
Enhancing CO2 hydrogenation to methanol via the synergistic effect of MoS2 interlayer spacing and sulfur vacancies†
Langlang
Qin
a,
Yunfei
Gao
 *b,
Caiyun
Han
a,
Minghui
Zhu
d and
Shuang
Wang
*ac
*b,
Caiyun
Han
a,
Minghui
Zhu
d and
Shuang
Wang
*ac
aCollege of Environmental Science and Engineering, Taiyuan University of Technology, Jinzhong 030600, Shanxi, P. R. China. E-mail: wangshuang@tyut.edu.cn
bInstitute of Clean Coal Technology, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: yunfeigao@ecust.edu.cn
cShanxi Key Laboratory of Gas Energy Efficient and Clean Utilization, Taiyuan University of Technology, Taiyuan 030024, Shanxi, P. R. China
dState Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, PR China
First published on 4th July 2024
Abstract
The hydrogenation of carbon dioxide (CO2) to methanol is an important reaction to convert CO2 into valuable products and reduce carbon emission. MoS2 is an effective catalyst for CO2 hydrogenation, but the synergistic effects of interlayer spacing expansion and surface sulfur vacancy strengthening have not been studied systematically. Here, this work reports hydrazine hydrate as an effective reducing agent for MoS2. The reducing agent can not only expand the interlayer spacing of MoS2, but also increase the concentration of sulfur vacancies through a simple treatment. More importantly, the synergistic effect between the interlayer spacing and sulfur vacancies of MoS2 significantly increases the methanol space-time yield (STY). At 220 °C, 4 MPa, and 8000 mL gcat−1 h−1, the MoS2–N2H4-4 catalyst exhibits 76.8% methanol selectively, 5.52% CO2 conversion, and a high methanol STY of up to 0.1214 g gcat−1 h−1 and lasts for at least 200 h. The structure–performance relationship was further studied using physio-chemical characterization and DFT. These results provide valuable insights into the development of highly efficient MoS2 catalysts for CO2 hydrogenation.
 Yunfei Gao | Yunfei Gao has been a professor at the East China University of Science and Technology (ECUST) since 2021. He graduated from North Carolina State University with a Ph.D. in 2019, and Tsinghua University in China as a bachelor in 2014. His work is mainly focused on CO2 utilization, hydrogen production and selective oxidative dehydrogenation of light alkanes using chemical looping technologies. He has published more than 40 papers in Science Advances, Nat. Commun., Energy Environ. Sci. and many other top journals. He is currently the project leader of several National Science Foundation Programs of China. |
1 Introduction
The rapid growth of the global economy has led to a large amount of carbon dioxide (CO2) emission, which has destroyed the ecological balance and caused various environmental problems.1,2 However, CO2 is also a cheap and readily available renewable carbon resource that can be converted into clean fuels and high-value chemical products (such as carbon monoxide, methanol, methane, etc.).3–5 Methanol exhibits favourable transportability and serves as fundamental feedstock for high-value chemicals (e.g., olefins and aromatics) as well as fuels (e.g., gasoline).6–8 Therefore, researchers have developed a strong interest in CO2 hydrogenation to methanol. At present, the main catalysts developed for CO2 hydrogenation to methanol are Cu-metal oxides,4 In2O3-based oxides,9,10 ZnO/ZrO2-solid solution,11 MoS2,12,13etc.In recent years, molybdenum disulfide (MoS2) has been explored in the process of CO2 hydrogenation to methanol and has shown excellent performance.12–15 There is a strong structure–activity relationship between the morphology of MoS2 and its CO2 hydrogenation performance.16,17 For example, Deng et al. prepared few-layer MoS2, which achieved a selectivity of 94.3% for methanol and a conversion of 12.5% of CO2 at 180 °C, attributed to the exposure of more active sites in the few-layer structure.18 Zhou et al. developed boxlike assemblies of quasi-single-layer MoS2 nanosheets (h-MoS2/ZnS), and the space-time yield (STY) of CO2 hydrogenation to methanol reached 0.93 gCH3OH gMoS2−1 h−1 at 260 °C.19 More importantly, highly dispersed molybdenum disulfide nanosheets with fewer layers can promote the generation of abundant active sites. They discovered that CO2 is decomposed into CO* on MoS2, leading to improved selectivity of methanol.18,19 In addition to the few-layer structure of MoS2, sulfur vacancies also play a key role in its catalytic ability. Fei et al. found that the presence of Sv caused an upshift in the d-band centre according to density functional theory (DFT), which enhanced the adsorption of reaction intermediates.20 They prepared a FL-MoS2-20 catalyst with a Sv concentration of 16.3% by a chemical reduction method, which showed excellent NH3 yield. Zhou et al. pointed out that the appropriate Sv concentration of MoS2 can greatly improve its proton adsorption/desorption capacity.21 From theoretical and experimental results, it can be seen that strengthening Sv while maintaining the few-layer structure has the potential to achieve the enhancement of catalytic performance.
The current techniques for Sv construction primarily include plasma treatment,22–25 heteroatom doping,14,17,26–28 chemical reduction,21,29–31etc., enabling precise control over the concentration of Sv. Jin et al. synthesized a NiS2 catalyst by using an argon plasma etching strategy; when the concentration of Sv was 5.9%, its hydrogen evolution reaction (HER) performance was greatly improved.32 Zhou et al. used hydrothermal doping of copper into MoS2 nanosheets, which doubled the number of Sv on MoS2, thereby increasing the STY of CO2 hydrogenation to methanol by 2.27 times.33 However, ion etching and heteroatom doping are relatively complex, and Ma et al. constructed a MoS2/C composite with Sv by chemical reduction, and used hydrazine hydrate to regulate the concentration of Sv to achieve efficient storage of Na+.34 The chemical reduction method is used to post-treat the catalyst, which is easy to operate and can accurately regulate Sv. Hence, the chemical reduction method is a simple and effective strategy for constructing Sv.
In this work, a series of MoS2 catalysts with different Sv concentrations and interlayer spacing were prepared by the chemical reduction method, and the Sv concentration in MoS2 was regulated by changing the concentration of hydrazine hydrate. By comparing different reductants, it was found that the MoS2–N2H4-4 catalyst had abundant Sv and large interlayer spacing. The characterization results showed that hydrazine hydrate successfully regulated both the concentration of Sv and interlayer spacing. Theoretical calculations show that the introduction of Sv can increase the electron density of Mo atoms, enhance the adsorption capacity of MoS2 to CO2, and increase the methanol STY. The synergistic effect of interlayer spacing and Sv plays a momentous role in tuning the activity of MoS2.
