
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZ-scheme water splitting utilizing CuLi1/3Ti2/3O2 as a hydrogen-evolving photocatalyst with photo-response up to 600 nm†
Shunya
Yoshino
a,
Tanya
Kurutach
ab,
Qingshan
Liu
ab,
Toshiki
Yamanaka
ab,
Shunsuke
Nozawa
c,
Makoto
Kobayashi
 d,
Hiromu
Kumagai
e and
Hideki
Kato
*a
d,
Hiromu
Kumagai
e and
Hideki
Kato
*a
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai 980-8577, Japan. E-mail: hideki.kato.e2@tohoku.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Tohoku University, Sendai 980-8579, Japan
cInstitute of Materials Structure Science, High Energy Accelerator Research Organization, Tsukuba 305-0801, Japan
dInstitute of Materials and Systems for Sustainability, Nagoya University, Nagoya 464-8601, Japan
eResearch Center for Advanced Science and Technology, The University of Tokyo, Tokyo 153-8904, Japan
First published on 5th February 2024
Abstract
CuLi1/3Ti2/3O2 (CLTO) is a visible-responsive photocatalyst, whose photo-response reaches up to 600 nm, for H2 evolution using sacrificial electron donors such as methanol and S2−. In this study, utilization of CLTO in Z-scheme water splitting (Z-WS) was investigated. The photocatalytic performance of Cr2O3/M/CLTO as a H2-evolving photocatalyst, which was prepared by sequential photodeposition of cocatalysts (M: Ru, Rh, Pd and Pt) and Cr2O3, was evaluated for Z-WS using BiVO4, an O2-evolving photocatalyst, and a Co(bpy)33+/2+ redox shuttle under visible light. Among the examined samples, Cr2O3/Ru/CLTO produced both H2 and O2 with meaningful rates. Thus, CLTO was first utilized in a visible responsive Z-scheme system for water splitting. The Cr2O3 layer played a significant role in the suppression of backward reactions, such as reduction of Co(bpy)33+. The activity of Cr2O3/Ru/CLTO for Z-WS was remarkably affected by the deposition conditions of the Ru cocatalyst. The activity for Z-WS was remarkably improved when the photodeposition of the Ru cocatalyst was conducted in a methanol solution of RuCl3. Unusually large plate Ru species with 100–200 nm sizes and about 30 nm thickness were present in the highly active sample. Characterization using X-ray photoelectron spectroscopy and X-ray absorption spectroscopy indicated that the Ru cocatalyst was deposited as mainly the oxyhydroxide of Ru. Z-WS also proceeded in the absence of Co(bpy)33+/2+ (the system based on interparticle electron transfer), however, the Z-scheme system using the Co(bpy)33+/2+ electron shuttle showed 10 times higher activity than the interparticle electron transfer system. The external quantum yield and efficiency of solar energy conversion to hydrogen were determined to be 0.5% at 430 nm and 0.029%, respectively.
Introduction
Solar water splitting is a promising reaction to convert solar energy, the most abundant renewable energy, to H2, a storable chemical fuel.1–3 Photocatalytic water splitting has been demonstrated to show high quantum yields exceeding 50% using ultraviolet (UV) responsive photocatalysts such as NaTaO3:La, Ga2O3:Zn and SrTiO3:Al.4–6 However, they are not eligible for practical use because their wide band gaps restrict the usable wavelength in the solar spectrum to very narrow ranges. Construction of photocatalytic Z-scheme systems is a beneficial approach to water splitting under visible light because Z-scheme water splitting (Z-WS) can be achieved by combining two kinds of photocatalysts incapable of water splitting when used alone.7–9 Thus, many Z-scheme systems have been reported for photocatalytic water splitting under visible light.7–18 SrTiO3:Rh and BiVO4, which have been developed for sacrificial H2 and O2 evolution under visible light,19,20 are frequently used in Z-scheme systems as a H2-evolving photocatalyst (HEP) and an O2-evolving photocatalyst (OEP), respectively. The SrTiO3:Rh-BiVO4 pair is one of the representative ones in the Z-scheme system and is available to systems with various routes of electron transfer from the OEP to HEP: redox electron mediators (Fe3+/2+, Co(bpy)33+/2+ and IO3−/I−), solid state electron mediators (reduced graphene oxide (RGO), carbon and metallic layer) and without any electron mediators (direct interparticle electron transfer).10,15,18,21–25 In particular, a photocatalyst sheet composed of SrTiO3:Rh, BiVO4 and a Au layer showed high performance in water splitting exceeding 1% efficiency of solar energy conversion to hydrogen (STH) and 30% external quantum yield (EQY) at 420 nm.25 This demonstrates the promising potential of the Z-scheme system for water splitting. However, use of photocatalysts with response to longer wavelengths is desired, because the absorption edges of SrTiO3:Rh and BiVO4, which lie at 540 and 520 nm respectively, are insufficient for utilization of sunlight.26Utilization of photocatalysts with response to longer wavelengths compared with SrTiO3:Rh and BiVO4 has been extensively studied. It has enlarged the library of long-wavelength responsive photocatalysts available for Z-scheme systems, for example, metal oxides such as CuLi1/3Ti2/3O2 (CLTO), ZnRh2O4, NaTaO3:Ir and BaTa2O6:Ir, (oxy)nitrides such as BaTaO2N, Ta3N5 and LaMg1/3Ta2/3O2N and (oxy)sulfides such as Sm2Ti2O5S2, Ga–La5Ti2CuO7S5 and CuGa0.8In0.2S2 as HEPs, as well as metal oxides such as Bi4V2O11, SrTiO3:Ir and KNbO3:Ir,Sb, (oxy)nitrides such as Ta3N5, LaTiO2N and La0.5Sr0.5Ta0.5Ti0.5O2N, oxychlorides such as Bi4NbO8Cl and natural photosystem II as OEPs.27–41 To improve the properties of Z-scheme systems, expanding the combination of HEPs and OEPs as well as enhancing the performance of photocatalysts is important. Among the aforementioned photocatalysts, CLTO is an attractive HEP for the topic of expanding combination in the Z-scheme system. CLTO, which is a delafossite-type Cu(I)-titanate discovered in 2015,42 produces H2 in the presence of sacrificial reagents such as methanol and a mixture of S2− and SO32− with photo-response up to 600 nm. Kudo et al. have achieved Z-WS employing Pt-deposited CLTO and RGO-modified TiO2,31 however excitation by UV light is indispensable for this system to excite the TiO2 component. Other Z-scheme systems utilizing CLTO have not been reported so far, thus CLTO has not been combined with visible-responsive OEPs nor redox electron shuttles. Utilization of TiO2 instead of the common visible responsive OEPs such as BiVO4 and WO3 in the previous report implies difficulties in the realization of Z-WS employing RGO-modified BiVO4 and WO3 with CLTO. Therefore, Z-scheme systems based on redox electron shuttles sound better than those with RGO-modified OEPs to achieve Z-WS by combining CLTO with a visible responsive OEP.
