
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing ene-type and stereochemistry to control reaction kinetics and network architecture in thiol–ene photopolymerizations using maleate and fumarate-derived monomers†
Rithwik
Ghanta
,
Ayaulym
Abilova
,
Cade
McAndrew
and
Alexa S.
Kuenstler
 *
*
Department of Chemical and Biomolecular Engineering, University of Illinois at, Urbana-Champaign, Urbana, IL 61801, USA. E-mail: akuenstl@illinois.edu
First published on 18th March 2025
Abstract
Herein we report photo-dose tunable crosslinking density in polymer networks by exploiting the relative rates of thiol–ene click chemistry and chain-growth homopolymerization in symmetric triene monomers. From biomass-derived diacids, these synthesized trienes incorporate terminal allyl ether groups and internal fumarate/maleate groups, providing varied reactivity. Through small-molecule monothiol addition, 1H-NMR results indicate fast preferential thiol addition to terminal allyl groups and slower stereochemistry-dependent homopolymerization of fumarate/maleate groups. Incorporating these monomers with dithiols and triallyl crosslinkers allows formation of polymer networks, using both thiol–ene addition and homopolymerization as photo-crosslinking mechanisms on differing timescales. In situ photo-rheology and dynamic mechanical analysis demonstrate impacts of the mixed-mechanism on light-dependent evolution of network architectures from initial gelation to increasing crosslinking density with prolonged exposure. Ultimately, the mixed-mechanism polymerization enables grayscale patterning and 3D printing, offering potential for in situ patterning of glasslike and rubbery regions within monolithic materials.
Introduction
Photopolymerized networks are widely used in diverse applications including dental materials,1,2 holographic materials,3,4 additive manufacturing,5–7 and microelectronics.8,9 These materials are most frequently made via chain-growth, free-radical polymerization of unsaturated functional groups including acrylates and vinyls.10 In these polymerizations, radical generation induces localized functional group conversion into ‘microgels’ of high crosslink density followed by coalescence of gelled regions into a network that spans the whole reaction media.11–13 This results in a spatially heterogeneous microstructure containing a distribution of both elastically active crosslinks that provide mechanical reinforcement and topological defects such as loops that nucleate cracks under load.14–16 Furthermore, molecular oxygen can inhibit free-radical polymerization by forming stable peroxy-radicals that do not re-initiate polymerization,17–19 leading to complicated kinetics and the persistence of unreacted monomers that can both compromise material mechanics and pose a leaching hazard. While living free-radical polymerizations can address some of these challenges,20–23 these can require stringent synthetic procedures and are known to result in the phase separation of polymers into complicated microstructures.24 Conversely, thiol–ene photopolymerizations suppress gelation until high monomer conversions by exploiting chain-transfer reactions across active carbon radicals produced by thiol addition.25 In the case of multi-functional thiols and alkenes (referred to commonly as “enes”), the regular alternation between propagation and chain transfer results in step-growth networks that produce significantly more uniform network structures compared to free-radical approaches.26–29Combining mixed chain-growth and step-growth mechanisms can produce networks with properties that are unachievable by either mechanism independently, where the relative rates of chain-growth and step-growth prescribe the final network structure and resulting bulk properties. This scheme is typically pursued via copolymerization between a thiol and either (1) an ene functionality, such as a (meth)acrylate,30 that can participate in both a thiol–ene step-growth polymerization and chain-growth homopolymerization; or (2) two distinct ene functionalities,31,32 one that can exclusively polymerize via a thiol–ene mechanism and one that can participate in both step- and chain-growth processes. In both cases, kinetic and coarse grain models33–35 and complementary experiments have revealed that small changes in monomer combinations translate into differences in gelation, crosslink density, and network structure. For example, the combination of thiol–acrylate step-growth reactions with acrylate homopolymerization can reduce oxygen inhibition and increase network homogeneity, resulting in a drastically narrowed glass transition temperature and decreased shrinkage stress.36–40 Ternary photopolymerization where purely step-growth monomers are added to thiol–acrylate systems such as allyl28 and vinyl ethers31,41,42 can further expand the accessible property space to control network architecture via the relative rates of step- and chain-growth polymerization.43 This presents the opportunity to control material property development in time and space, where control over network structure enables programming of bulk mechanical properties over expanded timescales compared to typical systems. The power of mixed-mechanism photopolymerizations is further evidenced by recent work beyond classic thiol–ene systems, where hybrid cationic/radical polymerizations44–46 and (semi) orthogonal mechanisms47–51 have enabled patterning of material properties across length scales.
To date, work on mixed-mechanism thiol–ene reactions has overwhelmingly focused on combining distinct functionalities on separate monomers. However, combining two different ene functionalities within a single reactant provides opportunities to further tune reaction kinetics and the resulting network structure, while reducing unreacted species within the final material. For example, Hoyle and Guymon showed that copolymerization of a thiol with a vinyl acrylate monomer mediated two separate free-radical processes with kinetics that strongly deviated from classic vinyl or acrylate homopolymerizations.52 Futhermore, incorporating 1,2-disubstituted enes is known to further augment reaction kinetics due to a combination of steric and electronic effects. In particular, internal 1,2-disubstituted enes have been shown to polymerize more sluggishly than terminal counterparts due to sterics and competing reversible thiol addition to less stable isomers.53,54 Finally, an additional motivation to understand the differential reactivity of multiple unsaturated units within the same monomer is to harness bio-based monomers for sustainable photopolymers.55 For example, natural products such as sugars and terpenes can be incorporated within thiol–ene networks,56–58 and are promising feedstocks to replace petroleum-derived monomers for more sustainable additive manufacturing.59–63
In this work, we demonstrate how the chemical functionality and stereochemistry of trienes can be harnessed to control polymerization kinetics and network structure. Specifically, using diallyl-functionalized maleate and fumarate-containing monomers, we show how reversible thiyl addition to internal enes mediates the relative rates of chain-growth and step-growth polymerization. These mechanistic changes are found to translate into differences in both the rate of gelation on short timescales and continued crosslinking on long timescales, enabling judicious control of thermomechanical properties via the extent of light exposure. Furthermore, we show that these kinetics can be tuned further with the thiol comonomer identity. Lastly, we demonstrate that these materials are compatible with digital light processing (DLP) additive manufacturing and offer insights into how the unique polymerization kinetics of these materials can be harnessed for future light-controlled manufacturing processes.
Experimental
Materials and chemicals
Diallyl maleate, diethyl maleate, hexyl mercaptan, methyl thioglycolate, methyl-3-mercaptopropionate, 1,6-hexanedithiol, ethylene bis(thioglycolate), and ethylene glycol bis(3-mercaptopropionate) were purchased from TCI Chemicals. Allyl alcohol and 1,3,5-triallyl-1,3,5-triazine-2,4,6-trione (TATO) were purchased from Sigma Aldrich. All chemicals were used as received. Common solvents used included toluene, ethyl acetate, dichloromethane, and hexanes. Diethyl fumarate, diallyl succinate, diallyl trans-3-hexendioate, and diallyl fumarate were synthesized through Fischer esterification of diacids as described below. The deuterated solvent CDCl3 was obtained from Cambridge Isotope Laboratories Inc. and dried over molecular sieves prior to use.Methods
 .
.
Results and discussion
Reactivity studies of small molecules
To understand the relative reactivities of terminal allyl groups and internal “enes” within a single monomer, we first considered the reaction of such molecules with commercial monofunctional thiols. Specifically, we chose maleic and fumaric monomers as model systems, which can be accessed from biomass66 and a one-step functionalization with allyl alcohol could be used to access trienes with different stereochemistry (Fig. 1A). A library of candidate monomers was synthesized via esterification of the appropriate diacid with allyl alcohol in toluene using an organic acid catalyst (see the Materials and Methods sections for more detailed discussions). Following purification, diallyl maleate (DM), and diallyl fumarate (DF) were successfully isolated with high purity as confirmed by NMR (Fig. S1-A-D†). As controls, the ethyl-terminated analogs diethyl maleate (DEM) and diethyl fumarate (DEF) were also prepared to isolate the role of allyl groups in photoreactions. Finally, diallyl succinate (DS) and diallyl trans-3-hexendioate (DT3HD) were designed and synthesized to probe the effect of the fumarate/maleate structure and stereochemistry on reaction kinetics. | ||
| Fig. 1 Molecular pool used for this study: (A) triene small molecules with colors and symbols used throughout the manuscript: diethyl fumarate (DEF), diethyl maleate (DEM), diallyl succinate (DS), diallyl maleate (DM), diallyl fumarate (DF); (B) mono- and di-thiols: hexyl mercaptan (HM), methyl thioglycolate (MTG), methyl 3-mercaptopropionate (M3M), 1,6-hexanedithiol (HDT), ethylene glycol bis(thioglycolate) (ETG), ethylene glycol bis(3-mercaptopropionate) (E3M); (C) 1,3,5-triallyl-1,3,5-triazine-2,4,6-trione (TATO) as the triallyl crosslinker and diphenyl(2,4,6-trimethylbenzoyl) phosphine oxide (TPO) as the photoinitiator. | ||
Model thiol–ene reactions between each unsaturated monomer were performed using 1-hexane thiol and 0.5 wt% diphenyl(2,4,6-trimethylbenzoyl) phosphine oxide (TPO) as the photoinitiator. We note that while TPO has raised cytotoxicity concerns in consumer products,67 we anticipate that many non-cytotoxic photoinitiators can easily be used as effective and safe replacements.68 Briefly, samples were mixed in defined ratios and irradiated with 405 nm light at 5 mW cm−2. Aliquots were removed at defined time points, mixed with CDCl3, and subjected to 1H NMR to monitor functional group conversion. Conversion as a function of time for each formulation is shown in Fig. 2A (Fig. 2B shows representative raw NMR spectra and the corresponding peak assignments; for all other spectra, see Fig. S2-D1†). We found that while allyl groups are rapidly and near-quantitatively consumed within ≈30 s for DS as expected for a pure thiol–ene addition,32,69,70 introduction of fumarate and maleate groups in DM and DF both slows allyl group consumption and limits overall conversion to ≈85% (Fig. 2A, left). These data suggest that ≈15% of the thiol adds to fumarate and maleate functionalities, thus reducing allyl conversion in the absence of cross-reactions between ene groups or allyl homopolymerization.71 Corresponding measurements of fumarate and maleate conversion reveal significant conversion that competes with allyl consumption on longer timescales (Fig. 2B). While these functional groups are expected to react with thiols via a step-growth thiol–ene pathway,69,72–75 as shown by diethyl control experiments with DEF and DEM (Fig. 2A, right, blue and gold symbols), the total ene conversion for both DF and DM normalized to available thiol (1.38 and 1.06, respectively) exceeds that of available thiol reactants, suggesting that chain-growth (i.e., ‘homopolymerization’) of fumarate and maleate groups occurs in addition to the step-growth thiol–ene mechanism. Furthermore, the presence of fumarate and maleate homopolymerization is supported by the formation of polymeric species as measured by SEC (Fig. 2C), as the addition of monofunctional thiols can only result in small molecule adducts per step-growth statistics. Importantly, control experiments show that carbonyl proximity to the internal ene is essential for this homopolymerization, as total ene conversion of DT3HD is consistent with quantitative thiol consumption (Fig. S2-A1†).
