DOI:
10.1039/D3TA08054D
(Paper)
J. Mater. Chem. A, 2024,
12, 10819-10837
A (μ-oxo) dicopper complex anchoring graphitized mesoporous carbon surface prepared by an in situ electrochemical method for bioinspired electrocatalytic reduction of nitrite to ammonia and sensing†
Received
27th December 2023
, Accepted 20th March 2024
First published on 20th March 2024
Abstract
The design and development of functional biomimetic systems that resemble natural enzymes is highly challenging. Cytochrome c nitrite reductase (ccNiR), a specific enzyme containing multiple heme sites, functions as a catalyst for converting nitrite to ammonium through a complex six-electron transfer reaction under physiological conditions. In the literature, only a few homogeneous biomimetic metal complexes catalyzing nitrite reduction were reported, all under non-physiological conditions (organic and acid media). Herein, we developed a bioinspired copper(II)–BPA(μ-oxo) complex (BPA = bis(pyrid-2-ylmethyl)amine), denoted as {Cu2(μ-O)2}, immobilized on a graphitized mesoporous carbon modified glassy carbon electrode, GCE/GMC@{Cu2(μ-O)2}, which was prepared by an in situ electrochemical reaction of a precursor complex, {Cu(BPA)Cl}. The as-prepared GCE/GMC@{Cu2(μ-O)2} worked as an efficient biomimetic heterogeneous electrocatalyst for the reduction of nitrite into ammonia, in an aqueous medium and at neutral pH. This bioinspired system showed a stable and well-defined redox system with Eo′ = −220 mV vs. Ag/AgCl, and a surface-excess value of 83.9 nmol cm−2. The peak potential is much lower (>500 mV) than that of the unmodified electrode. In the presence of nitrite, a well-defined irreversible reduction peak at −0.250 V vs. Ag/AgCl is developed, which was assigned to the electrocatalytic reduction of nitrite. The molecular reaction and product formation were confirmed by SEM, TEM, Raman, IR, electrochemical quartz crystal microbalance (EQCM), and ESI-MS. A strong π–π interaction between the graphitic sp2 carbons and the aromatic sp2 in the BPA-pyridyl moiety of the metal complex favors good stability. To prove its functional application, the electroanalytical performance and bulk electrolysis conversion of nitrite into ammonia (faradaic efficiency of 92 ± 1%) were demonstrated.
1. Introduction
Nitrite (NO2−) is ubiquitous in natural terrestrial and aquatic environments where it functions as an intermediate in the biological reduction of nitrate (NO3−) into molecular nitrogen (N2) or ammonia (NH3), throughout the denitrification and ammonification branches of the nitrogen cycle, respectively. It also partakes in the reverse direction, i.e., the oxidation of ammonia back to nitrate, through the nitrification pathway.1–4 All these processes occur in a highly articulated manner, though, in intensively managed environments, the local biological N-cycle can be perturbed to the point that its ecological functioning is compromised. In particular, the excessive load of nitrite in soils and water bodies, directly or indirectly through modern agricultural practices and some industrial activities, can put the ecosystems and human health at risk.5–7 The ecological and environmental importance of the microbial transformations of nitrite within the N-cycle has thus gained much attention and driven extensive research over the last five decades (Besson et al. provide a detailed account of this topic).8 Major progress was made in the characterization of the bacterial enzymes performing these reactions and their catalytic mechanisms,3,8 as well as on potential applications in radioactive waste conversion,9 clinical diagnoses,10 biosensors,11–15 electrocatalysis,16–22 and electrochemical sensing.23–32
In this work, we will focus on the direct nitrite reduction into ammonium, NO2− + 8H+ + 6e− → NH4+ + 2H2O (Eo′ = 0.103 vs. Ag/AgCl at pH 7), which is considered a challenging step in the global N-cycle.33,34 This complex six-electron and eight-proton transfer reaction is catalyzed by the well-characterized cytochrome c nitrite reductase (ccNiR), also coined as NrfA, a bacterial enzyme containing five c-type heme sites.8 ccNiR catalyzes the nitrite to ammonium conversion via intramolecular binding of nitrite on a low-valent aquo-ligand containing iron site Fe(II), through a multi-electron-transfer pathway.33,34 This reaction has been studied by electrochemical techniques using enzyme-modified electrodes,35,36 which have also been explored for bioelectroanalytical applications37–39 where the selectivity and sensitivity of detection have been shown to be major advantages. However, their mass production has been hindered by issues such as the cost of enzyme purification and the biosensor shelf-life.
Several authors have been trying to mimic the bioelectrocatalytic reduction of nitrite, but this has proved to be a challenging task. Biomimicking molecular systems based on MoS2,40 Cu-tris(triazolyl)borate,41 Cu-bearing four-carboxylate ligand (6-chloromethylpyridine-2-carboxylic acid),42 cobalt-tripeptide complex,43 and the Fe-pentadentate macrocycle 2,13-dimethyl-3,6,9,1,2,18-pentaazabicyclo[12.3.1]octadeca-1(18),2,12,14,16-pentaene complex19 have been developed as molecular electrocatalysts. However, the procedures involve complicated synthetic routes,43 non-physiological medium (non-aqueous solutions),19,41–44 homogeneous conditions19,41–44 and require a high-over potential (η > 500 mV) for nitrite reduction.19,42–44 Meanwhile, there have been many reports on metal and metal oxide based nitrite reduction with non-biomimetic approaches.45–48
Herein, we report a copper(I) bis(pyrid-2-ylmethyl)amine-(μ-oxo) complex, {Cu2(μ-O)2}, for the highly efficient biomimicking electrocatalytic reduction of nitrite into ammonia, at physiological pH and lower over-potentials (−0.250 V vs. Ag/AgCl). The complex was obtained by a simple and quick electrochemical method, which anchored it to graphitized mesoporous carbon. The as-prepared heterogeneous electrocatalyst was characterized using Raman, FTIR, a transmission electron microscope (TEM), an electrochemical quartz crystal microbalance (EQCM), and ESI-MS, which revealed that an authentic-active site has been involved in the electrocatalytic reaction. By using scanning-electrochemical microscopy (SECM), active electrochemical spots on the electrocatalytic surface were visualized. The electrochemical kinetics of the nitrite reduction reaction was monitored using the rotating disc electrode (RDE) technique. The number of electrons involved in the reaction was calculated by CV, RDE, and bulk electrolysis techniques. As functional applications, the bulk electrolytic conversion of nitrite to ammonia, and the electrocatalytic sensing of nitrite by amperometric i–t have been demonstrated.
2. Experimental section
2.1. Chemicals and reagents
Carbon black, N330 grade, was obtained from Phillips Carbon Black Ltd Kochi, India as a gift sample. Pristine multiwalled carbon nanotubes (MWCNTs) with a purity of ∼90 wt% (carbon basis), 10–15 nm outer diameter, 2–6 nm inner diameter and 0.1–10 μm length; double-walled carbon nanotubes (DWCNTs) with a purity of ∼90 wt% (carbon basis), 3–4 nm diameter and 1–10 μm length; single-walled carbon nanotubes (SWCNTs) with a purity of ∼70 wt% (carbon basis) and 0.7–1.1 nm diameter; graphitized mesoporous carbon (GMC) with purity assay of >99.95% and <50 nm pore size; and activated charcoal (AC) with purity assay >99.95% and 2–12 μm diameter were obtained from Merck, India. All other analytical grade chemicals were used as received. Aqueous solutions were prepared using deionized and alkaline KMnO4 double distilled water (DDW). A phosphate buffer solution (PBS) at pH 7 was used as a supporting electrolyte. Unless stated otherwise, all solutions were N2-gas purged for 20 minutes before measurements.
2.2. Apparatus
The cyclic voltammetric experiment was performed using a FRA2 μAutolab Type III potentiostat/galvanostat from Metrohm Autolab, Netherlands with an electrochemical cell volume of 10 mL. The three-electrode system consists of a glassy carbon electrode (GCE, working electrode) with a geometrical surface area of 0.0707 cm2, an Ag/AgCl, 3 M KCl (reference electrode) and a platinum wire (counter electrode). Raman spectroscopy analysis was conducted using an AZILTRON PRO 532 (USA) with a 532 nm laser excitation source. FTIR analysis was done using a JASCO 4100 spectrophotometer and the KBr method. A Nano Lc-ESi-QTOF Qstar Elite (ABSciex Pte Ltd) instrument was adopted for ESI-mass analysis. In situ CV-EQCM experiments were carried out using a gold single crystal electrode (EQCM-Au) with a geometric surface area of 0.196 cm2. SECM analysis was carried out using a PAR (Princeton Applied Research, USA) Versascan bipotentiostat through a feedback mode operation. A GCE with a geometric area of 0.0707 cm2 (3 mm diameter) and 25 μm Pt-tip ultramicroelectrode under bipotentiostat conditions was used for the SECM surface morphology studies. Prior to the analysis, the position of the Pt-tip against the modified electrode substrate was fixed from an approach curve in the feedback mode, obtained by moving the Pt-tip position at a speed of 2 μm s−1 in the z-axis direction, at a fixed applied potential of 0.36 V vs. Ag/AgCl, and substrate potential of 0.53 V vs. Ag/AgCl, at which the diffusion-controlled redox cycling of Fe3+/Fe2+ occurs.
2.3. Synthesis of bis(pyrid-2-ylmethyl)amine (L1)
The ligand was synthesized using the previously reported procedure.49 In brief, 20 mL methanolic solutions of N-pyridylamine (0.74 g, 10 mmol) and pyridine-2-carboxaldehyde (1.10 g, 10 mmol) were mixed and stirred overnight to get a bright yellow solution. To this, NaBH4 (0.57 g, 15 mmol) was added, and the solution was stirred for another day, followed by rotary-evaporation to dry. The resulting solid was dissolved in water, wherein the organic layer was extracted with CH2Cl2 and dried with anhydrous sodium sulphate. The CH2Cl2 layer was rotary-evaporated to get bis(pyrid-2-ylmethyl)amine (BPA) as a yellow oil. The compound's structural features were confirmed through the 1H NMR data (200 MHz, CDCl3): δ 8.54–7.36 (m, 8H), 3.91 (s, 4H), 2.28 (t, 2H), 1.32 (m, 2H), 1.11 (t, 3H). ESI-MS m/z = 241: C15H19N3+.
2.4. Preparation of the copper(II) L1-complex, {Cu(BPA)Cl}
The reported procedure was used to prepare this complex.50 To a 10 mL methanolic solution of the ligand BPA (0.17 g, 1 mmol), a 5 mL solution of CuCl2·2H2O (0.170 g, 1 mmol) in methanol was added, stirred well, and cooled. The pale blue crystalline precipitate obtained was filtered off, washed with small amounts of cold methanol, and vacuum dried, resulting in a 75% yield (0.22 g). The electronic spectral data of the ligand – two peaks at 664 nm (124 M−1 cm−1) and 263 nm (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 850 M−1 cm−1) – matched the reported values.49 The ESI-MS analysis indicated a peak at m/z 297.06, which corresponds to the complex {Cu(L1)Cl}.
850 M−1 cm−1) – matched the reported values.49 The ESI-MS analysis indicated a peak at m/z 297.06, which corresponds to the complex {Cu(L1)Cl}.
