A high-capacity dual-electrolyte aluminum/air electrochemical cell†
Lei Wanga,
Fude Liu*a,
Wentao Wanga,
Guandong Yanga,
Dawei Zhenga,
Zhuangchun Wub and
Michael K. H. Leungc
aDepartment of Mechanical Engineering, The University of Hong Kong, Hong Kong, China. E-mail: fordliu@hku.hk; Fax: +852-2858-5415; Tel: +852-2859-2631
bSchool of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
cAbility R&D Energy Research Centre, School of Energy and Environment, City University of Hong Kong, Hong Kong, China
First published on 4th July 2014
Abstract
A novel dual-electrolyte aluminum/air cell (DEAAC), consisting of an aluminum metal anode in an organic anolyte, an anion polymer exchange membrane, and an air electrode in an aqueous alkaline catholyte, has been investigated. The anion membrane separates the organic anolyte from the aqueous catholyte, while allowing hydroxide ions to pass through. The DEAAC exhibited an open circuit voltage (VOC) of 1.6 V and a short current density (JSC) of 65 mA cm−2. With kitchen aluminum foil as the fuel, the DEAAC achieved an anodic capacity of 6000 mA h cm−3 at a discharge current density of 30 mA cm−2, which is much higher than the lithium's theoretical capacity of 2060 mA h cm−3. The anodic capacity of the DEAAC increased by 30–50 folds at different discharge current densities compared with that of a traditional alkaline Al/air cell (AAC). Overall, the DEAAC is promising as an electrochemical energy storage device because it has no detrimental hydrogen generation problem and exhibits very high anodic capacity.
1. Introduction
To realize a sustainable modern society, batteries have seen ever more demanding challenges in diverse applications ranging from portable electronics and electrical vehicles to grid-level energy storage.1–4 Metal/air electrochemical cells lie at the intersection of classical battery design and the modern fuel cell concept while combining advantages of both.5 With a reactive anode coupled with an air-breathing cathode, a metal/air cell takes in oxygen from the inexhaustible air and, in some cases, exhibits very high gravimetric and volumetric energy densities.6 Among various metal candidates such as lithium, magnesium, calcium, zinc and iron, aluminum has been considered as a highly promising energy carrier, given its earth abundance, low price, environmental benignity, high capacity and energy density, and strong recyclability.7–10 The theoretical volumetric capacity of aluminum (8046 mA h cm−3) is approximately four times higher than that of lithium (2062 mA h cm−3), which makes it more attractive for small portable electronics. Although the lithium/air cell possesses the highest theoretical energy density, it is still in the early laboratory development and most reported studies utilize pure oxygen rather than air as the oxidant to prevent possible parasitic reactions.11 Sodium has been explored as an alternative to the costly and scarce lithium but with compromised capacity and energy density.12–14 Recently, Duan's group has broadened the classical metal/air concept to the metalloids and demonstrated high capacity silicon/air and alkaline silicide/air batteries.15,16 Yet these cells exhibit low discharge current densities (≤1 mA cm−2) at normal operation voltages due to the poor conductivity and reactivity of Si and silicide in alkaline. Moreover, these cells still have the anodic self-corrosion problem as in typical alkaline metal/air batteries.Research on Al/air cells has spanned over fifty years since the 1960s and their applications have been limited to niche markets so far.17 The anodic self-corrosion problem is the major challenge faced by traditional Al/air batteries, which increases the hydrogen explosion possibility and causes unacceptably high energy losses.7,8 Researchers have attempted to inhibit the hydrogen generation reaction by alloying aluminum with other elements7,18,19 or modifying the electrolyte using certain additives.20–22 However, these efforts have shown limited success, and oftentimes have increased the material cost and complexity of the battery system. Recently, we demonstrated a hybrid Al/H2/air cell system to turn the parasitic hydrogen-evolution reaction into a beneficial process through hydrogen utilization.23 However, to really solve the hydrogen evolution problem, aluminum oxidation should take place in a nonaqueous environment with high aluminum anode activity whilst reducing the corrosion rate to a low level.24 Licht et al. conducted a series of studies on the electrochemical behavior of aluminum in organic electrolytes and achieved improved aluminum coulombic efficiency.25–28 Other approaches, including employing gel electrolytes, membranes or ionic liquids, have also been proposed to tackle the self-corrosion problem.17,29 Herein we developed a novel dual-electrolyte Al/air cell (DEAAC) with a structure of Al anode | organic electrolyte || anion exchange membrane || aqueous electrolyte | air cathode. In the DEAAC, a nonaqueous anolyte and an aqueous catholyte are separated by an anion exchange membrane so that the aluminum oxidation reaction occurs in an organic-solvent-based alkaline environment. The hydrogen evolution reaction is thus suppressed to a minimal level and a very high cell capacity is achieved. In addition, the dual-electrolyte configuration provides more flexibility in choosing anolyte and catholyte. This primary cell can also be mechanically recharged by replenishing the aluminum anode.