Use of redox electron shuttles in Z-WS, such as IO3−/I−, Fe3+/2+ and Co(bpy)33+/2+, often brings unfavourable backward reactions involving the redox couple; that is, reduction of the oxidant occurs on the HEP and oxidation of the reductant occurs on the OEP.17,43–45 It has been reported that adsorption of redox species on a Pt cocatalyst effectively suppresses the backward reactions on Pt in Z-WS using IO3−/I− and Fe3+/2+.17,43,44 Li et al. have reported that a photodeposited Cr2O3 layer also hinders reduction of IO3− on the Pt cocatalyst.45 The universal effects of the Cr2O3 modification are expected in Z-WS using other redox shuttles except under acidic conditions, for which it is necessary to use Fe3+/2+ and VO2+/VO2+.
Based on the aforementioned background, we are attracted to Z-WS employing CLTO, visible responsive OEPs and redox shuttles. In particular, BiVO4, which functions as an OEP in various kinds of Z-scheme systems, is a good candidate for the OEP. A redox couple of Co(bpy)33+/2+, which works well with BiVO4 around neutral pH, is a strong candidate for the redox shuttle in this study. In this work, we examined Z-WS utilizing CLTO, BiVO4 and Co(bpy)33+/2+, and especially the influences of cocatalysts and Cr2O3 modification upon the performance of Z-WS were investigated.
Experimental
Preparation of photocatalysts
Powders of CLTO were synthesized by a reaction between Li4Ti5O12 and Cu2O with the help of a CuCl flux referring to the literature.42 Li4Ti5O12 was prepared by a polymerizable complex (PC) method from Li2CO3 (Wako Pure Chemical, 99.0%) and titanium tetrabutoxide (Kanto Chemical, 97.0%) as raw materials using citric acid (Wako Pure Chemical, 98%) and propylene glycol (Kanto Chemical, 99.0%) as chelating and esterification reagents. The obtained precursor was heated at 800 °C for 5 h in air to obtain a crystalline Li4Ti5O12 powder. The Li4Ti5O12 powder was subjected to milling treatment using a pot mill rotator (As One, PM-001S). Briefly, 1.5 g of Li4Ti5O12 was put in a 250 mL plastic bottle with 30 mL of distilled water and 60 g of stabilized zirconia balls (ϕ10 mm), then the bottle was rotated at 250 rpm for 20 h. This milling treatment was effective to reduce the size of secondary particles of Li4Ti5O12 obtained by the PC method as shown in Fig. S1.† Synthesis of CLTO from milled Li4Ti5O12 and Cu2O was performed at 600 and 700 °C for 10 h in a vacuumed silica tube. After the heat treatment, the remaining Cu2O and CuCl were washed with 2 M NH3 solution. BiVO4 was prepared from Bi2O3 (Wako Pure Chemical, 99.9%) and V2O5 (Kanto Chemical, 99.0%) by a liquid solid reaction method in 0.8 M HNO3 at 80 °C according to the literature.46 SrTiO3:Rh used as a reference photocatalyst was prepared by a spray drying method as previously reported.47For a cocatalyst survey, M (0.05 wt%)/CLTO (M: Ru, Rh, Pd and Pt) was prepared by a photodeposition method in an aqueous 10 vol% methanol solution using RuCl3·nH2O (Wako Pure Chemical, 99.9%), Rh(NO3)3 (Wako Pure Chemical, 95%), PdCl2 (Wako Pure Chemical, 99.0%) and H2PtCl6·6H2O (Kanto Chemical, 99.0%) as precursors. CLTO (0.3 g) was dispersed in a 10 vol% methanol solution containing the cocatalyst source, whose pH was adjusted to 10 by adding an aqueous NaOH solution. The suspension was irradiated with visible light (λ >420 nm) using a 300 W Xe-arc lamp (Excelitas, Cermax PE300BF) with a long pass filter. Cr2O3 modification for M/CLTO (0.2 g) was also performed by a photodeposition method using K2CrO4 (Kanto Chemical, 99.0%) in a 10 vol% methanol solution,48 where the concentration of K2CrO4 was 0.02 mM corresponding to 1.2 wt% of Cr2O3 if the whole chromium was deposited. For the Ru cocatalyst, the deposition conditions including an impregnation method were investigated in more detail. For photodeposition of the Ru cocatalyst, two kinds of reactant solutions, 100% methanol and an aqueous solution containing 0.1 M Na2S and 0.5 M Na2SO3, were examined in addition to the 10 vol% methanol solution. These samples were denoted as Ru(10% MeOH), Ru(100% MeOH) and Ru(S2−). In the impregnation method, CLTO was put in an aqueous RuCl3 solution, then water was evaporated by heating. The dried powder was subjected to heat treatment at 200 °C in N2, 200 °C under vacuum and 150 °C in H2, the samples thus obtained were denoted as Ru(N2), Ru(evac) and Ru(H2), respectively. It should be noted that treatment in air was not conducted because of easy oxidation of Cu+ in CLTO.