2 Experimental
2.1 Preparation of catalysts
Initially, 3 mmol of sodium molybdate (Na2MoO4) and 3 mmol of thioacetamide (TAA) were dissolved in 60 mL of deionized water. The resultant mixture was vigorously stirred for one hour and then transferred to a polytetrafluoroethylene (PTFE)-lined stainless steel high-pressure reactor. Subsequently, the solution was heated to 180 °C and maintained at this temperature for 24 hours. Upon cooling to room temperature, the precipitate was repeatedly washed with water and ethanol, and the resultant black precipitate was collected using centrifugation at 5000 rpm. Then, the collected material was vacuum-dried overnight at 70 °C, resulting in the MoS2 sample. Subsequently, X mL (X = 2, 4, and 8) of N2H4·H2O was added to 100 mg of the MoS2 sample. The reaction lasted for 3 hours, leading to different Sv concentrations, denoted as MoS2–N2H4-X (X = 2, 4, and 8). MoS2–NH3 and MoS2–NaBH4 were produced by the same procedure except using the corresponding reductants (NH3·H2O and NaBH4). A detailed list of the materials, measurements, and calculation parameters are provided in the ESI.†2.2 DFT calculations
See the ESI† for specific information on model building and calculations.3 Results and discussion
3.1 Structural and morphological properties of MoS2 after reductant treatment
The preparation route of MoS2 materials treated with different reductants is presented in Fig. 1a. During the progression of the hydrothermal reaction, the molybdenum source and sulfur sources react to generate MoS2. Three different reductants (N2H4·H2O, NH3·H2O, and NaBH4) were used to post-process MoS2 based on their different chemical reduction and molecular intercalation abilities to obtain MoS2 samples with varying interlayer spacing and Sv concentrations.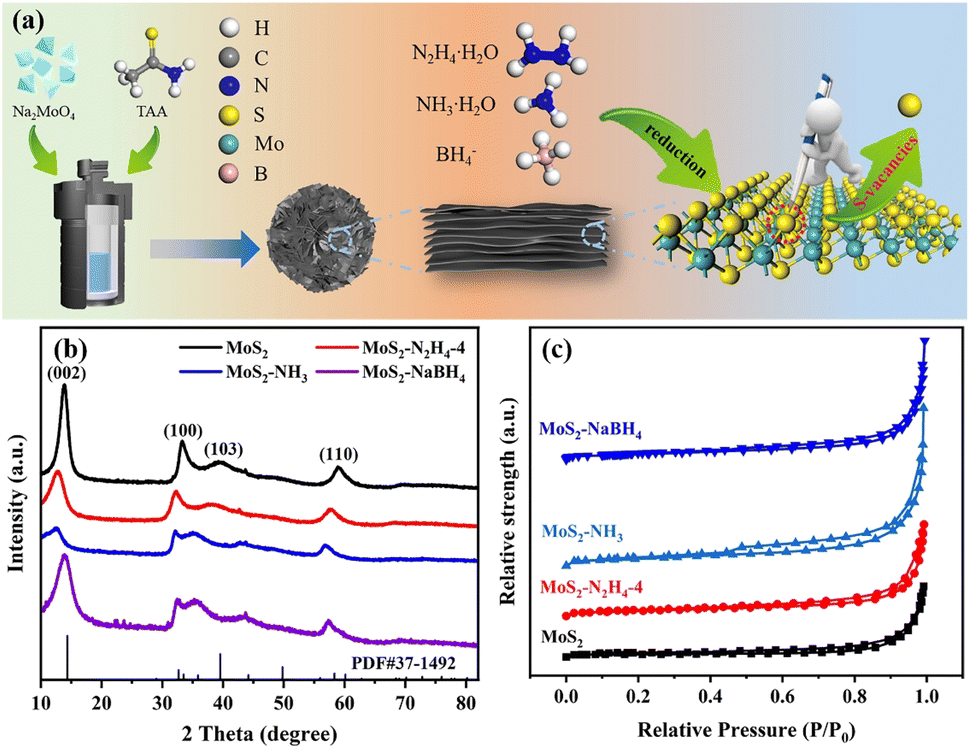 | ||
| Fig. 1 (a) Sketch of preparation of MoS2 with varying interlayer spacing and Sv concentrations via the chemical reduction method. (b) XRD patterns and (c) N2 adsorption–desorption isotherms of MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 catalysts. | ||
The composition of the sample was analyzed by X-ray diffraction (XRD) and the resulting XRD data were compared with the standard spectra of 2H-MoS2. As shown in Fig. 1b and S1,† all diffraction peaks were observed to coincide with those of 2H-MoS2, indicating that the MoS2 phase was successfully prepared in the sample. Upon chemical reduction, the XRD spectra of all samples revealed diffraction peaks characteristic of MoS2, which were observed to be consistent with the peak positions documented in PDF-37-1492. Specifically, the diffraction peaks at approximately 33.5° and 58.3° corresponded to the (100) and (110) planes, respectively, while the most prominent peak of the (002) plane was located at 14.3°. Observably, the peak intensity of the (002) crystal plane decreased for all three catalysts, and the peak intensity of MoS2–NH3 was the lowest, which was theoretically attributed to the monolayer or the few-layer structure of MoS2.35 Interestingly, with the increase of hydrazine hydrate addition, the (002) reflection peak of MoS2–N2H4-8 appeared to be almost unidentifiable, indicating the stacking of only a few MoS2 layers in the c-direction. Furthermore, no additional peaks were observed after chemical reduction treatment, demonstrating that the chemical reduction reaction utilizing reductants did not produce other phases or alter the crystal structure.
N2 adsorption/desorption experiments were carried out at 77 K to investigate the pore properties and specific surface area of the prepared catalyst. As shown in Fig. 1c and S2,† N2 physisorption isotherms indicated that these samples all exhibit typical type-IV curves and a distinct H3-type hysteresis loop, a distinctive characteristic of mesoporous materials. The Brunauer–Emmett–Teller (BET) specific surface areas of MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 were determined to be 16.1, 26.1, 37.5 and 26.5 m2 g−1 (Table S1†), respectively, indicating an increased specific surface area after chemical treatment.