2. Experimental
2.1 Experimental setup
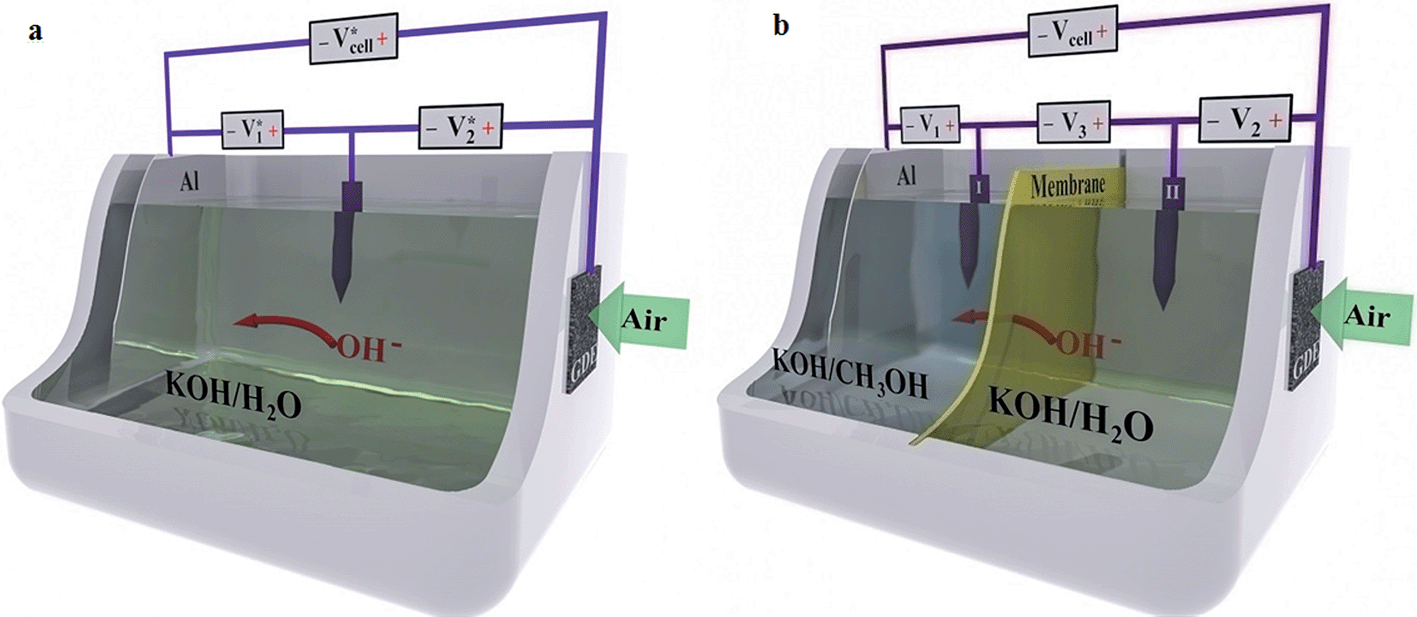 | ||
| Fig. 1 Schematics and experimental setups of two cells (not drawn to scale). (a) Traditional AAC. (b) DEAAC. | ||
2.2 Electrochemical tests
For testing the traditional AAC (Fig. 1a), a three-electrode configuration was used by placing a reference electrode (Hg/HgO/1 M NaOH; 0.14 V vs. SHE at 25 °C) into the electrolyte. For the DEAAC (Fig. 1b), a four-electrode configuration was used by inserting one Hg/HgO reference electrode in the anolyte and the other one in the catholyte. Excess electrolyte (60 mL aqueous electrolyte for the AAC; 30 mL anolyte and 30 mL catholyte for the DEAAC) was injected into the cell to minimize the concentration change due to cell reactions and carbonate formation related to atmospheric carbon dioxide. The DEAAC was left idle for twenty minutes to reach ionic balance across the membrane before inserting the aluminum anode. Aluminum was inserted into the cell immediately before use to avoid self-corrosion under stand-by condition. The current–voltage polarization curves were acquired with steady-state potentiostatic techniques using an electrochemical station (Reference 3000™, Gamry Instruments). The cell potential was stepped from 0 V to the open circuit voltage (VOC) by a 0.2 V increment with each discharge current recorded over three minutes until steady state conditions were reached. The average value of the current data (I–t) was used to represent the cell current at a certain voltage. The anodic polarization, cathodic polarization, and potential drop across membrane were simultaneously recorded in situ, as shown in Fig. 1a and b. The voltage across each individual component was monitored and recorded with digital multimeters (Agilent 34401A). For the AAC, V*1(= ϕAl − ϕRE) is the potential difference between the Al anode and Hg/HgO reference electrode; V*2(= ϕGDE − ϕRE) is the potential difference between the air cathode (GDE) and Hg/HgO reference electrode; V*cell(= ϕGDE − ϕAl = V*2 − V*1) is the potential difference between the cathode and anode or the cell voltage. For the DEAAC, V1(= ϕAl − ϕRE1) is the potential difference between the Al anode and Hg/HgO reference electrode I; V2(= ϕGDE − ϕRE2) is the potential difference between the air cathode (GDE) and Hg/HgO reference electrode II; V3(= ϕRE1 − ϕRE2) is the potential difference between the two reference electrodes I and II across the anion membrane, i.e., the membrane overpotential; Vcell(= ϕGDE − ϕAl = V2 − V1 − V3) is the cell voltage. Each cell capacity was evaluated by galvanostatically discharging the full cell using a two-electrode setup until the aluminum foil was totally consumed and the cell ceased functioning. All tests were conducted under standard ambient temperature and pressure (25 °C and 1 atm).3. Results and discussion
