
Synthesis, characterization and antimicrobial studies of some new trifluoromethyl quinoline-3-carbohydrazide and 1,3,4-oxadiazoles†
Abstract
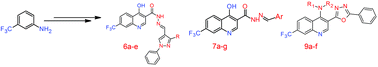
The present paper describes the synthesis of two new series of 7-(trifluoromethyl)-4-hydroxy substituted quinoline carbohydrazide derivatives (6a–e and 7a–g) and N-alkyl-3-(5-phenyl-1,3,4-oxadiazol-2-yl)-7-(trifluoromethyl) quinolin-4-amine derivatives (9a–f). Newly synthesized compounds were characterized by spectral studies. The structure of 9a was evidenced by X-ray crystallographic study. Synthesized compounds were screened for their antibacterial performance against Mycobacterium smegmatis and Pseudomonas aeruginosa. Antifungal activity was also carried out on the fungal stains Candida albicans and Penicillium chrysogenum. Compounds 7a and 9c showed significant antimicrobial activity against all the tested microorganisms. Among all the compounds, 6d and 6e showed the lowest MIC value of 6.25 μg mL−1 against Mycobacterium smegmatis indicating these compounds can be possible future antituberculosis agents.
 Please wait while we load your content...
Please wait while we load your content...