To determine whether the reduction by the three reductants has disrupted the original morphology, all the samples were observed under an electron microscope (SEM) (Fig. 2a1–d1 and S3†). Overall, all the samples were spherical in shape, composed of tiny nanosheets forming a nanoflower morphology, with particle diameters ranging from 300 nm to 500 nm, and their appearance remained unchanged following the chemical reduction reaction with hydrazine hydrate. The well-defined structure with a nanoflower was further confirmed by the TEM technique (Fig. 2a2–d2). Moreover, the sample exhibits stacked spheres with a diameter of approximately 300–350 nm. There were many transparent layers in the nanoflower of MoS2–N2H4-4 and MoS2–NH3, which confirmed that the two catalysts had fewer layers, consistent with the results of XRD analysis. In addition, the high-resolution transmission electron microscopy (HRTEM) images (Fig. 2a3–d3) showed a typical lamellar structure with a well-resolved d-spacing. It can be clearly seen that the original MoS2 nanoflower has more edges that exhibit a layer-to-layer spacing value of 0.64 nm, which is consistent with the XRD results. Besides, there are lattice fringes observed in the MoS2–N2H4-4 and MoS2–NH3 nanostructures that correspond to the (002) plane of MoS2 with an enlarged interlayer spacing of 0.74 nm. This is because the intercalation of ammonium expands the interlayer spacing of MoS2 during synthesis, thus favoring the exposure of more active sites.36,37 In contrast, sodium borohydride has a lower reducing ability. The layer spacing does not change and remained at 0.64 nm.
 | ||
| Fig. 2 (a1–d1) SEM, (a2–d2) TEM and (a3–d3) HRTEM images of (a1–a3) MoS2, (b1–b3) MoS2–N2H4-4, (c1–c3) MoS2–NH3 and (d1–d3) MoS2–NaBH4 catalysts. | ||
The d(002) of MoS2 was analyzed and measured using Bragg's law (2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ) for XRD data (Fig. 1b and S4†). The calculation results (Table S1†) show that the d(002) of MoS2–N2H4-4 is close to that of MoS2–NH3 and larger than that of MoS2 and MoS2–NaBH4, which is consistent with the TEM test results.
sinθ = nλ) for XRD data (Fig. 1b and S4†). The calculation results (Table S1†) show that the d(002) of MoS2–N2H4-4 is close to that of MoS2–NH3 and larger than that of MoS2 and MoS2–NaBH4, which is consistent with the TEM test results.
3.2 Sulfur vacancy of MoS2 after reductant treatment
To investigate the sulfur vacancies after reductant treatment, X-ray photoelectron spectroscopy (XPS) was utilized to study the chemical state and electronic structure of the different samples (Fig. 3). From the survey spectrum (Fig. 3a and S5†), it can be determined that all samples contain Mo and S elements. In Fig. 3b, the two peaks at 229.5 and 232.5 eV correspond to the 3d5/2 and 3d3/2 of Mo, while the short peak at 226.6 eV represents the 2 s electron of S. Specifically, the 3d peak of Mo can be resolved into two components, Mo4+ and Moδ+.19,38 According to the spectrogram, after chemical reduction treatment, the Moδ+ content of all catalysts was significantly higher than that of common MoS2, showing more Sv, because the small shoulder observed near Moδ+ (δ < 4) with low binding energy was due to the formation of Mo sites with unsaturated ligands.39 According to the ratio of the Moδ+ peak area, the order of Sv concentration can be determined as: MoS2–NaBH4 > MoS2–N2H4-4 > MoS2–NH3 > MoS2. And the Mo characteristic peak shifted towards lower binding energy. Despite its abundant sulfur vacancies, MoS2–NaBH4 was the worst performing in CO2 hydrogenation to methanol. Thus, having sulfur vacancies alone does not increase methanol production. | ||
| Fig. 3 (a) The XPS survey spectra and XPS spectra of (b) Mo 3d and (c) S 2p states in MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 catalysts. (d) Raman spectra of MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 catalysts. | ||
It was validated that Sv concentration can be further increased by chemical reduction treatment with hydrazine hydrate. According to Fig. 3b and S6a,† it can be found that with the increase of the dosage of hydrazine hydrate, the characteristic Mo peak moves towards lower binding energy and the Moδ+ peak area increases, which may be due to the reduction of hydrazine hydrate. In the S 2p XPS spectra of all the samples (Fig. 3c), the two peaks of S 2p3/2 and S 2p1/2 at 162.2 and 163.4 eV can be clearly distinguished in MoS2. Compared with the S 2p XPS spectra of MoS2, MoS2–N2H4-4 and MoS2–NaBH4 sample characteristic peak shift occurs, and this may be related to the generation of Sv.40 In contrast, ammonia liquor can only expand the interlayer spacing, and the two peaks of S 2p did not change. In addition, significant peak shifts were also observed in the S 2p XPS spectra of MoS2–N2H4-2 and MoS2–N2H4-8 (Fig. S6b†). With the increase of hydrazine hydrate addition, the peak shift was more obvious, indicating the presence of more Sv.41,42
The structural characteristics of Sv in these catalysts were investigated by Raman spectroscopy in this experiment. As shown in Fig. 3d, all samples exhibited the main characteristic Raman vibration peaks of MoS2 at 360–420 cm−1, corresponding to the in-plane Mo–S phonon mode (E12g) and out-of-plane Mo–S mode (A1g), respectively.43,44 The A1g and E12g vibration modes have changed for the expanded interlayer MoS2 materials as compared to MoS2.45 In detail, the A1g peaks blue shift from 408.2 cm−1 for MoS2 to 404.4 cm−1 for MoS2–N2H4-4 and to 406 cm−1 for MoS2–NH3 and to 405.4 cm−1 for MoS2–NaBH4, while the E12g peaks shift from 376 cm−1 for MoS2 to 378.2 cm−1 for MoS2–N2H4-4 and to 375 cm−1 for MoS2–NH3 and to 373 cm−1 for MoS2–NaBH4. It was worth noting that the E12g and A1g peak spread of MoS2–N2H4-4 and MoS2–NaBH4 was wider than that of MoS2. This is due to the Sv perturbation in the 2H lattice resulting in the reduction of the Mo–S bond and the weakening of the energy of the vibration mode, resulting in the melting of the Mo–S phonon mode in the principal plane.46–48 The above results are in agreement with our results using the XPS method. In addition, the distance between the E12g and A1g peaks of MoS2–N2H4-4 and MoS2–NH3 was reduced, indicating a diminished interaction between adjacent MoS2 layers, because the interlayer spacing of MoS2–N2H4-4 and MoS2–NH3 was expanded compared to the original MoS2. MoS2–N2H4-2 and MoS2–N2H4-8 also conform to the above rules (Fig. S7†); with the increase of the amount of hydrazine hydrate, the peak position is shifted, and the peak spread becomes wider, indicating that the Sv concentration increases (Table 1).49
In order to visually demonstrate the concentration changes of Sv in catalysts, MoS2, MoS2–N2H4, MoS2–NH3, and MoS2–NaBH4 were characterized by electron paramagnetic resonance (EPR).50–53 The signal at ∼330 mT (g = 2.0) in Fig. S8† indicated the concentration of unsaturated sites with unpaired electrons, which is proportional to the Sv in the sample. The analysis of the data revealed that original MoS2 exhibited some EPR signals, indicating that the MoS2 catalyst can generate some Sv after H2 activation. MoS2–NaBH4 exhibited the highest EPR signal, suggesting the strongest charge compensation effect and thus the highest Sv concentration. However, this contradicts the significant increase in methanol production with increasing Sv. This phenomenon is attributed to the change in the nature of the defects. Under the appropriate reducing agent's reduction ability, “point” defects are formed. Excessive reduction ability causes S atoms to detach, resulting in larger defects,54,55 and exposed edge S vacancies are not conducive to methanol production. Additionally, the peak area and intensity of MoS2–N2H4 were significantly higher than those of MoS2 and MoS2–NH3, indicating that MoS2 expands the interlayer spacing while undergoing reduction, leading to more sulfur vacancies. Therefore, both the reducing ability of the reducing agent and the enlargement of the interlayer distance contribute to increasing the number of Sv. Combining TEM, XPS and EPR test results, it is found that the enriched Sv may also contribute to the enlarged interlayer spacing.