3.1 Polarization and power characteristics
The anodic and cathodic reactions in an Al/air cell are shown as follows:17| Anodic reaction: Al + 4OH− → Al(OH)−4 + 3e−, E0 = −2.4 V vs. Hg/HgO | (1) |
| Cathodic reaction: O2 + 2H2O + 4e− → 4OH−, E0 = 0.3 V vs. Hg/HgO | (2) |
In aqueous media, aluminum will also be consumed through a parasitic reaction to generate hydrogen:
| Parasitic reaction: 2Al + 6H2O + 2OH− → 2Al(OH)−4 + 3H2↑ | (3) |
As shown in Fig. 2a, the benchmark AAC exhibits a linear polarization curve with a VOC of 1.5 V, a short-circuit current density (JSC) of 241 mA cm−2, and an internal resistance of 6.2 Ω cm2 (= ΔV/ΔJ = 1.5 V ÷ 241 mA cm−2). The maximum power density reaches 85 mW cm−2 at 107 mA cm−2. In comparison, the DEAAC has a slightly higher VOC of 1.58 V but lower JSC of 65 mA cm−2 and peak power density of 28 mW cm−2 (Fig. 2b). In addition, the DEAAC has a higher internal resistance of 24.3 Ω cm2 (= 1.58 V ÷ 65 mA cm−2), because of the less-conductive organic electrolyte and increased ionic resistance across the anion membrane.
 | ||
| Fig. 2 Polarization curves and power density plots. (a) Traditional AAC. (b) DEAAC. | ||
In situ single electrode characterization techniques were adopted to study each component of the cells by properly placing Hg/HgO reference electrode(s) into the electrolyte, and the results are shown in Fig. 3. For the AAC (Fig. 3a), both anode and cathode experienced a linear voltage drop with current increase, indicating that electrochemical reactions on both electrodes are dominated by ohmic losses. Over the entire current range, the cathode voltage (V*2) dropped significantly by 1.21 V from 0.06 to −1.15 V, contributing to four-fifth of the overall cell voltage drop with a corresponding ohmic resistance of 5.0 Ω cm2 (= ΔV*2/ΔJ = 1.21 V ÷ 241 mA cm−2). In contrast, the anode voltage (V*1) changed slightly by 0.29 V from −1.44 to −1.15 V, corresponding to an ohmic resistance of 1.2 Ω cm2 (= 0.29 V ÷ 241 mA cm−2). The aluminum anode exhibited, however, a substantial activation loss of ∼1.0 V, considering the aluminum's open circuit potential of −1.44 V and its theoretical potential of −2.4 V. This activation loss is mainly due to the parasitic reaction and the resultant mixed-potential.23 Overall for the AAC, its VOC is mainly limited by the anodic activation loss and its JSC by the cathodic ohmic loss.