2.5. Preparation of the GCE/GMC@{Cu2(μ-O)2} chemically modified electrode
First, 2 mg of GMC was dispersed in 500 μL ethanol and sonicated for 15 minutes, to obtain a suspension. The GCE was first cleaned mechanically by polishing it with 0.5 μm alumina powder, followed by sonication in double distilled water for 5 minutes. Then the polished GCE was electrochemically pretreated by potentially cycling in 0.1 M PBS, pH 7, within a potential window of −0.2 V to +1 V vs. Ag/AgCl, at a scan rate (ν) of 50 mV s−1 for ten continuous cycles. Over this pretreated GCE, 5 μL of GMC dispersed in ethanol was drop-cast and air-dried for 5 ± 1 minutes, followed by an electrochemical pretreatment in the potential window of −0.7 V to +0.2 V vs. Ag/AgCl, for ten cycles, in N2 purged in 0.1 M PBS, pH 7, at a ν of 50 mV s−1. Then, 5 μL of the precursor complex, {Cu(BPA)Cl}, dispersed in ethanol, was drop-cast over the GCE/GMC and air-dried for 15 ± 2 minutes, followed by drop-casting 5 μL of a dilute, 0.6% Nafion dispersed in ethanol and air-dried for 15 ± 2 minutes (denoted as GCE/GMC@{Cu(BPA)Cl}ads, where ads = adsorbed). A perfluoro cation-exchange polymer, Nafion, was used to protect the complex modified electrode. All other chemically modified electrodes were prepared similarly. The prepared GCE/GMC@{Cu(BPA)Cl}ads was electrochemically cycled in the potential window of −0.7 V to +0.2 V vs. Ag/AgCl, in nitrogen-purged 0.1 M PBS, pH 7, at ν = 50 mV s−1. The “as prepared” chemically modified electrode was used for further analysis.
The faradaic efficiency (FE) from bulk electrolysis was calculated using the following expression:51
| |  | (1) |
where
n = number of electrons involved in the electrocatalytic reaction,
F = Faraday constant (96
500 C mol
−1),
c = concentration of the product,
V = volume of the analyte, and
Q = total charge.
2.6. Identification of the product reaction
To authenticate the product identity, the bulk electrolysis of nitrite (5 mM) was carried out at an applied potential of −0.3 V vs. Ag/AgCl, in 0.1 M PBS, pH 7. The final product of the reaction was tested with Nessler's reagent (9 mM K2[HgI4] in 2.5 M KOH), using nitrite and ammonia solutions as controls. Further, an indophenol based colorimetric method combined with an ammonia standard addition approach, involving the electrolyzed sample in addition to the standard concentration of ammonia spiked, were also performed for the confirmation of NH3 formation as a reaction product.52
3. Results and discussion
3.1. Electrochemical behaviour of the Cu(BPA)Cl adsorbed GMC modified electrode
First, the {Cu(BPA)Cl} complex adsorbed on the GCE surface, GCE/{Cu(BPA)Cl}ads, was analyzed through electrochemical techniques in 0.1 M PBS, pH 7 (Scheme 1). As seen in Fig. 1A, the GCE-adsorbed form of the complex (curve a) gives no faradaic response, which is why most of the inorganic complexes developed so far could not be used for heterogeneous-electrochemical applications in aqueous electrolytes. Interestingly, when the same CV experiment was repeated on a GMC-modified GCE (Nafion covered), i.e., GCE/GMC@{Cu(BPA)Cl}ads, a well-defined pair of anodic (Epa) and cathodic peaks (Epc) was observed at a formal reduction potential (Eo′) of −220 mV vs. Ag/AgCl, estimated as the average mean of peak potentials ((Epa + Epc)/2). After an initial current decay (first 10 cycles), the voltammetric response was very stable (Fig. 1B). The calculated peak-to-peak separation, ΔEp, is 214 ± 5 mV at v = 50 mV s−1, and the surface-excess (ΓCuBPA) of the active site is 83.9 × 10−9 mol cm−2 (Fig. 1C; ESI Fig. S1†), and the relative standard deviation value for the last 5 cycles is 4.7%. The plot of peak current (ipa and ipc) vs. scan rate is linear up to v = 150 mV s−1 (Fig. 2A & B), indicating an adsorption-controlled-electron-transfer kinetic behavior at low scan rates, and a mixed-adsorption and diffusion-controlled (possibly an electron-hopping type mechanism) electron-transfer pathway at high scan rates (>150 mV s−1). The double logarithmic plot of ipvs. scan rate, where a slope of 0.5 relates to a diffusion-controlled reaction and a slope of 1 relates to an adsorption-controlled reaction mechanism,53 showed slope values of 0.730 for both the anodic and cathodic responses, supporting the mixed-diffusion and adsorption-controlled electron-transfer mechanism (Fig. 2C).54 The diffusion of the electrolyte within the modified electrode film and its kinetic limitation is the most likely reason for this observation. Based on the Laviron model equation,53–55 which is applicable for surface-confined electron-transfer reactions with a high peak-to-peak separation (>200 mV s−1), kinetic parameters such as the transfer coefficient, α, and heterogeneous electron-transfer rate constant, ks, can be calculated as follows:| | | logks = αlog(1 − α) + (1 − α)logα − log[RT/nFv] − α(1 − α)nFΔEp/2.303RT | (3) |
where Sa (0.223 V dec−1) and Sb (0.235 V dec−1) are the anodic slope and cathodic slope values of the plots of Epa and Epcvs. logv (Fig. 2D), v is 50 mV s−1, and ΔEp is 214 mV. The calculated value of α is thus 0.49, which is close to the ideal value of 0.5, denoting a symmetrical energy barrier for the electron-transfer reaction. The obtained ks value, 1.19 s−1, is comparable with those reported for hemin (1.3 s−1) and hematin (1.34 s−1) complexes.56,57
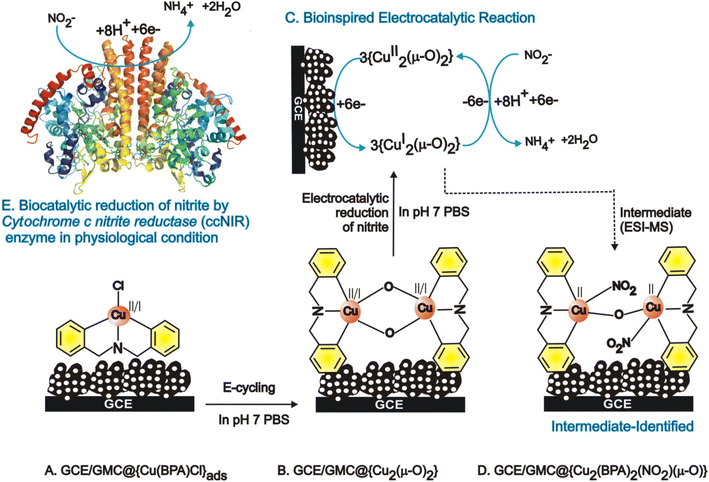 |
| | Scheme 1 Illustration of the [Cu(BPA)Cl]+ adsorbed GCE/GMC modified electrode (A) and the in situ electrochemical conversion of the (μ-O) copper complex modified electrode system by simple potential cycling, in pH 7 PBS (B) and the mechanism for the electrocatalytic reduction of nitrite into ammonia (C). Structure of the active intermediate involved in the nitrite reduction reaction obtained during the bulk electrolysis of nitrite (D). (E) Structure of cytochrome c nitrite reductase enzyme and its selective nitrite reduction to ammonia function. | |
 |
| | Fig. 1 (A) Cyclic voltammetric responses of (a) GCE@{Cu(BPA)Cl}ads; (b) GCE/GMC@{Cu(BPA)Cl}ads in nitrogen purged 0.1 M PBS, pH 7, at scan rate (ν) = 50 mV s−1 (n = 20 cycles) and (B) last five CVs; (C) comparative plots of surface excess values of different carbon nanomaterials used. | |
 |
| | Fig. 2 (A) Effect of scan rate on GCE/GMC@{Cu(BPA)Cl}-redox from 5 to 500 mV s−1; plots of (B) ipvs. v, (C) logipvs. logv and (D) Epvs. logv; (E) effect of various pHs (3–11) on GCE/GMC@{Cu(BPA)Cl}-redox. Electrolyte: in N2 purged 0.1 M PBS, pH 7. | |
The “as prepared” modified electrode was tested in different pH media, ranging from pH 3 to pH 11. As shown in Fig. 2E, the GCE/GMC@{Cu(BPA)Cl}ads has a nearly pH-independent redox feature. In general, metal(aquo)-based complexes show a strong-proton-coupled electron-transfer mechanism, with an Epavs. pH slope of ca. −60 mV pH−1 (Nernstian value).58 A molecular transformation of the {Cu(BPA)Cl} complex to a proton-independent redox complex under electrified conditions is a possible reason for this observation. Further experiments were carried out to characterize and identify the truly active species of the Cu-complex. It is noteworthy that since the amount of entrapped Cu-complex on the modified electrode is very low (about nanogram level) and the electrogenerated complex may not be stable outside the matrix, this is a challenging task. For the sake of clarity, hereafter, the electrochemically treated GCE/GMC@{Cu(BPA)Cl} is denoted as GCE/GMC@{Cu(BPA)Cl}-redox, where {Cu(BPA)Cl}-redox stands for the electrochemically transformed molecular complex.
3.2. Physicochemical characterization of GMC@{Cu(BPA)Cl}-redox
Since our primary focus is to characterize the copper complex as immobilized on the electrode surface, for comparison purposes, we also analyzed an electrode modified with GMC@{Cu(BPA)Cl}-redox but without the Nafion layer on top. From a qualitative perspective, both Nafion-modified and unmodified electrodes showed similar voltammetric responses (ESI Fig. S2†). Fig. 3A & B show the typical TEM images of GMC and GMC@{Cu(BPA)Cl}-redox showing a porous material-like structure and islands of agglomerated cloudy spots, respectively. Presumably, a strong non-covalent bond, like a π–π interaction between the aromatic electrons of the BPA ligand and the sp2 carbon in the graphitic structure of GMC, resulted in a cloudy image. Noteworthily, a similar effect was reported for an inorganic enzyme analog composed of Nafion/lead–ruthenium oxide pyrochlore.59Fig. 3C & D are typical FESEM images of GMC and GMC@{Cu(BPA)Cl}-redox showing globular structures like those seen by TEM. The presence of Cu and N atoms was confirmed by the EDAX results (ESI Fig. S3†).
 |
| | Fig. 3 TEM (A & B) and FESEM (C & D) images of GMC and GMC@{Cu(BPA)Cl}-redox, respectively. | |
The modified electrode surface was also subjected to a Raman spectroscopic analysis, and the data was compared to that of the unmodified GMC (Fig. 4A). Well-defined peaks corresponding to the D-band (graphitic disorder structure, sp3 bonding) and G-band (ordered graphitic structure, sp2 bonding) at 1350 and 1550 cm−1 were observed in both systems but with different relative intensities. The intensity ratio ID/IG, considered as a measure of the structural modification on the surface,53 increased from 0.62 to 0.95, in the GMC and GMC@{Cu(BPA)Cl}-redox electrode configurations, respectively. This suggests that there is a significant addition of sp3 bonds on the GMC surface upon its modification with the {Cu(BPA)Cl}-redox complex. Fig. 4B compares the FTIR spectra of a naked {Cu(BPA)Cl} complex (curve a), GMC@{Cu(BPA)Cl}-redox (curve b) and GMC (curve c); the peaks at 1620.2 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1458.6 cm−1 (–CHCH–) and 1049.2 cm−1 (–C–N) are characteristic of the complex. After the immobilization of the latter on GMC, one can observe a significant reduction in the peak intensities along with a marked shift in their position. Besides the above-mentioned peaks, the Cu-complex also shows a ν(Cu–N)-stretching vibrational peak at 500.9 cm−1 and ν(Cu–O)-stretching signal at 469.7 cm−1, whose intensity enhanced upon the immobilization of the complex on the GMC surface.60 These observations support the occurrence of molecular interactions between the functional groups of the {Cu(BPA)Cl}-redox complex and the underlying graphitic structure of the GMC. Furthermore, the {Cu(BPA)Cl} complex and an isolated ethanolic extract of GMC@{Cu(BPA)Cl}-redox were analyzed by UV-Vis spectroscopy, as shown in ESI Fig. S4.† The {Cu(BPA)Cl} complex gave peaks at 263 nm and 664 nm owing to the π–π* and d-transitions, whereas {Cu(BPA)Cl}-redox showed distinct peaks at 218, 289 nm, and 477 nm, which are very different from those of the precursor complex (at 263 and 664 nm) and closely match the spectroscopic signature of the dinuclear (μ-O) complex {Cu2(μ-O)2}.60 So, at this point, we propose that the surface-bound {Cu(BPA)Cl} complex is transformed into a {Cu2(μ-O)2} like complex due to an electrochemical reaction.