Characterization of photocatalysts
The crystal phases of the samples were analyzed by X-ray diffraction using Cu Kα radiation (XRD; Bruker AXS, D2 phaser). The samples were observed using scanning electron microscopes (SEM; Hitachi, SU1510 and S-4800) and a transmission electron microscope (TEM; Jeol, JEM-2100F/HK) with 200 kV acceleration voltage. The diffuse reflectance spectra of the samples were obtained using an ultraviolet-visible-near infrared spectrometer (Jasco, V-770) equipped with an integrating sphere and were converted by the Kubelka–Munk method. The specific surface areas (SBET) of the samples were determined by N2 adsorption at 77 K and BET analysis (MicrotracBEL, BELSORP-miniII). X-ray photoelectron spectra (XPS) were obtained using Kratos apparatus (ESCA-3400) with Mg Kα radiation as the excitation source. Measurements of X-ray absorption fine structure (XANES and EXAFS) at the Ru K-edge were carried out in a fluorescence mode at the NW10A beamline of the Photon Factory Advanced Ring in the High Energy Accelerator Research Organization (Tsukuba, Japan). The incident X-ray was monochromatized by a Si(311) double-crystal monochromator.Photocatalytic reactions
Sacrificial H2 evolution and Z-WS were conducted using a top-window reaction vessel connected to a gas-closed circulation system. Appropriate amounts of samples were dispersed in 160 mL of a reactant solution. Solutions of a 10 vol% methanol, 0.1 M Na2S and 0.5 M Na2SO3 mixture and 0.5 mM Co(bpy)3SO4 were used for H2 evolution. For Z-WS, CLTO (50 mg) and BiVO4 (50 mg) were dispersed in a 0.1 mM Co(bpy)3SO4 solution. For general experiments, 10 kPa of Ar was introduced into the system after deaeration. The suspension was irradiated with visible light (λ >420 nm) using a 300 W Xe-arc lamp with a long pass filter. In addition, monochromatic lights at 430 and 560 nm emitted from light-emitting diodes (LED; Asahi Spectra, CL-H1-430-9-1-B and CL-H1-568-9-1-B) attached with band pass filters (Asahi Spectra, HMX0430 and MX0560), whose power was 68.5 mW for 430 nm and 11.2 mW for 560 nm, were also used to evaluate EQY and photo-response. To evaluate the efficiency of solar energy conversion to hydrogen (STH), a solar simulator with 100 mW cm−2 incident density and 16 cm2 irradiation area (Asahi spectra, HAL-320) was also used. The evolved gas was analyzed using an online gas chromatograph (Shimadzu, GC-8A with MS-5A column, TCD detector and Ar carrier).Electrochemical measurements
Cyclic voltammogram (CV) measurements were performed using bare and Cr2O3-coated fluorine-doped tin oxide (FTO) electrodes. Cr2O3/FTO was prepared by electrical reduction of K2CrO4 at −0.2 V vs. RHE for 3 min in 0.1 M K2SO4 containing 0.1 mM K2CrO4. Measurements were performed in an aqueous solution containing 0.1 M K2SO4 and 0.1 mM Co(bpy)3SO4 under an Ar atmosphere. A platinum wire and Ag/AgCl in saturated KCl were used as counter and reference electrodes, respectively. Electric potential was swept at a rate of 20 mV s−1.Results and discussion
Synthesis of CLTO
There are two polymorphs of CLTO belonging to trigonal and hexagonal systems which are regarded as low- and high-temperature phases, respectively. The CLTO sample prepared at 600 °C was a mixture of trigonal and hexagonal phases whereas that at 700 °C was obtained as a hexagonal phase as reported previously (Fig. S2a†).42 Li4Ti5O12, the precursor used in the synthesis of CLTO, remained in both samples. Both samples showed the same absorption onset at 600 nm wavelength, being consistent with previous reports (Fig. S2b†).31,42 As shown in Fig. S3,† the particle sizes of the CLTO samples synthesized at 600 and 700 °C were 2–5 and 3–10 μm, respectively, thus higher synthesis temperature remarkably promoted crystal growth. Accordingly, SBET of CLTO decreased from 0.6 to 0.2 m2 g−1 upon increasing the synthesis temperature from 600 to 700 °C. The photocatalytic performance of the CLTO samples was evaluated for sacrificial H2 evolution from an aqueous solution containing 0.1 M Na2S and 0.5 M Na2SO3 with an in situ photodeposited Ru cocatalyst (Fig. S4†). Both samples produced H2 by visible light excitation, and the 600 °C sample containing tri-CLTO as the majority phase exhibited higher activity than the 700 °C sample containing hex-CLTO with a trace of Li4Ti5O12. The Li4Ti5O12 phase, which remained in both samples, gave no photocatalytic activity under visible light because of its wide band gap (3.7 eV), indicating that CLTO phases were responsible for the activity under visible light. Kudo et al. reported that both trigonal and hexagonal phases were active and the activity of the hexagonal phase (143 μmol h−1) was higher than that of the trigonal one (103 μmol h−1).31 Interestingly, our CLTO showed the opposite tendency where the activity of the tri-CLTO rich sample (402 μmol h−1) was much higher than that of the hex-CLTO rich sample (117 μmol h−1). This indicates that not only crystal phases but also other properties such as defects and particle size, which are influenced by synthetic conditions, strongly affect the photocatalytic performance of CLTO. The EQY of the 600 °C sample for this sacrificial H2 evolution was determined to be 2.5% and 2.2% at 430 and 560 nm, respectively. Thus, the successful synthesis of CLTO exhibiting moderate photocatalytic activity at long wavelength was confirmed. The 600 °C sample was used for further experiments.Cocatalyst survey for Z-WS