To summarize, N2H4·H2O, NH3·H2O and NaBH4 treatments all lead to changes in the original MoS2 in terms of the structure, morphology and sulfur vacancies. But the effects are different due to their distinct chemical reduction and interlayer expansion capabilities. In short, NaBH4 can lead to the enhancement of sulfur vacancies but did not expand the interlayer spacing substantially. NH3·H2O expands the interlayer spacing but did not lead to the enhancement of sulfur vacancies due to its lack of reducing capabilities. Meanwhile, N2H4·H2O leads to both enhanced sulfur vacancies and increased interlayer spacing. The effects of these structure, morphology and sulfur vacancy changes will be elaborated in the following section.
3.3 Catalytic performance evaluation for CO2 hydrogenation
To illustrate the advantage of the synergistic effect between the interlayer spacing and sulfur vacancies, the MoS2 catalysts and three catalysts treated with chemical reduction were initially compared through the reaction of CO2 hydrogenation over a temperature range of 180–260 °C at a gaseous hourly space velocity (GHSV) of 8000 mL gcat−1 h−1. Within this reaction system, methanol was the desired product, and CO and CH4 were identified as by-products in all tested scenarios. Fig. 4 shows the CO2 space-time yield of methanol (STYCH3OH), conversion, and methanol selectivity on MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 catalysts. It can be seen from Fig. 4a that under the same reaction conditions, MoS2–N2H4-4 has the best performance. Under the optimal reaction conditions, at 220 °C, the STYCH3OH of the MoS2–N2H4-4 catalyst can reach 0.1214 g gcat−1 h−1, the CO2 conversion rate is 5.52%, and the methanol selectivity is 76.8%. The STYCH3OH of the MoS2–N2H4-4 catalyst is 2.43 times higher than that of the MoS2 catalyst, and the optimal temperature is 20 °C lower. This is because MoS2–N2H4-4 has the highest methanol selectivity and high CO2 conversion at any reaction temperature (Fig. 4b and c), which confirmed that the Sv can indeed improve the performance of the catalyst. At low temperature (180 °C), the MoS2–NH3 catalyst exhibits excellent methanol properties. Under the same reaction conditions, at 200 °C, the STYCH3OH of the MoS2–NH3 catalyst can reach 0.0772 g gcat−1 h−1, the CO2 conversion rate is 3.65%, and the methanol selectivity is 73.99%. However, as the temperature increased (>200 °C), the methanol production began to decline, and beyond 240 °C, the performance of the catalyst was not even as good as that of the original MoS2. This may be attributed to the fact that although the CO2 conversion rate increased with the increase of temperature, which was comparable to that of MoS2–N2H4-4, the methanol selectivity decreased, resulting in methane becoming the main product. This may be because the active sites exposed by the catalyst are the edge sulfur vacancies. The results in Fig. 4 show that the methanol yield, CO2 conversion and methanol selectivity of the MoS2–NaBH4 catalyst are lower than those of the original MoS2 catalyst. This may be because the reducibility of sodium borohydride is too strong, which leads to a decrease in the dissociation ability of MoS2 to hydrogen. Some studies have shown that the CO2 conversion is not only related to the catalyst's ability to activate CO2, but also related to the catalyst's ability to activate H2.2 When CO2 was adsorbed to the surface of the catalyst and CO was produced, due to the lack of sufficient hydrogen ions for the hydrogenation reaction, CO cannot be further reduced and was desorbed, so the main product of the MoS2–NaBH4 catalyst was CO. According to the experimental results, with the increase of reaction temperature, the CO2 conversion increases, but the selectivity of methanol decreases. This phenomenon can be attributed to the exothermic nature of the methanol production reaction (CO2 + H2 → CH3OH + H2O, ΔĤ298 K = −49.5 kJ mol−1), so it has obvious disadvantages in thermodynamics at high temperature. Alternatively, with the increase of reaction temperature, the improved CO selectivity is related to the reverse water–gas shift reaction (RWGS, CO2 + H2 → CO + H2O, ΔĤ298 K = 41.2 kJ mol−1), which is a main side reaction of CO2 hydrogenation. It was also found that the best reaction temperature of MoS2–NaBH4 catalysts was 240 °C, lower than that of the original MoS2. The decrease of the optimal reaction temperature of the MoS2 catalyst may be related to the expansion of MoS2 interlayer spacing. | ||
| Fig. 4 (a) STY of CH3OH and (b) CO2 conversion on MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 catalysts. (c) Product selectivity of MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 catalysts. Reaction conditions: VCO2/H2 = 1/3, GHSV = 8000 mL gcat−1 h−1, and P = 4.0 MPa. | ||
According to the above performance comparison, it is found that hydrazine hydrate treated MoS2 has the best performance. Therefore, the added amount of hydrazine hydrate was optimized, and the optimized results are shown in Fig. S9 and S10.† Under the conditions of 4 MPa, 220 °C and GHSV = 8000 mL gcat−1 h−1, the methanol STY initially increased with the increase of the addition of hydrazine hydrate, and when the addition of hydrazine hydrate reached a certain amount (4 mL), the performance of the catalyst was stable and no longer increased, which indicated that the performance of the catalyst may be limited by other factors. After analysis, it was found that MoS2–N2H4-2, MoS2–N2H4-2 and MoS2–N2H4-8 had similar product selectivity, but MoS2–N2H4-2 had lower CO2 conversion, resulting in lower performance. In contrast, the CO2 conversion rates of MoS2–N2H4-4 and MoS2–N2H4-8 were almost equal. In conclusion, the addition amount of hydrazine hydrate can affect the conversion rate of CO2, but its effect gradually decreases after reaching a certain amount. Therefore, MoS2–N2H4-4 is considered to be the most appropriate catalyst for the addition of hydrazine hydrate.
