
Mechanisms into dehydroaromatization of bio-derived limonene to p-cymene over Pd/HZSM-5 in the presence and absence of H2†
Huimei Cui,
Jingjing Zhang,
Zhicheng Luo and
Chen Zhao*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: czhao@chem.ecnu.edu.cn
First published on 7th July 2016
Abstract
The mechanisms of dehydroaromatization of limonene to p-cymene are intrinsically investigated over Pd/HZSM-5 under different N2/H2 atmospheres using the mathematical tool of Matlab. It is found that the dehydroaromatization reaction network starts with the isomerization step, and is followed by the sequential dehydrogenation in the presence of N2 or H2 at the selected system. The addition of hydrogen in the atmosphere would not change this reaction pathway, but leads to lower selectivity of p-cymene due to the accelerated hydrogenation rates on the double bonds. Besides, the additional hydrogen speeds up the overall reaction by facilitating the isomerization step on limonene while impeding its reverse reaction, as isomerization of limonene is proved to be the determining step of the whole dehydroaromatization reaction. Furthermore, the presence of hydrogen dramatically decreases the apparent and true activity energy of the target dehydroaromatization reaction and reduces the impact of temperatures to such processes compared to that with a N2 gas carrier.
Introduction
Cellulose,1 lignin,2 lipid,3 and chitin4 biomass can be used as abundant sustainable raw materials to produce valuable chemicals and fuels. Limonene is a kind of available mono-terpene biomass that can release aromatics and hydrogen via the dehydroaromatization reaction in the presence and absence of H2.5 p-Cymene, a valuable aromatic produced from terpene is used as an important industrial intermediate for synthesis of p-cresol and is also a kind of food flavor.6 The in situ generated H2 from limonene may catalyze the crucial hydrodeoxygenation reaction for upgrading of biomass to fuels and valuable chemicals, providing a feasible solution for partially providing the required H2 source in a bio-refinery.Recently, we have reported a novel route using limonene as H-donor to provide inherent aromatics p-cymene and H2. While with in situ generated H2, the one-pot hydrodeoxygenation of palm oil is simultaneously facilitated. The combination of the hydrocarbon products from two biomass resources (palm oil and terpene) helps to construct the perfect bio-jet fuel ingredients in a highly integrated process.7 However, it is difficult to draw a clear mechanism involving into individual steps and precise reaction pathways of the limonene dehydroaromatization, due to the fast hydrogen shift in the framework of the limonene structure with two double-bonds that are anchoring to in- and outside of the cyclic ring.8
One of the proposed pathway for limonene dehydroaromatization undergoes disproportionation into p-cymene and methane,9 with p-menthene or p-cymenyl as side-product (shown as route 1 in Scheme 1).5 This mechanism reflects the reaction of cyclohexene disproportionation to benzene and cyclohexane on metallic Pt at mild conditions.10 C. Catrinescu et al. reported that there are three competitive reaction pathways (isomerization, disproportionation, and polymerization). And the disproportionation of limonene in the dienes occurred as the main reaction route, as almost equal amounts of p-cymene and p-menthene have been detected.11
 | ||
| Scheme 1 Plausible mechanisms for dehydroaromatization of bio-derived limonene to p-cymene. | ||
The proposed second mechanism starts with the initial isomerization of limonene to terpinenes and terpinolenes, and then followed by further dehydroaromatization to p-cymene (shown as route 2 in Scheme 1).8 To support this conclusion, P. Lesage et al. provided an evidence that adding α-olefin as a hydrogen acceptor could significantly increase the yield of p-cymene (up to 92%), which is essentially incompatible with the disproportionation mechanism.12 As a result, the produced saturated menthane may be generated from the complete hydrogenation with p-cymene in presence of in situ generated H2. Luengo et al.13 and Nguyen et al.14 reported that the full-hydrogenation product menthane was not detected when tracking the transformation process with the chromatograph analysis coupled with mass spectrometer, indirectly negating the disproportionation mechanism.
Apart from these two mechanisms, the theory of isomerization following the π-allylic route or Lewis acid-induced process, together with the route of hydrogenation/dehydrogenation are extensively studied and proposed as well (shown as route 3 in Scheme 1). However, an agreement with respect to the reliable visual evidence, especially incorporated with mathematical theory inference and kinetics deduction has not been reached. Herein, in this work we will dedicatedly investigate the roles of support acidity, gas atmosphere, temperature, as well as their cooperative action in the conversion of limonene with Pd/HZSM-5 in n-dodecane. In addition, the mathematical tool of Matlab is intensively applied for simulating the intrinsic mechanism into the complex dehydroaromatization reaction, concerning on the shift of two double bonds locating at the cyclic-ring.