 | ||
Fig. 2 (A) 1H NMR conversion of allyl (left) and maleate/fumarate (right) functional groups in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio* photoreactions shows that consumption of maleate/fumarate groups occurs along with thiol addition to allyl ethers. The solid-line fits to first order kinetics are shown for pure thiol–ene mechanisms and omitted for others for clarity; see the ESI† for other fits. (B) The extent of photoreaction is monitored via integration of characteristic peaks of the 1H NMR spectra, as shown here for DS used in a 1:1 ratio* photoreaction from 0–1800 s. Peaks corresponding to the terminal carbon of 1-hexanethiol was used for normalization. (C) SEC traces of 1:1 ratio* photoreactions of DM and DF after 30 minutes reveal formation of higher molecular-weight adducts, suggesting homopolymerization of maleate/fumarate groups. Molecular weights are estimates determined from polystyrene standards. *The ratio is allyl:thiol or fumarate/maleate:thiol for DEF and DEM. :1 ratio* photoreactions shows that consumption of maleate/fumarate groups occurs along with thiol addition to allyl ethers. The solid-line fits to first order kinetics are shown for pure thiol–ene mechanisms and omitted for others for clarity; see the ESI† for other fits. (B) The extent of photoreaction is monitored via integration of characteristic peaks of the 1H NMR spectra, as shown here for DS used in a 1:1 ratio* photoreaction from 0–1800 s. Peaks corresponding to the terminal carbon of 1-hexanethiol was used for normalization. (C) SEC traces of 1:1 ratio* photoreactions of DM and DF after 30 minutes reveal formation of higher molecular-weight adducts, suggesting homopolymerization of maleate/fumarate groups. Molecular weights are estimates determined from polystyrene standards. *The ratio is allyl:thiol or fumarate/maleate:thiol for DEF and DEM. | ||
While fumarate groups are established to homopolymerize by a free-radical mechanism,76 this pathway is typically suppressed in maleate groups except in the presence of an isomerization catalyst.77–80 These catalysts, usually based on amines,81–83 enable polymerization by catalyzing cis–trans isomerization of maleate to the active fumarate followed by chain propagation. However, our NMR studies reveal that maleates are capable of homopolymerization in the presence of thiols, albeit at a slower rate and with reduced conversion compared to the trans fumarate group (Fig. 2A, right). This suggests that thiyl radicals play a key role in facilitating homopolymerization of maleate groups. Thiyl radicals are known to facilitate isomerization of cis unsaturated units via an addition–elimination pathway that yields a new, energetically favorable trans stereoisomer (Fig. 3C).70,84–86 This isomerization phenomenon is observed in this system as evidenced by the simultaneous disappearance of characteristic 1H NMR peaks indicating maleate consumption and the appearance of downfield peaks corresponding to fumarate. Time-resolved experiments show that these isomerization events precede the subsequent internal ene conversion, while the reverse trans-to-cis isomerization is not observed (Fig. S2-D†). The importance of this pathway in the addition of thiol to the internal ene is further demonstrated by control experiments with DEM, where isomerization into DEF occurs before any significant bond conversion and thiol adduct formation (Fig. 3B). Furthermore, in DM systems, although cis content reaches a steady state when the overall allyl conversion plateaus and the thiol reactant is presumably consumed, trans content slowly continues to decrease over longer times (Fig. 3A). Collectively, these data suggest that homopolymerization of the diallyl maleate follows an isomerization–polymerization mechanism where reversible thiyl radical addition mediates maleate isomerization to the active fumarate that can homopolymerize, while the associated cis–trans equilibria significantly slows the polymerization rate.
 | ||
| Fig. 3 (A) Conversion of internal cis maleate is higher than total internal ene conversion as trans fumarate is formed. This isomerization of DM to DF is observed during photoreactions with thiols and is followed by conversion of DF, suggesting an isomerization–polymerization mechanism that drives maleate homopolymerization. (B) Control experiments show that thiol catalyzes the transformation of DEM into DEF via a thiol addition–elimination reaction. All points are normalized to total internal ene functionality. (C) The isomerization–polymerization mechanism. | ||
We next turned our attention to understanding how thiol structure impacts these reactions given the well-established effect of thiol type on the relative rates of chain-transfer and propagation in thiol–ene polymerizations.87–89 To determine the effect of thiol structure on reactivity in these systems, we repeated the same photoreactions with methyl thioglycolate (MTG) and methyl 3-mercaptopropionate (M3M) in addition to 1-hexane thiol (HT) (see Fig. S2A-I† for structures and corresponding raw data). Furthermore, reactions with M3M were monitored by real time Fourier transform infrared (FTIR) spectroscopy to corroborate quantitative thiol conversions and similar conversions of ene functionalities (Fig. S2-I†). Photoreactions with M3M resulted in both higher extents and rates of reactions for all functionalities including the cis maleate of DM, implying that faster thiol addition across this group increases cis–trans isomerization, therefore leading to a higher overall conversion of the disubstituted cis ene (Fig. 4). This is consistent with precedents in the literature, which suggest that the increase in thiol reactivity is due to the weakening of the S–H bond from hydrogen bonding to the carbonyl group and a corresponding increase in chain transfer rate.90,91 However, reactions with MTG are shown to be slower than the reactions with M3M or HT despite a similar hydrogen bonding effect. This can be partially explained by having a comparatively low thiyl radical stability to M3M that slows chain transfer,92 which is the rate limiting step for thiol-addition to allyl, fumarate, and maleate functional groups.33 Nevertheless, trends of thiol reactivity are self-consistent among ene functionalities. Specifically, we found that higher thiol reactivity catalyzes an increased rate of allyl consumption, which is accompanied by an accelerated rate and extent of internal ene consumption (Fig. 4). These observations highlight the role of thiols in the homopolymerization mechanism of diallyl maleate and fumarate, and suggest that higher thiol reactivity will act to both increase the rate of propagation of radicals on the trans fumarate and rate of cis–trans isomerization.
 | ||
| Fig. 4 Functional group conversion as a function of time for 1:1 ratios of small molecules with different monothiols show that reaction rate can be tuned by thiol structure. (A) Allyl and maleate conversion of DM plotted with controls DS and DEM (left and right, respectively). (B) Allyl and fumarate conversion of DF plotted with controls DS and DEF (left and right, respectively). | ||
Beyond chemical functionality, thiol–ene reactions are known to be highly sensitive to changes in stoichiometry. In particular, in mixed-mechanism systems, even small changes in reactant stoichiometry can have a profound effect in biasing the preferred reaction pathway and resulting material properties.32,93 Therefore, we repeated the same experiments as discussed previously but with excess ene groups in the system to understand how stoichiometry influences the extent of chain-growth in these systems. We found that while allyl groups in DS rapidly react with thiols to reach ≈50% conversion when reacted in a 2:1 allyl:thiol ratio, introduction of maleate and fumarate groups both slows the reaction and leads to additional conversion (Fig. 5). Furthermore, we observed the formation of an insoluble solid under extended illumination times (>30 minutes), suggesting the cross-reaction between allyl and fumarate groups at low thiol concentrations.94,95 These results are consistent with our prior observations as lower thiol concentration decreases chain transfer reactions, increasing the likelihood of allyl groups participating in the chain-growth reaction. However, the absence of homopolymerization in DS with off-stoichiometric amounts of thiol (Fig. 5, pink data) suggests that cross-reactions between allyls and fumarates/maleates occur at a much slower rate than homopolymerization of maleate/fumarate groups. In addition, control molecules DEF and DEM (Fig. 5, gold and red data) show a more sluggish and incomplete conversion with thiols, which is consistent with a faster rate of thiol–ene addition to terminal allyl groups. Collectively, these data suggest that the two photoreactive mechanisms observed here proceed at differing rates: fast thiol–ene addition to terminal allyl groups, and slower chain growth of fumarate/maleate groups. This provides a potential photopolymerization mechanism where at low irradiation levels, thiol–ene step-growth facilitates initial polymerization and prolonged light exposure furthers chain growth.