 | ||
| Fig. 3 Single-electrode polarization curves. (a) Traditional AAC. (b) DEAAC. Also included is the membrane polarization curve (J–V3). | ||
For the DEAAC case (Fig. 3b), besides potential drops at the anode (V1) and cathode (V2), there is also a potential drop across membrane (V3), which is related to both chemical and electrostatic potentials that drive hydroxide ions to flow through.30,31 The aluminum anode in the DEAAC exhibits a slightly higher open circuit potential of −1.57 V than that in the AAC, but its ohmic overpotential (from −1.57 to −0.57 V) plays a dominant role for the overall cell voltage drop, as opposed to the AAC case (from −1.44 to −1.15 V). V1, V2 and V3 contribute a voltage drop of 1.0, 0.292 and 0.286 V, respectively, over the entire current range. The anodic ohmic resistance of the DEAAC is 15.4 Ω cm2 (= 1.0 V ÷ 65 mA cm−2) (in contrast, the value for the AAC is 1.2 Ω cm2), contributing nearly three-fifth to the overall internal resistance (24.3 Ω cm2). As expected, the cathodic ohmic resistance is 4.5 Ω cm2 (= 0.292 V ÷ 65 mA cm−2), similar to the AAC case. Interestingly, the membrane exhibits a satisfactory ionic conductivity of 4.4 Ω cm2 (= 0.286 V ÷ 65 mA cm−2) so that the membrane overpotential can be relatively small. In fact, by further reducing the membrane thickness (450 μm) to the level of Nafion membranes (50–175 μm), an even lower overpotential V3 and thus better cell performance can be attained. Therefore, the low current and power densities of the DEAAC, compared with the AAC case, were mainly resulted from aluminum's lower electrochemical activity in the organic electrolyte than in the aqueous media25 and the associated higher anodic ohmic loss. Similar dual-electrolyte configuration has been adopted in a Li/air battery.32,33 However, the high internal resistance of the solid lithium-ion conducting electrolyte seriously impairs the conversion efficiency and power density of dual-electrolyte Li/air cells.34
3.2 Discharge characteristics
The discharge curves of traditional AAC (Fig. 4a) and DEAAC (Fig. 4b) reveal that aluminum can exhibit much more efficient coulombic oxidation in organic electrolytes than in aqueous ones, which is in consistent with findings by Licht et al.25,26 In fact, the hydrogen generation was barely observed in the DEAAC, indicating that the parasitic reaction was almost completely suppressed. Wang et al.24 have reported a similar phenomenon that the self-corrosion rate of aluminum in KOH–CH3OH solutions is less than 5% of that in KOH–H2O solutions. Both AAC and DEAAC can maintain flat voltage plateaus, which is consistent with the polarization results in Fig. 2. At a discharge current density of 10 mA cm−2, the AAC achieved a capacity of 89 mA h cm−3 (Fig. 4a), which is only ∼1.1% of aluminum's theoretical capacity. In contrast, the DEAAC exhibited a much superior capacity of 4310 mA h cm−3 at the same current density (see the red curve in Fig. 4b), which is ∼53.6% of the theoretical value. At 20 and 30 mA cm−2, the AAC and DEAAC exhibit capacities of 145 and 195 mA h cm−3, and 4881 and 5952 mA h cm−3, respectively. Although the AAC exhibited a slightly higher voltage at each discharge current density, its coulombic efficiency reaches only 2.4% at 30 mA cm−2, which is significantly lower than the 74.0% efficiency of the DEAAC achieved at the same current density. Even though operating at a higher current density could help the AAC achieve a higher capacity (∼1700 mA h cm−3 at 100 mA cm−2 and a small voltage of ∼0.85 V), its capacity and energy density are still much lower than those of the DEAAC (see Fig. S2 in the ESI† for details). Compared with the AAC case, the DEAAC's anodic capacity increases by 33 and 31 folds at 20 and 30 mA cm−2, respectively. The extraordinary capacity improvement is the major merit of the dual-electrolyte configuration with organic anolyte. Researchers have investigated the aluminum's electrochemical behaviors in alkaline alcohol solutions and found that it undergoes a similar reaction mechanism as in the aqueous alkaline solution but with the parasitic reaction significantly suppressed.24,35,36 This is consistent with our experimental observation that the self-corrosion rate of aluminum in methanol alkali under open circuit condition is only ∼2.5% of that in aqueous alkali (see the ESI† for details). So it is reasonable that the DEAAC has much higher capacities than the AAC. | ||
| Fig. 4 Typical discharge curves at different current densities. (a) Traditional AAC. (b) DEAAC. | ||
With increased discharge current density, Al/air systems (both AAC and DEAAC) achieved higher capacities, as opposed to typical Li-ion batteries whose capacity declines. This is due to the competition between the parasitic reaction (eqn (3)) that consumes electrons to generate hydrogen and the main anodic reaction (eqn (1)) that releases electrons to the external circuit. When discharged at higher current densities, because the aluminum oxidation rate increases dramatically and the hydrogen generation rate increases slightly, a higher proportion of electrons flow to the external circuit. Therefore the cell exhibits higher coulombic efficiencies and capacities at higher current densities. This is consistent with our previous findings23 and other reports35,37 on the aluminum's electrochemical behavior in organic electrolytes.