The influence of GHSV was also investigated using the MoS2–N2H4-4 catalyst at 220 °C (Fig. 5a). As the GHSV increased, the conversion of CO2 decreased, while the selectivity of methanol and STYCH3OH increased. This indicates that at high GHSV, the residence time of the reactants is short and the RWGS reaction is limited. When the reaction pressure increases from 3 MPa to 5 MPa, both CO2 conversion and methanol selectivity are improved (Fig. S11†). Considering the CO2 conversion rate and the mild experimental conditions, we determined the optimized GHSV of 8000 mL gcat−1 h−1 and pressure of 4 MPa, as the basis for this study. According to the Arrhenius formula, the apparent activation energy (Ea) of the three catalysts (MoS2–N2H4, MoS2–NH3, and MoS2–NaPH4) was calculated (Fig. S12†), and the results showed that MoS2–N2H4 had the lowest Ea, indicating that it had the greatest advantage in reaction kinetics. However, MoS2–NaPH4 has the lowest Ea and the slowest reaction rate, which conforms to the performance law.
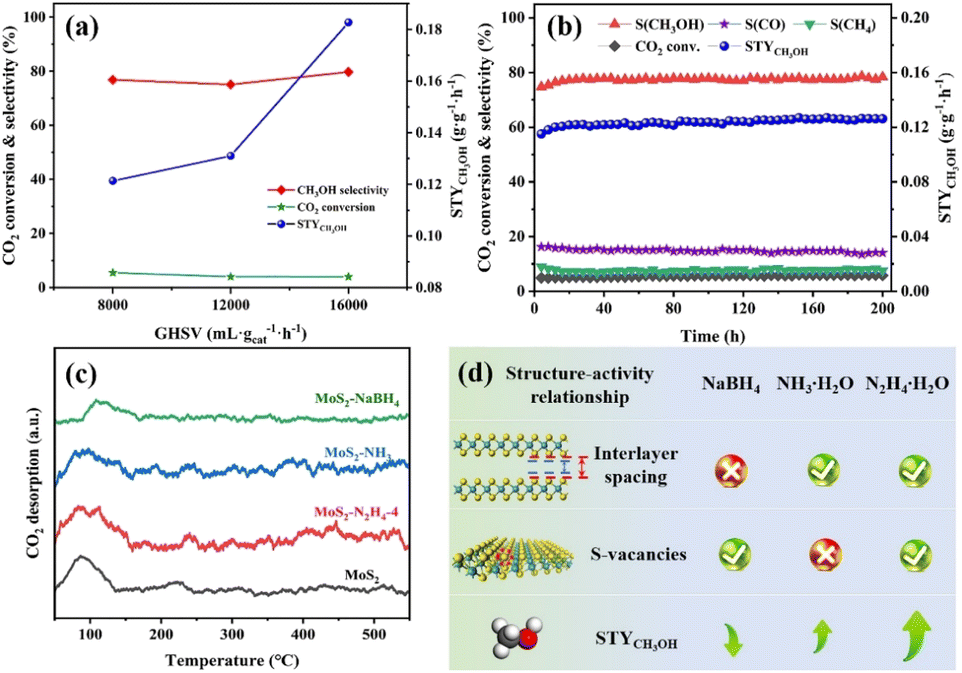 | ||
| Fig. 5 (a) Effect of GHSV on CO2 hydrogenation over the MoS2–N2H4-4 catalyst. Reaction conditions: T = 220 °C, VCO2/H2 = 1/3, and P = 4.0 MPa. (b) Long-term test of the MoS2–N2H4-4 catalyst. Reaction conditions: T = 220 °C, VCO2/H2 = 1/3, GHSV = 8000 mL gcat−1 h−1, and P = 4.0 MPa. (c) CO2-TPD profiles of MoS2, MoS2–N2H4-4, MoS2–NH3 and MoS2–NaBH4 catalysts. (d) Structure–activity relationship diagram between different chemical reductants and the STY, interlayer spacing and sulfur vacancies of MoS2. | ||
The long-term stability of the MoS2–N2H4-4 catalyst was tested for a total of 200 h at 220 °C (Fig. 5b). In the initial phase of CO2 conversion using the MoS2–N2H4-4 catalyst, the CO2 conversion and methanol selectivity showed a slow upward trend and stabilized at about 5.8% and 77%, respectively, after 20 hours. This suggests that the reducing environment of the reaction contributes to Sv formation. As the reaction time was extended to 200 h, the CO2 conversion rate, methanol selectivity and STYCH3OH showed no sign of weakening, indicating that MoS2–N2H4-4 possesses excellent stability. According to the SEM results (Fig. S13†), the nanoflower structure of MoS2–N2H4-4 remained good after long-term testing. The XRD pattern of the catalyst after the reaction (Fig. S14†) shows that the diffraction peak is almost the same as that of the fresh catalyst before the test, which proves that the MoS2–N2H4-4 catalyst has excellent structural stability. Comparing the XPS spectra of the catalyst MoS2–N2H4-4 before and after the reaction (Fig. S15†), it was found that the proportion of the Moδ+ peak area increased slightly, confirming that the reducing environment has increased Sv, consistent with the long-term stability performance test (Fig. 5b). The comparison of MoS2–N2H4-4 catalysts with reported methanol synthesis (Table S2†) showed that the prepared catalysts in this study have good CO2 hydrogenation performance.