 | ||
| Fig. 5
1H NMR conversion of allyl (left) and maleate/fumarate (right) in 2:1 ratio* photoreactions show a self-limiting reaction for DS, whereas combined maleate/fumarate and allyl consumption exceeds the number of available thiols. Combined with the observation of insoluble solids at long reaction times, this suggests some cross-reaction of allyls and maleates/fumarates is possible at low thiol concentration. *The ratio is allyl:thiol or fumarate/maleate:thiol for DEF and DEM. | ||
Gelation and thermomechanical properties
Mixed-mechanism thiol–ene polymerizations provide access to a wide range of thermomechanical properties due to changes in network topology and structure.96–99 Thus, we sought to understand how differences in DF and DM reactivity translate into macromolecular network development and the resulting bulk properties. Based on the small molecule studies presented above, we hypothesized that when copolymerized with a crosslinker, the relative propensity for thiol–ene addition versus homopolymerization will control the photo-dose required for gelation. Additionally, we anticipated that because fumarate and maleate groups are consumed more slowly, their incorporation within network strands could provide an additional handle to tune crosslinking density with additional light exposure over longer timescales. To study this, model polymer network formulations were prepared using dithiol and 1,3,5-triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TATO) as a triallyl crosslinker. To gain further insight on thiol structures in network architectures, we utilized dithiol analogs of the monothiols used in the NMR studies: hexanedithiol (HDT), ethylene bis(thioglycolate) (ETG), and ethylene glycol bis(3-mercaptopropionate) (E3M). Network polymer resins were formed in an equal allyl to thiol ratio with TATO contributing 5% of total allyl functionality and 0.5 wt% TPO as photoinitiator.To understand how this chemistry impacts network formation, we monitored conversion of ene and thiol functionalities throughout the gelation process with real-time transmission FTIR and Raman spectroscopy. Using the peaks associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches (1660–1630 cm−1) and S–H stretches (2600–2500 cm−1) in real-time FTIR, allyl ether groups of DS and thiols are observed to be quickly and quantitatively consumed within 30 s (Fig. S4A†). These data are consistent with the expected ideal thiol–ene step-growth polymerization. However, while quantitative conversions of thiol groups are observed, the same CC stretch shows incomplete disappearance within DF and DM systems (Fig. 6A). This is attributed to the persistence of the internal ene functionality, which has a characteristic IR peak that overlaps with the allyl ether stretch.100 While symmetric disubstituted functional groups are often infrared inactive due to the presence of centrosymmetric vibrations,101 the trans–cis ester conformations of maleic and fumaric compounds activate the IR stretch.102,103 Furthermore, since maleic functionalities enable more trans–cis ester vibrations, the maleate peak shows a higher absorbance stretch than fumarate, limiting direct comparison of peak area. For better comparison of total ene conversion, we turned to Raman spectroscopy where all centrosymmetric groups are active and absorbance of maleic, fumaric, and allyl ether groups are similar in magnitude, despite overlap. These experiments further support rapid (≈30s) and quantitative conversion of allyl ether groups in DS as evidenced by the disappearance of the “ene” peak at 1646 cm−1. In DM and DF, this ene peak shows a significant reduction that is accompanied by a decrease in the ester carbonyl group (1735 cm−1) peak during the reaction as conjugation onto the surrounding ester groups is lost (Fig. 6B).104 In addition, the cis maleate groups show limited reactivity compared to the trans fumarate counterparts. Taken together, these data echo the small-molecule NMR studies presented earlier, and suggest a mechanism whereby allyl ether groups are rapidly consumed by thiol addition while fumarate/maleate groups continue to react after thiol consumption.
C stretches (1660–1630 cm−1) and S–H stretches (2600–2500 cm−1) in real-time FTIR, allyl ether groups of DS and thiols are observed to be quickly and quantitatively consumed within 30 s (Fig. S4A†). These data are consistent with the expected ideal thiol–ene step-growth polymerization. However, while quantitative conversions of thiol groups are observed, the same CC stretch shows incomplete disappearance within DF and DM systems (Fig. 6A). This is attributed to the persistence of the internal ene functionality, which has a characteristic IR peak that overlaps with the allyl ether stretch.100 While symmetric disubstituted functional groups are often infrared inactive due to the presence of centrosymmetric vibrations,101 the trans–cis ester conformations of maleic and fumaric compounds activate the IR stretch.102,103 Furthermore, since maleic functionalities enable more trans–cis ester vibrations, the maleate peak shows a higher absorbance stretch than fumarate, limiting direct comparison of peak area. For better comparison of total ene conversion, we turned to Raman spectroscopy where all centrosymmetric groups are active and absorbance of maleic, fumaric, and allyl ether groups are similar in magnitude, despite overlap. These experiments further support rapid (≈30s) and quantitative conversion of allyl ether groups in DS as evidenced by the disappearance of the “ene” peak at 1646 cm−1. In DM and DF, this ene peak shows a significant reduction that is accompanied by a decrease in the ester carbonyl group (1735 cm−1) peak during the reaction as conjugation onto the surrounding ester groups is lost (Fig. 6B).104 In addition, the cis maleate groups show limited reactivity compared to the trans fumarate counterparts. Taken together, these data echo the small-molecule NMR studies presented earlier, and suggest a mechanism whereby allyl ether groups are rapidly consumed by thiol addition while fumarate/maleate groups continue to react after thiol consumption.
 | ||
| Fig. 6 (A) RT-FTIR-normalized peak area of thiol (left) and ene groups (right) in the photoreaction of DS, DM, and DF with HDT and 5% TATO. The thiol peak is quickly and quantitatively consumed along with the allyl peak of DS. However, the remaining peaks in DM and DF are associated with an overlapping internal ene of differing absorptions. (B) Raman spectroscopy shows conversion of ene groups and conversion of internal ene groups by the decrease in carbonyl peak intensity with the reduction in unsaturated conjugation. | ||
Complementary in situ photorheological studies show that changes in reaction mechanism are manifested as differences in gelation kinetics (Fig. 7A). Upon illumination, all formulations show a rapid increase in storage (G′) and loss (G′′) moduli until reaching a plateau set by the stiffness limits of the rheometer (G′ ≈ 1 MPa). Compared to pure step-growth systems, systems with maleate and fumarate groups reach the gelation point (defined as the point where G′ first exceeds G′′) at a lower critical photo-dose for all thiols studied, suggesting that even small amounts of chain-growth homopolymerization dramatically accelerates network percolation (Fig. 7C). This trend is perhaps most clearly apparent for ETG, where small-molecule analogs showed that allyl conversion for all monomers was nearly equal in rate but the addition of a small amount of chain growth via fumarate/maleate homopolymerization (Fig. 7B) results in a near order in magnitude reduction in the required gelation dose. These data are both consistent with classic gelation theories for chain-growth systems where even low conversions can lead to network formation,105 as well as previous predictions for thiol–acrylate polymerizations where suppression of acrylate homopolymerization delays gelation.106 Relatively modest changes in gelation dose for HDT and E3M between diallyl monomers can likely be explained by the distinct differences in rate of thiol addition to allyl groups (see small molecule analogs in Fig. S2-A1 and A3†), where the decrease in allyl activity of DM/DF acts to counter the rate of gelation for otherwise fast chain-growth reactions. This is further evidenced by trends for ETG, where DM gels faster than DF due to the increased rate of thiol-to-allyl addition (Fig. S2-A3†). Finally, while a large increase in viscosity was observed for E3M/DS (Fig. S3-C3†), full gelation is not reached despite full conversion of allyl groups (Fig. S3-A†). This suggests changes in chain conformation or phase behavior during polymerization that biases chain connectivity towards topologies that form individual clusters that do not percolate into a full network, highlighting the limits of pure reaction kinetics alone in predicting gelation processes.29,107
 | ||
| Fig. 7 (A) Photorheology traces that depict rapid increases in modulus and crossover between storage and loss modulus during gelation in resins with HDT. (B) Photorheology shows the importance of the chain-growth mechanism on gelation kinetics in resins with EBTG. (C) Photo-dose for gelation obtained by the product of crossover time and light intensity for resins of different dithiols. | ||
Since only a small proportion of the maleate/fumarate groups are consumed during the initial gelation process, a large fraction is preserved within network strands, providing a handle to tune crosslink density under increased light intensity as shown by FTIR and Raman measurements (Fig. S3-A and B†). As evidence of this, temperature-dependent DMA traces under fixed strain amplitude and frequency of network formulations incorporating HDT with DM and DF are shown in Fig. 8A and B. The absence of thermally driven curing during testing was confirmed through unchanged FTIR spectra and mechanical properties after heating a 30 s-cured specimen at 100 °C for 1 hour (Fig. S3-D4†). These data reveal two key trends as a function of illumination time: (1) a longitudinal increase in the E′ rubbery plateau, and (2) a concurrent shift to higher temperatures and broadening of tan(δ). Additionally, control experiments with DS (Fig. S3-D1†) reveal that DF/DM polymerization leads to much higher E′ plateaus and tan(δ) peaks, demonstrating that the increased conversion of fumarate and maleate groups via a chain-growth mechanism translates into the formation of additional elastically active crosslinks in the network.108 The association of these crosslinks with chain-growth polymerization is further corroborated by the widening of the tan(δ) glass transition peak or full width at half maximum (Fig. 8C),109–111 which is attributed to the incorporation of inhomogeneities into the network via localized regions of increased polymerization. Furthermore, stereochemistry-dependent reactivity differences of maleic and fumaric groups is observed to impact network architecture evolution. With increased illumination dose, DF polymer networks increase by an order of magnitude in stiffness and become increasingly glassy. However, the reduced reactivity of DM due to early thiol consumption that prevents further isomerization limits the increase in crosslinking density, keeping the materials rubbery at higher light doses. Thus, by varying the internal stereochemistry of the small-molecule precursors, bulk mechanical properties of the polymer networks can be rationally controlled (Fig. 9). Additionally, similar variations in network properties using DM and DF systems with varying thiol were observed, demonstrating the amenability of this chemistry to dithiol structure (Fig. S3-D†). This provides a mechanism to tune both gelation kinetics and longitudinal thermomechanical property development via light.