Surveying effective cocatalysts was conducted for construction of Z-scheme systems utilizing CLTO as an HEP. Fig. 1a shows H2 evolution from a 10 vol% methanol solution over in situ photodeposited M/CLTO. It should be noted that the Ru/CLTO sample in this section corresponded to Ru(10% MeOH)/CLTO in the following sections. The bare CLTO was not active for H2 evolution, therefore cocatalyst modification was indispensable for H2 evolution by CLTO. The highest activity was obtained with a Pt cocatalyst. A Ru cocatalyst also exhibited relatively high activity while Rh and Pd cocatalysts were less effective. H2 evolution rates were remarkably decreased upon deposition of Cr2O3 irrespective of the kinds of cocatalysts (Fig. 1b). The order of effectiveness of cocatalysts in sacrificial H2 evolution using methanol was not changed by deposition of Cr2O3. The Cr2O3/M/CLTO samples were examined for Z-WS combined with BiVO4 and Co(bpy)3SO4. Both H2 and O2 were evolved in all cases as shown in Fig. 2. This fact indicates that cocatalyst-modified CLTO functions as an HEP using Co(bpy)32+ because BiVO4 is incapable of H2 evolution. The reactions started with Co(bpy)32+, which worked as an electron donor, therefore the occurrence of O2 evolution proved the achievement of the Z-scheme mechanism between CLTO and BiVO4.23,39 BiVO4 oxidized water accompanied with the reduction of Co(bpy)33+ which was the representative of holes generated in CLTO. Among those examined, Cr2O3/Pt and Cr2O3/Ru exhibited similar activity in the early stage of reaction although Cr2O3/Pt showed higher activity than Cr2O3/Ru in sacrificial H2 evolution using methanol. The lower activity of Cr2O3/Pt in Z-WS would be due to water formation from H2 and O2. Water formation in the dark was examined for Ru/CLTO and Cr2O3/M/CLTO (M: Rh, Pd and Pt) to see the effects of Cr2O3 deposition (Fig. S5†). Only Cr2O3/Pt/CLTO showed the decrease of H2 and O2 with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio attributed to water formation. It has been reported that deposition of Cr2O3 suppresses water formation on noble metal cocatalysts due to covering the noble metal cores with a thin Cr2O3 layer.6,48 However, the result indicated that a part of the Pt surface was still exposed after Cr2O3 deposition. Thus, the cocatalyst survey revealed that the Ru cocatalyst was the most effective cocatalyst among those examined. Gradual decreases in gas evolution rates during Z-WS were observed for all cocatalysts. Although water formation would be one reason for the deactivation in the reaction using Cr2O3/Pt/CLTO as described above, reduction of O2 over the surface of CLTO and/or exposed cocatalysts mainly caused the deactivation as discussed later. Modification with Cr2O3 seemed to be not important for the Ru cocatalyst in consideration of the inactivity for water formation. However, remarkable effects of Cr2O3 deposition were observed in Z-WS (Fig. S6†). Although Z-WS took place even with Ru/CLTO, Cr2O3/Ru/CLTO exhibited 8.5 times higher activity than Ru/CLTO. Cr2O3/Ru/CLTO also showed higher activity in H2 evolution from the Co(bpy)3SO4 solution (Fig. S7†), which was a half reaction of Z-WS. Thus, the modification with Cr2O3 was crucial in the present Z-scheme system using the Co(bpy)33+/2+ redox shuttle. CV measurements using bare FTO and Cr2O3/FTO clearly proved the significant role of Cr2O3 (Fig. S8†). The bare FTO showed a pair of reversible currents ascribed to the oxidation and reduction of the Co complex. The current due to the redox of the Co complex was suppressed over the Cr2O3/FTO electrode. Thus, Cr2O3 suppressed the reduction of Co(bpy)33+, which is an undesired backward reaction in Z-WS. It should be noted that the H2 evolution rate in Z-WS was much higher than that in the half reaction. This suggested that the competitive reduction reaction strongly suppressed H2 production. In the half reaction, Co(bpy)33+ is simultaneously formed by oxidation of Co(bpy)32+ by holes when electrons are consumed by H2 production. If the electrons preferably reduce Co(bpy)33+ rather than H+, H2 evolution should be remarkably suppressed. The reduction of Co(bpy)33+ was possible on the surface of CLTO even after the Cr2O3 modification. In the Z-scheme system, the coexistent OEP pushes Co(bpy)33+ back to Co(bpy)32+ in addition to O2 production and keeps the low quantity of Co(bpy)33+ if the OEP functions well. Thus, Z-WS with H2 evolution rates higher than those in the half reaction is possible. Indeed, Abe and coworkers have reported similar phenomena in the IO3−/I−-based Z-scheme system.27
