597–599 CrossRef CAS
- M. Lupacchini, A. Mascitti, V. Canale, L. Tonucci, E. Colacino, M. Passacantando, A. Marrone and N. d'Alessandro, Catal. Sci. Technol., 2019, 9, 3036–3046 RSC
- X. Li, D. Wu, T. Lu, G. Yi, H. Su and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4200–4204 CrossRef CAS
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS PubMed
- X. Li and Y. Zhang, ChemSusChem, 2016, 9, 2774–2778 CrossRef CAS
- E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS
- Y. Amada, H. Watanabe, Y. Hirai, Y. Kajikawa, Y. Nakagawa and K. Tomishige, ChemSusChem, 2012, 5, 1991–1999 CrossRef CAS
- S. Qu, Y. Dang, M. Wen and Z.-X. Wang, Chem. – Eur. J., 2013, 19, 3827–3832 CrossRef CAS PubMed
- L. Lai, M. Liu, J. Liu, W. Li, W. Miao, Z. Sun, Z. Wang, Y. Wang, H. Shi, C. Chen, X. Jin and C. Yang, ACS Sustainable Chem. Eng., 2023, 11, 15851–15864 CrossRef CAS
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Lukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC
- C. Crestini, P. Pro, V. Neri and R. Saladino, Bioorg. Med. Chem., 2005, 13, 2569–2578 CrossRef PubMed
- C. Crestini, M. C. Caponi, D. S. Argyropoulos and R. Saladino, Bioorg. Med. Chem., 2006, 14, 5292–5302 CrossRef PubMed
- B. Zhang, C. Li, T. Dai, G. W. Huber, A. Wang and T. Zhang, RSC Adv., 2015, 5, 84967–84973 RSC
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS PubMed
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS PubMed
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, ChemSusChem, 2015, 8, 628–635 CrossRef CAS
- M. Tamura, N. Yuasa, J. Cao, Y. Nakagawa and K. Tomishige, Angew. Chem., Int. Ed., 2018, 57, 8058–8062 CrossRef CAS PubMed
- J. R. Bernardo, M. C. Oliveira and A. C. Fernandes, Mol. Catal., 2019, 465, 87–94 CrossRef CAS
- M. Chia, B. J. O'Neill, R. Alamillo, P. J. Dietrich, F. H. Ribeiro, J. T. Miller and J. A. Dumesic, J. Catal., 2013, 308, 226–236 CrossRef CAS
- S. Avramescu, C. D. Ene, M. Ciobanu, J. Schnee, F. Devred, C. Bucur, E. Vasile, L. Colaciello, R. Richards, E. M. Gaigneaux and M. N. Verziu, Catal. Sci. Technol., 2022, 12, 167–180 RSC
- E. Blanco, A. B. Dongil, J. L. García-Fierro and N. Escalona, Appl. Catal., A, 2020, 599, 117600 CrossRef
- P. Sirous-Rezaei, J. Jae, J.-M. Ha, C. H. Ko, J. M. Kim, J.-K. Jeon and Y.-K. Park, Green Chem., 2018, 20, 1472–1483 RSC
- I. T. Ghampson, G. Pecchi, J. L. G. Fierro, A. Videla and N. Escalona, Appl. Catal., B, 2017, 208, 60–74 CrossRef CAS
- F. Yang, D. Liu, H. Wang, X. Liu, J. Han, Q. Ge and X. Zhu, J. Catal., 2017, 349, 84–97 CrossRef
- X. Wang, W. Zhou, Y. Wang, S. Huang, Y. Zhao, S. Wang and X. Ma, Catal. Today, 2021, 365, 223–234 Search PubMed
- C. W. Park, J. W. Kim, H. U. Kim, Y.-K. Park, S. S. Lam, J.-M. Ha and J. Jae, Int. J. Energy Res., 2021, 45, 16349–16361 CrossRef
- L. Dong, L. Lin, X. Han, X. Si, X. Liu, Y. Guo, F. Lu, S. Rudić, S. F. Parker, S. Yang and Y. Wang, Chem, 2019, 5, 1521–1536 Search PubMed
- S. Totong, W. Laosiripojana, N. Laosiripojana and P. Daorattanachai, Ind. Eng. Chem. Res., 2022, 61, 215–223 CrossRef