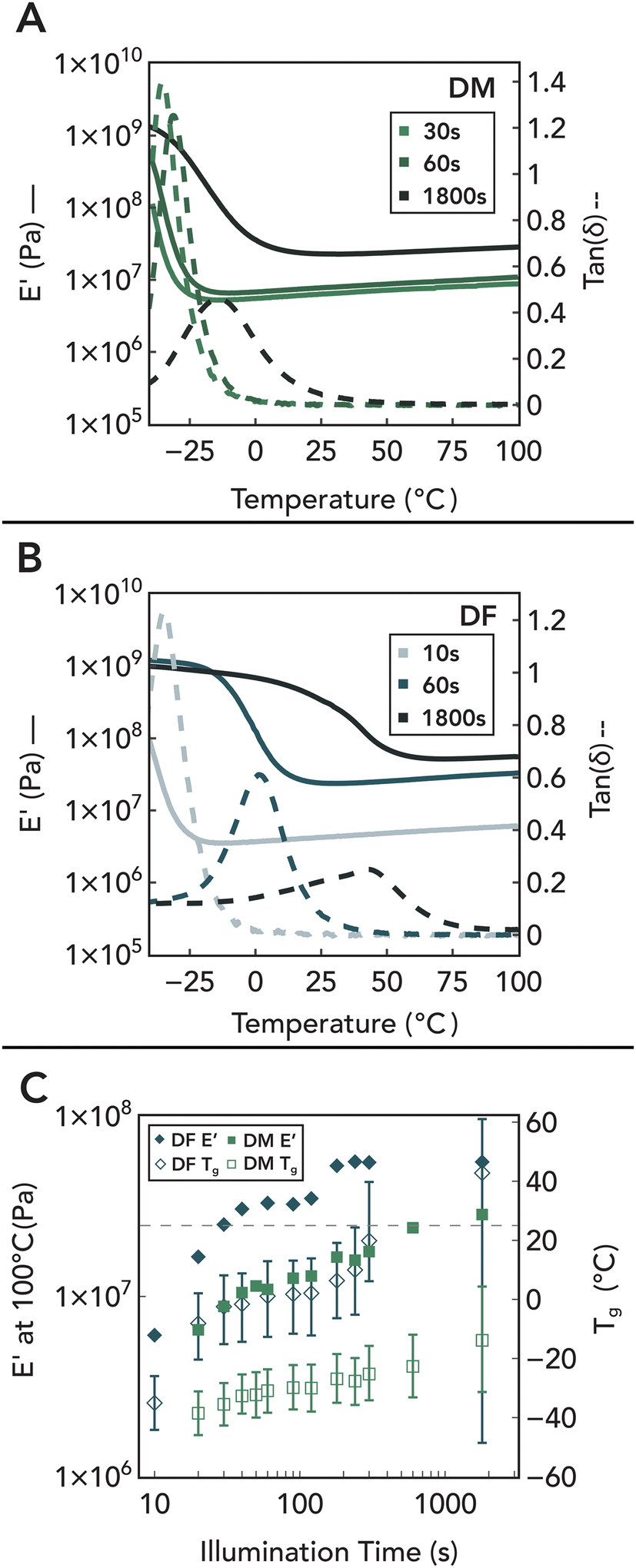 | ||
| Fig. 8 (A) DMA traces of DM network polymers illuminated for 30 s, 60 s, and 1800 s. (B) DMA traces of DF network polymers illuminated for 10 s, 60 s, and 1800 s. (C) E′ at 100 °C and Tg of DF and DM with error bars representing full width at half maximum of the glass transition. Collectively, these results suggest an increase in crosslinking density and network heterogeneity with continued light exposure. | ||
 | ||
| Fig. 9 Under first illumination, initial gelation from the polymer network occurs largely from thiol–ene addition and is advanced with slow chain-growth homopolymerization. Over prolonged light exposure, the chain-growth homopolymerization forms additional crosslinks. | ||
Patterning crosslinking and 3D printing of candidate resins
Photosensitive polymer network resins can be harnessed to simultaneously control local material properties and global geometry within 3D printed parts through spatially defined photo-doses.112,113 Most commonly, this is done via spatial variation in monomer conversion, and thereby crosslinking density, with light intensity during digital light processing (DLP) additive manufacturing.114 Since crosslinking density has a linear relationship with stiffness for amorphous thermosets, these objects can achieve magnitude differences in elastic modulus over small length-scales.115,116 In addition, the use of single-material chemistry creates covalent bonds between contrasting regions, lessening interfacial failure and brittleness found in classical examples of multimaterials.117 However, this method typically produces materials with residual monomers and is impractical for common uses. In the chemical system described herein, crosslinking density can be perturbed within polymer networks after full incorporation of monomers.To demonstrate the utility of this chemistry for spatial patterning, candidate network formulations of DS, DM, and DF with HDT were exposed to varying light patterns via photolithography using 2D photomasks. After initial illumination of the full film for 20 seconds under UV light (405 nm, 5 mW cm−2) to trigger gelation, a photomask depicting the block Illinois ‘I’ was placed over the sample and exposure was continued for 30 additional minutes to reach near full conversion (Fig. 10A). Due to differences in refractive index with crosslinking density, the block ‘I’ is visible within the film despite optical clarity (Fig. 10B). For further visualization, films were placed in acetone with 0.45% fluoresceinamine as fluorescent dye overnight and washed off with acetone. Due to additional crosslinks, areas exposed to higher photo-dose levels should imbibe less dye due to lower swelling extent and therefore fluoresce more weakly.116 This behavior was successfully observed in films formed from DF and is partially attributed to the increase in Tg above room temperature (Fig. 10C) as this was not observed in films formed from the less-reactive DM resins (Fig. S4-A1†). Even so, the ability to photo-pattern images implies the applicability of this material chemistry to be used in grayscale methods for stiffness gradients within polymer networks after complete monomer incorporation.
 | ||
| Fig. 10 (A) To photo-pattern UIUC's block-I, an initial network polymer film was formed under low light-dose levels, a photomask was placed on top, and the remaining film was illuminated with an additional light dose. (B) DM photo-pattern visible due to changes in optical properties with crosslinking density. (C) The DF polymer film swollen with fluorescent dye to visually show differences in crosslinking density with “grayscale” illumination. | ||
Finally, this chemistry is amenable to 3D printing with an off-the-shelf DLP printer. Thiol–ene chemistry is a particularly useful material system for additive manufacturing due to its rapid kinetics, oxygen tolerance, and low shrinkage stress that prevents warpage of manufactured objects.5,114 While much work has been focused on pure thiol–ene step-growth resin systems,118–121 combinations of step-growth and chain-growth modalities have been shown to greatly expand this toolbox.122–124 As a proof-of-concept with this chemistry, we developed a printing resin containing the studied DM network formulation with 0.05 wt% pyrogallol as a photo-absorber to prevent overcuring (Fig. 11A). This formulation remained stable for several weeks when stored in the dark at room temperature. Printing conditions were selected by determining the optimum balance between printing speed, feature resolution, and fidelity via construction of a Jacobs working curve (Fig. S4-B1†). Candidate prints were produced using a 10 second exposure time and a 50 μm layer thickness, followed by post-curing through flood illumination. A variety of high-resolution prints with complex, overhanging features were produced (Fig. 11B), including the canonical 3D Benchy model and the Alma Mater statue of the University of Illinois (Fig. 11C). Prints were observed to closely match CAD file inputs. While beyond the scope of this work, this resin chemistry is promising for extension to grayscale printing, where internal ene polymerization is expected to generate a larger difference in crosslinking density, opening prospects for fabricating graded materials in a single-vat 3D printing process.
 | ||
| Fig. 11 (A) Additive manufacturing is done by extending the chemistry used for polymer network studies and incorporating a photo-absorber for increased fidelity. (B) To benchmark the 3D printing ability of our resin, we printed a 3D Benchy model, a readily used benchmark print. (C) Demonstrating the capability of this chemistry, we printed UIUC's Alma Mater statue. | ||
Conclusion
This work extends the paradigm of mixed-mechanism thiol-X chemistries, where the relative rates of chain-growth and step-growth mechanisms define the overall network evolution and final properties. The chemical system illustrated here incorporates two ene functionalities within biomass-derived triene monomers to undergo both step-growth thiol–ene addition and chain-growth homopolymerization: (1) terminal allyl ether groups and (2) internal maleate/fumarate groups. We demonstrate that thiol addition quickly and preferentially occurs across the terminal allyl groups, leaving the internal ene to homopolymerize over longer durations. In addition, chain-growth homopolymerization of the internal ene moieties is shown to be sensitive to stereochemistry as cis maleate groups undergo an isomerization–polymerization mechanism facilitated by thiol addition. These reactivities are also shown to be additionally tunable by the thiol structure. By utilizing these diallyl monomers in polymer networks, initial network formation predominantly by thiol–ene addition incorporates fumarate and maleate moieties into network strands, which act as reactive handles for additional crosslink formation. The described mixed mechanism impacts both gelation kinetics and network architecture development. We show that glass transition and crosslinking density can be controlled by photo-dose levels within fully formed network polymers. Finally, this mixed-mechanism photopolymerization allows for dose-dependent patterning of material properties and 3D printing. In the future, we anticipate that insights from this work could find applications in polymer functionalization,125 new resins for additive manufacturing,115,126 and resist chemistries for photolithography.127Author contributions
R.G. and A.S.K conceptualized the general concept and designed the experiments. R.G. and C.M. synthesized the monomers and performed NMR experiments. R.G. conducted all thermomechanical measurements. R.G. and A.A. executed all patterning and 3D printing experiments. All authors contributed to analyzing the data. R.G., A.A., and A.S.K. wrote and revised the manuscript. A.S.K. supervised the research.Data availability
The data supporting this article have been included as part of the ESI†.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by an ARO STIR award (W911NF2410159) and start-up funding from the University of Illinois. We thank the Simon Rogers lab for access to photorheology and the Damien Guironnet lab for SEC use. We thank Neil Pearse for providing the Blender file of the Alma Mater. Major funding for the 500 MHz Bruker CryoProbe was provided by the Roy J. Carver Charitable Trust (Muscatine, Iowa; Grant #15-4521) to the School of Chemical Sciences NMR Lab.References
- A. Della Bona, V. Cantelli, V. T. Britto, K. F. Collares and J. W. Stansbury, 3D Printing Restorative Materials Using a Stereolithographic Technique: A Systematic Review, Dent. Mater., 2021, 37(2), 336–350, DOI:10.1016/j.dental.2020.11.030.