:1 ratio attributed to water formation. It has been reported that deposition of Cr2O3 suppresses water formation on noble metal cocatalysts due to covering the noble metal cores with a thin Cr2O3 layer.6,48 However, the result indicated that a part of the Pt surface was still exposed after Cr2O3 deposition. Thus, the cocatalyst survey revealed that the Ru cocatalyst was the most effective cocatalyst among those examined. Gradual decreases in gas evolution rates during Z-WS were observed for all cocatalysts. Although water formation would be one reason for the deactivation in the reaction using Cr2O3/Pt/CLTO as described above, reduction of O2 over the surface of CLTO and/or exposed cocatalysts mainly caused the deactivation as discussed later. Modification with Cr2O3 seemed to be not important for the Ru cocatalyst in consideration of the inactivity for water formation. However, remarkable effects of Cr2O3 deposition were observed in Z-WS (Fig. S6†). Although Z-WS took place even with Ru/CLTO, Cr2O3/Ru/CLTO exhibited 8.5 times higher activity than Ru/CLTO. Cr2O3/Ru/CLTO also showed higher activity in H2 evolution from the Co(bpy)3SO4 solution (Fig. S7†), which was a half reaction of Z-WS. Thus, the modification with Cr2O3 was crucial in the present Z-scheme system using the Co(bpy)33+/2+ redox shuttle. CV measurements using bare FTO and Cr2O3/FTO clearly proved the significant role of Cr2O3 (Fig. S8†). The bare FTO showed a pair of reversible currents ascribed to the oxidation and reduction of the Co complex. The current due to the redox of the Co complex was suppressed over the Cr2O3/FTO electrode. Thus, Cr2O3 suppressed the reduction of Co(bpy)33+, which is an undesired backward reaction in Z-WS. It should be noted that the H2 evolution rate in Z-WS was much higher than that in the half reaction. This suggested that the competitive reduction reaction strongly suppressed H2 production. In the half reaction, Co(bpy)33+ is simultaneously formed by oxidation of Co(bpy)32+ by holes when electrons are consumed by H2 production. If the electrons preferably reduce Co(bpy)33+ rather than H+, H2 evolution should be remarkably suppressed. The reduction of Co(bpy)33+ was possible on the surface of CLTO even after the Cr2O3 modification. In the Z-scheme system, the coexistent OEP pushes Co(bpy)33+ back to Co(bpy)32+ in addition to O2 production and keeps the low quantity of Co(bpy)33+ if the OEP functions well. Thus, Z-WS with H2 evolution rates higher than those in the half reaction is possible. Indeed, Abe and coworkers have reported similar phenomena in the IO3−/I−-based Z-scheme system.27
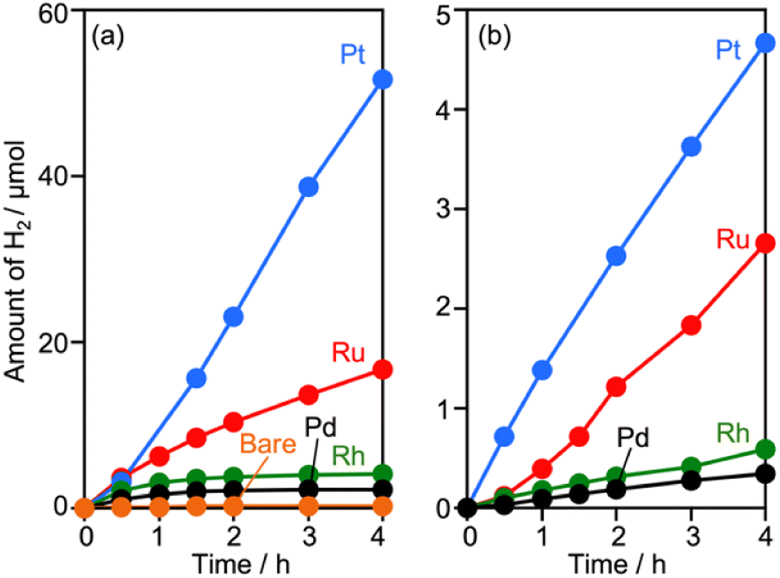 | ||
| Fig. 1 Sacrificial H2 evolution from a 10% methanol solution over (a) M (0.05 wt%)/CLTO and (b) Cr2O3(1.2 wt%)/M(0.05 wt%)/CLTO. Photocatalyst: 0.3 g for (a) and 0.2 g for (b); reactant solution: 10% methanol solution, 160 mL; light source: 300 W Xe lamp (λ >420 nm). | ||
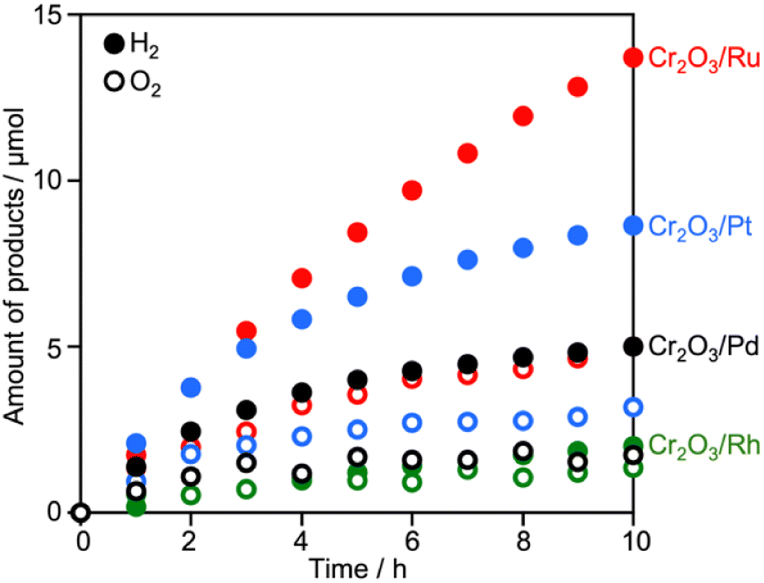 | ||
| Fig. 2 Z-WS by the Cr2O3/M/CLTO-BiVO4-Co(bpy)3SO4 system. Photocatalyst: 0.05 g each; reactant solution: 0.1 mM Co(bpy)3SO4, 160 mL; light source: 300 W Xe lamp (λ >420 nm). | ||
Deposition of Ru cocatalysts under different conditions
Deposition conditions were examined for Ru/CLTO because they frequently affect photocatalytic performance.49,50 In the aforementioned experiments, Ru(10% MeOH)/CLTO prepared by photodeposition in a 10% methanol solution was used. However, the activity of Ru/CLTO for sacrificial H2 evolution in 10% methanol was negligible in comparison with that in the reaction using Na2SO3 and Na2S (Fig. 1 and S4†). This indicated the poor ability of diluted methanol in scavenging photogenerated holes in CLTO. The state of photodeposited Ru cocatalysts would be influenced by conditions with different abilities in scavenging holes. Thus, Ru(100% MeOH) and Ru(S2−), which were prepared by photodeposition in 100% methanol and a mixed solution of Na2S and Na2SO3, were examined for H2 evolution from the 10% methanol solution with in situ deposition of Cr2O3 and for Z-WS. In H2 evolution (Fig. 3a), Cr2O3/Ru(S2−)/CLTO showed very low activity (1.8 μmol h−1) although its parent Ru(S2−)/CLTO produced H2 with a 402 μmol h−1 rate from the solution containing Na2S and Na2SO3 as shown in Fig. S4.† Cr2O3/Ru(10% MeOH) and Cr2O3/Ru(100% MeOH) deposited CLTO showed higher activity than Cr2O3/Ru(S2−)/CLTO by 3.6 and 17.3 times, respectively. In Z-WS (Fig. 3b), Cr2O3/Ru(10% MeOH) and Cr2O3/Ru(100% MeOH) produced H2 and O2, and the latter exhibited higher activity. In contrast, Cr2O3/Ru(S2−)/CLTO was inactive. The order of activity in Z-WS showed the same trend as that in H2 evolution using 10% methanol. Thus, the photodeposition conditions of the Ru cocatalyst gave remarkable differences in photocatalytic activity.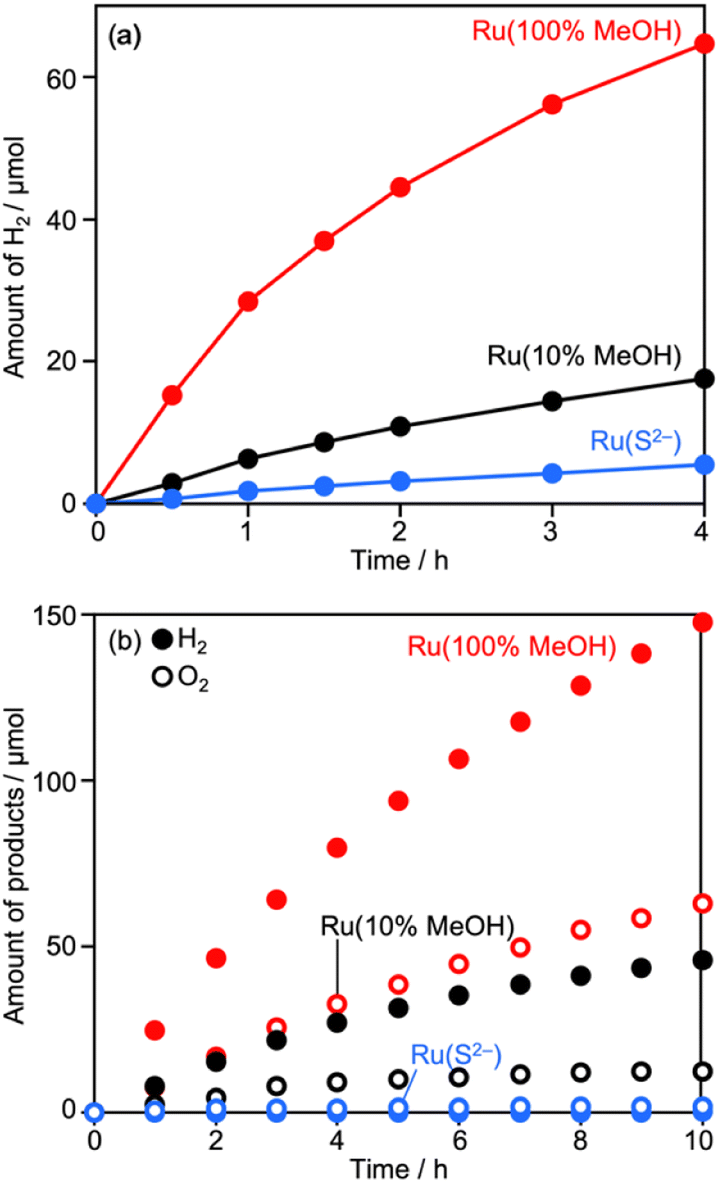 | ||