Throughout the entire discharge process, almost no hydrogen bubbles were observed in the DEAAC while there was a violent hydrogen evolution in the AAC. This further confirms that the high discharge capacity of the DEAAC was due to the suppressed self-discharge reaction in the organic anolyte. We also verified no water or methanol crossover through the membrane with an impermeability test that was conducted by filling one chamber of the DEAAC with corresponding electrolyte while leaving the other one empty; no liquid leakage was found on the dry side of the membrane over 24 hours. A further crossover check was conducted: a droplet of methyl red was added into one chamber to make it yellow, and no yellow color was observed in the other chamber over 48 hours of cell discharge. The absence of crossover can also be confirmed by the discharge curves of the DEAAC shown in Fig. 4b where we see flat voltage plateaus. If there is methanol crossover to the air cathode, a voltage drop will be observed because of the parasitic methanol oxidation reaction on the GDE cathode. If there is water crossover to the anode, a voltage rise will be expected under the galvanostatic discharge since the aluminum has higher reaction rate in the aqueous solution.
3.3 Performance comparison of the DEAAC with other metal/air cells
The performance of the DEAAC was compared with that of reported high-performance metal/air cells (Table 1). The DEAAC has a gravimetric energy density of 2081 W h kg−1, which is over four folds higher than that of aqueous alkaline Al/air cells (300–500 W h kg−1) reported in the literature.8,38 Saline Al/air cells have lower corrosion rates than alkaline Al/air cells, and can achieve an energy density up to 800 W h kg−1 which is still much lower than that of the DEAAC.39 In fact, due to the low solubility of Al(OH)3 in the neutral electrolyte (e.g., 12 wt% sodium chloride solution), a large amount of electrolyte is required to avoid Al(OH)3 precipitation and consequently the system energy density of saline cells could be even lower than that of alkaline cells.17 With high energy densities, zinc/air batteries are so far the only commercialized metal/air devices for niche markets, for example, hearing aids. Dai's group40 in 2013 reported a Zn/air cell that has a gravimetric energy density of 741 W h kg−1 with advanced hybrid electrocatalysts and outperforms most commercially available Zn/air batteries (200–300 W h kg−1).38,41 Compared to Dai's Zn/air cell,40 the DEAAC exhibits three-fold gravimetric capacity and energy density, and has slightly higher volumetric capacity and energy density. Recently, silicon has emerged as a new anode candidate given its high theoretical capacity (3817 mA h g−1 and 8890 mA h cm−3) and energy density (8359 W h kg−1 and 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 469 W h L−1) (see Table S1 in ESI†).15,16,42–44 Silicon undergoes a similar reaction mechanism as aluminum and zinc in alkaline electrolytes. The DEAAC outperforms the reported high-capacity alkaline Si/air cell15 in terms of energy density (both gravimetric and volumetric) and discharge current density (10–30 mA cm−2 vs. 0.01–0.1 mA cm−2); its volumetric capacity (5950 mA h cm−3 at 30 mA cm−2) is more than twice as high as that of the Si/air cell (2809 mA h cm−3 at 0.1 mA cm−2). As Li-ion batteries are approaching their maximum potential energy density and are unable to meet the long-term needs, researchers have recently paid tremendous efforts to Li/air cells owing to their considerably 3–5 times higher anodic gravimetric energies than Li-ion batteries.45 Jung et al.46 have developed an improved rechargeable Li/air cell. After detailed calculations (see ESI†), the cell discharge current density is actually 0.5 mA cm−2 at 2.7 V with a limited anodic capacity of 234 mA h cm−3 (i.e., 11% of the lithium's theoretical capacity). The results of this Li/air cell are actually consistent with the well-known statement47 that current densities (0.05–1 mA cm−2) of Li/air cells so far are too low for most of practical applications. Although this Li/air cell is rechargeable, the DEAAC exhibits at least one order of magnitude higher capacities and energy densities and its anode can be mechanically replaced. Hayashi et al.14 have investigated a mixed aqueous/aprotic Na/air primary cell that has a similar cell structure as the DEAAC (see Table 1). Compared to the DEAAC, the Na/air cell has a higher VOC (2.85 V vs. 1.58 V), but exhibits a remarkably lower peak power density (5 mW cm−2 vs. 28 mW cm−2). While the Na/air cell exhibits a similar gravimetric energy density as the DEAAC, its volumetric energy density and capacity are much lower than those of the DEAAC. The authors attribute the cell's inferior performance to the high internal resistance of ∼440 Ω associated mainly with the NASICON ceramic membrane (65 Ω) and the aqueous electrolyte (200 Ω).14 In contrast, with the highly conductive polymer membrane and high-concentration KOH electrolyte, the DEAAC has a much lower internal resistance of ∼24.3 Ω and hence has significantly less power and energy losses. Note that more than enough electrolyte has been used in the DEAAC to make sure that Al(OH)3 will not precipitate. Also, the weight of the electrolyte and the cell case has not been taken into consideration (which will reduce the overall energy density of course). So in our future work the amount of electrolyte will be optimized so that an appropriate evaluation of the overall cell energy density may be achieved.