- W. M. Palin, J. G. Leprince and M. A. Hadis, Shining a Light on High Volume Photocurable Materials, Dent. Mater., 2018, 34(5), 695–710, DOI:10.1016/j.dental.2018.02.009.
- F.-K. Bruder, T. Fäcke and T. Rölle, The Chemistry and Physics of Bayfol® HX Film Holographic Photopolymer, Polymers, 2017, 9(10), 472, DOI:10.3390/polym9100472.
- R. Fernández, S. Bleda, S. Gallego, C. Neipp, A. Márquez, Y. Tomita, I. Pascual and A. Beléndez, Holographic Waveguides in Photopolymers, Opt. Express, 2019, 27(2), 827–840, DOI:10.1364/OE.27.000827.
- J. J. Schwartz, Additive Manufacturing: Frameworks for Chemical Understanding and Advancement in Vat Photopolymerization, MRS Bull., 2022, 47(6), 628–641, DOI:10.1557/s43577-022-00343-0.
- B. Narupai and A. Nelson, 100th Anniversary of Macromolecular Science Viewpoint: Macromolecular Materials for Additive Manufacturing, ACS Macro Lett., 2020, 9(5), 627–638, DOI:10.1021/acsmacrolett.0c00200.
- M. Ahmadi, K. Ehrmann, T. Koch, R. Liska and J. Stampfl, From Unregulated Networks to Designed Microstructures: Introducing Heterogeneity at Different Length Scales in Photopolymers for Additive Manufacturing, Chem. Rev., 2024, 124(7), 3978–4020, DOI:10.1021/acs.chemrev.3c00570.
- C. K. Ober, F. Käfer and C. Yuan, Recent Developments in Photoresists for Extreme-Ultraviolet Lithography, Polymer, 2023, 280, 126020, DOI:10.1016/j.polymer.2023.126020.
- J. Cen, Z. Deng and S. Liu, Emerging Trends in the Chemistry of Polymeric Resists for Extreme Ultraviolet Lithography, Polym. Chem., 2024, 15(45), 4599–4614, 10.1039/D4PY00957F.
- H. Lai, X. Peng, L. Li, D. Zhu and P. Xiao, Novel Monomers for Photopolymer Networks, Prog. Polym. Sci., 2022, 128, 101529, DOI:10.1016/j.progpolymsci.2022.101529.
- K. S. Anseth and C. N. Bowman, Kinetic Gelation Model Predictions of Crosslinked Polymer Network Microstructure, Chem. Eng. Sci., 1994, 49(14), 2207–2217, DOI:10.1016/0009-2509(94)E0055-U.
- K. S. Anseth, C. M. Wang and C. N. Bowman, Kinetic Evidence of Reaction Diffusion during the Polymerization of Multi(Meth)Acrylate Monomers, Macromolecules, 1994, 27(3), 650–655, DOI:10.1021/ma00081a004.
- K. S. Anseth, K. J. Anderson and C. N. Bowman, Radical Concentrations, Environments, and Reactivities during Crosslinking Polymerizations, Macromol. Chem. Phys., 1996, 197(3), 833–848, DOI:10.1002/macp.1996.021970306.
- A. Arora, T.-S. Lin, H. K. Beech, H. Mochigase, R. Wang and B. D. Olsen, Fracture of Polymer Networks Containing Topological Defects, Macromolecules, 2020, 53(17), 7346–7355, DOI:10.1021/acs.macromol.0c01038.
- M. Zhong, R. Wang, K. Kawamoto, B. D. Olsen and J. A. Johnson, Quantifying the Impact of Molecular Defects on Polymer Network Elasticity, Science, 2016, 353(6305), 1264–1268, DOI:10.1126/science.aag0184.
- C. W. Barney, Z. Ye, I. Sacligil, K. R. McLeod, H. Zhang, G. N. Tew, R. A. Riggleman and A. J. Crosby, Fracture of Model End-Linked Networks, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(7), e2112389119, DOI:10.1073/pnas.2112389119.
- C. Decker and A. D. Jenkins, Kinetic Approach of Oxygen Inhibition in Ultraviolet- and Laser-Induced Polymerizations, Macromolecules, 1985, 18(6), 1241–1244, DOI:10.1021/ma00148a034.
- C. Decker and K. Moussa, A New Method for Monitoring Ultra-Fast Photopolymerizations by Real-Time Infra-Red (RTIR) Spectroscopy, Die Makromol. Chemie, 1988, 189(10), 2381–2394, DOI:10.1002/macp.1988.021891016.
- E. Andrzejewska, M. B. Bogacki, M. Andrzejewski and M. Janaszczyk, Termination Mechanism during the Photo-Induced Radical Cross-Linking Polymerization in the Presence and Absence of Oxygen, Phys. Chem. Chem. Phys., 2003, 5(12), 2635, 10.1039/b301849k.
- N. Ide and T. Fukuda, Nitroxide-Controlled Free-Radical Copolymerization of Vinyl and Divinyl Monomers. Evaluation of Pendant-Vinyl Reactivity, Macromolecules, 1997, 30(15), 4268–4271, DOI:10.1021/ma9700946.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Development of Branching in Living Radical Copolymerization of Vinyl and Divinyl Monomers, Macromolecules, 2006, 39(22), 7483–7492, DOI:10.1021/ma061811b.
- J. Cuthbert, S. V. Wanasinghe, K. Matyjaszewski and D. Konkolewicz, Are RAFT and ATRP Universally Interchangeable Polymerization Methods in Network Formation?, Macromolecules, 2021, 54(18), 8331–8340, DOI:10.1021/acs.macromol.1c01587.
- H. Gao and K. Matyjaszewski, Synthesis of Functional Polymers with Controlled Architecture by CRP of Monomers in the Presence of Cross-Linkers: From Stars to Gels, Prog. Polym. Sci., 2009, 34(4), 317–350, DOI:10.1016/j.progpolymsci.2009.01.001.
- A. Z. Dookhith, Z. Zhang, V. Ganesan and G. E. Sanoja, Impact of Reversible Deactivation Radical Copolymerizations (RDRPs) on Gelation, Phase Separation, and Mechanical Properties of Polymer Networks, Macromolecules, 2024, 57(18), 8698–8711, DOI:10.1021/acs.macromol.4c00905.
- C. E. Hoyle and C. N. Bowman, Thiol – Ene Click Chemistry, Angew. Chem., Int. Ed., 2010, 49, 1540–1573, DOI:10.1002/anie.200903924.
- A. R. Kannurpatti, J. W. Anseth and C. N. Bowman, A Study of the Evolution of Mechanical Properties and Structural Heterogeneity of Polymer Networks Formed by Photopolymerizations of Multifunctional (Meth)Acrylates, Polymer, 1998, 39(12), 2507–2513, DOI:10.1016/S0032-3861(97)00585-5.
- H. Lu, J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Investigations of Step-Growth Thiol-Ene Polymerizations for Novel Dental Restoratives, Dent. Mater., 2005, 21(12), 1129–1136, DOI:10.1016/j.dental.2005.04.001.
- A. F. Senyurt, H. Wei, C. E. Hoyle, S. G. Piland and T. E. Gould, Ternary Thiol−Ene/Acrylate Photopolymers: Effect of Acrylate Structure on Mechanical Properties, Macromolecules, 2007, 40(14), 4901–4909, DOI:10.1021/ma062534b.
- S. Grube and W. Oppermann, Inhomogeneity in Hydrogels Synthesized by Thiol–Ene Polymerization, Macromolecules, 2013, 46(5), 1948–1955, DOI:10.1021/ma302520p.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Thiol−Ene Photopolymerization Mechanism and Rate Limiting Step Changes for Various Vinyl Functional Group Chemistries, Macromolecules, 2003, 36(21), 7964–7969, DOI:10.1021/ma034667s.
- H. Wei, A. F. Senyurt, S. Jönsson and C. E. Hoyle, Photopolymerization of Ternary Thiol–Ene/Acrylate Systems: Film and Network Properties, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(5), 822–829, DOI:10.1002/pola.21844.
- S. K. Reddy, N. B. Cramer and C. N. Bowman, Thiol−Vinyl Mechanisms. 2. Kinetic Modeling of Ternary Thiol−Vinyl Photopolymerizations, Macromolecules, 2006, 39(10), 3681–3687, DOI:10.1021/ma0600097.