- J. Hu, S. Zhang, R. Xiao, X. Jiang, Y. Wang, Y. Sun and P. Lu, Bioresour. Technol., 2019, 279, 228–233 CrossRef PubMed
- P. Sirous-Rezaei, D. Creaser and L. Olsson, Appl. Catal., B, 2021, 297, 120449 CrossRef CAS
- M. Tudorache, C. Opris, B. Cojocaru, N. G. Apostol, A. Tirsoaga, S. M. Coman, V. I. Parvulescu, B. Duraki, F. Krumeich and J. A. van Bokhoven, ACS Sustainable Chem. Eng., 2018, 6, 9606–9618 CrossRef CAS
- C. Opris, B. Cojocaru, N. Gheorghe, M. Tudorache, S. M. Coman, V. I. Parvulescu, B. Duraki, F. Krumeich and J. A. van Bokhoven, ACS Catal., 2017, 7, 3257–3267 CrossRef CAS
- G. Guo, W. Li, X. Dou, A. T. Ogunbiyi, T. Ahmed, B. Zhang and M. Wu, Bioresour. Technol., 2021, 321, 124443 CrossRef CAS PubMed
- C. Herrera, I. T. Ghampson, K. Cruces, C. Sepúlveda, L. Barrientos, D. Laurenti, C. Geantet, R. Serpell, D. Contreras, V. Melin and N. Escalona, Fuel, 2020, 259, 116245 CrossRef CAS
- L. Kong, L. Zhang, J. Gu, L. Gou, L. Xie, Y. Wang and L. Dai, Bioresour. Technol., 2020, 299, 122582 CrossRef PubMed
- X. She, H. M. Brown, X. Zhang, B. K. Ahring and Y. Wang, ChemSusChem, 2011, 4, 1071–1073 CrossRef PubMed
- M. Rimoldi, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 35067–35074 Search PubMed
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC
- T. J. Korstanje, J. I. van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef PubMed
- H. S. Broadbent, G. C. Campbell, W. J. Bartley and J. H. Johnson, J. Org. Chem., 1959, 24, 1847–1854 CrossRef
- B. Rozmysłowicz, A. Kirilin, A. Aho, H. Manyar, C. Hardacre, J. Wärnå, T. Salmi and D. Y. Murzin, J. Catal., 2015, 328, 197–207 CrossRef
- Y. Takeda, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2012, 2, 2221–2223 RSC
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2015, 5, 7034–7047 CrossRef
- S. Liu, T. Simonetti, W. Zheng and B. Saha, ChemSusChem, 2018, 11, 1446–1454 CrossRef PubMed
- K. Ralphs, G. Collins, H. Manyar, S. L. James and C. Hardacre, ACS Sustainable Chem. Eng., 2022, 10, 6934–6941 CrossRef
- X. Cao, J. Zhao, F. Long, P. Liu, X. Jiang, X. Zhang, J. Xu and J. Jiang, Appl. Catal., B, 2022, 312, 121437 CrossRef
- L. Zhou, W. Lin, K. Liu, Z. Wang, Q. Liu, H. Cheng, C. Zhang, M. Arai and F. Zhao, Catal. Sci. Technol., 2020, 10, 222–230 RSC
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed
- X. Di, Z. Shao, C. Li, W. Li and C. Liang, Catal. Sci. Technol., 2015, 5, 2441–2448 RSC
- U. G. Hong, H. W. Park, J. Lee, S. Hwang, J. Yi and I. K. Song, Appl. Catal., A, 2012, 415–416, 141–148 CrossRef
- B. K. Ly, D. P. Minh, C. Pinel, M. Besson, B. Tapin, F. Epron and C. Especel, Top. Catal., 2012, 55, 466–473 CrossRef
- K. H. Kang, U. G. Hong, Y. Bang, J. H. Choi, J. K. Kim, J. K. Lee, S. J. Han and I. K. Song, Appl. Catal., A, 2015, 490, 153–162 CrossRef
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Catal. Sci. Technol., 2016, 6, 5668–5683 RSC
- X. Di, C. Li, G. Lafaye, C. Especel, F. Epron and C. Liang, Catal. Sci. Technol., 2017, 7, 5212–5223 RSC