N), 1458.6 cm−1 (–CHCH–) and 1049.2 cm−1 (–C–N) are characteristic of the complex. After the immobilization of the latter on GMC, one can observe a significant reduction in the peak intensities along with a marked shift in their position. Besides the above-mentioned peaks, the Cu-complex also shows a ν(Cu–N)-stretching vibrational peak at 500.9 cm−1 and ν(Cu–O)-stretching signal at 469.7 cm−1, whose intensity enhanced upon the immobilization of the complex on the GMC surface.60 These observations support the occurrence of molecular interactions between the functional groups of the {Cu(BPA)Cl}-redox complex and the underlying graphitic structure of the GMC. Furthermore, the {Cu(BPA)Cl} complex and an isolated ethanolic extract of GMC@{Cu(BPA)Cl}-redox were analyzed by UV-Vis spectroscopy, as shown in ESI Fig. S4.† The {Cu(BPA)Cl} complex gave peaks at 263 nm and 664 nm owing to the π–π* and d-transitions, whereas {Cu(BPA)Cl}-redox showed distinct peaks at 218, 289 nm, and 477 nm, which are very different from those of the precursor complex (at 263 and 664 nm) and closely match the spectroscopic signature of the dinuclear (μ-O) complex {Cu2(μ-O)2}.60 So, at this point, we propose that the surface-bound {Cu(BPA)Cl} complex is transformed into a {Cu2(μ-O)2} like complex due to an electrochemical reaction.
 |
| | Fig. 4 Comparative (A) Raman and (B) FTIR spectra of GMC, {Cu(BPA)Cl} and GMC@{Cu(BPA)Cl}-redox. | |
After exploring various carbon nanomaterials, including carbon black, double-walled carbon nanotubes, single-walled carbon nanotubes, activated charcoal (AC), and graphene-modified carbon (GMC), it was observed that the GMC-based material exhibited the most favorable electrochemical response. This superiority can be attributed to its optimized pore size and graphitic structure, facilitating the effective immobilization of the {Cu2(μ-O)2} complex (refer to ESI Fig. S1†).
To identify the true molecular species, a clear ethanolic extract of the GMC@{Cu(BPA)Cl}-redox complex was analyzed through ESI-MS. As seen in ESI Fig. S5,† two major m/z peaks were observed at 379.59 and 663.93, which correspond to the ions {Cu(BPA)(H2O)2}+ (MW ca. 379.07) and {Cu2(BPA)2s(μ-O)2}4+ (MW ca. 662.03), respectively. As a control, a dilute ethanolic solution of {Cu(BPA)Cl} (MW ca. 297.01) was also examined by ESI-MS (ESI Fig. S6†); the peaks with m/z 261.06, 297.06, and 361.02 were respectively assigned to the following structural forms {Cu(BPA)} (MW ca. 262.04), {Cu(BPA)Cl} (MW ca. 297.01), and {Cu(BPA)(H2O)(OCH3)}+ (MW ca. 361.06). These data indicate that a {Cu(μ-O)}-complex has been formed on the modified electrode surface, which can explain the proton-independent electron-transfer reaction previously seen in Fig. 2E (the new oxo compound has no acidic protons). Therefore, from now on, the GCE/GMC@{Cu(BPA)Cl}-redox will be denoted as GCE/GMC@{Cu2(μ-O)2}.
3.3.
In situ electrochemical EQCM analysis
To further support the occurrence of the molecular transformation GCE/GMC@{Cu(BPA)Cl} → GCE/GMC@{Cu2(μ-O)2} on the electrode interface, the precursor electrode GCE/GMC@{Cu(BPA)Cl} was subjected to an in situ EQCM analysis. This technique provides detailed information about mass changes (Δm/g) throughout the electrochemical reaction. Fig. 5A and B present the continuous CV and EQCM responses of the EQCM-Au/GMC@{Cu(BPA)Cl}ads modified electrode in N2 purged 0.1 M PBS, pH 7, showing a regular increase in the Δm during the potential cycles (based on the relation MW = F × Δm/ΔQ, where Q = charge consumed, MW = molar mass (g mol−1) per electron (n = 1), and F = Faraday constant, one can evaluate the molar mass involved in the intermediate region).61–64Fig. 5C and D are typical plots of Δm vs. E/V vs. Ag/AgCl and Δm vs. ΔQ for the EQCM-Au/GMC@{Cu(BPA)Cl}ads modified electrode, whilst Fig. 6E zooms in the latter plot in several potential regions. Based on the slope value (Δm/ΔQ) obtained in different potential windows, the calculated Mw values were 107.2 ± 0.3 g mol−1 (0.2 V to −0.1 V – forward sweep) and 93.9 ± 1 g mol−1 (−0.1 V to 0.2 V – reverse sweep), which correspond to the addition of the molecular species 6H2O and PO4− within the modified electrode upon potential cycling. Although the exact molecular mechanism is unclear, the intake of the H2O molecule likely helps the conversion of {Cu(BPA)Cl} into the {Cu2(μ-O)2} complex, at the electrified interface.
 |
| | Fig. 5 (A and B) In situ CV-EQCM responses of GCE/GMC@{Cu(BPA)Cl}ads in N2 purged 0.1 M PBS, pH 7, at a scan rate of 10 mV s−1. Inset of (A) shows the molecules involved in the redox reaction. Corresponding derivative plots of (C) Δm vs. E and Δm vs. ΔQ plots of (D) 1–10 cycles and (E) second cycle. (F) Picture of an EQCM-Au electrode. | |
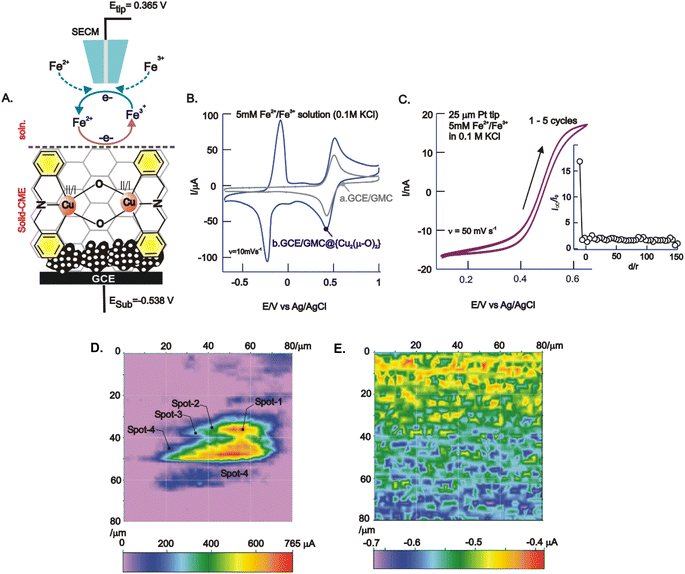 |
| | Fig. 6 (A) Schematic illustration of SECM feedback mode operation for GCE/GMC@{Cu2(μ-O)2} using the Fe2+/Fe3+ redox mediator. Cyclic voltammetric response of GCE/GMC (B, curve a) and GCE/GMC@{Cu2(μ-O)2} (B, curve b) and Pt-tip (C). SECM responses of (D) GCE/GMC@{Cu2(μ-O)2} and (E) GCE/GMC (control) images. Conditions: Etip = 0.365 V and Esub = −0.538 V vs. Ag/AgCl in 5 mM Fe2+/Fe3+ solution containing 0.1 M KCl. | |
3.4.
In situ SECM imaging of electroactive sites
The surface morphology of GCE/GMC@{Cu2(μ-O)2} was visualized using an ultramicroelectrode as an SECM probe, under a feedback current approach (Fig. 6A). Herein, the redox couple is first electro-oxidized to a high-valent species (Fe3+) on the underlying surface (substrate) and subsequently reduced to a low-valent species (Fe2+) continuously on the Pt-tip (moved in the x–y direction) throughout the electrochemical reactions, so that the variation in the electronic conductivity of the surface can be viewed simultaneously (Fig. 6A). Fig. 6B compares the CV responses of GCE/GMC@{Cu2(μ-O)2} and GCE/GMC in the presence of 5 mM Fe3+ and Fe2+ each, in a 0.1 M KCl solution, at v = 10 mV s−1. The current intensity of both cathodic and anodic peaks increased with the GCE/GMC@{Cu2(μ-O)2} modified electrode, due to changes in the electronic conductivity at several surface sites. Before the surface analysis, the Pt-tip position was properly placed using the approach curve experiment, as displayed in Fig. 6C (inset). Fig. 6C is a typical CV response of the Pt-tip in an Fe3+/Fe2+ solution (5 mM each) in 0.1 M KCl, showing an “S” shaped curve due to the electrochemical reaction's radial diffusion mechanism (a mandatory condition in SECM imaging). Fig. 6D is a typical SECM image of the GCE/GMC@{Cu2(μ-O)2} showing a cloud-like electro-active spot with a size of 60 ± 5 μm on the modified electrode surface. This observation is supported by the TEM results (Fig. 3B), but one should note that because the TEM study was carried out using a diluted test solution, the observed image is only an approximation of the real surface morphology. Under optimal working conditions, the unmodified electrode, GCE/GMC, was also analyzed through SECM, delivering a featureless image (Fig. 6E); here we can see a highly heterogeneous catalytic surface, with 4 different spots showing a stepwise increase in the current signals (spot-1 > 2 > 3 > 4). Based on the electron-microscopy images, spot-1 can be correlated to an active {Cu2(μ-O)2} complex staged on the GMC via π–π interaction (assigned to a black-colored spot in the TEM). On the other hand, spot-2 relates to the adsorbed {Cu2(μ-O)2} complex on porous cavities without any specific interaction. Similarly, spots 3 and 4 are due to the adsorbed complex with poor or weak π–π interactions. This is the reason why multiple redox peaks were detected in the CVs of the GCE/GMC@{Cu2(μ-O)2} electrode (Fig. 1A).
3.5. Biomimetic nitrite reduction by GCE/GMC@{Cu2(μ-O)2}
In nature, the direct 6-electron reduction of nitrite into ammonium is effectively performed by the multiheme nitrite reductase ccNiR. The binding of NO2− by replacing the aquo-ligand has been referred to as a key step for the overall conversion reaction.41,44 Although several synthetic analogues have been chemically prepared, their functional application to monitor the nitrite reduction reaction in real-time is very limited (some of them failed to catalyze the electrochemical nitrite reduction).40,42,43 In this work, we explored GMC@{Cu2(μ-O)2} as a biomimetic functional analogue of a nitrite reductase enzyme. Fig. 7A shows the CV response of GCE/GMC@{Cu2(μ-O)2} in 0.1 M PBS, pH 7, with and without nitrite. In the absence of nitrite (Fig. 7, curve a), a well-defined pair of peaks is observed at the same formal potential (Eo′). Following the addition of 50 μM nitrite (Fig. 7, curve b), the cathodic peak increases its intensity and shifts to a more negative potential (E1/2 = −270 ± 10 mV vs. Ag/AgCl), while the intensity of the anodic peak decreases, which should be due to the irreversible electrocatalytic reduction of nitrite, according to an EC′ mechanism, i.e., an electrochemical reaction coupled with a homogeneous irreversible chemical reaction (Scheme 2). As demonstrated in section 3.6, the final product of the reaction is ammonium.
 |
| | Fig. 7 (A) Comparative CV responses of GCE/GMC@{Cu2(μ-O)2} without (curve a) and with 50 μM NO2− (curve b), GCE/GMC (curve c), at scan rate (ν) = 10 mV s−1; and inset is a Tafel plot. (B) Effect of scan rate (ν) on the GCE/GMC@{Cu2(μ-O)2} from 5 to 500 mV s−1, in the presence of 50 μM NO2−; plots of (C) ipcvs. ν1/2, (D) ipc/ν1/2vs. ν and (E) Epcvs. log(ν). Electrolyte: nitrogen purged 0.1 M PBS, pH 7. | |
 |
| | Scheme 2 Reaction mechanism on the GMC@{Cu2(μ-O)2} for the electrocatalytic reduction of nitrite to ammonium under electrochemical (step 3) and chemical (steps 1 & 2) conditions. [GMC@{CuI2 (μ-O)2}⋯NO2−] is an intermediate species similar to Scheme 1D. | |
As a control experiment, the bare GCE/GMC was also tested in the presence of nitrite (Fig. 7A, curve c), but it failed to develop significant catalytic currents throughout the entire potential window (+0.4 to −1.0 V vs. Ag/AgCl), demonstrating the ineffectiveness of the bare GCE/GMC as a nitrite electrocatalytic reducer.