- O. Okay and C. N. Bowman, Kinetic Modeling of Thiol-Ene Reactions with Both Step and Chain Growth Aspects, Macromol. Theory Simul., 2005, 14(4), 267–277, DOI:10.1002/mats.200500002.
- S. K. Reddy, O. Okay and C. N. Bowman, Network Development in Mixed Step-Chain Growth Thiol−Vinyl Photopolymerizations, Macromolecules, 2006, 39(25), 8832–8843, DOI:10.1021/ma060249m.
- J. J. Karnes, T. H. Weisgraber, C. C. Cook, D. N. Wang, J. C. Crowhurst, C. A. Fox, B. S. Harris, J. S. Oakdale, R. Faller and M. Shusteff, Isolating Chemical Reaction Mechanism as a Variable with Reactive Coarse-Grained Molecular Dynamics: Step-Growth versus Chain-Growth Polymerization, Macromolecules, 2023, 56(6), 2225–2233, DOI:10.1021/acs.macromol.2c02069.
- S. K. Reddy, K. S. Anseth and C. N. Bowman, Modeling of Network Degradation in Mixed Step-Chain Growth Polymerizations, Polymer, 2005, 46(12), 4212–4222, DOI:10.1016/j.polymer.2005.02.050.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Degradable Thiol-Acrylate Photopolymers: Polymerization and Degradation Behavior of an in Situ Forming Biomaterial, Biomaterials, 2005, 26(22), 4495–4506, DOI:10.1016/j.biomaterials.2004.11.046.
- T. S. Hebner, H. E. Fowler, K. M. Herbert, N. P. Skillin, C. N. Bowman and T. J. White, Polymer Network Structure, Properties, and Formation of Liquid Crystalline Elastomers Prepared via Thiol–Acrylate Chain Transfer Reactions, Macromolecules, 2021, 54(23), 11074–11082, DOI:10.1021/acs.macromol.1c01919.
- M. Roquart, A. Kharlamova, L. Marcos Celada, S. Norvez, R. Nicolaÿ and L. Corté, PEG-Based Photo-Cross-Linked Networks with Adjustable Topologies and Mechanical Properties, Biomacromolecules, 2023, 24(10), 4454–4464, DOI:10.1021/acs.biomac.2c01265.
- Q. Thijssen and S. V. Vlierberghe, Thiol-Mediated Chain Transfer as a Tool to Improve the Toughness of Acrylate Photo-Crosslinked Poly(ε-Caprolactone), Macromol. Mater. Eng., 2022, 307(3), 2100754, DOI:10.1002/mame.202100754.
- N. B. Cramer and C. N. Bowman, Kinetics of Thiol–Ene and Thiol–Acrylate Photopolymerizations with Real-Time Fourier Transform Infrared, J. Polym. Sci., Part A: Polym. Chem., 2001, 39(19), 3311–3319, DOI:10.1002/pola.1314.
- M. Sahin, S. Ayalur-Karunakaran, J. Manhart, M. Wolfahrt, W. Kern and S. Schlögl, Thiol-Ene versus Binary Thiol–Acrylate Chemistry: Material Properties and Network Characteristics of Photopolymers, Adv. Eng. Mater., 2017, 19(4), 1600620, DOI:10.1002/adem.201600620.
- K. C. McCleary-Petersen, K. R. Wiegand, M. T. Taleff and D. Guironnet, Engineering Polymer Architecture Through Reaction Rates, Macromolecules, 2025, 58(1), 18–31, DOI:10.1021/acs.macromol.4c01662.
- E. Hasa, J. P. Scholte, J. L. P. Jessop, J. W. Stansbury and C. A. Guymon, Kinetically Controlled Photoinduced Phase Separation for Hybrid Radical/Cationic Systems, Macromolecules, 2019, 52(8), 2975–2986, DOI:10.1021/acs.macromol.9b00177.
- T. L. Grover and C. A. Guymon, Controlling Network Morphology in Hybrid Radical/Cationic Photopolymerized Systems, Polym. Chem., 2023, 14(2), 126–136, 10.1039/D2PY01288J.
- I. Calvez, C. R. Szczepanski and V. Landry, Effect of Copolymer on the Wrinkle Structure Formation and Gloss of a Phase-Separated Ternary Free-Radical/Cationic Hybrid System for the Application of Self-Matting Coatings, Polymers, 2022, 14(12), 2371, DOI:10.3390/polym14122371.
- M. J. Allen, H.-M. Lien, N. Prine, C. Burns, A. K. Rylski, X. Gu, L. M. Cox, F. Mangolini, B. D. Freeman and Z. A. Page, Multimorphic Materials: Spatially Tailoring Mechanical Properties via Selective Initiation of Interpenetrating Polymer Networks, Adv. Mater., 2023, 35(9), 2210208, DOI:10.1002/adma.202210208.
- B. R. Nelson, B. E. Kirkpatrick, C. E. Miksch, M. D. Davidson, N. P. Skillin, G. K. Hach, A. Khang, S. N. Hummel, B. D. Fairbanks, J. A. Burdick, C. N. Bowman and K. S. Anseth, Photoinduced Dithiolane Crosslinking for Multiresponsive Dynamic Hydrogels, Adv. Mater., 2024, 36(43), 2211209, DOI:10.1002/adma.202211209.
- T. R. Heyl, J. M. Beebe, A. J. Silvaroli, A. Perce, D. Ahn, S. Mangold, V. Mazure, K. R. Shull and M. Wang, In Situ Investigations of Microstructure Formation in Interpenetrating Polymer Networks, Macromolecules, 2024, 57(5), 1950–1961, DOI:10.1021/acs.macromol.3c02097.
- S. Huang, S. M. Adelmund, P. S. Pichumani, J. J. Schwartz, Y. Mengüç, M. Shusteff and T. J. Wallin, One-Pot Ternary Sequential Reactions for Photopatterned Gradient Multimaterials, Matter, 2023, 6(7), 2419–2438, DOI:10.1016/j.matt.2023.05.040.
- B. E. Kirkpatrick, G. K. Hach, B. R. Nelson, N. P. Skillin, J. S. Lee, L. P. Hibbard, A. P. Dhand, H. S. Grotheer, C. E. Miksch, V. Salazar, T. S. Hebner, S. P. Keyser, J. T. Kamps, J. Sinha, L. J. Macdougall, B. D. Fairbanks, J. A. Burdick, T. J. White, C. N. Bowman and K. S. Anseth, Photochemical Control of Network Topology in PEG Hydrogels, Adv. Mater., 2024, 36(46), 2409603, DOI:10.1002/adma.202409603.
- T. Y. Lee, T. M. Roper, C. A. Guymon, E. S. Jonsson and C. E. Hoyle, Copolymerization Mechanism of Photoinitiator Free Thiol—Vinyl Acrylate Systems, in Film Formation, ACS Symposium Series, American Chemical Society, 2006, vol. 941, pp. 17–28. DOI:10.1021/bk-2006-0941.ch002.
- T. M. Roper, T. Y. Lee, C. A. Guymon and C. E. Hoyle, In Situ Characterization of Photopolymerizable Systems Using a Thin-Film Calorimeter, Macromolecules, 2005, 38(24), 10109–10116, DOI:10.1021/ma051586i.
- T. M. Roper, C. A. Guymon, E. S. Jönsson and C. E. Hoyle, Influence of the Alkene Structure on the Mechanism and Kinetics of Thiol–Alkene Photopolymerizations with Real-Time Infrared Spectroscopy, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(24), 6283–6298, DOI:10.1002/pola.20452.
- H. Lai, J. Zhang and P. Xiao, Renewable Photopolymers: Transformation of Biomass Resources into Value-Added Products Under Light, ACS Sustainable Chem. Eng., 2023, 11(46), 16365–16406, DOI:10.1021/acssuschemeng.3c05257.
- E. A. Prebihalo, M. Johnson and T. M. Reineke, Bio-Based Thiol–Ene Network Thermosets from Isosorbide and Terpenes, ACS Macro Lett., 2024, 13(5), 586–591, DOI:10.1021/acsmacrolett.4c00078.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Terpene-Based Renewable Monomers and Polymers via Thiol–Ene Additions, Macromolecules, 2011, 44(18), 7253–7262, DOI:10.1021/ma201544e.
- M. Claudino, M. Jonsson and M. Johansson, Utilizing Thiol–Ene Coupling Kinetics in the Design of Renewable Thermoset Resins Based on d -Limonene and Polyfunctional Thiols, RSC Adv., 2014, 4(20), 10317–10329, 10.1039/C3RA47922F.
- A. C. Weems, K. R. D. Chiaie, J. C. Worch, C. J. Stubbs and A. P. Dove, Terpene- and Terpenoid-Based Polymeric Resins for Stereolithography 3D Printing, Polym. Chem., 2019, 10(44), 5959–5966, 10.1039/C9PY00950G.
- M. K. Porwal, M. M. Hausladen, C. J. Ellison and T. M. Reineke, Biobased and Degradable Thiol–Ene Networks from Levoglucosan for Sustainable 3D Printing, Green Chem., 2023, 25(4), 1488–1502, 10.1039/D2GC04185E.
- E. Constant, O. King and A. C. Weems, Bioderived 4D Printable Terpene Photopolymers from Limonene and β-Myrcene, Biomacromolecules, 2022, 23(6), 2342–2352, DOI:10.1021/acs.biomac.2c00085.
- T. O. Machado, C. J. Stubbs, V. Chiaradia, M. A. Alraddadi, A. Brandolese, J. C. Worch and A. P. Dove, A Renewably Sourced, Circular Photopolymer Resin for Additive Manufacturing, Nature, 2024, 629(8014), 1069–1074, DOI:10.1038/s41586-024-07399-9.