Results and discussion
Catalyst characterization
The metallic and acidic properties of the used parent HZSM-5 and Pd/HZSM-5 (SiO2/Al2O3 ratio: 258, Pd content: 1 wt%, as analyzed by ICP) were characterized. The nitrogen sorption results showed that HZSM-5 had a surface area of 438 m2 g−1, with a pore volume of 0.57 cm3 g−1 and a pore width of 52 Å. After 1 wt% Pd incorporation, the specific surface area was decreased to ca. 364 m2 g−1, and the pore volume was increased by 20% with an average pore width of 47 Å. Scanning electron microscope image (Fig. 1a) suggested that HZSM-5 was consisted of numbers of round shape particle (ca. 200 nm). The Pd particle size of Pd/HZSM-5 was 4.8 ± 1.9 nm, as demonstrated by transmission electron microscope image (Fig. 1b). The Pd dispersion was determined to be 46%, as obtained by CO chemisorption measurement. The XRD patterns (Fig. 1c) showed that Pd/HZSM-5 contained face-centered cubic (fcc) Pd (111) phase. The acid sites were analyzed by IR spectra of adsorbed pyridine (Fig. 1d), which demonstrated that Pd/HZSM-5 contained 3 μmol g−1 Brönsted acid sites (BAS) and 3 μmol g−1 Lewis acid sites (LAS). An obvious decrease on BAS and on LAS compared to parent HZSM-5 implies that the Pd incorporation occupies parts of acid sites of HZSM-5 (Table 1). | ||
| Fig. 1 Characterization with (a) SEM image of Pd/HZSM-5, (b) TEM image of Pd/HZSM-5, (c) XRD patterns, and (d) IR spectra of adsorbed pyridine of HZSM-5 and Pd/HZSM-5. | ||
Dehydroaromatization of limonene with different acid concentrations of Pd/HZSM-5
Limonene can be converted into p-cymene through three main routes, i.e. isomerization–dehydrogenation, hydrogenation–dehydrogenation, and dehydroaromatization.8 In the first step, the acid concentrations of HZSM-5 support is optimized to achieve the highest activity in dehydroaromatization of limonene.Taking the system without hydrogen as an example, with the increasing of Si/Al ratios from 5 to 43, 197, and 258, the selectivity of p-cymene and the overall conversion increased until reaching an equilibrium state with a selectivity of p-cymene as high as 83% (i.e. from 27%, 40%, 83% to 82%) and 100% conversion (i.e. from 91%, 81%, 100% to 100%) (Table 2). The dehydrogenation is catalyzed by the Pd nanoparticles, and the reactions of isomerization are mainly catalyzed by acidic sites. The observed isomerization products (i.e., 4-isopropylidene-1-methyl-cyclohexene, 1-isopropyl-4-methyl-cyclohexa-1,3-diene, 1-isopropyl-4-methyl-cyclohexa-1,4-diene, and 5-isopropyl-2-methyl-cyclohexa-1,3-diene) decreased with higher Si/Al ratios accordingly, while the Si/Al ratios were inversely proportional to the acidic concentrations.
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | 8 bar N2 | 2 bar H2 and 6 bar N2 | ||||||||||
| Conv. (%) | Yield (%) | Pb (bar) | Conv. (%) | Yield (%) | Pb (bar) | ||||||||
| A | B | C | D | A | B | C | D | ||||||
| a Reaction condition: 10 mL limonene, 0.2 g Pd/HZSM-5 (1 wt%), 80 mL n-dodecane, 260 °C, 2 h, stirring at 600 rpm.b This records the pressure for reactions lasting 2 h at working temperature. | |||||||||||||
| 1 | Pd/HZSM-5 (5) | 91 | 11 | 27 | 53 | 18 | 91 | 34 | 49 | 5 | 13 | ||
| 2 | Pd/HZSM-5 (43) | 81 | 15 | 40 | 26 | 15 | 78 | 32 | 1 | 42 | 3 | 13 | |
| 3 | Pd/HZSM-5 (197) | 100 | 16 | 83 | 23 | 100 | 29 | 69 | 15 | ||||
| 4 | Pd/HZSM-5 (258) | 100 | 16 | 82 | 22 | 100 | 32 | 66 | 16 | ||||

Apart from the effect of acidity, the decrease of isomerization products (i.e. from 53%, 26%, and 0 to 0) might be resulted from the faster rates of sequential dehydrogenation reaction via the isomerization intermediates, which agreed well with the result obtained by A. F. Thomas et al.9 It can be demonstrated by our experimental data that the yield of dehydroaromatization products increased (i.e. from 27% to 82%) with a decrease in isomerization products, which suggests that the dehydrogenation step follows the initial isomerization step.
Besides, it was observed that an increase in pressure (i.e. from 18, 15, and 23 to 22 bar) was attained during the reaction with increased Si/Al ratios of Pd/HZSM-5. The high efficiency of dehydroaromatization generated more hydrogen, and then the higher partial pressure of hydrogen catalyzed further conversion of partial-hydrogenation products into full-hydrogenation products (i.e. from 0, 0, 16% to 16%).
It was clearly showed that lower acidity made a contribution to the high selectivity of p-cymene, as it modified the rates of individual steps involved into the reaction network, i.e., partial hydrogenation, full hydrogenation, dehydroaromatization, and isomerization. Therefore, Pd/HZSM-5 (258) was chosen as the best catalyst into next use for its high performances in selectivity and activity.
Table 2 showed that the existence of H2 had an influence on the selectivity of p-cymene as well. There was no doubt that the addition of H2 would increase the fraction of partial hydrogenation and full hydrogenation products. For example, partial hydrogenation products took up more than twice of those without hydrogen with four Pd/HZSM-5 catalysts. And full hydrogenation product also showed an significant increase with 0, 1%, 29%, 32% as a function of Si/Al ratios, compared with those (0, 0, 16%, and 16%) in the system without hydrogen. Partial hydrogenation products (i.e. 34%, 32%, and 0 to 0) were converted into full hydrogenation product (i.e. 0, 1%, 29% and 32%) gradually.
According to equilibrium principle, the addition of hydrogen causes less p-cymene. And hydrogen also has the capability to hydrogenate the p-cymene which leads to further decease of the yield of p-cymene. It is evidenced by the lower pressure compared with the hydrogen-free system. The yields of p-cymene catalyzed with Pd/HZSM-5 (197) and Pd/HZSM-5 (258) in the system with 2 bar hydrogen were 69% and 66% respectively. When changing into in the system without hydrogen, the yields become lower (i.e. 83% and 82%). But when accounting for the yields of p-cymene catalyzed with Pd/HZSM-5 (5) and Pd/HZSM-5 (43), the yields in the hydrogen-addition system were higher, which was probably caused by the faster rates of dehydroaromatization reaction. And the reason for the increase when adding hydrogen into the system would be discussed with the help of Matlab software. The fast rates would transform the isomerization products into p-cymene and lead to higher yield of p-cymene. The fact that the sum of isomerization product and p-cymene were still lower in the system with additional hydrogen could support this hypothesis.