The CO2-TPD technique was used to compare the CO2 adsorption capacity of all catalysts to explain the increased catalytic activities. To elucidate the reasons for the change of the CO2 conversion rate, the CO2 desorption peak in the 200–500 °C region was analysed, and the results are shown in Fig. 5c and S16.† It was found that the peak area of the catalyst decreased successively: MoS2–N2H4-4 > MoS2–NH3 > MoS2 > MoS2–NaBH4, which was consistent with the trend of the CO2 conversion rate in Fig. 4b. Therefore, the high CO2 conversion rate of the MoS2–N2H4-4 catalyst was due to the production of a large number of alkaline sites, which may be caused by the Sv of the catalyst, indicating that the reduction of hydrazine hydrate can enhance the adsorption capacity of CO2. The CO2 adsorption peak of the MoS2–NaBH4 catalyst appears last, indicating that the MoS2–NaBH4 catalyst had the weakest CO2 adsorption/activation capacity. It can be seen that the reductant with stronger reducing capacity is not more favorable to the CO2 hydrogenation reaction. In addition, with the increase of hydrazine hydrate addition, the CO2 adsorption area of MoS2–N2H4-2 and MoS2–N2H4-8 also gradually expanded (Fig. S16†).
The structure–performance relationship of MoS2 treated with different chemical agents is summarized in Fig. 5d, where expanded interlayer spacing and enhanced sulfur vacancies lead to the highest methanol STY. It can also be inferred that by increasing Sv concentration and interlayer spacing, the adsorption of CO2 can be promoted, and the conversion rate of CO2 can be improved. MoS2–N2H4 can release NH4+ to expand the interlayer spacing, and the reducing agent is more conducive to enter the interlayer for reduction, resulting in in-plane Sv, thereby increasing the methanol yield. The methanol yield of expanding the interlayer spacing alone or increasing the Sv alone is lower than that of the combination of the two. Therefore, there is a synergistic effect between the interlayer spacing and the Sv.
3.4 Density functional theory studies
Density functional theory (DFT) was employed to investigate pristine MoS2 and MoS2-Sv, focusing on the impact of S vacancies on the adsorption of CO2 in the context of CO2 hydrogenation to methanol. The DFT models constructed were MoS2, MoS2-Sv1, MoS2-Sv2, and MoS2-Sv3 (Fig. S17 and S18†). The S-vacancy samples showed better electron transfer capacity in terms of charge density difference diagrams (Fig. 6a) during the CO2 adsorption process compared with the vacancy-free sample. Furthermore, it was evident that an increase in sulfur vacancies results in the Mo atom gaining more electrons, leading to enhanced electron density and subsequently improving CO2 adsorption. Based on the calculated total density of states (TDOS) (Fig. 6b), the samples with sulfur vacancies exhibited slightly smaller MoS2 bandgaps compared to the vacancy-free sample. The increasing presence of sulfur vacancies brought the conduction band closer to the Fermi level, resulting in enhanced electron transport.34 This enhancement facilitated the conversion of CO2 into methanol and improved selectivity. The adsorption energy (Eads) for CO2 adsorption was calculated using the equation Eads = ECO2+surface − ECO2 − Esurface.13 As shown in Fig. 6c, the adsorption energy of MoS2 with sulfur vacancies on CO2 was significantly higher than that of the original MoS2 catalyst, indicating that sulfur vacancies can significantly enhance the CO2 adsorption and thus promote the CO2 conversion.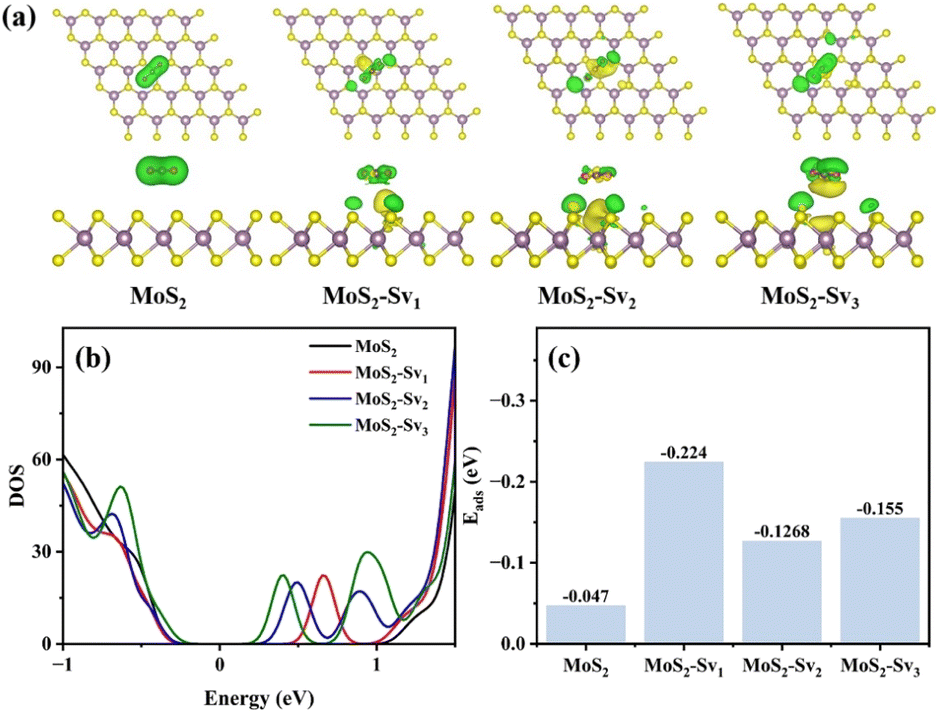 | ||
| Fig. 6 (a) DFT calculated charge density difference diagrams of CO2 adsorption, (b) total density of states, and (c) adsorption energy (Eads) of CO2. | ||
4 Conclusions
In summary, a series of MoS2 catalysts with different Sv concentrations and interlayer spacing were synthesized via a simple chemical reduction method. It was worth noting that when hydrazine hydrate was employed as a reductant in preparing the MoS2–N2H4-4 catalyst, it not only resulted in larger interlayer spacing but also led to an increased number of sulfur vacancies. Under their combined influence, methanol STY was significantly enhanced. DFT calculations revealed that sulfur vacancies in MoS2 increased electron density around Mo atoms, bringing them closer to the Fermi level and making active sites more reactive and catalytically efficient in CO2 hydrogenation towards methanol production. In summary, this work presents a new insight to resolve the reaction of MoS2 to catalyze CO2 valorization, highlighting its promising potential in efficient CO2 valorization or in other catalytic systems.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Langlang Qin: conceptualization, investigation, methodology, data curation, writing – original draft, validation, and visualization. Yunfei Gao: conceptualization, formal analysis, validation, supervision, writing – review & editing, and funding acquisition. Caiyun Han: writing – review & editing. Minghui Zhu: writing – review & editing. Shuang Wang: conceptualization, formal analysis, writing – review & editing, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work is financially supported by the National Natural Science Foundation of China (grant numbers 22078215 and 22208104) and Research Project by Shanxi Scholarship Council of China (grant number 2021-055).References
- S. De, A. Dokania, A. Ramirez and J. Gascon, ACS Catal., 2020, 10, 14147–14185 CrossRef CAS.