Table 1 summarizes the works available in the literature16,43,65,66,72–77 reporting the electrochemical reduction of nitrite into ammonium. One should emphasize that very few studies were based on molecular heterogeneous systems for the nitrite reduction reaction. Typically, the reduction potentials are high; for example [FeIII(H2O)(TMPyP)]5+(TMPyP = meso-tetrakis(N-methyl-4-pyridyl)porphine dication)16 works at −1.0 V vs. Ag/AgCl at pH 2.87 and the cobalt-tripeptide complex (CoGGH)43 is active at −0.95 V vs. Ag/AgCl at pH 7. Additionally, the operational conditions are non-physiological, not mimicking the biological nitrite reduction into ammonium. To the best of the authors' knowledge, the GCE/GMC@{Cu2(μ-O)2} is the first synthetic complex that can perform the heterogeneous electrocatalytic reduction of nitrite into ammonium under physiological-like conditions.
Table 1 Works reported in the literature on the electrocatalytic conversion of nitrite into ammoniaa
|
|
Molecular system |
Condition |
pH |
E/V |
Remarks |
Ref. |
|
GCE: glassy carbon electrode; HMDE: hanging mercury drop electrode; JDC: cobalt nanoparticle decorated biomass Juncus derived carbon; GF: graphite felt.
|
| 1 |
[FeIII(H2O)(TMPyP)]5+ |
Heterogeneous |
2.87 |
−0.9 V vs. SCE (−0.85 V vs. Ag/AgCl) |
(i) Non-physiological medium |
16
|
| WE: GCE |
| 2 |
Cobalt-tripeptide complex (CoGGH) |
Homogeneous |
7.2 |
−0.65 V vs. Ag/AgCl |
(i) Hazardous working electrode |
43
|
| WE: HMDE |
| 3 |
PEDOT–BIPY–Fe film |
Homogeneous |
4.4 |
−0.65 V vs. Ag/AgCl |
(i) Non-physiological medium; (ii) poor redox response |
65
|
| 4 |
VIVO(SB)-CPE |
Homogeneous |
4 |
0.8 V vs. Ag/AgCl |
(i) Non-physiological medium; (ii) overpotential; (iii) non-molecular system |
66
|
| 5 |
[(tpy)(bpy)M(NO2)]3+ |
Homogeneous |
3.97 |
0.89 V vs. SSCE (0.93 V vs. Ag/AgCl) |
(i) Non-physiological medium |
72
|
| WE: mercury pool |
| 6 |
Thin-film diamond electrode |
Homogeneous |
13 |
−2 V vs. SCE (−1.96 V vs. Ag/AgCl) |
(i) Non-physiological medium; (ii) overpotential; (iii) non-molecular system |
73
|
| 7 |
{(TpRu)2(μ-pz)} (Tp = HB(pyrazol-1-yl)3) |
Homogeneous |
[nBu4N][PF6] |
Not available |
(i) Non-physiological medium |
74
|
| WE: platinum disk |
| 8 |
Ag@NiO/CC |
Heterogeneous |
0.1 M NaOH |
−0.15 V vs. RHE (−0.35 V vs. Ag/AgCl) |
(i) Non-physiological medium; (ii) non-molecular system |
75
|
| WE: Ag nanoarray using a NiO nanosheet array on carbon cloth |
| 9 |
Co@JDC/GF |
Heterogeneous |
0.1 M NaOH |
−0.2 V vs. RHE (−0.4 V vs. Ag/AgCl) |
(i) Non-physiological medium |
76
|
| 10 |
CoP NA/TM |
Heterogeneous |
7 |
−0.2 V vs. RHE (−0.4 V vs. Ag/AgCl) |
(i) Non-molecular system |
77
|
| WE: CoP nanoarray supported on titanium mesh |
| 11 |
GMC@{Cu2(μ-O)2} |
Heterogeneous |
7 |
−0.25 V vs. Ag/AgCl |
(i) Molecular system, (ii) physiological medium, (iii) good redox response and (iv) less overpotential |
This work |
| WE: GCE |
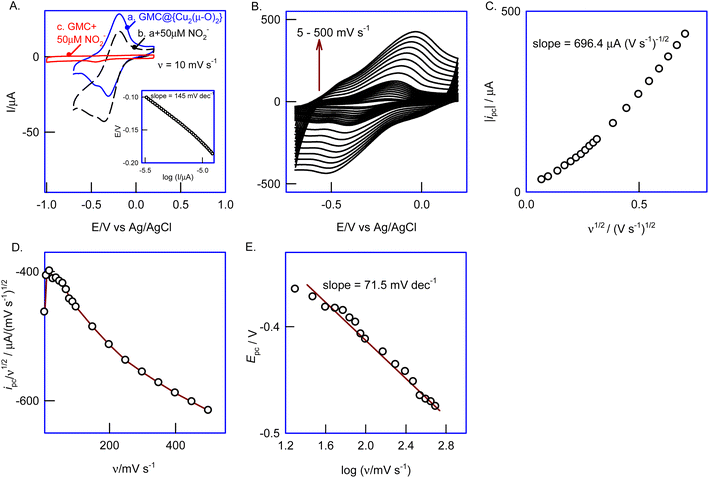
To understand the reaction kinetics and mechanism, we studied the effect of the scan rate (5–500 mV s−1) on the GCE/GMC@{Cu2(μ-O)2} CVs in the presence of NO2− (Fig. 7B). As expected, the current of both peaks rose with the scan rate, and the electrocatalytic effect disappeared due to the increase in the heterogeneous electron transfer rate. As depicted in Fig. 7C, the plot of the cathodic peak current (ipc) against the square root of the scan rate (ν1/2) is linear starting from the origin, with a slope of 696.4 μA (V s−1)−1/2. Fig. 7D shows the plot of the catalytic peak intensity (ipc/ν1/2) against the scan rate which gives a parabolic curve, indicating a higher current function at a lower scan rate (<50 mV s−1), and a lower current function at a higher scan rate (>50 mV s−1). This is also a characteristic response of an EC′ reaction mechanism53 (Scheme 2).
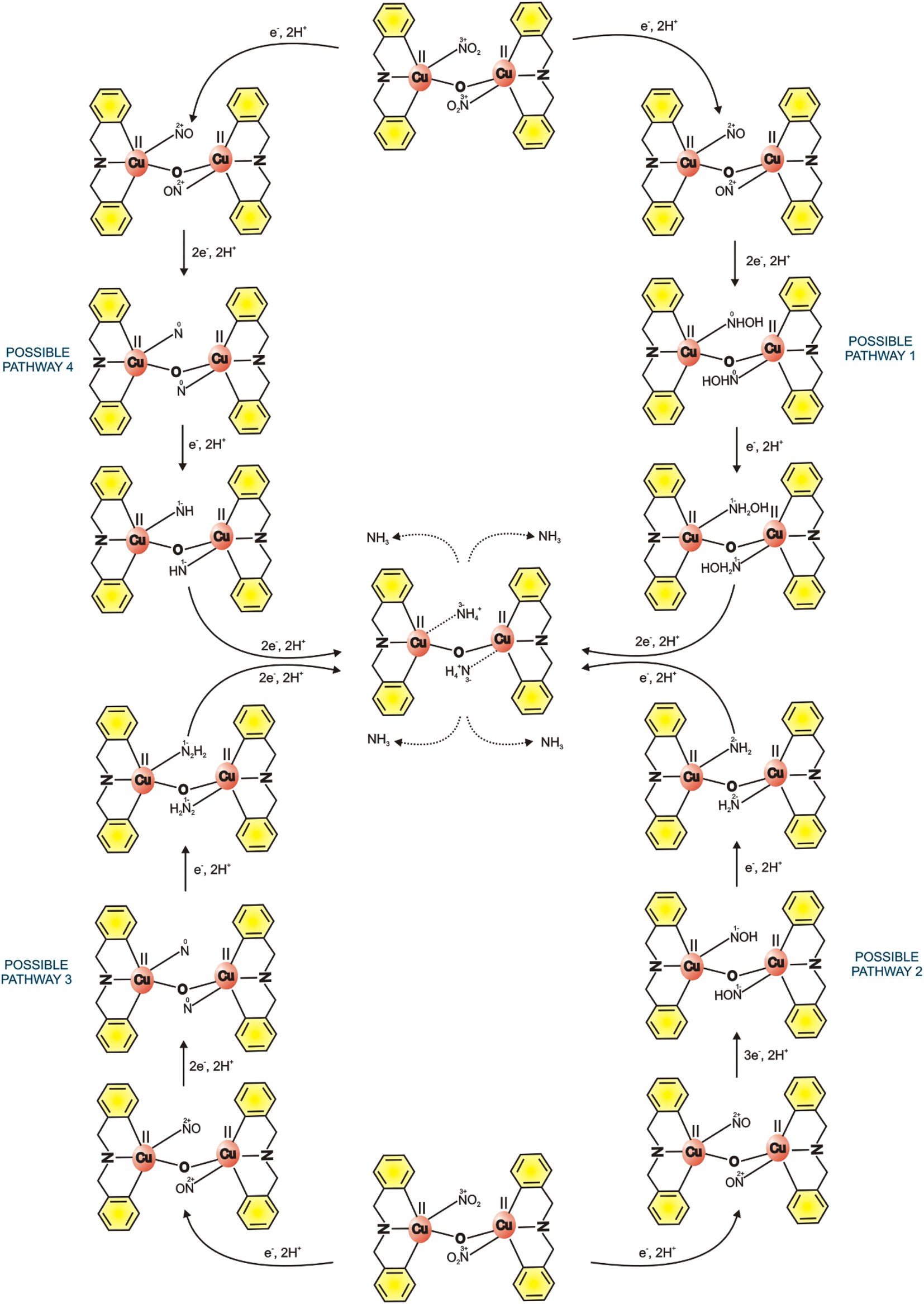
In addition, Scheme 3 illustrates the potential reaction pathways for the electrochemical reduction of nitrite. During the electrochemical process, adsorbed nitrite undergoes conversion to NO, which subsequently transforms into NH3via four distinct pathways. In pathway 1, sequential hydrogenation (NO → NHOH) and deoxygenation (NHOH → NH2OH → NH3) of the nitrogen and oxygen atoms occur. Pathway 2 involves deoxygenation (NO → NOH) followed by hydrogenation (NOH → NH2 → NH3). Pathways 3 and 4 entail hydrogenation of NO (NO → N) followed by sequential deoxygenation of uncoordinated oxygen (N → NH or N2H2 → NH3). The precise details of the correct pathway for this mechanism are currently unknown to us.
 |
| | Scheme 3 Possible electrochemical reduction reaction mechanism pathway of GMC@{Cu2(μ-O)2} on nitrite to form ammonium. The adsorbed NO2− is first reduced to NO and its conversion to NH3 occurs via four pathways: pathway 1: NO → NHOH → NH2OH → NH3; pathway 2: NO → NOH → NH3; pathway 3: NO → N → NH3; pathway 4: NO → N → NH → NH3. The oxidation states of nitrogen are depicted at each step. | |
Fig. 7E shows a typical Tafel plot (E vs. logI), obtained in the presence of 50 μM nitrite. For a totally irreversible diffusion-controlled reaction, the number of electrons involved in the rate-determining step (nc′) and transfer coefficient (α′) values can be deduced from the Tafel equation:
| | | Epc = K + (bc/2)logν | (4) |
where the Tafel slope
bc is given by
and
K,
R,
T, and
F are constants. The calculated Tafel slope (∂
Epc/∂log
ν) is 145 V dec
−1. A similar value of 71.5 mV dec
−1 was also obtained indirectly from the plot of
Epavs. log
v, from which the
bc (2 × slope) was estimated to be 143 mV dec
−1. Furthermore, using the Randles–Sevcik equation, the total number of electrons involved in the electrocatalytic nitrite reduction (
n′) for an irreversible diffusion-controlled electron process can be calculated using
eqn (6):
53| | | ipc = 2.99 × 105n′[(1 − α′)nc′]1/2AgeoCNO2−DNO2−½ν½ | (6) |
where (1 −
α′) = 0.34,
Ageo = 0.0707 cm
2,
CNO2− = 1.5 × 10
−6 mol cm
−3, and
DNO2− = 3.7 × 10
−5 cm
2 s
−1.