- B. K. Ly, B. Tapin, M. Aouine, P. Delichere, F. Epron, C. Pinel, C. Especel and M. Besson, ChemCatChem, 2015, 7, 2161–2178 CrossRef
- J. M. Keels, X. Chen, S. Karakalos, C. Liang, J. R. Monnier and J. R. Regalbuto, ACS Catal., 2018, 8, 6486–6494 CrossRef
- B. Tapin, B. Khanh Ly, C. Canaff, F. Epron, C. Pinel, M. Besson and C. Especel, Mater. Chem. Phys., 2020, 252, 123225 CrossRef
- M. Sajid, U. Farooq, G. Bary, M. M. Azim and X. Zhao, Green Chem., 2021, 23, 9198–9238 RSC
- W.-P. Xu, X.-F. Chen, H.-J. Guo, H.-L. Li, H.-R. Zhang, L. Xiong and X.-D. Chen, J. Chem. Technol. Biotechnol., 2021, 96, 3009–3024 Search PubMed
- R. Bassi, P. Baeza, C. Sepulveda, I. T. Ghampson, E. Camu, A. Brückner, U. Bentrup, J. L. G. Fierro and N. Escalona, Appl. Catal., A, 2021, 625, 118328 CrossRef
- D. Lee, H. U. Kim, J. R. Kim, Y.-K. Park, J.-M. Ha and J. Jae, J. Ind. Eng. Chem., 2024, 131, 490–502 CrossRef
- R. Bacchiocchi, J. De Maron, T. Tabanelli, D. Bianchi and F. Cavani, Sustainable Energy, 2023, 7, 671–681 Search PubMed
- L. Luo, Y. Ma, Y. He, J. Wang, T. Xue, H. Wu, Y. Guan and P. Wu, Fuel, 2023, 344, 128028 CrossRef CAS
- S. T. Thompson and H. H. Lamb, ACS Catal., 2016, 6, 7438–7447 CrossRef CAS
- F. Toledo, I. T. Ghampson, C. Sepúlveda, R. García, J. L. G. Fierro, A. Videla, R. Serpell and N. Escalona, Fuel, 2019, 242, 532–544 CrossRef
- K. Tomishige, Y. Nakagawa and M. Tamura, Chin. Chem. Lett., 2020, 31, 1071–1077 CrossRef
- Y. Liu, B. Zhong and A. Lawal, RSC Adv., 2022, 12, 27997–28008 RSC
- Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2010, 94, 318–326 CrossRef
- Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2011, 105, 117–127 CrossRef
- Y. Amada, H. Watanabe, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, J. Phys. Chem., 2012, 116, 23503–23514 CAS
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS
- C. Deng, X. Duan, J. Zhou, D. Chen, X. Zhou and W. Yuan, Catal. Today, 2014, 234, 208–214 Search PubMed
- D. D. Falcone, J. H. Hack, A. Y. Klyushin, A. Knop-Gericke, R. Schlögl and R. J. Davis, ACS Catal., 2015, 5, 5679–5695 CrossRef
- V. Korpelin, G. Sahoo, R. Ikonen and K. Honkala, J. Catal., 2023, 422, 12–23 CrossRef
- S. Liu, M. Tamura, Z. Shen, Y. Zhang, Y. Nakagawa and K. Tomishige, Catal. Today, 2018, 303, 106–116 CrossRef
- K. S. Vargas, J. Zaffran, M. Araque, M. Sadakane and B. Katryniok, Mol. Catal., 2023, 535, 112856 CrossRef
- T. J. Korstanje, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, ChemSusChem, 2010, 3, 695–697 CrossRef PubMed
- T. J. Korstanje, E. F. de Waard, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, ACS Catal., 2012, 2, 2173–2181 CrossRef CAS
- A. L. Denning, H. Dang, Z. Liu, K. M. Nicholas and F. C. Jentoft, ChemCatChem, 2013, 5, 3567–3570 CrossRef CAS
- L. Sandbrink, E. Klindtworth, H.-U. Islam, A. M. Beale and R. Palkovits, ACS Catal., 2016, 6, 677–680 CrossRef CAS
- J. H. Jang, H. Sohn, J. Camacho-Bunquin, D. Yang, C. Y. Park, M. Delferro and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2019, 7, 11438–11447 CrossRef CAS