| Fig. 3 Photocatalytic (a) H2 evolution and (b) Z-WS using Cr2O3 (1.2 wt%)/Ru (0.5 wt%)/CLTO prepared under different photodeposition conditions. Photocatalyst: 0.2 g for (a) and 0.05 g each for (b); reactant solution: 10 vol% methanol for (a) and 0.1 mM Co(bpy)3SO4 for (b), 160 mL; light source: 300 W Xe lamp (λ >420 nm). | ||
Characterization of Ru/CLTO prepared under different photodeposition conditions was conducted to clarify the differences. Fig. 4a–c show the results of XAFS analysis carried out at the Ru K-edge. The XANES spectra of Ru(10% MeOH) and Ru(100% MeOH) were similar to that of RuOx(OH)y, whereas the spectrum of Ru(S2−)/CLTO was slightly shifted to the low energy side in comparison with that of Ru(10% MeOH). The EXAFS oscillations of Ru(10% MeOH) and Ru(100% MeOH) appeared to be similar to that of RuOx(OH)y. On the other hand, Ru(S2−) showed different features from RuOx(OH)y in both XANES and EXAFS. The oscillation period in EXAFS of Ru(S2−) was shorter than that of RuOx(OH)y, resulting in a longer bonding distance than that of RuOx(OH)y in the Fourier transforms of EXAFS. This EXAFS feature of Ru(S2−) was similar to that of RuS2. The samples were also analyzed by XPS (Fig. 4d–f). At Ru 3d, Ru(10% MeOH) and Ru(100% MeOH) showed a similar peak at 281.8 eV attributed to RuOx(OH)y.50 Ru(S2−) showed a peak at 280.6 eV which was remarkably low binding energy in comparison with Ru(10% MeOH) and Ru(100% MeOH). In S 2p, Ru(S2−) gave two obvious peaks at 162.0 and 167.6 eV attributed to S2− and SO32− species, respectively. The bare CLTO was treated in solutions of Na2S and Na2SO3 in the dark to evaluate the influences of S2− and SO32− upon the CLTO surface. No sulfur signals were observed in the Na2SO3-treated sample, whereas a weak peak attributed to S2− was observed in the Na2S-treated sample. Thus, the CLTO surface suffered from partial sulfurization due to its strong affinity to S2−. The signal intensity of S2− in the Ru(S2−) sample was stronger than that in the Na2S-treated sample, indicating that the S2− signal in Ru(S2−) was not due to the sulfurized CLTO surface but the sulfide-like Ru species. In Cl 2p, Ru(100% MeOH) showed a very weak peak whereas Ru(10% MeOH) and Ru(S2−) showed no peaks. The results of XAFS and XPS revealed the changes in chemical states of the Ru cocatalyst with the photodeposition conditions; RuOx(OH)y in Ru(10% MeOH), RuOx(OH)yClz in Ru(100% MeOH) and sulfide-like Ru species adsorbing SO32− in Ru(S2−). However, the state of Ru(10% MeOH) and Ru(100% MeOH) could be considered to be almost the same because the Cl content in Ru(100% MeOH) was very low. Ru(100% MeOH) and Ru(S2−) as well as Ru(10% MeOH) were also inactive for water formation as depicted in Fig. S9.† None of the Ru cocatalysts were in the metallic state but oxyhydroxide or sulfide-like states, giving the inactivity for water formation. Thus, the different photocatalytic activities of Ru/CLTO in Z-WS shown in Fig. 3 were not influenced by water formation over the Ru cocatalysts. As described above, XPS analysis revealed that the CLTO surface was partly sulfurized in the S2− solution. The partly sulfurized surface would be inactive for oxidation of methanol and Co(bpy)32+, resulting in the very low activity of the Ru(S2−) sample as depicted in Fig. 3.
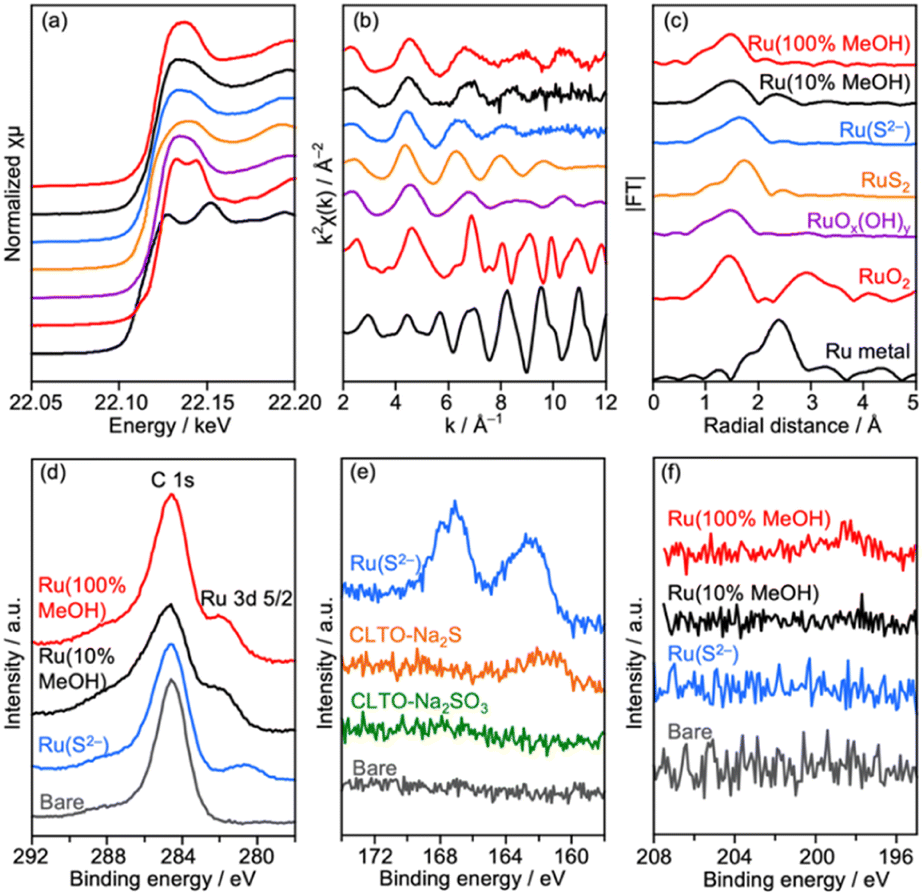 | ||
| Fig. 4 (a) XANES spectra, (b) EXAFS oscillations, (c) their Fourier transforms and XPS at (d) Ru 3d, (e) S 2p and (f) Cl 2p of Ru/CLTO prepared under different photodeposition conditions. | ||
SEM observation was conducted to examine the particle morphology of the photodeposited Ru cocatalysts (Fig. 5). In Ru(10% MeOH) and Ru(S2−), several tens of nm of particles were deposited on the smooth CLTO surface, and aggregation occurred. In contrast, Ru(100% MeOH) showed a greatly different feature. Large plate objects with 100–200 nm sizes and about 30 nm thickness were deposited in addition to fine particles with 10 nm size. It was suspected that the large plates were caused by damage from photoexcitation in 100% methanol. However, STEM-EDS analysis proved that the plate object did not contain Cu and Ti but Ru (Fig. 6). Thus, the characterization revealed that the Ru species in Ru(10% MeOH) and Ru(100% MeOH) was in a similar chemical state, but Ru(100% MeOH) had the characteristic morphology. The unusually large Ru(100% MeOH) provides two positive factors compared with aggregated fine Ru(10% MeOH). One is difference in contact between the Ru particle and CLTO. The fine Ru particles loosely deposited on CLTO while the large Ru plates were in close contact on CLTO. Another is the difference in electron migration in the Ru cocatalyst particle. Electrons moved to the Ru cocatalyst poorly migrate in the aggregated fine particles due to the grain boundary, while smooth migration occurs in Ru(100% MeOH) despite the large sizes. The close contact on CLTO and poor grain boundary in Ru(100% MeOH) improved electron extraction from CLTO and electron migration in the Ru cocatalyst, resulting in the highest activity of Cr2O3/Ru(100% MeOH)/CLTO in H2 evolution and Z-WS.