- V. Chiaradia, E. Pensa, T. O. Machado and A. P. Dove, Improving the Performance of Photoactive Terpene-Based Resin Formulations for Light-Based Additive Manufacturing, ACS Sustainable Chem. Eng., 2024, 12(18), 6904–6912, DOI:10.1021/acssuschemeng.3c08191.
- B. T. Worrell, M. K. McBride, G. B. Lyon, L. M. Cox, C. Wang, S. Mavila, C.-H. Lim, H. M. Coley, C. B. Musgrave, Y. Ding and C. N. Bowman, Bistable and Photoswitchable States of Matter, Nat. Commun., 2018, 9(1), 2804, DOI:10.1038/s41467-018-05300-7.
- P. F. Jacobs, Rapid Prototyping & Manufacturing: Fundamentals of Stereolithography, Society of Manufacturing Engineers, 1992 Search PubMed.
- Q. Li and J. Xing, Production of 1,4-Diacids (Succinic, Fumaric, and Malic) from Biomass, in Production of Platform Chemicals from Sustainable Resources, ed. Z. Fang, R. L. Smith Jr and X. Qi, Springer, Singapore, 2017, pp. 231–262. DOI:10.1007/978-981-10-4172-3_8.
- Commission Delegated Regulation (EU) 2024/197 of 19 October 2023 Amending Regulation (EC) No 1272/2008 as Regards the Harmonised Classification and Labelling of Certain Substances.
- J. Yin, Y. Zhang, B. Graff, C. Dietlin, M. Schmitt, F. Morlet-Savary, T. Petithory, L. Pieuchot, J. Zhang, Y. Xu, J.-M. Becht, J. Lalevée and P. Xiao, 2,4,6−Trimethylbenzoyldiphenylphosphine Oxide (TPO) Analog: A Non-Cytotoxic Type-I Photoinitiator for Free Radical Photopolymerization, Green Chem., 2025, 27(5), 1451–1461, 10.1039/D4GC04127E.
- Y. B. Kim, H. K. Kim, H. C. Choi and J. W. Hong, Photocuring of a Thiol-Ene System Based on an Unsaturated Polyester, J. Appl. Polym. Sci., 2005, 95(2), 342–350, DOI:10.1002/app.21248.
- T. M. Roper, C. A. Guymon, E. S. Jönsson and C. E. Hoyle, Influence of the Alkene Structure on the Mechanism and Kinetics of Thiol–Alkene Photopolymerizations with Real-Time Infrared Spectroscopy, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(24), 6283–6298, DOI:10.1002/pola.20452.
- T. Y. Lee, Z. Smith, S. K. Reddy, N. B. Cramer and C. N. Bowman, Thiol−Allyl Ether−Methacrylate Ternary Systems. Polymerization Mechanism, Macromolecules, 2007, 40(5), 1466–1472, DOI:10.1021/ma062494b.
- T. R. Yeazel-Klein, A. G. Davis and M. L. Becker, Thiol-Ene-Based 3D Printing of Bioresorbable Fumarate-Based ABA Triblock Copolyester Elastomers, Adv. Mater. Technol., 2023, 8(23), 2201904, DOI:10.1002/admt.202201904.
- D. A. Echeverri, V. Cádiz, J. C. Ronda and L. A. Rios, Synthesis of Elastomeric Networks from Maleated Soybean-Oil Glycerides by Thiol-Ene Coupling, Eur. Polym. J., 2012, 48(12), 2040–2049, DOI:10.1016/j.eurpolymj.2012.09.004.
- S. L. Brooks, E. J. Constant, O. M. King and A. C. Weems, Stereochemistry and Stoichiometry in Aliphatic Polyester Photopolymers for 3D Printing Tailored Biomaterial Scaffolds, Polym. Chem., 2022, 13(14), 2048–2056, 10.1039/D1PY01405F.
- A. Kirillova, T. R. Yeazel, K. Gall and M. L. Becker, Thiol-Based Three-Dimensional Printing of Fully Degradable Poly(Propylene Fumarate) Star Polymers, ACS Appl. Mater. Interfaces, 2022, 14(34), 38436–38447, DOI:10.1021/acsami.2c06553.
- Z. Kassenova, S. Kozhabekov, A. Zhubanov and A. Galymzhan, Overview of Synthesis, Properties, and Applications of Alkyl Fumarate Polymers, Polym.-Plast. Technol. Mater., 2024, 63(17), 2397–2417 CAS.
- W. Ken Busfield, I. D. Jenkins and K. Heiland, Free Radical Initiation Mechanisms in the Polymerisation of Diethyl Fumarate and Diethyl Maleate Studied by the Nitroxide Trapping Technique, Eur. Polym. J., 1994, 30(11), 1259–1267, DOI:10.1016/0014-3057(94)90136-8.
- T. Otsu, O. Ito and N. Toyoda, Polymers from 1,2-Disubstituted Ethylenic Monomers, 3. Homopolymers from Alkyl Maleates: Further Results of Monomer-Isomerization Radical Polymerization, Die Makromolekulare Chemie, Rapid Communications, 1981, 2(12), 729–732, DOI:10.1002/marc.1981.030021205.
- T. Otsu, O. Ito and N. Toyoda, Polymers from 1,2-Disubstituted Ethylenic Monomers. V. Radical Polymerization of Dimethyl Maleate in the Presence or Absence of Isomerization Catalyst, J. Macromol. Sci., Part A, 1983, 19(1), 27–39, DOI:10.1080/00222338308069420.
- S. Choi, W. Frank and H. Ritter, Novel Polymerization of Diethyl Fumarate and Maleate in Aqueous Media via Cyclodextrin-Complexes, React. Funct. Polym., 2006, 66(1), 149–156, DOI:10.1016/j.reactfunctpolym.2005.07.013.
- T. Otsu and K. Shiraishi, Monomer-Isomerization Radical Polymerization of Di-Tert-Butyl Maleate to High-Molecular Weight Poly(Di-Tert-Butyl Fumarate), Macromolecules, 1985, 18(9), 1795–1796, DOI:10.1021/ma00151a028.
- T. Otsu and N. Toyoda, Monomer-Isomerization Polymerization of Dialkyl Maleates by Radical Mechanism, Die Makromolekulare Chemie, Rapid Communications, 1981, 2(1), 79–81, DOI:10.1002/marc.1981.030020116.
- N. Toyoda, M. Yoshida and T. Otsu, Polymers from 1,2-Disubstituted Ethylenic Monomers VI. Monomer-Isomerization Radical Polymerization of Diethyl Maleate, Polym J., 1983, 15(4), 255–260, DOI:10.1295/polymj.15.255.
- C. Walling and W. Helmreich, Reactivity and Reversibility in the Reaction of Thiyl Radicals with Olefins1, J. Am. Chem. Soc., 1959, 81(5), 1144–1148, DOI:10.1021/ja01514a032.
- C. Chatgilialoglu, A. Altieri and H. Fischer, The Kinetics of Thiyl Radical-Induced Reactions of Monounsaturated Fatty Acid Esters, J. Am. Chem. Soc., 2002, 124(43), 12816–12823, DOI:10.1021/ja027428d.
- A. B. Lowe, Thiol-Ene “Click” Reactions and Recent Applications in Polymer and Materials Synthesis, Polym. Chem., 2010, 1(1), 17–36, 10.1039/B9PY00216B.
- K. F. Long, N. J. Bongiardina, P. Mayordomo, M. J. Olin, A. D. Ortega and C. N. Bowman, Effects of 1°, 2°, and 3° Thiols on Thiol–Ene Reactions: Polymerization Kinetics and Mechanical Behavior, Macromolecules, 2020, 53(14), 5805–5815, DOI:10.1021/acs.macromol.0c00369.
- K. Griesbaum, Problems and Possibilities of the Free-Radical Addition of Thiols to Unsaturated Compounds, Angew. Chem., Int. Ed. Engl., 1970, 9(4), 273–287, DOI:10.1002/anie.197002731.
- A. A. Oswald, K. Griesbaum, D. N. Hall and W. Naegele, Allene Chemistry., VIII. Diaddition of Thiol Compounds to Allene, Can. J. Chem., 1967, 45(11), 1173–1184, DOI:10.1139/v67-196.
- C. R. Morgan, F. Magnotta and A. D. Ketley, Thiol/Ene Photocurable Polymers, J. Polym. Sci., Polym. Chem. Ed., 1977, 15(3), 627–645, DOI:10.1002/pol.1977.170150311.
- C. E. Hoyle, T. Y. Lee and T. Roper, Thiol–Enes: Chemistry of the Past with Promise for the Future, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(21), 5301–5338, DOI:10.1002/pola.20366.
- N. J. Bongiardina, S. M. Soars, M. Podgorski and C. N. Bowman, Radical-Disulfide Exchange in Thiol–Ene–Disulfidation Polymerizations, Polym. Chem., 2022, 13(27), 3991–4003, 10.1039/D2PY00172A.
- N. B. Cramer, T. Davies, A. K. O'Brien and C. N. Bowman, Mechanism and Modeling of a Thiol−Ene Photopolymerization, Macromolecules, 2003, 36(12), 4631–4636, DOI:10.1021/ma034072x.
- K. Urushido, A. Matsumoto and M. Oiwa, Polymerization of Allyl Esters of Unsaturated Acids. V. Cyclopolymerization of Methyl Allyl Fumarate, J. Polym. Sci., Polym. Chem. Ed., 1980, 18(6), 1771–1785, DOI:10.1002/pol.1980.170180611.
- T. Y. Lee, J. Carioscia, Z. Smith and C. N. Bowman, Thiol−Allyl Ether−Methacrylate Ternary Systems. Evolution Mechanism of Polymerization-Induced Shrinkage Stress and Mechanical Properties, Macromolecules, 2007, 40(5), 1473–1479, DOI:10.1021/ma0624954.