Impact of reaction temperatures towards limonene dehydroaromatization
At Scheme 2, the formula of limonene dehydroaromatization to form p-cymene and H2 was displayed. Calculated from the HSC software, the parameter changes of enthalpy (18.6 kJ), entropy (232.9 J K−1), and Gibbs free energy (−105.4 kJ) were listed in Table 3 for the process of limonene dehydroaromatization at 260 °C. In addition, the theoretical equilibrium constant (Ksp) was determined to be 2.26 × 1010. Consequently, the limonene concentration was 0.05919 mol in the theoretical equilibrium (the starting limonene was 0.05919 mol with 0.2 g catalyst, and the detailed calculation was listed in ESI†). In comparison, in the experimental limonene dehydroaromatization system using Pd/HZSM-5 at 260 °C, the limonene concentration was 0.0438 mol after achieving equilibrium in our experiment. The relative 26% error was probably resulted from the side reactions in the complex limonene dehydroaromatization network. | ||
| Scheme 2 The formula for limonene dehydroaromatization to p-cymene and hydrogen over Pd/HZSM-5 catalyst. | ||
| ΔH (kJ) | ΔS (J K−1) | ΔG (kJ) | Ksp | log (Ksp) |
|---|---|---|---|---|
| 18.6 | 232.9 | −105.4 | 2.26 × 1010 | 10.4 |
In the hydrogen-free system for limonene dehydroaromatization with Pd/HZSM-5, the yield of p-cymene was directly measured as a function of time at 240, 260, 280, 300 °C (Fig. 2a). Since dehydroaromatization is a kind of endothermic reaction, a temperature increase is beneficial for the equilibrium shift (i.e. from 3%, 6%, 9% and 33%) according to thermodynamics principles. High temperature accelerates the molecular collision as well, and the initial rates of dehydroaromatization at 300 °C (187 mmol g−1 h−1, equals to turnover frequency of 1120 mmol molPd site−1 h−1) which is twenty-three times faster than that at 240 °C (8 mmol g−1 h−1, equals to turnover frequency of 48 mmol molPd site−1 h−1) (Table 4).
 | ||
| Fig. 2 Dehydroaromatization of limonene over Pd/HZSM-5 at a function of time within the temperature range of 240, 260, 280, 300 °C (a) without hydrogen and (b) with hydrogen. (c) The Arrhenius plot towards the change of dehydroaromatization rate within the selected temperature range. (d) The relationship between Ea, Etrue, and ΔH for limonene dehydroaromatization over Pd/HZSM-5 in presence and absence of H2. | ||
| Gas | 8 bar N2 | 2 bar H2 and 6 bar N2 | ||||||
|---|---|---|---|---|---|---|---|---|
| T (°C) | 240 | 260 | 280 | 300 | 240 | 260 | 280 | 300 |
| a General conditions: 10 mL limonene, 0.02 g 1 wt% Pd/HZSM-5 (258), 80 mL n-dodecane, 260 °C, 2 h, stirring at 600 rpm. | ||||||||
| Yield of p-cymene (%) | 3 | 6 | 9 | 33 | 17 | 31 | 50 | 57 |
| Dehydroaromatization rate (mmol g−1 h−1) | 8 | 17 | 49 | 187 | 117 | 255 | 335 | 478 |
| Dehydroaromatization TOF (mmol molPd site−1 h−1) | 48 | 102 | 294 | 1120 | 701 | 1528 | 2007 | 2864 |
On addition of 2 bar hydrogen, the rate for formation of p-cymene was highly enhanced, e.g. it was increased from 8 mmol g−1 h−1 in N2 to 117 mmol g−1 h−1 with 2 bar hydrogen. And the rates and yields were all tested far away from reaching the equilibrium during the tested time. As a whole, the introduction of H2 (2 bar) speeded up the dehydroaromatization rate, and resulted in higher yield of p-cymene (initiation stage). Generally, the rate of dehydroaromatization on limonene is much slower with N2 than that with additional H2 at the temperature range from 240 to 300 °C (Table 4).
Based on Arrhenius plot, the calculated apparent activation energy (39 kJ mol−1, with N2) was lower than the system with additional hydrogen (126 kJ mol−1) (Fig. 2c). This result suggested that the impact of temperature on the rate of p-cymene with H2 was much less than that without H2. This was probably resulted from the fact that the added hydrogen brings additional exothermic hydrogenation reactions, reducing the impact towards the rate variation as a function of the temperature.
The observed apparent activation energy can be expressed as:15
| Eaobs. = Eatrue − ΔHΔn |
As the adsorption heat is considered to be comparable in two systems in presence and absence of H2 with the same reactant, and the reaction order Δn is usually considered to be 1. It is indicated that the enhanced observed activation energy is attributed to the different true activation energy in two systems with and without H2 (Fig. 2d). The additional of extra hydrogen could lower the true reaction energy barrier, as displayed at Fig. 2d. Additionally, the extra hydrogen also accelerates the reaction rates, which is of great significance to the industrial application. D. Buhl et al.16 also reported that hydrogen introduction lowered the deactivation rates of the catalysts (by coking), and the appropriate hydrogen addition into the system would lead to a better activity on cost of the selectivity, which agreed with our observed results.