 | ||
| Fig. 5 SEM images of Ru/CLTO prepared under different photodeposition conditions. | ||
 | ||
| Fig. 6 (a) TEM image and (b–d) STEM-EDS images of Ru(100% MeOH)/CLTO. | ||
The optimal amount of the Ru(100% MeOH) cocatalyst was investigated for Z-WS (Fig. S10†). Activity was remarkably enhanced as the amount of the Ru cocatalyst increased up to 0.3 wt%. The evolution rate of the 0.5 wt% sample was similar to that of the 0.3 wt% sample in the early stage of the reaction, however the evolution rate decreased after 3 h of reaction time. Further deposition of Ru (1 wt%) gave lower activity.
Deposition of the Ru cocatalyst was also performed by an impregnation method under three heat-treatment conditions as described in the experimental section. No remarkable changes were seen in XRD for samples treated at 200 °C in N2 (Ru(N2)) and under vacuum (Ru(evac)), while diffraction peaks attributed to metallic Cu obviously appeared after treatment in H2 at 150 °C (Ru(H2)) despite the relatively low treatment temperature (Fig. S11†). Accordingly, numerous particles with 50–100 nm diameters were observed on the surface of CLTO (Fig. S12†), and these particles seemed to be metallic Cu as proved in XRD analysis. Thus, CLTO underwent the reductive decomposition by H2 treatment at 150 °C. XAFS analysis revealed that the state of Ru cocatalysts was also changed by the heat treatment conditions (Fig. 7). XANES and EXAFS clarified that metallic Ru was present in the Ru(H2) sample while the samples of Ru(N2) and Ru(evac) seemed to be RuOx(OH)y in consideration of the similarity in spectra between the samples and RuOx(OH)y. No remarkable differences were observed in the morphology of the Ru catalyst except for Ru(H2) as shown in Fig. S12.†
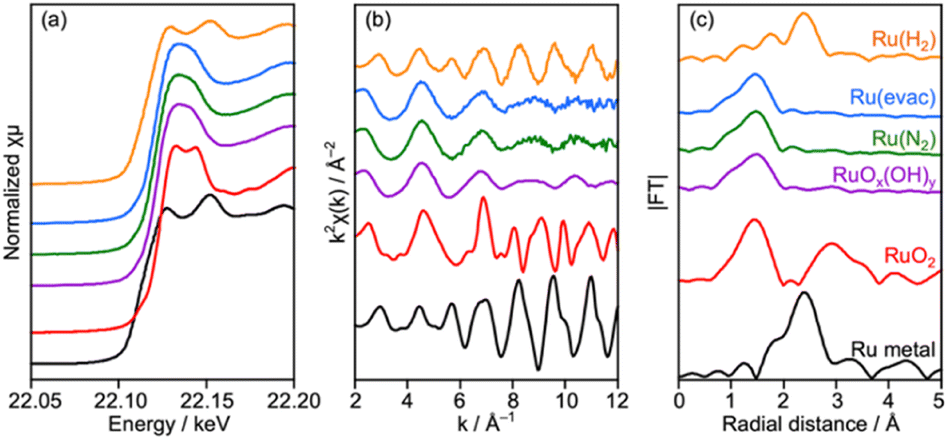 | ||
| Fig. 7 (a) XANES spectra, (b) EXAFS oscillation and (c) its Fourier transforms analyzed at the Ru K-edge for Ru/CLTO prepared by the impregnation method under different heat treatment conditions. | ||
The photocatalytic performance of Cr2O3/Ru/CLTO prepared via the impregnation method was evaluated for Z-WS after the Cr2O3 modification (Fig. S13†). The Ru(H2) sample which suffered from the reductive decomposition was excluded for this evaluation. The Ru(evac) and Ru(N2) samples were also active for Z-WS. Their activities were only comparable with that of the Ru(10% MeOH) sample prepared by the photodeposition method shown in Fig. 3b. Thus, it has been concluded that the photodeposition in 100% methanol is the best condition for Ru/CLTO to obtain high activity in Z-WS. The STH value of the present Z-WS was determined to be 0.029% using the optimized sample. The Ru(100% MeOH) samples were used for further experiments.