- N. B. Cramer and C. N. Bowman, Kinetics of Thiol–Ene and Thiol–Acrylate Photopolymerizations with Real-Time Fourier Transform Infrared, J. Polym. Sci., Part A: Polym. Chem., 2001, 39(19), 3311–3319, DOI:10.1002/pola.1314.
- A. F. Senyurt, H. Wei, C. E. Hoyle, S. G. Piland and T. E. Gould, Ternary Thiol−Ene/Acrylate Photopolymers: Effect of Acrylate Structure on Mechanical Properties, Macromolecules, 2007, 40(14), 4901–4909, DOI:10.1021/ma062534b.
- K. Jin, N. Wilmot, W. H. Heath and J. M. Torkelson, Phase-Separated Thiol–Epoxy–Acrylate Hybrid Polymer Networks with Controlled Cross-Link Density Synthesized by Simultaneous Thiol–Acrylate and Thiol–Epoxy Click Reactions, Macromolecules, 2016, 49(11), 4115–4123, DOI:10.1021/acs.macromol.6b00141.
- Z. Chen, B. J. Chisholm, R. Patani, J. F. Wu, S. Fernando, K. Jogodzinski and D. C. Webster, Soy-Based UV-Curable Thiol–Ene Coatings, J. Coat. Technol. Res., 2010, 7(5), 603–613, DOI:10.1007/s11998-010-9241-x.
- M. Johansson, E. Malmström and A. Hult, Synthesis, Characterization, and Curing of Hyperbranched Allyl Ether–Maleate Functional Ester Resins, J. Polym. Sci., Part A: Polym. Chem., 1993, 31(3), 619–624, DOI:10.1002/pola.1993.080310304.
- I. Fleming and D. Williams, Spectroscopic Methods in Organic Chemistry, Springer, Cham, Switzerland, Seventh edition, 2019 Search PubMed.
- D. A. C. Compton, W. O. George and A. J. Porter, Conformations of Some Aβ-Unsaturated Carbonyl Compounds. Part VII. Assignments of Raman and Infrared Spectra of Dimethyl, Diethyl, and Di-n-Butyl Fumarates and Maleates, J. Chem. Soc., Perkin Trans. 2, 1975,(4), 400–404, 10.1039/P29750000400.
- H.-G. Buge, P. Reich and E. Steger, Vibrational Spectra and Molecular Conformation of Diethyl Fumarate and Especially the Maleate, J. Mol. Struct., 1976, 35(2), 175–179, DOI:10.1016/0022-2860(76)82037-6.
- E. D. Schmid, P. Schlenker and R. R. M. Brand, Raman Intensity and Conjugation. IV-Determination of the Conjugation between a Phenyl Ring and a Carbonyl Group by Raman Intensity, J. Raman Spectrosc., 1977, 6(6), 314–318, DOI:10.1002/jrs.1250060610.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953 Search PubMed.
- O. Okay and C. N. Bowman, Kinetic Modeling of Thiol-Ene Reactions with Both Step and Chain Growth Aspects, Macromol. Theory Simul., 2005, 14(4), 267–277, DOI:10.1002/mats.200500002.
- M. C. Burroughs, T. H. Schloemer, D. N. Congreve and D. J. Mai, Gelation Dynamics during Photo-Cross-Linking of Polymer Nanocomposite Hydrogels, ACS Polym. Au, 2023, 3(2), 217–227, DOI:10.1021/acspolymersau.2c00051.
- C. Creton, 50th Anniversary Perspective: Networks and Gels: Soft but Dynamic and Tough, Macromolecules, 2017, 50(21), 8297–8316, DOI:10.1021/acs.macromol.7b01698.
- K. L. Ngai and R. W. Rendell, From Conformational Transitions in a Polymer Chain to Segmental Relaxation in a Bulk Polymer, J. Non-Cryst. Solids, 1991, 131–133, 942–948, DOI:10.1016/0022-3093(91)90706-C.
- C. M. Roland’ and K. L. Ngai, Segmental Relaxation and the Correlation of Time and Temperature Dependencies in Poly(Viny1 Methyl Ether)/Polystyrene Mixtures, Macromolecules, 1992, 25(1), 363–367, DOI:10.1021/ma00027a056.
- J. Roovers and P. M. Toporowski, Microheterogeneity in Miscible Blends of 1,2-Polybutadiene and 1,4-Polyisoprene, Macromolecules, 1992, 25(13), 3454–3461, DOI:10.1021/ma00039a023.
- J. J. Schwartz and A. J. Boydston, Multimaterial Actinic Spatial Control 3D and 4D Printing, Nat. Commun., 2019, 10(1), 791, DOI:10.1038/s41467-019-08639-7.
- Z. Zhao, J. Wu, X. Mu, H. Chen, H. J. Qi and D. Fang, Origami by Frontal Photopolymerization, Sci. Adv., 2017, 3(4), e1602326, DOI:10.1126/sciadv.1602326.
- U. Shaukat, E. Rossegger and S. Schlögl, A Review of Multi-Material 3D Printing of Functional Materials via Vat Photopolymerization, Polymers, 2022, 14(12), 2449, DOI:10.3390/polym14122449.
- L. Yue, S. M. Montgomery, X. Sun, L. Yu, Y. Song, T. Nomura, M. Tanaka and H. Jerry Qi, Single-Vat Single-Cure Grayscale Digital Light Processing 3D Printing of Materials with Large Property Difference and High Stretchability, Nat. Commun., 2023, 14(1), 1251, DOI:10.1038/s41467-023-36909-y.
- X. Kuang, J. Wu, K. Chen, Z. Zhao, Z. Ding, F. Hu, D. Fang and H. J. Qi, Grayscale Digital Light Processing 3D Printing for Highly Functionally Graded Materials, Sci. Adv., 2019, 5(5), eaav5790, DOI:10.1126/sciadv.aav5790.
- N. D. Dolinski, E. B. Callaway, C. S. Sample, L. F. Gockowski, R. Chavez, Z. A. Page, F. Eisenreich, S. Hecht, M. T. Valentine, F. W. Zok and C. J. Hawker, Tough Multimaterial Interfaces through Wavelength-Selective 3D Printing, ACS Appl. Mater. Interfaces, 2021, 13(18), 22065–22072, DOI:10.1021/acsami.1c06062.
- A. S. Kuenstler, J. J. Hernandez, M. Trujillo-Lemon, A. Osterbaan and C. N. Bowman, Vat Photopolymerization Additive Manufacturing of Tough, Fully Recyclable Thermosets, ACS Appl. Mater. Interfaces, 2023, 15(8), 11111–11121, DOI:10.1021/acsami.2c22081.
- J. J. Schwartz, D. H. Porcincula, C. C. Cook, E. J. Fong and M. Shusteff, Volumetric Additive Manufacturing of Shape Memory Polymers, Polym. Chem., 2022, 13(13), 1813–1817, 10.1039/D1PY01723C.
- P. Marx, A. Romano, I. Roppolo, A. Chemelli, I. Mühlbacher, W. Kern, S. Chaudhary, T. Andritsch, M. Sangermano and F. Wiesbrock, 3D-Printing of High-κ Thiol-Ene Resins with Spiro-Orthoesters as Anti-Shrinkage Additive, Macromol. Mater. Eng., 2019, 304(12), 1900515, DOI:10.1002/mame.201900515.
- D. G. Sycks, T. Wu, H. S. Park and K. Gall, Tough, Stable Spiroacetal Thiol-Ene Resin for 3D Printing, J. Appl. Polym. Sci., 2018, 135(22), 1–12, DOI:10.1002/app.46259.
- D. Ahn, L. M. Stevens, K. Zhou and Z. A. Page, Additives for Ambient 3D Printing with Visible Light, Adv. Mater., 2021, 33(44), 2104906, DOI:10.1002/adma.202104906.
- U. Shaukat, E. Rossegger and S. Schlögl, Thiol–Acrylate Based Vitrimers: From Their Structure–Property Relationship to the Additive Manufacturing of Self-Healable Soft Active Devices, Polymer, 2021, 231, 124110, DOI:10.1016/j.polymer.2021.124110.
- L. Shahzadi, F. Maya, M. C. Breadmore and S. C. Thickett, Functional Materials for DLP-SLA 3D Printing Using Thiol–Acrylate Chemistry: Resin Design and Postprint Applications, ACS Appl. Polym. Mater., 2022, 4(5), 3896–3907, DOI:10.1021/acsapm.2c00358.
- R. J. Chethalen, E. J. Fastow, E. B. Coughlin and K. I. Winey, Thiol–Ene Click Chemistry Incorporates Hydroxyl Functionality on Polycyclooctene to Tune Properties, ACS Macro Lett., 2023, 12(1), 107–112, DOI:10.1021/acsmacrolett.2c00670.
- G. I. Peterson, J. J. Schwartz, D. Zhang, B. M. Weiss, M. A. Ganter, D. W. Storti and A. J. Boydston, Production of Materials with Spatially-Controlled Cross-Link Density via Vat Photopolymerization, ACS Appl. Mater. Interfaces, 2016, 8(42), 29037–29043, DOI:10.1021/acsami.6b09768.
- Q. Wang, H. Cui, X. Wang, Z. Hu, P. Tao, M. Li, J. Wang, Y. Tang, H. Xu and X. He, Exceptional Light Sensitivity by Thiol–Ene Click Lithography, J. Am. Chem. Soc., 2023, 145(5), 3064–3074, DOI:10.1021/jacs.2c11887.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01361a |
| This journal is © The Royal Society of Chemistry 2025 |