469 W h L−1) (see Table S1 in ESI†).15,16,42–44 Silicon undergoes a similar reaction mechanism as aluminum and zinc in alkaline electrolytes. The DEAAC outperforms the reported high-capacity alkaline Si/air cell15 in terms of energy density (both gravimetric and volumetric) and discharge current density (10–30 mA cm−2 vs. 0.01–0.1 mA cm−2); its volumetric capacity (5950 mA h cm−3 at 30 mA cm−2) is more than twice as high as that of the Si/air cell (2809 mA h cm−3 at 0.1 mA cm−2). As Li-ion batteries are approaching their maximum potential energy density and are unable to meet the long-term needs, researchers have recently paid tremendous efforts to Li/air cells owing to their considerably 3–5 times higher anodic gravimetric energies than Li-ion batteries.45 Jung et al.46 have developed an improved rechargeable Li/air cell. After detailed calculations (see ESI†), the cell discharge current density is actually 0.5 mA cm−2 at 2.7 V with a limited anodic capacity of 234 mA h cm−3 (i.e., 11% of the lithium's theoretical capacity). The results of this Li/air cell are actually consistent with the well-known statement47 that current densities (0.05–1 mA cm−2) of Li/air cells so far are too low for most of practical applications. Although this Li/air cell is rechargeable, the DEAAC exhibits at least one order of magnitude higher capacities and energy densities and its anode can be mechanically replaced. Hayashi et al.14 have investigated a mixed aqueous/aprotic Na/air primary cell that has a similar cell structure as the DEAAC (see Table 1). Compared to the DEAAC, the Na/air cell has a higher VOC (2.85 V vs. 1.58 V), but exhibits a remarkably lower peak power density (5 mW cm−2 vs. 28 mW cm−2). While the Na/air cell exhibits a similar gravimetric energy density as the DEAAC, its volumetric energy density and capacity are much lower than those of the DEAAC. The authors attribute the cell's inferior performance to the high internal resistance of ∼440 Ω associated mainly with the NASICON ceramic membrane (65 Ω) and the aqueous electrolyte (200 Ω).14 In contrast, with the highly conductive polymer membrane and high-concentration KOH electrolyte, the DEAAC has a much lower internal resistance of ∼24.3 Ω and hence has significantly less power and energy losses. Note that more than enough electrolyte has been used in the DEAAC to make sure that Al(OH)3 will not precipitate. Also, the weight of the electrolyte and the cell case has not been taken into consideration (which will reduce the overall energy density of course). So in our future work the amount of electrolyte will be optimized so that an appropriate evaluation of the overall cell energy density may be achieved.
| DEAAC | Zn/air40 | Si/air15 | Li/air46 | Na/air14 | |
|---|---|---|---|---|---|
| a Capacity and energy density values are based on the anode alone. All data is reported on primary cells expect the Li/air that is rechargeable. Detailed derivations are included in the ESI. | |||||
| Cell structure | (See Fig. 1b) | Zn | 6 M aqueous KOH | O2 | Si nanowire | 0.6 M aqueous KOH | air | Li | TEGDME–LiCF3SO3 | O2 | Na | organic electrolyte || ceramic membrane || aqueous NaOH | O2 |
| Discharge voltage (V) | 1.15 | 1.3 | 0.85 | 2.7 | 2.5 |
| Discharge current density (mA cm−2) | 20 | 10 | 0.1 | 0.5 (500 mA gcarbon−1) | 0.63 |
| Gravimetric capacity (mA h g−1) | 1810 | 570 | 1206 | 234 (5000 mA h gcarbon−1) | 835 |
| Volumetric capacity (mA h cm−3) | 4886 | 4070 | 2809 | 125 | 808 |
| Gravimetric energy density (W h kg−1) | 2081 | 741 | 1025 | 631.8 | 2087 |
| Volumetric energy density (W h L−1) | 5819 | 5291 | 2387 | 337 | 2020 |
With enough electrolytes, the DEAAC could run at the same performance level by simply replenishing the aluminum anode. Recently, an Al/air battery system has been demonstrated to power electric vehicles with driving ranges and acceleration similar to gasoline powered cars.38,48 The battery system can be quickly refueled with new aluminum plates and fresh electrolyte. Our DEAAC is suitable for such a mechanically rechargeable battery system. By increasing the aluminum effective area to fully utilize the GDE's current capacity and properly connecting cells into stack, we believe the DEAAC also promises to be a reliable and scalable stationary power backup.