- S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- C. J. Huang, S. A. Zhang, W. B. Wang, H. Z. Zhou, Z. L. Shao, L. Xia, H. Wang and Y. H. Sun, ACS Catal., 2024, 14, 1324–1335 CrossRef CAS.
- Y. Wang, Y. Zhu, X. Zhu, J. Shi, X. Ren, L. Zhang and S. Li, ACS Catal., 2022, 13, 714–724 CrossRef.
- T. Biswal, K. P. Shadangi, P. K. Sarangi and R. K. Srivastava, Chemosphere, 2022, 298, 134299 CrossRef CAS.
- R. Sen, A. Goeppert and G. K. Surya Prakash, Angew. Chem., Int. Ed., 2022, 61, e202207278 CrossRef CAS.
- C. F. Shih, T. Zhang, J. H. Li and C. L. Bai, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- X. H. Lin, S. B. Wang, W. G. Tu, Z. B. Hu, Z. X. Ding, Y. D. Hou, R. Xu and W. X. Dai, Catal. Sci. Technol., 2019, 9, 731–738 RSC.
- Y. H. Wang, Y. Liu, L. Tan, X. H. Lin, Y. X. Fang, X. F. Lu, Y. D. Hou, G. G. Zhang and S. B. Wang, J. Mater. Chem. A, 2023, 11, 26804–26811 RSC.
- Z. D. Feng, C. Z. Tang, P. F. Zhang, K. Li, G. N. Li, J. J. Wang, Z. C. Feng and C. Li, J. Am. Chem. Soc., 2023, 145, 12663–12672 CrossRef CAS.
- S. H. Zhou, M. Kosari and H. C. Zeng, J. Am. Chem. Soc., 2024, 146, 10032–10043 CrossRef CAS.
- G. Wang, X.-L. Jiang, Y.-F. Jiang, Y.-G. Wang and J. Li, ACS Catal., 2023, 13, 8413–8422 CrossRef CAS.
- H.-Y. Su, X. Ma, C. Sun and K. Sun, Catal. Sci. Technol., 2021, 11, 3261–3269 RSC.
- Z. F. Wang, Y. R. Kang, J. T. Hu, Q. Q. Ji, Z. X. Lu, G. L. Xu, Y. T. Qi, M. Zhang, W. W. Zhang, R. Huang, L. Yu, Z. Q. Tian and D. H. Deng, Angew. Chem., Int. Ed., 2023, 62, e202307086 CrossRef CAS.
- S. Kanuri, S. Roy, C. Chakraborty, S. P. Datta, S. A. Singh and S. Dinda, Int. J. Energy Res., 2021, 46, 5503–5522 CrossRef.
- Y. Yuan, L. Qi, Z. Gao, T. Guo, D. Zhai, Y. He, J. Ma and Q. Guo, Molecules, 2023, 28, 5796 CrossRef CAS PubMed.
- J. T. Hu, L. Yu, J. Deng, Y. Wang, K. Cheng, C. Ma, Q. H. Zhang, W. Wen, S. S. Yu, Y. Pan, J. Z. Yang, H. Ma, F. Qi, Y. K. Wang, Y. P. Zheng, M. S. Chen, R. Huang, S. H. Zhang, Z. C. Zhao, J. Mao, X. Y. Meng, Q. Q. Ji, G. J. Hou, X. W. Han, X. H. Bao, Y. Wang and D. H. Deng, Nat. Catal., 2021, 4, 242–250 CrossRef CAS.
- S. Zhou and H. C. Zeng, ACS Catal., 2022, 12, 9872–9886 CrossRef CAS.
- H. Fei, R. Liu, J. Wang, T. Guo, F. Liu, Z. Wu and D. Wang, Chem. Eng. J., 2023, 476, 146895 CrossRef CAS.
- Y. Zhou, C. Li, Y. Zhang, L. Wang, X. Fan, L. Zou, Z. Cai, J. Jiang, S. Zhou, B. Zhang, H. Zhang, W. Li and Z. Chen, Adv. Funct. Mater., 2023, 33, 2304302 CrossRef CAS.
- J. W. Guo, H. B. Zhao, Z. W. Yang, L. W. Wang, A. Z. Wang, J. Zhang, L. H. Ding, L. F. Wang, H. Liu and X. Yu, Adv. Funct. Mater., 2024, 2315714 CrossRef CAS.
- C. Li, R. Z. Xu, S. X. Ma, Y. H. Xie, K. G. Qu, H. F. Bao, W. W. Cai and Z. H. Yang, Chem. Eng. J., 2021, 415, 129018 CrossRef CAS.
- A. Y. Lu, X. L. Yang, C. C. Tseng, S. X. Min, S. H. Lin, C. L. Hsu, H. N. Li, H. C. Idriss, J. L. Kuo, K. W. Huang and L. J. Li, Small, 2016, 12, 5530–5537 CrossRef CAS PubMed.
- L. Tian, Z. J. Huang, X. H. Lu, T. J. Wang, W. J. Cheng, H. M. Yang, T. Z. Huang, T. X. Li and Z. Li, Inorg. Chem., 2023, 62, 1659–1666 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- C. H. Yan, E. S. Han, X. H. Yang, K. S. Hu, H. T. Xu, Y. D. Li, Y. Z. He and S. Lu, Ceram. Int., 2023, 49, 14155–14165 CrossRef CAS.
- S. Q. Ding, Z. J. Li, X. Dai, C. L. Sun and A. L. Meng, Chem. Eng. J., 2021, 417, 129328 CrossRef CAS.