Exploration of the intrinsic pathways into dehydroaromatization of limonene under different gas (H2/N2) atmosphere
Combining the possible products (i.e., 4-isopropyl-1-methyl-cyclohexene, 1-isopropenyl-4-methyl-cyclohexane, p-cymene and four kinds of isomerization products) allows us to formulate all of the plausible reaction pathways for conversion of limonene under different gas atmosphere. Shown from Scheme 3, the process involves the initial isomerization of limonene (route 1), and further dehydroaromatization to produce p-cymene (route 2). According to different isomerization products in the process of limonene isomerization at 260 °C, the parameter changes of enthalpy (from −19.2 to 21.6 kJ), entropy (from −11.8 to 25.7 J K−1), and Gibbs free energy (from −21.5 to 8.0 kJ) calculated from the HSC software were listed in Table 5. It can be deduced from Table 5 that route 1 is a reversible reaction, and route 8 (reverse route 1) may exist based on the gained values of Ksp, as the equilibrium constants Ksp range from 0.2 to 127. | ||
| Scheme 3 Summarized plausible pathways for conversion of limonene under different (H2/N2) gas atmospheres in the overall reaction network. | ||
| Isomer | ΔH (kJ) | ΔS (J K−1) | ΔG (kJ) | Ksp | log (Ksp) |
|---|---|---|---|---|---|
| Terpinolene | −17.1 | −11.8 | −10.9 | 11.4 | 1.1 |
| α-Phellandrene | −9.8 | 14.9 | −17.6 | 54.9 | 1.7 |
| α-Terpinene | 21.6 | 25.7 | 8.0 | 0.2 | −0.8 |
| γ-Terpinene | −19.2 | 4.4 | −21.5 | 127 | 2.1 |
The isomerized intermediate can be hydrogenated into 1-isopropyl-4-cyclohexane and 4-isopropyl-1-methyl-cyclohexene (routes 6, 7, and 9), as well. The former compound is also the product from full hydrogenation of limonene (route 5), while the latter compound is the partial hydrogenation product from limonene (route 3). In parallel, p-cymene can be formed from the partial hydrogenation production of limonene (route 4), followed by sequential dehydrogenation. In addition, the route via full hydrogenation, subsequent dehydrogenation to p-cymene has been excluded.17
In the hydrogen-free system, isomerization products were the major products (i.e. 17% showed in Table S1†), so “isomerization–dehydrogenation” pathway (routes 1 and 2) seemed to be the major reaction path. As small amount of partial hydrogenation products was observed, routes 3 and/or 9 should exist. But only with the simulated reaction path in line 2 (routes 1, 2, 8 and 9) could we gain the smallest resnorm (i.e. 0.0016) without ignoring the small amount of partial hydrogenation product (Table 6 in line 1 to line 3). Additionally, we also studied whether the conversion from 4-isopropyl-1-methyl-cyclohexene to p-cymene occurred with the simulated reaction paths (1, 2, 8, 9 and 4). In such case, k4 fitted to 0, illustrating that only with the path “isomerization–dehydrogenation” p-cymene could be formed in the system without hydrogen. Consequently, paths 1, 2, 8 and 9 participated in the simulated hydrogen-free reaction network, and the individual reaction equations are displayed in eqn (1).
| H2 pressure | Simulation order | Simulated reaction path | Resnorm | Reason of exclusion |
|---|---|---|---|---|
| 0 bar | 1 | 1 2 3 8 | 0.0016 | k3 = 0 |
| 2 | 1 2 9 8 | 0.0016 | ||
| 3 | 1 2 9 8 3 | 0.0016 | k3 = 0 | |
| 4 | 1 2 9 8 4 | 0.0016 | k4 = 0 | |
| 2 bar | 1 | 1 2 9 8 7 | 6.13 × 10−4 | k7 = 0 |
| 2 | 1 2 3 8 7 | 4.73 × 10−4 | ||
| 3 | 1 2 3 9 8 7 | 3.60 × 10−4 | ||
| 4 | 1 2 3 9 8 6 | 3.59 × 10−4 | ||
| 5 | 1 2 3 9 8 5 | 3.59 × 10−4 | k5 = 0 | |
| 6 | 1 2 3 9 8 5 6 | 3.56 × 10−4 | k6 < 0 | |
| 7 | 1 2 3 9 8 6 7 | 3.56 × 10−4 | k6 < 0 | |
| 8 | 1 2 3 9 8 5 7 | 3.57 × 10−4 | k7 < 0 | |
| 9 | 1 2 3 9 8 5 6 7 | 3.57 × 10−4 | k7 < 0 | |
| 10 | 1 2 9 8 6 | 6.08 × 10−4 | ||
| 11 | 1 2 3 8 6 | 4.74 × 10−4 | ||
| 12 | 1 2 3 9 8 6 4 | 2.54 × 10−4 | k4 < 0 | |
| 6 bar | 1 | 1 2 3 6 8 9 | 0.0024 | k3 = 0, k8 = 0 |
| 2 | 1 2 9 6 | 0.0024 | ||
| 3 | 1 2 3 6 8 | 0.0054 | ||
| 4 | 1 2 6 9 5 7 | 0.0024 | k5 < 0, k6 < 0 | |
| 5 | 1 2 9 7 | 0.0025 | ||
| 6 | 1 2 9 5 | 0.0025 | ||
| 7 | 1 2 9 6 4 | 0.0024 | k4 = 0 | |
| 10 bar | 1 | 1 2 9 3 8 6 | 0.0041 | k3 = 0 |
| 2 | 1 2 9 8 6 | 0.0041 | ||
| 3 | 1 2 3 8 6 | 0.0049 | ||
| 4 | 1 2 9 8 5 | 0.0043 | ||
| 5 | 1 2 9 8 7 | 0.0043 | ||
| 6 | 1 2 9 8 6 4 | 0.0043 | k4 < 0 | |
| 14 bar | 1 | 1 2 3 9 6 8 | 0.0042 | k3 = 0, k8 = 0 |
| 2 | 1 2 9 6 | 0.0042 | ||
| 3 | 1 2 3 6 | 0.0053 | ||
| 4 | 1 2 9 5 | 0.0105 | ||
| 5 | 1 2 9 7 | 0.0099 | ||
| 6 | 1 2 9 6 4 | 0.0044 | k4 < 0 |
Reaction equations in the simulated hydrogen-free system.
 | (1) |
When adding 2 bar hydrogen into the system, it was reasonable to obtain more partial hydrogenation products (15% compare with 1% in the system without hydrogen in Table S1†) and a few full hydrogenation production (1%). Therefore, we firstly assumed that the complete hydrogenation production could only be converted through path 7, and then verified if the partial hydrogenation products were transformed from path 3, 9 or combination. Consequently, the simulation results showed that both paths 3 and 9 were possible to contribute to the partial hydrogenation products according to the Matlab mathematical tool (line 1 to line 3 in Table 6).