58 Substituting the slope of (
ipc/
ν½), 696.4 × 10
−6 A (V s
−1)
−1/2, and the above-listed values in
eqn (6), the value of
nc′ was calculated to be 6.19, confirming the occurrence of a six-electron reduction reaction.
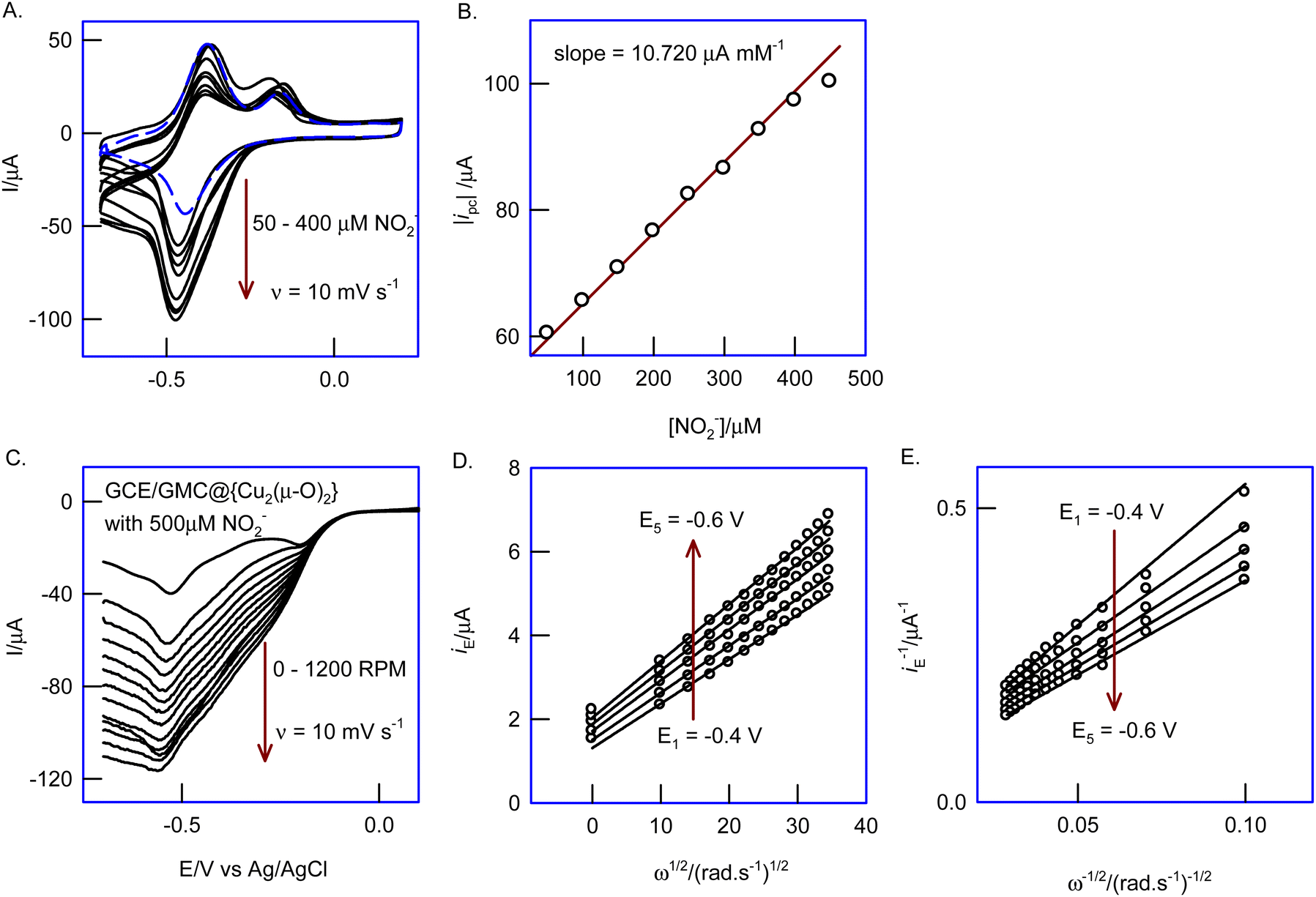
Fig. 8A shows the CVs of the modified electrode with increasing nitrite concentrations in the range 50–400 μM. A linear increase of icat with the concentration of NO2− was observed, with slope and regression values of 10.72 μA mM−1 and 0.9964, respectively (Fig. 8B). The GMC@{Cu2(μ-O)2} modified electrode was also studied with the rotating disc electrode (RDE) technique; the linear sweep voltammetry response was recorded in 500 μM NO2−, at a scan rate of 10 mV s−1, with a systematic increase of the hydrodynamic rotation speed from 0 to 1200 rpm. As shown in Fig. 8C, as the rotating speed increased, the hydrodynamic reduction current also increased. The system obeyed the Levich equation, as follows:67
| | | iE = [0.62nFAD2/3ν−1/6CNO2−]ω1/2 | (7) |
where
iE denotes the hydrodynamic currents at different potentials. The plot of
iEvs. ω1/2 shown in
Fig. 8D gives linear responses denoting the diffusion-controlled electron transfer behavior of the electrocatalytic reduction of NO
2−. Using the Koutecky–Levich (KL) equation, the kinetic parameter of the electrochemical reduction reaction was calculated according to
eqn (8) and
(9):
| | | 1/iE = 1/ik + 1/0.62ncFAD2/3ν−1/6ω−1/2CNO2− | (8) |
and
where
ik is the kinetic current and
kh = heterogeneous rate constant for the electrocatalytic reduction of NO
2−. As shown in
Fig. 8E, a linear response was observed for the plot of
iE−1vs. ω−1/2, from which the respective
ik values were obtained from the intercept. Substituting the latter in
eqn (9), the
kh value was estimated to be 4.97 ± 0.5 × 10
2 mol
−1 cm
3 s
−1, which is comparable to other nitrite reduction systems (5.9 × 10
2 mol
−1 cm
3 s
−1 and 8.4 × 10
3 mol
−1 cm
3 s
−1).
68,69 Based on the Levich slope (135.5 nA (rad s
−1)
−1), the calculated average number of electrons for the nitrite reduction reaction is 6.3 ± 0.5, which is close to the value obtained from the CVs (
n = 6.19).
 |
| | Fig. 8 (A) Effect of increasing the concentration of NO2− (0.5–4 mM) on GCE/GMC@{Cu2(μ-O)2} in N2 purged 0.1 M PBS, pH 7, at ν = 10 mV s−1; plots of (B) |ipc| vs. CNO2−, (C) RDE response of GCE/GMC@{Cu2(μ-O)2} in 500 μM of NO2−, at different potentials, −0.4, −0.45, −0.5, −0.55, −0.6 V vs. Ag/AgCl; (D) Levich and (E) Koutecky–Levich plots. | |
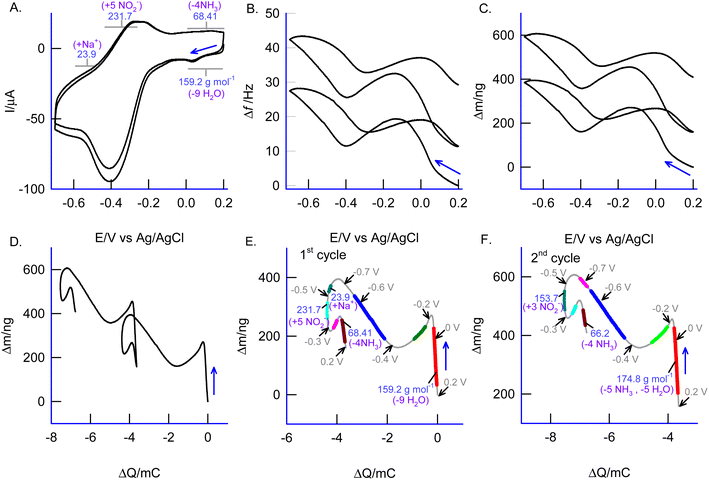
An in situ EQCM experiment was carried out for the electrochemical nitrite reduction reaction on the EQCM-Au/GMC@{Cu2(μ-O)2} complex modified electrode in 0.1 M PBS, pH 7. As seen in Fig. 9A & B, there was a sudden increment in the mass of about 231.7 g mol−1, corresponding to the formation of five NO2− molecules (Mw = 46.01 g mol−1) at the beginning of the potential cycling experiments. Similarly, at the positive potential of 0.1 V vs. Ag/AgCl, an Mw of 68.41 ± 0.4 was observed, corresponding to the release of four NH3 (Mw = 17.03 g mol−1) from the matrix (Fig. 9E). These observations revealed that a specific electrochemical reaction took place at the negative potential, which was followed by chemical steps that led to the overall nitrite conversion into ammonium. In the electrochemical process, the reduced form of the complex, GMC@{CuI2(μ-O)2}, catalyzes the nitrite reduction reaction, thus recycling the oxidized form of the complex, {CuII2(μ-O)2}. Under the electrochemical dynamic conditions, the latter is regenerated back to the reduced Cu-complex, {CuI2(μ-O)2}, which can then be involved in a new turnover cycle, as shown in Schemes 2 & 3.