- Y. H. Ma, D. F. Leng, X. M. Zhang, J. J. Fu, C. R. Pi, Y. Zheng, B. A. Gao, X. G. Li, N. Li, P. K. Chu, Y. S. Luo and K. F. Huo, Small, 2022, 18, 2203173 CrossRef CAS.
- Z. S. Zhang, C. L. Mao, D. M. Meira, P. N. Duchesne, A. A. Tountas, Z. Li, C. Y. Qiu, S. L. Tang, R. Song, X. Ding, J. C. Sun, J. F. Yu, J. Y. Howe, W. G. Tu, L. Wang and G. A. Ozin, Nat. Commun., 2022, 13, 1512 CrossRef CAS PubMed.
- Y. Zhang, L. Ye, J. H. Guo, Y. Y. Shang, F. M. Guo, Y. J. Zhang and J. Xu, J. Mater. Sci., 2021, 56, 9368–9381 CrossRef CAS.
- J. Jin, X. Wang, Y. Hu, Z. Zhang, H. Liu, J. Yin and P. Xi, Nano-Micro Lett., 2024, 16, 63 CrossRef CAS.
- S. Zhou, W. Ma, U. Anjum, M. Kosari, S. Xi, S. M. Kozlov and H. C. Zeng, Nat. Commun., 2023, 14, 5872 CrossRef CAS.
- X. Ma, L. Diao, Y. Wang, L. Zhang, Y. Lu, D. Li, D. Yang and X. She, Chem. Eng. J., 2023, 457, 141116 CrossRef CAS.
- H. F. Li, Q. Yang, F. N. Mo, G. J. Liang, Z. X. Liu, Z. J. Tang, L. T. Ma, J. Liu, Z. C. Shi and C. Y. Zhi, Energy Storage Mater., 2019, 19, 94–101 CrossRef.
- J. W. Tan, W. B. A. Zhang, Y. J. Shu, H. Y. Lu, Y. Tang and Q. S. Gao, Sci. Bull., 2021, 66, 1003–1012 CrossRef CAS PubMed.
- T. L. Yan, Y. S. Jia, K. G. Hou, Q. S. Gao, Y. H. Zhang and Y. Tang, iScience, 2024, 27, 109824 CrossRef CAS PubMed.
- L. Li, Z. D. Qin, L. Ries, S. Hong, T. Michel, J. Yang, C. Salameh, M. Bechelany, P. Miele, D. Kaplan, M. Chhowalla and D. Voiry, ACS Nano, 2019, 13, 6824–6834 CrossRef CAS.
- R. Liu, L. N. Ma, G. D. Niu, X. L. Li, E. Y. Li, Y. Bai and G. H. Yuan, Adv. Funct. Mater., 2017, 27, 1701635 CrossRef.
- F. Zhou, S. Xin, H. W. Liang, L. T. Song and S. H. Yu, Angew. Chem., Int. Ed., 2014, 53, 11552–11556 CrossRef CAS PubMed.
- P. Shen, X. C. Li, Y. J. Luo, Y. L. Guo, X. L. Zhao and K. Chu, ACS Nano, 2022, 16, 7915–7925 CrossRef CAS PubMed.
- L. R. L. Ting, Y. L. Deng, L. Ma, Y. J. Zhang, A. A. Peterson and B. S. Yeo, ACS Catal., 2016, 6, 861–867 CrossRef CAS.
- T. Zhu, W. Shen, X. Y. Wang, Y. F. Song and W. Wang, Chem. Eng. J., 2019, 378, 122159 CrossRef CAS.
- G. H. Yang, L. Zhao, G. Q. Huang, Z. P. Liu, S. Y. Yu, K. W. Wang, S. S. Yuan, Q. W. Sun, X. T. Li and N. Li, ACS Appl. Mater. Interfaces, 2021, 13, 21474–21481 CrossRef CAS.
- X. Li, S. Guo, J. Su, X. Ren and Z. Fang, ACS Appl. Mater. Interfaces, 2020, 12, 28474–28483 CrossRef CAS.
- C. Tsai, H. Li, S. Park, J. Park, H. S. Han, J. K. Norskov, X. L. Zheng and F. Abild-Pedersen, Nat. Commun., 2017, 8, 15113 CrossRef.
- C. Hu, Z. Z. Jiang, W. D. Zhou, M. M. Guo, T. Yu, X. F. Luo and C. L. Yuan, J. Phys. Chem. Lett., 2019, 10, 4763–4768 CrossRef CAS.
- P. Thangasamy, S. Oh, S. Nam and I.-K. Oh, Carbon, 2020, 158, 216–225 CrossRef CAS.
- A. T. Garcia-Esparza, S. Park, H. Abroshan, O. A. P. Mellone, J. Vinson, B. Abraham, T. R. Kim, D. Nordlund, A. Gallo, R. Alonso-Mori, X. L. Zheng and D. Sokaras, ACS Nano, 2022, 16, 6725–6733 CrossRef CAS.
- X. Wang, Y. Zhang, H. Si, Q. Zhang, J. Wu, L. Gao, X. Wei, Y. Sun, Q. Liao, Z. Zhang, K. Ammarah, L. Gu, Z. Kang and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 4298–4308 CrossRef CAS PubMed.
- F. Wei, W. Xue, Z. Yu, X. F. Lu, S. Wang, W. Lin and X. Wang, Chin. Chem. Lett., 2024, 35, 108313 CrossRef CAS.
- Y. Liu, W. Xue, X. Liu, F. Wei, X. Lin, X. F. Lu, W. Lin, Y. Hou, G. Zhang and S. Wang, Small, 2024, 2402004 CrossRef.
- J. W. Tan, J. J. Shao, Y. H. Shi, W. B. Zhang and Q. S. Gao, ACS Sustainable Chem. Eng., 2022, 10, 13525 CrossRef CAS.
- L. Li, Z. Qin, L. Ries, S. Hong, T. Michel, J. Yang, C. Salameh, M. Bechelany, P. Miele, D. Kaplan, M. Chhowalla and D. Voiry, ACS Nano, 2019, 13, 6824–6834 CrossRef CAS.
- Y. H. Shi, W. B. Zhang, J. W. Tan, T. L. Yan, Y. S. Jia, Z. Y. Wang, Y. Tang and Q. S. Gao, Adv. Mater. Interfaces, 2022, 9, 2200505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02861a |
| This journal is © The Royal Society of Chemistry 2024 |