When considering about the complete hydrogenation products, they could be produced from paths 5, 6, 7 or combinations. These possibilities were added with paths 1, 2, 3, 9 and 8. We could get the best simulation result (i.e. resnorm: 3.59 × 10−4 from line 3 to line 8) only with path 6. As expected, we could also verify again through line 4, line 9 and line 10 to conclude that hydrogenation of limonene and the isomerization intermediate contribute to 4-isopropyl-1-methyl-cyclohexene. And 4-isopropyl-1-methyl-cyclohexane was more likely to be transformed from the intermediate with only one double bond. Finally, we also tried to verify the existence of the partial hydrogenation pathway. The results showed that k4 < 0, proving that this path did not exist. As a viewpoint of the simulation process, paths of 1 2 3 6 8 9 participated in the formation of the network in this system when adding 2 bar hydrogen and its individual reaction equations were showed in eqn (2).
Reaction equations in the simulated system with 2 bar hydrogen.
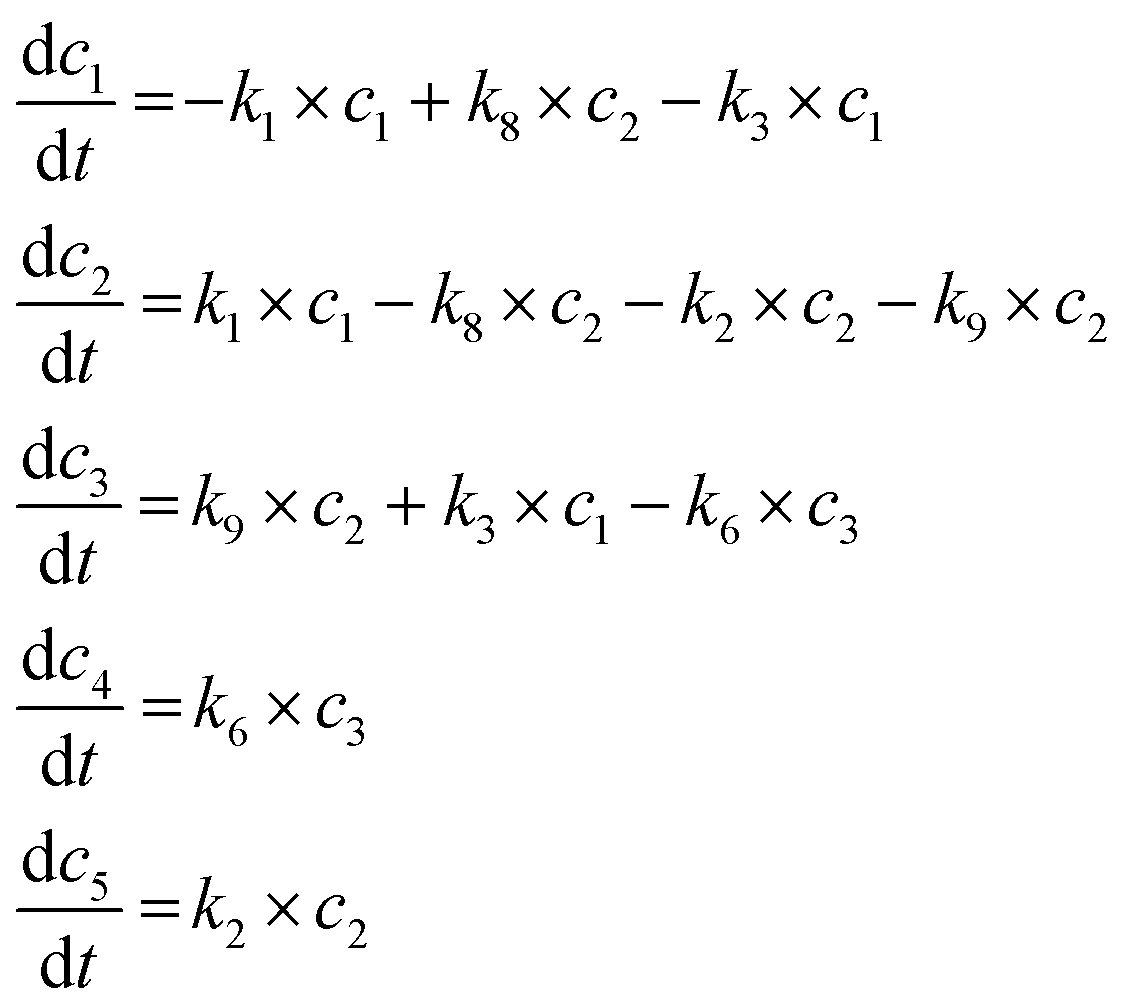 | (2) |
The other simulation processes (for reactions conducted at 6, 10, 14 bar hydrogen) were all followed by the similar logic. As showed in Table 6, it was through paths 1, 2, 9 and 6 when adding 6 bar hydrogen, paths 1, 2, 9, 8 and 6 when adding 10 bar hydrogen, and paths 1, 2, 6 and 9 when adding 14 bar hydrogen. The reaction equations are displayed in Table S2† and Fig. 3 presented the fitting curves for dehydroaromatization of limonene over Pd/HZSM-5.