- I. Meiners, Y. Louven and R. Palkovits, ChemCatChem, 2021, 13, 2393–2397 CrossRef CAS
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 3213–3226 CrossRef CAS
- J. Cao, M. Tamura, R. Hosaka, A. Nakayama, J.-y. Hasegawa, Y. Nakagawa and K. Tomishige, ACS Catal., 2020, 10, 12040–12051 CrossRef CAS
- K. Yamaguchi, Y. Nakagawa, C. Li, M. Yabushita and K. Tomishige, ACS Catal., 2022, 12, 12582–12595 CrossRef CAS
- K. Yamaguchi, J. Cao, M. Betchaku, Y. Nakagawa, M. Tamura, A. Nakayama, M. Yabushita and K. Tomishige, ChemSusChem, 2022, 15, e202102663 CrossRef CAS
- Y. Nakagawa, S. Tazawa, T. Wang, M. Tamura, N. Hiyoshi, K. Okumura and K. Tomishige, ACS Catal., 2017, 8, 584–595 CrossRef
- T. Wang, S. Liu, M. Tamura, Y. Nakagawa, N. Hiyoshi and K. Tomishige, Green Chem., 2018, 20, 2547–2557 RSC
- T. Wang, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2019, 12, 3615–3626 CrossRef CAS PubMed
- T. Wang, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, React. Chem. Eng., 2020, 5, 1237–1250 RSC
- M. Gu, L. Liu, Y. Nakagawa, C. Li, M. Tamura, Z. Shen, X. Zhou, Y. Zhang and K. Tomishige, ChemSusChem, 2021, 14, 642–654 CrossRef CAS PubMed
- E. M. Virgilio, M. E. Sad and C. L. Padró, Appl. Catal., A, 2022, 643, 118691 CrossRef CAS
- E. M. Virgilio, C. L. Padró and M. E. Sad, ChemCatChem, 2023, 15, e202201618 CrossRef CAS
- J. Cao, S. Larasati, M. Yabushita, Y. Nakagawa, J. Wärnå, D. Y. Murzin, D. Asada, A. Nakayama and K. Tomishige, ACS Catal., 2024, 14, 1663–1677 CrossRef CAS
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017–11020 RSC
- W. Deng, L. Yan, B. Wang, Q. Zhang, H. Song, S. Wang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 4712–4719 CrossRef CAS
- J. H. Jang, I. Ro, P. Christopher and M. M. Abu-Omar, ACS Catal., 2021, 11, 95–109 CrossRef CAS
- J. Chuseang, R. Nakwachara, M. Kalong, S. Ratchahat, W. Koo-amornpattana, W. Klysubun, P. Khemthong, K. Faungnawakij, S. Assabumrungrat, V. Itthibenchapong and A. Srifa, Sustainable Energy, 2021, 5, 1379–1393 CAS
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Green Chem., 2014, 16, 617–626 RSC
- K. Chen, K. Mori, H. Watanabe, Y. Nakagawa and K. Tomishige, J. Catal., 2012, 294, 171–183 CrossRef
- S. Liu, S. Dutta, W. Zheng, N. S. Gould, Z. Cheng, B. Xu, B. Saha and D. G. Vlachos, ChemSusChem, 2017, 10, 3225–3234 CrossRef
- K. Chen, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, ChemCatChem, 2010, 2, 547–555 CrossRef
- B. Xiao, M. Zheng, X. Li, J. Pang, R. Sun, H. Wang, X. Pang, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 2175–2184 RSC
- X. Zhang, L. Shang, Z. Yang and T. Zhang, ChemPlusChem, 2021, 86, 1635–1639 CrossRef
- X.-Z. Yue, Y.-C. Liu, B.-A. Lu, X. Du, W. Lei, Z.-Y. Liu, S.-S. Yi and C. Lu, Energy Environ. Sci., 2024, 17, 5892–5900 RSC
- D. Zubenko, S. Singh and B. A. Rosen, Appl. Catal., B, 2017, 209, 711–719 CrossRef
- J. Qi, J. Finzel, H. Robatjazi, M. Xu, A. S. Hoffman, S. R. Bare, X. Pan and P. Christopher, J. Am. Chem. Soc., 2020, 142, 14178–14189 CrossRef
| This journal is © The Royal Society of Chemistry 2024 |