 |
| | Fig. 9 (A and B) In situ CV-EQCM responses of GCE/GMC@{Cu2(μ-O)2} with 500 μM NO2− in N2 purged pH 7 PBS at a scan rate of 10 mV s−1. Corresponding derivative plots of (C) Δm vs. E and Δm vs. ΔQ of (D) 1–2 cycles, (E) the first cycle and (F) second cycle. | |
3.6. Identification of the product of nitrite reduction by GCE/GMC@{Cu2(μ-O)2}
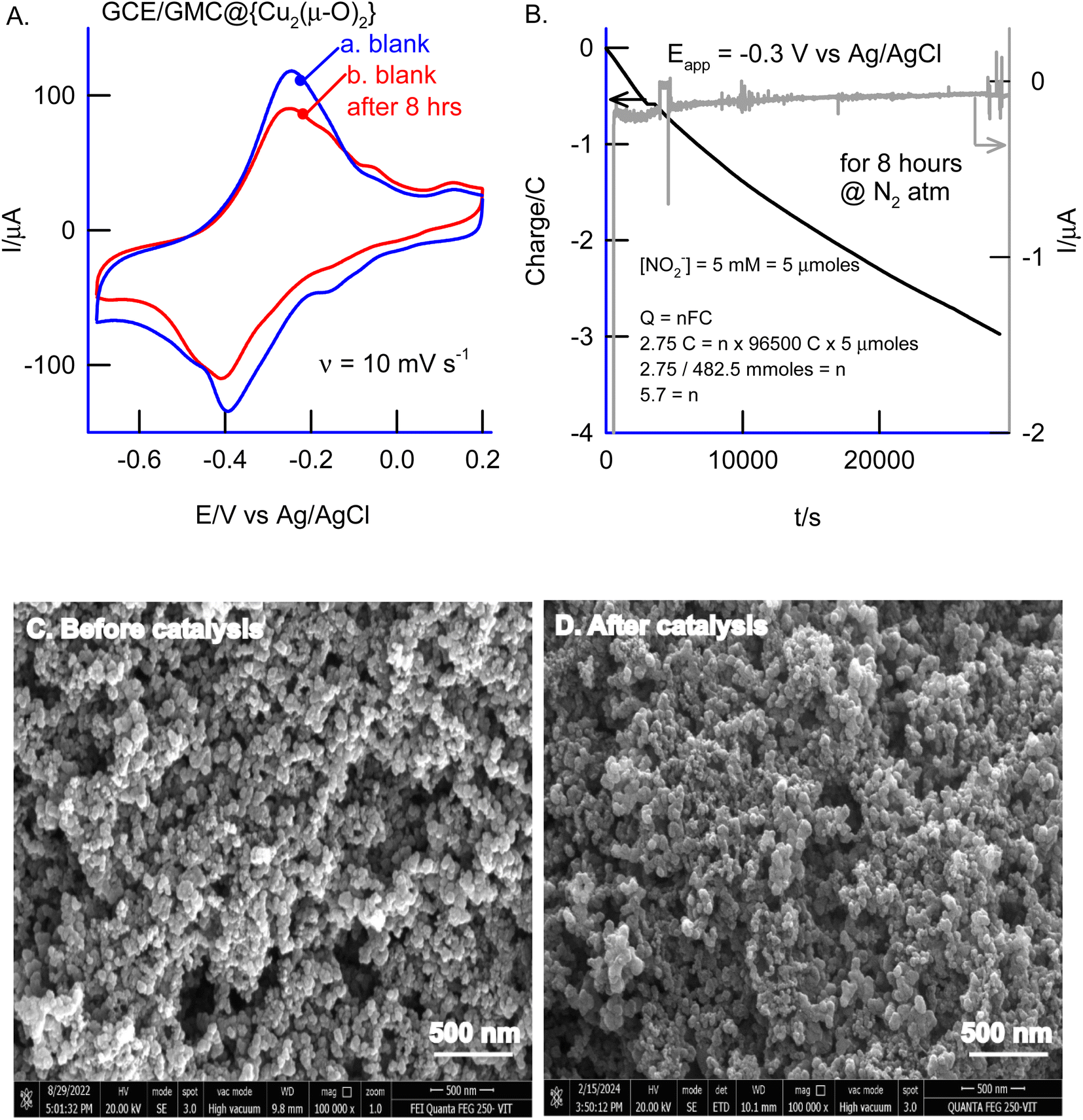
To confirm the identity of the reaction product, we conducted bulk electrolysis of nitrite (5 mM) at −0.3 V vs. Ag/AgCl (Fig. 10A & B). The final product was tested using Nessler's reagent, resulting in an immediate appearance of a pale orange color, indicating the formation of HgO·Hg(NH2)I due to the production of an ammonium salt (ESI Fig. S7†). Additionally, we employed an indophenol-based colorimetric method, along with standard addition of ammonia, to authenticate the formation of NH3 as a reaction product (ESI Fig. S8†). The results confirmed the selective formation of NH3. Furthermore, we hypothesized that N2 reduction and hydrazine formation might occur in this process. To investigate this, we conducted an independent study on the electrochemical oxidation reaction using a chemically modified electrode, both in the presence and absence of N2-purged pH 7 PBS. However, minimal change in the faradaic response was observed. Additionally, if hydrazine were formed during the reaction, it would be detectable through specific oxidation at an oxidation potential of 0.6 V vs. Ag/AgCl on the chemically modified electrode (ESI Fig. S9†). However, our experiments did not yield such a specific signal, refuting the notion of hydrazine formation as an intermediate or product of the reaction.
 |
| | Fig. 10 (A) CV responses of GCE/GMC@{Cu2(μ-O)2} before and after the bulk electrolysis of 5 mM NO2− at an applied potential of −0.3 V vs. Ag/AgCl in N2 purged pH 2 HCl–KCl. (B) Bulk electrolysis response (inset: number of electrons calculation). Comparison of GCE/GMC@{Cu2(μ-O)2} before (C) and after (D) electrocatalytic nitrite reduction. | |
Due to the stability of the modified electrode towards electrocatalytic nitrite reduction, the catalyst can facilitate long-term ammonia production. A faradaic efficiency of approximately 80 ± 1% can be anticipated for a duration of around 50 hours. Following bulk electrolysis, the catalyst underwent FESEM analysis to assess its morphology, revealing that the catalyst remained undamaged despite the adsorption of nitrite (Fig. 10C & D). This finding supports the notion of catalyst stability.
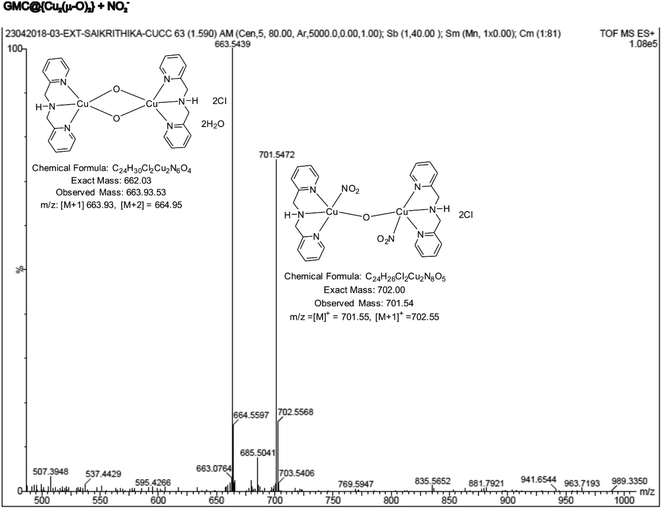
The active metal complex involved in the electrochemical nitrite reduction reaction was analysed by ESI-MS using an ethanolic extract of {Cu2(μ-O)2}, obtained at the middle of the electrochemical reaction. Fig. 11 shows a typical ESI-MS spectrum where the peak Mw = 701.54 is assigned to the intermediate {Cu2(μ-O)2} where one of the oxo-bridges was cleaved due to NO2− binding, resulting in {Cu2(BPA)2s(NO2)2(μ-O)}2+ (Scheme 1D). This complex intermediate formation is similar to the catalytic mechanism of ccNiR.70,71 Overall, the biomimicking of the heterogeneous electrocatalytic reduction of nitrite into ammonium, at neutral pH and low overpotential, was observed for the first time, owing to the in situ generation of a GMC@{Cu2(μ-O)2} complex.
 |
| | Fig. 11 ESI-MS data of an ethanolic extract of GCE/GMC@{Cu2(μ-O)2}, upon NO2− electrocatalysis. | |
3.7. Electroanalytical application
As a proof of concept, the biomimetic electrode GCE/GMC@{Cu2(μ-O)2} was applied in the amperometric sensing of nitrite (Eapp = −0.4 V vs. Ag/AgCl), by successively spiking 500 μM (Fig. 12A) or 50 μM nitrite (Fig. 12B) to a 0.1 M PBS, pH 7, electrolyte solution, under hydrodynamic conditions. As seen in Fig. 12C, the current increases linearly upon continuous addition of 500 μM and 50 μM NO2−, yielding sensitivity values of 16.14 μA mM−1 (curve a) and 15.54 μA mM−1 (curve b), respectively. In a control assay, the unmodified electrode GCE/GMC showed a featureless response (Fig. 12A, curve b). Furthermore, possible electrochemical and biochemical interferents, namely, ascorbic acid (AA), uric acid (UA), dopamine (DA), citric acid (CA), glucose (G), hydrogen peroxide (H2O2), cysteine (CySH) and oxygen (O2), were tested under identical conditions. Except for oxygen, none of the other compounds were found to interfere with the electrocatalytic reduction of nitrite, like a natural enzyme under physiological conditions (Fig. 12D).
 |
| | Fig. 12 Comparative amperometric i–t responses of GCE/GMC@{Cu2(μ-O)2} with continuous spikes of (A) 500 μM (curve b is the control response of the GCE/GMC) and (B) 50 μM NO2−; (C) calibration plots of ipcvs. [NO2−] in different NO2− concentration regions. (D) Effect of interference of various analytes. (E) Analysis of a tap water sample. | |
The nitrite content of a real water sample was assessed by the standard addition method, showing a recovery value of ∼100 ± 3%, thereby attesting to the applicability of this new electroanalytical system in a practical application (Fig. 12E).
4. Conclusions
The in situ electrochemical conversion of the {Cu(BPA)Cl} complex into {Cu2(μ-O)2} has been demonstrated on a GMC-modified glassy carbon electrode by simple potential cycling of the precursor complex, {Cu(BPA)Cl}, as chemically adsorbed on the GCE. A control experiment with the non-electrochemically modified {Cu(BPA)Cl} complex has failed to provide any faradaic response. In contrast, the {Cu2(μ-O)2} chemically modified electrode, GCE/GMC@{Cu2(μ-O)2}, showed a well-defined redox peak at Eo′ = −0.25 V vs. Ag/AgCl. The as-prepared electrochemically modified electrode was characterized through various physicochemical techniques and by an in situ EQCM study. In addition, an ethanolic extract of the chemically modified electrode was subjected to ESI-MS analysis. The experimental observations revealed that the {Cu(BPA)Cl} precursor complex was transformed into a {Cu2(μ-O)2} complex, as a surface-confined molecular species on the GMC surface. By using the SECM technique, the electro-active sites involved in the electronic conductivity of the Cu-complex modified electrode were visualized. The surface morphology features are in good agreement with the observations from the TEM analysis. The GCE/GMC@{Cu2(μ-O)2} heterogeneous system showed a highly efficient electrocatalytic reduction of nitrite at a much lower potential than the values reported in the literature for inorganic electrocatalysts. By using cyclic voltammetry and rotating disc electrode studies, electrokinetic parameters such as the Tafel slope (145 mV dec−1), the transfer coefficient (0.49), and the number of electrons involved in the rate-determining step (nc′ = 1) and the overall process (n = 6; nitrite to ammonia formation) have been evaluated. A possible reaction mechanism for the nitrite reduction reaction was probed using in situ EQCM analysis showing specific binding of nitrite to the complex, for the enzyme-based reaction systems. To validate the nature of the electrosynthesized product, a bulk electrolysis experiment was carried out in a 500 μM nitrite solution, using the Cu-complex chemically modified electrode; the final product was tested with Nessler's reagent, confirming the ammonium formation.
The novel GCE/GMC@{Cu2(μ-O)2} biomimicking catalytic system has important practical applications. First, it potentially offers an alternative route for the synthesis of ammonium, a very important fertilizer whose industrial production relies on the Haber–Bosch process, an energy-intensive method with a huge carbon footprint.78 Second, it enables the selective chrono-amperometric sensing of nitrite at neutral pH.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors acknowledge the Department of Science and Technology – Science and Engineering Research Board (DST/CRG/2021/001048) scheme for financial support.
References
- M. M. M. Kuypers, H. K. Marchant and B. Kartal, The Microbial Nitrogen-Cycling Network, Nat. Rev. Microbiol., 2018, 16, 263–276 CrossRef CAS PubMed.
- X. Zhang, B. B. Ward and D. M. Sigman, Global Nitrogen Cycle: Critical Enzymes, Organisms, and Processes for Nitrogen Budgets and Dynamics, Chem. Rev., 2020, 120, 5308–5351 CrossRef CAS PubMed.
-
W. Zumft and H. Bothe, Nitrous Oxide Reductases, in Biology of Nitrogen Cycle, ed. Bothe, H., Ferguson, S. J. and Newton, W. E., Elsevier, Amsterdam, 2006, pp. 67–81 Search PubMed.
-
E. Kandeler, C. Poll, W. T. Frankenberger Jr and M. A. Tabatabai, Nitrogen Cycle Enzymes, Methods of Soil Enzymology, 2011, vol. 9, pp. 211–245 Search PubMed.
- O. Van Cleemput and A. H. Samater, Nitrite in soils: accumulation and role in the formation of gaseous N compounds, Fert. Res., 1995, 45, 81–89 CrossRef CAS.
- J. W. Erisman, J. N. Galloway, S. Seitzinger, A. Bleeker, N. B. Dise, A. M. R. Petrescu, A. M. Leach and W. de Vries, Consequences of human modification of the global nitrogen cycle, Philos. Trans. R. Soc., B, 2013, 368, 20130116 CrossRef PubMed.