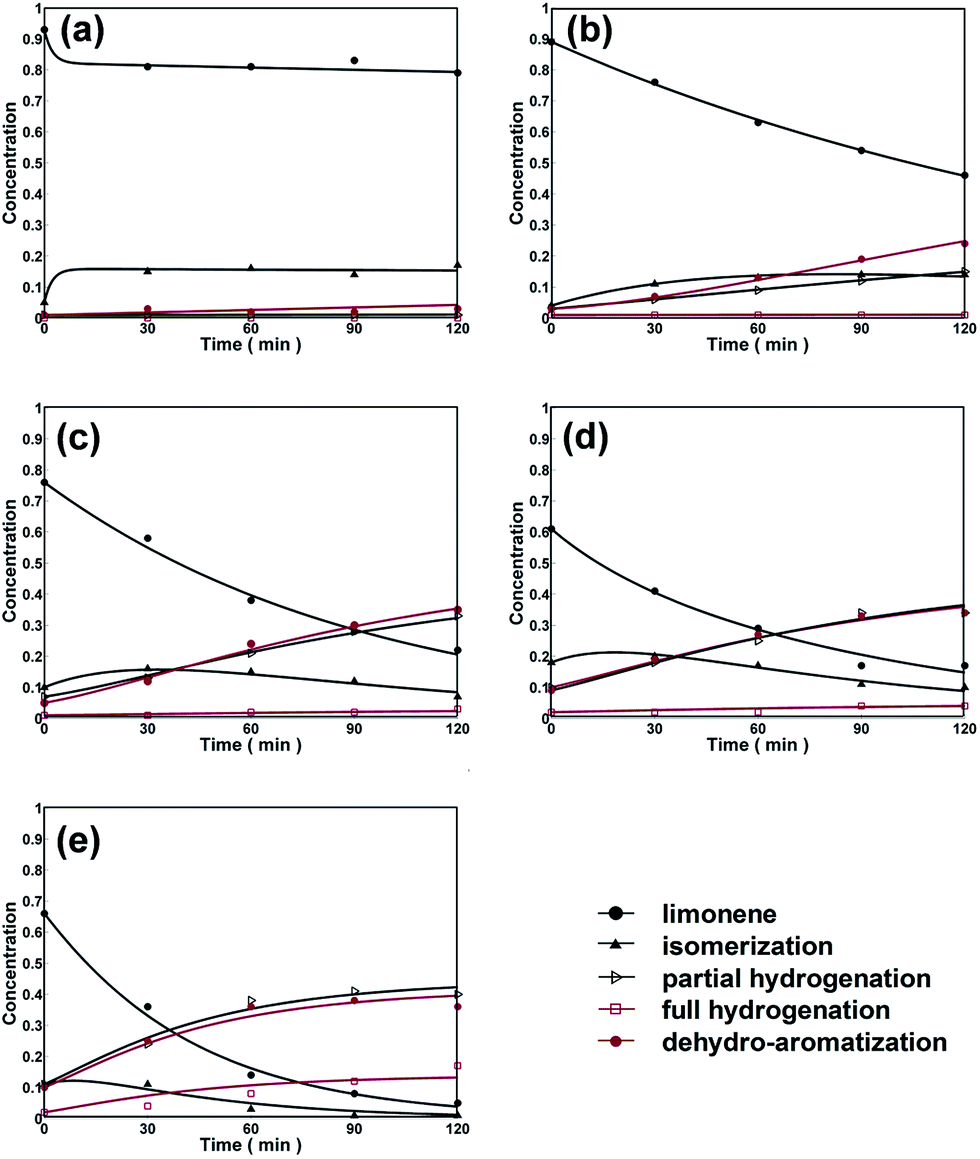 | ||
| Fig. 3 Dehydroaromatization of limonene over Pd/HZSM-5 as a function of time at 260 °C, (a) in hydrogen-free system, (b) with 2 bar hydrogen; (c) with 6 bar hydrogen; (d) with 10 bar hydrogen; (e) with 14 bar hydrogen. | ||
It could be clearly observed that with more hydrogen added into the system, the rate of dehydroaromatization into p-cymene was increased, but the yields of the hydrogenation products were also increased, which led to lower p-cymene selectivity.
From Fig. 4, it could be manifested that the yields of partial hydrogenation products, the full hydrogenation products, and dehydroaromatization products increased as a function of time with an increase in hydrogen pressure from 0 to 14 bar. But the changing yield of isomerization intermediate followed an interesting law, which was increased to a maximal value and then decreased gradually. The initial increase of isomerization products indicates that isomerization products are primary products from limonene conversion. But the further decrease in isomerization products is attributed to the consumption by sequential dehydrogenation reaction to p-cymene. Under different atmosphere with H2/N2, the different catalytic performance (Fig. 4a–d) might be guidance on controlling this consecutive reaction.
 | ||
| Fig. 4 The yields of (a) isomerization, (b) partial hydrogenation, (c) full hydrogenation, (d) dehydroaromatization from limonene, and (e) the remaining limonene concentrations after catalyzed over Pd/HZSM-5 at 260 °C as a function of time with 0, 2, 6, 10, and 14 bar hydrogen. | ||
In can be concluded that the major pathway for limonene dehydroaromatization follows the reaction steps of isomerization (route 1)–dehydrogenation (route 2)”, as depicted in Scheme 4. For all of isomers from limonene (terpinolene, α-phellandrene, α-terpinene, and γ-terpinene), Ksp range from 0.2 to 127 (see Table 5). Such equilibrium values are far below the complete reaction requirement (Ksp ≈ 105), indicating that the transformation of limonene and isomers occur easily. Thus, route 8 should be considered as a reversible reaction of route 1 (limonene isomerization). The limonene isomer mixture can be considered as a unified compound (labelled as compound 2 in Scheme 3 in the manuscript) due to the fast shift of each other.