4. Conclusions
In summary, we investigated a novel high-capacity dual-electrolyte Al/air semi-fuel cell with an anion exchange membrane separating the organic anolyte and the aqueous catholyte. The dual-electrolyte structure is a novel approach to solving the parasitic self-corrosion problem in traditional alkaline metal/air batteries. The DEAAC successfully suppressed the parasitic reaction and hence achieved superior anodic capacities compared with traditional alkaline Al/air cells. The measured anode capacity of the DEAAC (6000 mA h cm−3 at 30 mA cm−2) is even greater than the lithium's theoretical capacity (2060 mA h cm−3). The DEAAC shows a high energy density of 2081 W h kg−1 (or 5819 W h L−1). The DEAAC can be mechanically rechargeable and has more flexibility in choosing appropriate anolyte, catholyte and membrane. With all these features, we believe that the DEAAC is a promising electrochemical power source in various applications such as electric vehicles and power backup.Acknowledgements
The authors thank the financial support from the Seed Funding Programme for Basic Research at HKU (Project codes 200910159016 and 201211159091), the University Development Fund (UDF) 2009–2010 (Second Round), and the HKU Initiative on Clean Energy & Environment (HKU-ICEE). We thank Frank Tse in the Department of Mechanical Engineering at HKU for helping with the cell fabrication using rapid prototyping. Also, we would like to give special thanks to Xiaochen Ren in the Department of Mechanical Engineering at HKU for helping with the 3D drawings of cells.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 CAS.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS.
- T. B. Reddy, Linden's Handbook of Batteries, McGraw-Hill, 2011, p. 33.31–33.32 Search PubMed.
- Q. F. Li and N. J. Bjerrum, J. Power Sources, 2002, 110, 1–10 CrossRef CAS.
- E. I. Shkolnikov, A. Z. Zhuk and M. S. Vlaskin, Renewable Sustainable Energy Rev., 2011, 15, 4611–4623 CrossRef CAS PubMed.
- W. Y. Li, C. S. Li, C. Y. Zhou, H. Ma and J. Chen, Angew. Chem., Int. Ed., 2006, 45, 6009–6012 CrossRef CAS PubMed.
- B. T. Hang, T. Watanabe, M. Egashira, I. Watanabe, O. Shigeto and J. Yamaki, J. Power Sources, 2006, 155, 461–469 CrossRef CAS PubMed.
- J. S. Lee, S. T. Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 CAS.
- P. Hartmann, C. L. Bender, M. Vracar, A. K. Durr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS PubMed.
- K. Hayashi, K. Shima and F. Sugiyama, J. Electrochem. Soc., 2013, 160, A1467–A1472 CrossRef CAS PubMed.
- X. Zhong, H. Zhang, Y. Liu, J. W. Bai, L. Liao, Y. Huang and X. F. Duan, ChemSusChem, 2012, 5, 177–180 CrossRef CAS PubMed.
- H. Zhang, X. Zhong, J. C. Shaw, L. Liu, Y. Huang and X. Duan, Energy Environ. Sci., 2013, 6, 2621–2625 CAS.
- D. R. Egan, C. Ponce de León, R. J. K. Wood, R. L. Jones, K. R. Stokes and F. C. Walsh, J. Power Sources, 2013, 236, 293–310 CrossRef CAS PubMed.
- D. Macdonald, K. Lee, A. Moccari and D. Harrington, Corrosion, 1988, 44, 652–657 CrossRef CAS.
- M. Doche, F. Novel-Cattin, R. Durand and J. Rameau, J. Power Sources, 1997, 65, 197–205 CrossRef CAS.
- A. Abdel-Gaber, E. Khamis, H. Abo-ElDahab and S. Adeel, Mater. Chem. Phys., 2008, 109, 297–305 CrossRef CAS PubMed.