- R. M. Martínez-Espinosa, J. A. Cole, D. J. Richardson and N. J. Watmough, Enzymology and ecology of the nitrogen cycle, Biochem. Soc. Trans., 2011, 39, 175–178 CrossRef PubMed.
- S. Besson, M. G. Almeida and C. M. Silveira, Nitrite reduction in bacteria: A comprehensive view of nitrite reductases, Coord. Chem. Rev., 2022, 464, 214560 CrossRef CAS.
- J. R. Thayer, J. H. Chasko, L. A. Swartz and N. J. Parks, Gut Reactions of Radioactive Nitrite After Intratracheal Administration in Mice, Science, 1982, 217, 151–153 CrossRef CAS PubMed.
- E. Hanff, M. Lützow, A. A. Kayacelebi, A. Finkel, M. Maassen, G. R. Yanchev, A. Haghikia, U. Bavendiek, A. Buck, T. Lücke, N. Maassen and D. Tsikas, Simultaneous GC-ECNICI-MS Measurement of Nitrite, Nitrate and Creatinine in Human Urine and Plasma in Clinical Settings, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1047, 207–214 CrossRef CAS PubMed.
- B. Strehlitz, B. Gründig, W. Schumacher, P. M. H. Kroneck, K.-D. Vorlop and H. Kotte, A Nitrite Sensor Based on a Highly Sensitive Nitrite Reductase Mediator-Coupled Amperometric Detection, Anal. Chem., 1996, 68, 807–816 CrossRef CAS PubMed.
- Q. Wu, D. Storrier, F. Pariente, Y. Wang, J. P. Shapleigh and H. D. Abruña, A Nitrite Biosensor Based on a Maltose Binding Protein Nitrite Reductase Fusion Immobilized on an Electropolymerized Film of a Pyrrole-Derived Bipyridinium, Anal. Chem., 1997, 69, 4856–4863 CrossRef CAS PubMed.
- A. S. Serra, S. Jorge, C. Silveira, J. J. G. Moura, E. Jubete, E. Ochoteco and M. G. Almeida, Cooperative use of Cytochrome cd1 Nitrite Reductase and its Redox Partner Cytochrome c552 to Improve the Selectivity of Nitrite Biosensing, Anal. Chim. Acta, 2011, 693, 41–46 CrossRef CAS PubMed.
- J. Wang, G. Zhan, X. Yang, D. Zheng, X. Li, L. Zhang, T. Huang and X. Wang, Rapid Detection of Nitrite Based on Nitrite-Oxidizing Bacteria Biosensor and its Application in Surface Water Monitoring, Biosens. Bioelectron., 2022, 215, 114573 CrossRef CAS PubMed.
- T. Madasamy, M. Pandiaraj, M. Balamurugan, K. Bhargava, N. K. Sethy and C. Karunakaran, Copper, Zinc Superoxide Dismutase and Nitrate Reductase Coimmobilized Bienzymatic Biosensor for the Simultaneous Determination of Nitrite and Nitrate, Biosens. Bioelectron., 2014, 52, 209–215 CrossRef CAS PubMed.
- M. H. Barley, M. R. Rhodes and T. J. Meyer, Electrocatalytic Reduction of Nitrite to Nitrous Oxide and Ammonia Based on The N-methylated, Cationic Iron Porphyrin Complex [FeIII(H2O)(TMPyP)]5+, Inorg. Chem., 1987, 26, 1746–1750 CrossRef CAS.
- J. A. Cox and A. F. Brajter, Selective Electrocatalytic Method for the Determination of Nitrite, Anal. Chem., 1979, 51, 2230–2232 CrossRef CAS.
- M. H. Barley and T. J. Meyer, Electrocatalytic Reduction of Nitrite to Ammonia Based on a Water-Soluble Iron Porphyrin, J. Am. Chem. Soc., 1986, 108, 5876–5885 CrossRef CAS PubMed.
- J. R. Stroka, B. Kandemir, E. M. Matson and K. L. Bren, Electrocatalytic Multielectron Nitrite Reduction in Water by an Iron Complex, ACS Catal., 2020, 10, 13968–13972 CrossRef CAS.
- H. Liu, J. Park, Y. Chen, Y. Qiu, Y. Cheng, K. Srivastava, S. Gu, B. H. Shanks, L. T. Roling and W. Li, Electrocatalytic Nitrate Reduction on Oxide-Derived Silver with Tunable Selectivity to Nitrite and Ammonia, ACS Catal., 2021, 11, 8431–8442 CrossRef CAS.
- L. Ouyang, L. Yue, Q. Liu, Q. Liu, Z. Li, S. Sun, Y. Luo, A. A. Alshehri, M. S. Hamdy, Q. Kong and X. Sun, Cu Nanoparticles Decorated Juncus-Derived Carbon for Efficient Electrocatalytic Nitrite-to-Ammonia Conversion, J. Colloid Interface Sci., 2022, 624, 394–399 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, Y. Wang, Y. Guo, X. Xie, Y. Yu and B. Zhang, Recent Advances in Electrocatalytic Nitrite Reduction, Chem. Commun., 2022, 58, 2777–2787 RSC.
- C. Xia, X. Yanjun and W. Ning, Hollow Fe2O3 Polyhedrons: One-Pot Synthesis and Their Use as Electrochemical Material for Nitrite Sensing, Electrochim. Acta, 2012, 59, 81–85 CrossRef.
- P. K. Rastogi, V. Ganesan and S. Krishnamoorthi, A Promising Electrochemical Sensing Platform Based on A Silver Nanoparticles Decorated Copolymer for Sensitive Nitrite Determination, J. Mater. Chem. A, 2014, 2, 933–943 RSC.
- U. P. Azad, S. Turllapati, P. K. Rastogi and V. Ganesan, Tris(1,10-phenanthroline)iron(II)-bentonite Film as Efficient Electrochemical Sensing Platform for Nitrite Determination, Electrochim. Acta, 2014, 127, 193–199 CrossRef CAS.
- P. Lin, F. Chai, R. Zhang, G. Xu, X. Fan and X. Luo, Electrochemical Synthesis of Poly(3,4-ethylenedioxythiophene) Doped with Gold Nanoparticles, and Its Application to Nitrite Sensing, Microchim. Acta, 2016, 183, 1235–1241 CrossRef CAS.
- R. Xi, S.-H. Zhang, l. Zhang, C. Wang, L.-J. Wang, J.-H. Yan and G.-B. Pan, Electrodeposition of Pd-Pt Nanocomposites on Porous GaN for Electrochemical Nitrite Sensing, Sensors, 2019, 19, 606 CrossRef PubMed.
- V. Sudha, S. M. S. Kumar and R. Thangamuthu, Simultaneous Electrochemical Sensing of Sulphite and Nitrite on Acid-Functionalized Multi-walled Carbon Nanotubes Modified Electrodes, J. Alloys Compd., 2018, 749, 990–999 CrossRef CAS.
- X. Zhao, N. Li, M. Jing, Y. Zhang, W. Wang, L. Liu, Z. Xu, L. Liu, F. Li and N. Wu, Monodispersed and Spherical Silver Nanoparticles/Graphene Nanocomposites from Gamma-Ray Assisted In-situ Synthesis for Nitrite Electrochemical Sensing, Electrochim. Acta, 2019, 295, 434–443 CrossRef CAS.
- S.-L. Meng, C. Zhang, C. Ye, J.-H. Li, S. Zhou, L. Zhu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Cobaloximes: selective nitrite reduction catalysts for tandem ammonia synthesis, Energy Environ. Sci., 2023, 16, 1590 RSC.
- X. Zhu, M. He, L. Xiao, H. Liu, M. Hu, S. Li, Q. Zhai, Y. Chen and Y. Jiang, Enzymatic biosensor for nitrite detection based on direct electron transfer by CPO-ILEMB/Au@MoS2/GC, J. Appl. Electrochem., 2022, 52, 979 CrossRef CAS.
- Z. Yang, Z. Gong, M. Fan, W. Zhang, H. Xue and W. Fang, Trimetallic zeolitic imidazolate framework-derived hollow structure as a sensing material for nitrite electrochemical detection, Colloids Surf., A, 2023, 678, 132513 CrossRef CAS.
- O. Einsle, A. Messerschmidt, R. Huber, M. H. Kroneck and F. Neese, Mechanism of The Six-Electron Reduction of Nitrite to Ammonia by Cytochrome c Nitrite Reductase, J. Am. Chem. Soc., 2002, 124, 11737–11745 CrossRef CAS PubMed.
- D. Bykov and F. Neese, Six-Electron Reduction of Nitrite to Ammonia by Cytochrome c Nitrite Reductase: Insights from Density Functional Theory Studies, Inorg. Chem., 2015, 54, 9303–9316 CrossRef CAS PubMed.
- M. G. Almeida, C. M. Silveira, B. Guigliarelli, P. Bertrand, J. J. Moura, I. Moura and C. Léger, A needle in a haystack: the active site of the membrane-bound complex cytochrome c nitrite reductase, FEBS Lett., 2007, 581, 284–288 CrossRef CAS PubMed.
- E. T. Judd, M. Youngblut, A. A. Pacheco and S. J. Elliott, Direct electrochemistry of Shewanella oneidensis cytochrome c nitrite reductase: evidence of interactions across the dimeric interface, Biochemistry, 2012, 51, 10175–10185 CrossRef CAS PubMed.
- T. Monteiro, P. Rodrigues, A. L. Gonçalves, J. J. G. Moura, E. Jubete, L. Añorga, B. Piknova, A. N. Schechter, C. M. Silveira and M. G. Almeida, Construction of Effective Disposable Biosensors for Point Of Care Testing of Nitrite, Talanta, 2015, 142, 246–251 CrossRef CAS PubMed.
- T. Monteiro, M. Moreira, S. B. R. Gaspar and M. G. Almeida, Bilirubin Oxidase as a Single Enzymatic Oxygen Scavenger for the Development of Reductase-Based Biosensors in the Open Air and its Application to a Nitrite Biosensor, Biosens. Bioelectron., 2022, 217, 114720 CrossRef CAS PubMed.
- X.-Y. Qiu, Y.-Y. Cheng, Q. Li, Y.-Y. Yu and X. Xiao, An In-Field Assembled Hierarchical Mesoporous Electroenzymatic Sensor for Sensitive and Real-Time Monitoring of Nitrite, J. Cleaner Prod., 2023, 426, 139102 CrossRef CAS.
- Y. Li, A. Yamaguchi, M. Yamamoto, K. Takai and R. Nakamura, Molybdenum Sulfide: A Bioinspired Electrocatalyst for Dissimilatory Ammonia Synthesis with Geoelectrical Current, J. Phys. Chem. C, 2017, 121, 2154–2164 CrossRef CAS.
- S. Siek, N. A. Dixon and E. T. Papish, Electrochemical Reduction of Ttz Copper(II) Complexes in the presence and Absence of Protons: Processes Relevant to Enzymatic Nitrite Reduction (TtzR,R' = tris(3-R, 5-R’-1,2,4-triazolyl)borate), Inorg. Chim. Acta, 2017, 459, 80–86 CrossRef CAS.
- X. Liu, T. Zhang, X. li, S. Ai and S. Zhou, Non-Enzymatic Electrochemical Sensor Based on An AuNPs/Cu-N-C Composite for Efficient Nitrite Sensing in Sausage Samples, New J. Chem., 2022, 46, 10415–10421 RSC.
- Y. Guo, J. R. Stroka, B. Kandemir, C. E. Dickerson and K. L. Bren, Cobalt Metallopeptide Electrocatalyst for the Selective Reduction of Nitrite to Ammonium, J. Am. Chem. Soc., 2018, 140, 16888–16892 CrossRef CAS PubMed.
- G. Cioncoloni, I. Roger, P. S. Wheatley, C. Wilson, R. E. Morris, S. Sproules and M. D. Symes, Proton-Coupled Electron Transfer Enhances the Electrocatalytic Reduction of Nitrite to NO in a Bioinspired Copper Complex, ACS Catal., 2018, 8, 5070–5084 CrossRef CAS.