 | ||
| Scheme 4 Summarized major pathway for conversion of limonene under different (H2/N2) gas atmospheres in the overall reaction network. | ||
To delineate the rate-determining step of the reaction pathways for dehydroaromatization of limonene under different gas atmosphere, the simulated rates of each reaction step were considered (in Table 7). Fig. 5 showed the changes of main individual rate constants in different N2/H2 atmosphere. It was reasonable to reach much higher rates for routes 6 and 9 (partial and full hydrogenation) when hydrogen pressure increased (Fig. 5), as hydrogenation reactions are more inclined to occur in high hydrogen pressure. The partial hydrogenation products were simulated to be formed from both isomerization intermediates and limonene hydrogenation, the full hydrogenation product was more likely to be converted from 4-isopropyl-1-methyl-cyclohexene (with one double bond), but not from hydrogenation of diene with conjugated double bonds.
| H2 pressure | k1 (min−1) | k2 (min−1) | k3 (min−1) | k6 (min−1) | k8 (min−1) | k9 (min−1) | Resnorm |
|---|---|---|---|---|---|---|---|
| 0 bar | 0.0719 | 0.0018 | 0.3704 | 0.0001 | 0.0016 | ||
| 2 bar | 0.0047 | 0.0150 | 0.0009 | 0.0001 | 0.0001 | 0.0035 | 0.0003 |
| 6 bar | 0.0108 | 0.0195 | 0.0006 | 0.0171 | 0.0024 | ||
| 10 bar | 0.0198 | 0.0144 | 0.0008 | 0.0148 | 0.0146 | 0.0047 | |
| 14 bar | 0.0229 | 0.0406 | 0.0039 | 0.0429 | 0.0042 |
 | ||
| Fig. 5 The main individual rate constants in selected catalysis systems with additional 2, 6, 10 and 14 bar hydrogen. | ||
According to the rate constants sum up in Table 7, p-cymene could be produced though the main “isomerization–dehydrogenation” pathway, but not via “partial hydrogenation–dehydrogenation” or “disproportion” route, because reaction 4 was not detected and the rate of determining-rate step of isomerization reaction was not twice of the rate of generation of 1-isopropenyl-4-methyl-cyclohexane.
By comparing main routes 1 with 2, route 1 was much slower than route 2 (Fig. 5), so route 1 (isomerization of limonene) is concluded to be the rate determining step. With added hydrogen in the system, both steps of isomerization of limonene and dehydroaromatization for p-cymene were accelerated, and the influence of N2/H2 atmosphere on the latter reaction was more obvious. This performance is beneficial for the whole reaction network, which was consistent with the results discussed-above (Table 2). This was mainly because that, in the hydrogen-free system, the reverse reaction of isomerization was quiet fast and depressed the further dehydrogenation reaction through lowering the concentration of isomerization intermediates. But hydrogen addition led to a fast isomerization rate, which benefited for further dehydrogenation reaction. And when increasing the pressure of hydrogen from 2 to 14 bar, the rate of isomerization increased, i.e. k1 increased from 0.0047, 0.0108, 0.0198, to 0.0229. Then the whole reaction rate increased, as such isomerization step was the rate-determining step.
Conclusions
Dehydroaromatization of bio-derived limonene to p-cymene is explored over Pd/HZSM-5 with varying acid concentrations under different atmospheres (N2/H2). It is showed to be a reaction network involving several individual steps. Among the modified Pd/HZSM-5, catalyst with lower acidity exhibits much higher conversion and selectivity. Hydrogen addition into the system not only decreases the apparent activation energy from 126 to 39 kJ mol−1 (calculated from the Arrhenius plot), but also accelerates the whole process. The explored intrinsic mechanisms of this process investigated by Matlab software also reveal that such acceleration effect of additional hydrogen is through facilitating the isomerization reaction (i.e. rate-determining step) and suppressing its reverse reaction at the same time. However, with more hydrogen in the atmosphere, the whole rates become much faster on the cost of the selectivity of p-cymene, due to the additional hydrogenation reaction. So there is an optimum value for the partial pressure of hydrogen. It is worthy noted that the addition of hydrogen (from 2 to 14 bar) would not change the dehydroaromatization path of the process. The reaction process still undergoes “isomerization–dehydrogenation” instead of “partial hydrogenation–dehydrogenation” or “disproportion” routes to produce p-cymene, as investigated by Matlab mathematic tool. Meanwhile the partial hydrogenation products are formed from hydrogenation of isomerization intermediates and limonene, while full hydrogenation product undergoes sequential two-steps of hydrogenation.Experimental
Chemicals
D-Limonene (Sinopec Co., Ltd., >95%), PdCl2 (R&K, >59.5% Pd), HCl (Sinopec Co., Ltd., >36%), and n-dodecane (Sinopec Co., Ltd.) were applied as received without any further purification. Air, H2 and N2 gases (99.999 vol%) were provided by Shanghai Pujiang Specialty Gases Co., Ltd.HZSM-5 with different Si/Al ratios were synthesized as follows: aluminum iso-propoxide (AIP, Sinopec Co., Ltd., containing 24.7 wt% Al2O3) was dissolved in a mixture of tetra-propyl-ammonium hydroxide (TPAOH, Sinopec Co., Ltd., 25 wt%) and distilled water with stirring at ambient temperature for 2 h until AIP was totally dissolved. Then tetra-ethylorthosilicate (TEOS, Sinopec Co., Ltd., containing 28.4 wt% SiO2) was added drop-wise into the mixed solution and kept stirring for another 4 h, leading to a gel which chemical composition was 1 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :x Al2O3:15 H2O:0.25 TPAOH (x refers to series of Si/Al2 mole ratio). Then the gel was transferred into a Teflon-lined stainless steel autoclave to perform crystallization process in an oven lasted 72 h at 170 °C. After cooled down to ambient temperature naturally, the solid product was washed by water, and sequentially centrifugated to remove supernatant liquid, and finally collected after drying at 80 °C overnight. Prior to use, it was calcined in flowing air (flowing rate: 100 mL min−1) at 550 °C for 6 h to remove TPAOH template.
