
Effect of ligand binding on a protein with a complex folding landscape†
 a
Gilad
Haran
*a
a
Gilad
Haran
*a
Abstract
Ligand binding to a protein can stabilize it significantly against unfolding. The variation of the folding free energy, ΔΔG0, due to ligand binding can be derived from a simple reaction scheme involving exclusive binding to the native state. One obtains the following expression: 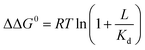 , where Kd is the ligand dissociation constant and L is its concentration, R is the universal gas constant and T is the temperature. This expression has been shown to correctly describe experimental results on multiple proteins. In the current work we studied the effect of ligand binding on the stability of the multi-domain protein adenylate kinase from E. coli (AKE). Unfolding experiments were conducted using single-molecule FRET spectroscopy, which allowed us to directly obtain the fraction of unfolded protein in a model-free way from FRET efficiency histograms. Surprisingly, it was found that the effect of two inhibitors (Ap5A and AMPPNP) and a substrate (AMP) on the stability of AKE was much smaller than expected based on Kd values obtained independently using microscale thermophoresis. To shed light on this issue, we measured the Kd for Ap5A over a range of chemical denaturant concentrations where the protein is still folded. It was found that Kd increases dramatically over this range, likely due to the population of folding intermediates, whose binding to the ligand is much weaker than that of the native state. We propose that binding to folding intermediates may dominate the effect of ligands on the stability of multi-domain proteins, and could therefore have a strong impact on protein homeostasis in vivo.
, where Kd is the ligand dissociation constant and L is its concentration, R is the universal gas constant and T is the temperature. This expression has been shown to correctly describe experimental results on multiple proteins. In the current work we studied the effect of ligand binding on the stability of the multi-domain protein adenylate kinase from E. coli (AKE). Unfolding experiments were conducted using single-molecule FRET spectroscopy, which allowed us to directly obtain the fraction of unfolded protein in a model-free way from FRET efficiency histograms. Surprisingly, it was found that the effect of two inhibitors (Ap5A and AMPPNP) and a substrate (AMP) on the stability of AKE was much smaller than expected based on Kd values obtained independently using microscale thermophoresis. To shed light on this issue, we measured the Kd for Ap5A over a range of chemical denaturant concentrations where the protein is still folded. It was found that Kd increases dramatically over this range, likely due to the population of folding intermediates, whose binding to the ligand is much weaker than that of the native state. We propose that binding to folding intermediates may dominate the effect of ligands on the stability of multi-domain proteins, and could therefore have a strong impact on protein homeostasis in vivo.

- This article is part of the themed collection: Complex molecular systems: supramolecules, biomolecules and interfaces
 Please wait while we load your content...
Please wait while we load your content...