- O. K. Abiola and J. Otaigbe, Corros. Sci., 2009, 51, 2790–2793 CrossRef CAS PubMed.
- T. Hirai, J. Yamaki, T. Okada and A. Yamaji, Electrochim. Acta, 1985, 30, 61–67 CrossRef CAS.
- L. Wang, W. Wang, G. Yang, D. Liu, J. Xuan, H. Wang, M. K. H. Leung and F. Liu, Int. J. Hydrogen Energy, 2013, 38, 14801–14809 CrossRef CAS PubMed.
- J.-B. Wang, J.-M. Wang, H.-B. Shao, J.-Q. Zhang and C.-N. Cao, J. Appl. Electrochem., 2007, 37, 753–758 CrossRef CAS PubMed.
- S. Licht, G. Levitin, C. Yarnitzky and R. Tel-Vered, Electrochem. Solid-State Lett., 1999, 2, 262–264 CrossRef CAS PubMed.
- S. Licht, R. Tel-Vered, G. Levitin and C. Yarnitzky, J. Electrochem. Soc., 2000, 147, 496–501 CrossRef CAS PubMed.
- S. Licht, G. Levitin, R. Tel-Vered and C. Yarnitzky, Electrochem. Commun., 2000, 2, 329–333 CrossRef CAS.
- G. Levitin, C. Yarnitzky and S. Licht, Electrochem. Solid-State Lett., 2002, 5, A160–A163 CrossRef CAS PubMed.
- Z. Zhang, C. Zuo, Z. Liu, Y. Yu, Y. Zuo and Y. Song, J. Power Sources, 2013, 251, 470–475 CrossRef PubMed.
- V. M. Aguilella, S. Mafe, J. A. Manzanares and J. Pellicer, J. Membr. Sci., 1991, 61, 177–190 CrossRef CAS.
- E. Volodina, N. Pismenskaya, V. Nikonenko, C. Larchet and G. Pourcelly, J. Colloid Interface Sci., 2005, 285, 247–258 CrossRef CAS PubMed.
- L. J. Li, X. S. Zhao and A. Manthiram, Electrochem. Commun., 2012, 14, 78–81 CrossRef CAS PubMed.
- J. P. Zheng, P. Andrei, M. Hendrickson and E. J. Plichta, J. Electrochem. Soc., 2011, 158, A43–A46 CrossRef CAS PubMed.
- L. J. Li and A. Manthiram, J. Mater. Chem. A, 2013, 1, 5121–5127 CAS.
- H. Shao, J. Wang, X. Wang, J. Zhang and C. Cao, Electrochem. Commun., 2004, 6, 6–9 CrossRef CAS PubMed.
- X. Chang, J. Wang, H. Shao, J. Wang, X. Zeng, J. Zhang and C. Cao, Acta Phys.-Chim. Sin., 2008, 24, 1620–1624 CrossRef CAS.
- J. B. Wang, J. M. Wang, H. B. Shao, X. T. Chang, L. Wang, J. Q. Zhang and C. N. Cao, Mater. Corros., 2009, 60, 269–273 CrossRef CAS.
- S. H. Yang and H. Knickle, J. Power Sources, 2002, 112, 162–173 CrossRef CAS.
- T. B. Reddy, Linden's Handbook of Batteries, McGraw-Hill, 2011, p. 33.30–33.32 Search PubMed.
- Y. G. Li, M. Gong, Y. Y. Liang, J. Feng, J. E. Kim, H. L. Wang, G. S. Hong, B. Zhang and H. J. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- G. Cohn, A. Altberg, D. D. Macdonald and Y. Ein-Eli, Electrochim. Acta, 2011, 58, 161–164 CrossRef CAS PubMed.
- G. Cohn and Y. Ein-Eli, J. Power Sources, 2010, 195, 4963–4970 CrossRef CAS PubMed.
- G. Cohn, D. Starosvetsky, R. Hagiwara, D. D. Macdonald and Y. Ein-Eli, Electrochem. Commun., 2009, 11, 1916–1918 CrossRef CAS PubMed.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 CAS.
- H. G. Jung, J. Hassoun, J. B. Park, Y. K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef CAS PubMed.
- A. Kraytsberg and Y. Ein-Eli, J. Power Sources, 2011, 196, 886–893 CrossRef CAS PubMed.
- B. Yirka, http://phys.org, http://phys.org/news/2013-03-phinergy-aluminum-air-battery-capable-fueling.html, accessed on 24 April, 2013.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05222f |
| This journal is © The Royal Society of Chemistry 2014 |