:x Al2O3:15 H2O:0.25 TPAOH (x refers to series of Si/Al2 mole ratio). Then the gel was transferred into a Teflon-lined stainless steel autoclave to perform crystallization process in an oven lasted 72 h at 170 °C. After cooled down to ambient temperature naturally, the solid product was washed by water, and sequentially centrifugated to remove supernatant liquid, and finally collected after drying at 80 °C overnight. Prior to use, it was calcined in flowing air (flowing rate: 100 mL min−1) at 550 °C for 6 h to remove TPAOH template.
Catalyst synthesis
In typical procedure, 1 wt% Pd/HZSM-5 (SiO2/Al2O3 ratio: 258) was prepared by incipient wetness impregnation. H2PdCl4 aqueous solution (6.25 mM, 30.1 mL) was added into 1.98 g HZSM-5 support gradually and hosted with continuous stirring at ambient temperature for a certain time. Then the catalysts were filtered, dried at 353 K overnight, and light yellow powders were obtained. Finally, catalysts were calcined in air at 350 °C for 4 h followed by reduced in flowing hydrogen (flowing rate: 100 mL min−1) at 350 °C for 4 h before reaction.Catalytic tests
A typical experiment was carried out as follows: limonene (10 mL), 1 wt% Pd/HZSM-5 catalyst (0.2 g), n-dodecane (80 mL) were charged into a batch autoclave (Parr Instrument, 300 mL). N2 was firstly purged at room temperature for three times to remove the air in the reactor, subsequently, N2 or H2 was flushed according to different reaction conditions. Then the reaction system brought up to 260 °C at which reaction started, with a stirring speed of 650 rpm. The liquid products were in situ sampled, and analyzed by a Shimadzu QP-2010 Ultra GC-MS equipped with a Rtx-5Sil MS capillary column (30 m × 0.25 mm × 0.25 μm).Catalyst characterization
The specific surface area and pore information were measured on a BELSORP-MAX instrument at −196 °C by N2 adsorption–desorption. Powder X-ray diffraction (XRD) patterns were performed on Rigaku Ultima IV X-ray diffractometer utilizing Cu-Kα radiation (λ = 1.5405 Å) operated at 35 kV and 25 mA. Scanning electron microscopy (SEM) was operated on a Hitachi S-4800 microscope, while transmission electron microscopy images (TEM) were performed on a FEI Tecnai G2 F30 microscope working at 300 kV. The IR spectra of adsorbed pyridine (IR-Py) were recorded with a Bruker VERTEX 70 spectrometer equipped with an in situ IR cell. The samples were activated in vacuum at 400 °C for 1 h before equilibrated with pyridine at 150 °C, then evacuated at 150 °C for 1 h. CO Chemisorption was used to obtain the dispersion of metal palladium by dynamic CO pulse chemisorption at 35 °C with an AutoChem 2950HP instrument, after the catalyst sample was reduced at 100 °C in flow hydrogen atmosphere for 2 h to ensure Pd was reduced.Acknowledgements
This research was supported by the Recruitment Program of Global Young Experts in China, National Natural Science Foundation of China (Grant No. 21573075), and Shanghai Pujiang Program (PJ1403500).Notes and references
-
(a) R. H. Atalla and D. L. Vanderhart, Science, 1984, 223, 283 CAS
; (b) A. C. O'Sullivan, Cellulose, 1997, 4, 173 CrossRef
-
(a) A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS PubMed
-
(a) B. Ma and C. Zhao, Green Chem., 2015, 17, 1692 RSC
-
(a) N. Yan and X. Chen, Nature, 2015, 524, 155 CrossRef CAS PubMed
-
(a) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed
-
(a) D. M. Roberge, D. Buhl, J. P. M. Niederer and W. F. Hölderich, Appl. Catal., A, 2001, 215, 111 CrossRef CAS
-
(a) J. J. Zhang and C. Zhao, Chem. Commun., 2015, 51, 17249 RSC
-
(a) R. J. Grau, P. D. Zgolicz, C. Gutierrez and H. A. Taher, J. Mol. Catal. A: Chem., 1999, 148, 203 CrossRef CAS
-
(a) A. F. Thomas and Y. Bessiere, Nat. Prod. Rep., 1989, 6, 291 RSC
-
(a) A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3454 CrossRef CAS PubMed
- C. Catrinescu, C. Fernandes, P. Castilho and C. Breen, Appl. Catal., A, 2006, 331, 172 CrossRef
- P. Lesage, J. P. Candy, C. Hirigoyen, F. Humblot and J. M. Basset, J. Mol. Catal. A: Chem., 1996, 112, 431 CrossRef CAS
-
(a) M. A. Martin-Luengo, M. Yates, E. S. Rojo, D. H. Arribas, D. Aguilar and E. R. Hitzky, Appl. Catal., A, 2010, 387, 141 CrossRef CAS
-
(a) A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982 RSC
- F. Kapteijn, J. A. Moulijn, R. A. van Santen and R. Wever, Stud. Surf. Sci. Catal., 1999, 123, 81 CrossRef CAS
-
(a) D. Buhl, P. A. Weyrich, W. M. H. Sachtler and W. F. Hölderich, Appl. Catal., A, 1998, 171, 1 CrossRef CAS
- C. Zhao, W. Gan, X. B. Fan, Z. P. Cai, P. J. Dyson and Y. Kou, J. Catal., 2008, 254, 244 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17159a |
| This journal is © The Royal Society of Chemistry 2016 |