- X. He, Z. Li, J. Yao, K. Dong, X. Li, L. Hu, S. Sun, Z. Cai, D. Zheng, Y. Luo, B. Ying, M. S. Hamdy, L. Xie, Q. Liu and X. Sun, High-Efficiency Electrocatalytic Nitrite Reduction Toward Ammonia Synthesis On CoP@TiO2 Nanoribbon Array, iScience, 2023, 26, 107100 CrossRef CAS PubMed.
- L. Yue, W. Song, L. Zhang, Y. Luo, Y. Wang, T. Li, B. Ying, S. Sun, D. Zheng, Q. Liu, A. Farouk, M. S. Hamdy, S. Alfaifi and X. Sun, Recent Advance in Heterogenous Electrocatalysts for Highly Selective Nitrite Reduction to Ammonia Under Ambient Condition, Small Struct., 2023, 4, 2300168 CrossRef CAS.
- L. Ouyang, X. Fan, Z. Li, X. He, S. Sun, Z. Cai, Y. Luo, D. Zheng, B. Ying, J. Zhang, A. A. Alshehri, Y. Wang, K. Ma and X. Sun, High-Efficiency Electroreduction Of Nitrite To Ammonia On A Cu@TiO2 Nanobelt Array, Chem. Commun., 2023, 59, 1625–1628 RSC.
- H. Wang, F. Zhang, M. Jin, D. Zhao, X. Fan, Z. Li, Y. Luo, D. Zheng, T. Li, Y. Wang, B. Ying, S. Sun, Q. Liu, X. Liu and X. Sun, V-doped TiO2 Nanobelt Array For High-Efficiency Electrocatalytic Nitrite Reduction To Ammonia, Mater. Today Phys., 2023, 30, 100944 CrossRef CAS.
- M. Velusamy, R. Mayilmurugan and M. Palaniandavar, Functional Models for Catechol Dioxygenases: Iron(III) Complexes of cis-facially Coordinating Linear 3N Ligands, J. Inorg. Biochem., 2005, 99, 1032–1042 CrossRef CAS PubMed.
- A. Raja, V. Rajendiran, P. U. Maheswari, R. Balamurugan, C. A. Kilner, M. A. Halcrow and M. Palaniandavar, Copper(II) Complexes of Tridentate pyridylmethylethylenediamines: Role of Ligand Steric Hindrance on DNA Binding and Cleavage, J. Inorg. Biochem., 2005, 99, 1717–1732 CrossRef CAS PubMed.
- Q. Gao, H. S. Pillai, Y. Huang, S. Liu, Q. Mu, X. Han, Z. Yan, H. Zhou, Q. He, H. Xin and H. Zhu, Breaking Adsorption-Energy Scaling Limitations of Electrocatalytic Nitrate Reduction on Intermetallic CuPd Nanocubes by Machine-Learned Insights, Nat. Commun., 2020, 13, 2338–2344 CrossRef PubMed.
- X. He, X. Li, X. Fan, J. Li, D. Zhao, L. Zhang, S. Sun, Y. Luo, D. Zheng, L. Xie, A. M. Asiri, Q. Liu and X. Sun, Ambient Electroreduction of Nitrite to Ammonia over Ni Nanoparticle Supported on Molasses-Derived Carbon Sheets, ACS Appl. Nano Mater., 2022, 5, 14246–14250 CrossRef CAS.
- A. S. Kumar, R. Shanmugam, N. Vishnu, K. C. Pillai and S. Kamaraj, Electrochemical Immobilization of Ellagic Acid Phytochemical on MWCNT Modified Glassy Carbon Electrode Surface and Its Efficient Hydrazine Electrocatalytic Activity in Neutral pH, J. Electroanal. Chem., 2016, 782, 215–224 CrossRef CAS.
- E. Laviron, General Expression of the Linear Potential Sweep Voltammogram in The Case of Diffusionless Electrochemical Systems, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2009 Search PubMed.
- K. Amreen, A. S. Kumar, V. Mani and S.-T. Huang, Axial Coordination Site-Turned Surface Confinement, Electron Transfer, and Bio-Electrocatalytic Applications of a Hemin Complex on Graphitic Carbon Nanomaterial-Modified Electrodes, ACS Omega, 2018, 3, 5435–5444 CrossRef CAS PubMed.
- K. Amreen and A. S. Kumar, Highly Redox-Active Hematin-Functionalized Carbon Mesoporous Nanomaterial for Electrocatalytic Reduction Applications in Neutral Media, ACS Appl. Nano Mater., 2018, 1, 2272–2283 CrossRef CAS.
- A. S. Kumar, B. Palani and K. C. Pillai, Highly Stable and Redox Active Nano Copper Species Stabilized Functionalized-Multiwalled Carbon Nanotube/Chitosan Modified Electrode for Efficient Hydrogen Peroxide Detection, Colloids Surf., A, 2012, 395, 207–216 CrossRef.
- J.-M. Zen and A. S. Kumar, A Mimicking Enzyme Analogue for Chemical Sensors, Acc. Chem. Res., 2001, 34, 772–780 CrossRef CAS PubMed.
- S. Layek, R. Ganguly and D.
D. Pathak, Unprecedented Formation of A m-Oxobridged Polymeric Copper(II) Complex: Evaluation of Catalytic Activity in Synthesis of 5-Substituted 1H-Tetrazoles, J. Organomet. Chem., 2018, 870, 16–22 CrossRef CAS.
- S. Sundaram and S. K. Annamalai, Selective Immobilization of Hydroquinone on Carbon Nanotube Modified Electrode via Phenol Electro-Oxidation Method and its Hydrazine Electro-Catalysis and Escherichia Coli Antibacterial Activity, Electrochim. Acta, 2012, 62, 207–217 CrossRef CAS.
- P. Gayathri, A. Senthil Kumar and S. Kamaraj, An Unusual Electrochemical Reductive Cleavage of Azo Dye into Highly Redox Active Copolymeric Aniline Derivatives on a MWCNT Modified Electrode Surface at Neutral pH and its Electroanalytical Features, J. Phys. Chem. C, 2015, 119, 7791–7801 CrossRef CAS.
- P. Swetha and A. S. Kumar, Selective Electrochemical Recognition of the α-Naphthol Isomer and in situ Immobilization of Naphthoquinones for Tunable Electrocatalysis, Chem.–Asian J., 2013, 8, 896–901 CrossRef CAS PubMed.
- K. S. S. Devi, S. Jacob and A. S. Kumar, In Situ Structural Elucidation and Selective Pb2+ Ion Recognition of Polydopamine Film Formed by Controlled Electrochemical Oxidation of Dopamine, Langmuir, 2018, 34, 7048–7058 CrossRef PubMed.
- O. W. Jensen and B. W. Jensen, Reduction of Nitrite to Ammonia on PEDOT-bipyridinium-Fe Complex Electrodes, Electrochem. Commun., 2014, 43, 98–101 CrossRef.
- M. A. Kamyabi and F. Aghajanloo, Electrocatalytic Oxidation and Determination of Nitrite on Carbon Paste Electrode Modified with Oxovanadium(IV)-4-methyl Salophen, J. Electroanal. Chem., 2008, 614, 157–165 CrossRef CAS.
- P. Mayuri, N. Saravanan and A. S. Kumar, A Bioinspired Copper 2,2-bipyridyl Complex Immobilized MWCNT Modified Electrode Prepared By A New Strategy For Elegant Electrocatalytic Reduction and Sensing of Hydrogen Peroxide, Electrochim. Acta, 2017, 240, 522–533 CrossRef CAS.
- A. G. Vargas, M. A. O. Tolentino, E. Lima and J. F. Morenod, Efficient Electrocatalytic Reduction of Nitrite Species on Zeolite Modified Electrode with Cu-ZSM-5, Electrochim. Acta, 2013, 108, 583–590 CrossRef.
- K. Calfumán, M. J. Aguirre, P. C. Rosales, S. Bolloc, R. Llusar and M. Isaacs, Electrocatalytic Reduction of Nitrite on Tetraruthenated Metalloporphyrins/Nafion Glassy Carbon Modified Electrode, Electrochim. Acta, 2011, 56, 8484–8491 CrossRef.
- H. Liu, K. Guo, J. Lv, Y. Gao, C. Duan, L. Deng and Z. Zhu, A Novel Nitrite Biosensor Based on the Direct Electrochemistry of Horseradish Peroxidase Immobilized on Porous Co3O4 Nanosheets and Reduced Graphene Oxide Composite Modified Electrode, Sens. Actuators, B, 2017, 238, 249–256 CrossRef CAS.
- H. J. Wijma, L. J. C. Jeuken, M. P. Verbeet, F. A. Armstrong and G. W. Canters, Protein Film Voltammetry of Copper-Containing Nitrite Reductase Reveals Reversible Inactivation, J. Am. Chem. Soc., 2007, 129, 8557–8565 CrossRef CAS PubMed.
- W. R. Murphy Jr, K. Takeuchi, M. H. Barley and T. J. Meyer, Mechanism of Reduction of Bound Nitrite to Ammonia, Inorg. Chem., 1986, 25, 1041–1053 CrossRef.
- C. Reuben, E. Galun, H. Cohen, R. Tenne, R. Kalish, Y. Muraki, K. Hashimoto, A. Fujishima, J. M. Butler and C. L. Clément, Efficient Reduction of Nitrite and Nitrate to Ammonia using Thin-Film B-doped Diamond Electrodes, J. Electroanal. Chem., 1995, 396, 233–239 CrossRef.
- Y. Arikawa, Y. Otsubo, H. Fujino, S. Horiuchi, E. Sakuda and K. Umakoshi, Nitrite Reduction Cycle on a Dinuclear Ruthenium Complex Producing Ammonia, J. Am. Chem. Soc., 2018, 140, 842–847 CrossRef CAS PubMed.
- Q. Liu, G. Wen, D. Zhao, L. Xie, S. Sun, L. Zhang, y. Luo, A. A. Alshehri, M. S. Hamdy, Q. Kong and X. Sun, Nitrite Reduction Over Ag Nanoarray Electrocatalyst for Ammonia Synthesis, J. Colloid Interface Sci., 2022, 623, 513–519 CrossRef CAS PubMed.
- J. Wang, J. Liang, P. Liu, Z. Yan, L. Cui, L. Yue, L. Zhang, Y. Ren, T. Li, Y. Luo, Q. Liu, X.-E. Zhao, N. Li, B. Tang, Y. Liu, S. Gao, A. M. Asiri, H. Hao, R. Gao and X. Sun, Biomass Juncus Derived Carbon Decorated with Cobalt Nanoparticles Enables High-Efficiency Ammonia Electrosynthesis by Nitrite Reduction, J. Mater. Chem. A, 2022, 10, 2842–2848 RSC.
- G. Wen, J. Liang, Q. Liu, T. Li, X. An, F. Zhang, A. A. Alshehri, K. A. Alzahrani, Y. Luo, Q. Kong and X. Sun, Ambient Ammonia Production Via Electrocatalytic Nitrite Reduction Catalyzed by A CoP Nanoarray, Nano Res., 2022, 15, 972–977 CrossRef CAS.
- J. Jose Osorio-Tejada, N. N. Tran and V. Hessel, Techno-environmental assessment of small-scale Haber-Bosch and plasma-assisted ammonia supply chains, Sci. Total Environ., 2022, 154162 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Effect of different carbon nanomaterials on the preparation of the modified electrode; role of Nafion; FESEM-EDAX results; UV-visible spectroscopy data; ESI-MS data of the {Cu(BPA)Cl} complex; Nessler's reagent test results. See DOI: https://doi.org/10.1039/d3ta08054d |
| ‡ Present address: Department of Chemistry, Aringnar Anna Govt. Arts & Science College, Karaikal-609603, India. |
|
| This journal is © The Royal Society of Chemistry 2024 |

 cd and
Annamalai Senthil
Kumar
cd and
Annamalai Senthil
Kumar