Characteristics of the Cr2O3/Ru/CLTO-BiVO4 Z-scheme system
As described above, water splitting was achieved by the combination of Cr2O3/Ru/CLTO and BiVO4 in the presence of the Co(bpy)33+/2+ redox shuttle. In the absence of Co(bpy)33+/2+, water splitting with significant activity was not achieved by the Cr2O3/Ru/CLTO-BiVO4 combination (Fig. 8). It has been reported that RGO-modification facilitates electron transfer from an OEP to HEP in Z-WS without redox couples.22 Water splitting, indeed, took place with higher gas evolution rates when RGO-modified BiVO4 was employed. Nevertheless, its activity was much lower than that of the system using Co(bpy)33+/2+. The results clearly prove the effectiveness of the Co(bpy)33+/2+ redox shuttle in the present Z-scheme system in spite of the redox couple causing unfavourable backward reactions.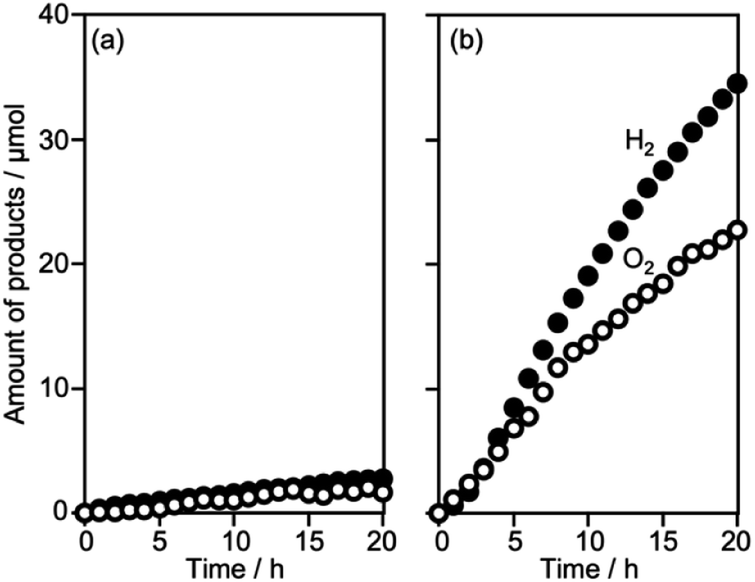 | ||
| Fig. 8 Time courses of gas evolution by Z-scheme systems without the Co(bpy)33+/2+ electron shuttle; (a) Cr2O3/Ru/CLTO-BiVO4 and (b) Cr2O3/Ru/CLTO-RGO/BiVO4. Photocatalyst: 0.05 g each; reactant solution: water, 160 mL; light source: 300 W Xe lamp (λ >420 nm). | ||
The vital role of the Cr2O3 modification in the suppression of reduction of Co(bpy)33+ was confirmed for Ru(10% MeOH)/CLTO as shown in Fig. S6 and S7.† The activity for Z-WS was remarkably improved when the Ru cocatalyst was changed from Ru(10% MeOH) to Ru(100% MeOH). We investigated the necessity of the Cr2O3 modification for the highly efficient Ru(100% MeOH) cocatalyst (Fig. 9a). H2 and O2 were evolved in the early stage of the reaction even without the Cr2O3 modification, however the gas evolution stopped soon. In contrast, the sample modified with Cr2O3 evolved H2 and O2 with a rate higher than that for without Cr2O3 modification and continued for a long time. It was confirmed that Cr2O3 was deposited around the Ru particles as shown in Fig. 9b. In the case of Ru/CLTO, reduction of Co(bpy)33+ easily occurred on the Ru cocatalyst. As proved in electrochemical measurements (Fig. S8†), the Cr2O3 layer hindered Co(bpy)33+ from accessing the Ru cocatalyst as reported for the IO3−/I− system,45 resulting in higher and stable water splitting. On the other hand, the activity of Cr2O3/Ru/CLTO gradually decreased with the reaction time and recovered in the second run performed after removal of gaseous products. This behaviour indicated that the performance of Cr2O3/Ru/CLTO did not collapse during the reaction. To check the stability of CLTO, XPS and XRD measurements were performed for the samples after photoirradiation (Fig. S14 and S15†). A new weak peak at 935.1 eV ascribed to Cu2+ appeared after deposition of Ru(100% MeOH). After deposition of Cr2O3 in 10% methanol, the Cu2+ peak increased while the Cu+ peak at 933.3 eV decreased significantly. The changes in XPS were due to oxidation of Cu+ during photoirradiation in 10% methanol. On the other hand, no changes in XRD patterns were seen after deposition of Ru and Cr2O3. Moreover, no changes except the presence of BiVO4 were observed in XPS and XRD after Z-WS. Thus, sufficient stability of the CLTO crystal was confirmed despite partial oxidation of Cu+ at the surface of CLTO. The Z-scheme system consisting of Ru/SrTiO3:Rh, BiVO4 and Co(bpy)33+/2+ did not show such deactivation during the reaction although the same OEP was employed as shown in Fig. S16.† In addition, water formation from H2 and O2 did not take place on Ru/CLTO as described before (Fig. S9†). We suspected from these results that the gradual decrease in the activity was due to the reduction of O2 over Cr2O3/Ru/CLTO. When 10 kPa of O2 was introduced into the reaction system instead of Ar, H2 evolution caused by water splitting was significantly suppressed (Fig. S17†). This indicated that some of the electrons photogenerated in CLTO reacted with O2 on the exposed Ru cocatalyst and/or the surface of CLTO.
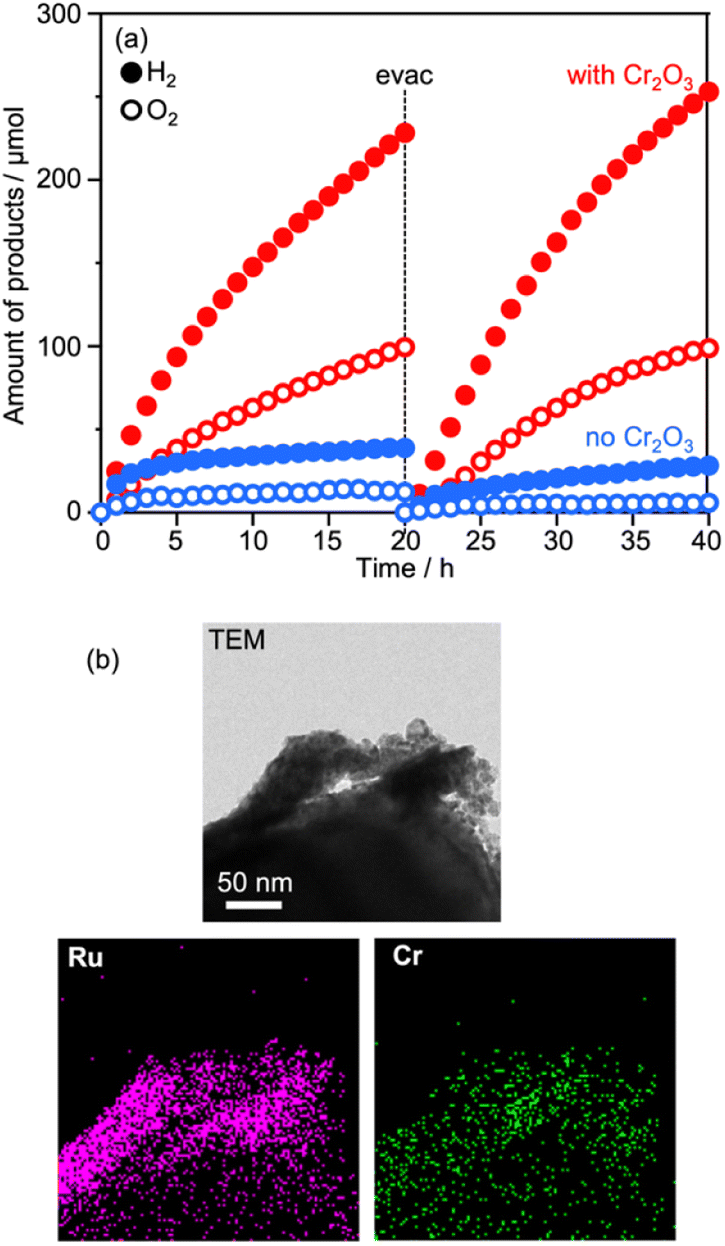 | ||
| Fig. 9 (a) Influences of Cr2O3 modification on Z-WS employing Ru(100% MeOH)/CLTO. (b) TEM and element mapping images of Cr2O3/Ru(100% MeOH)/CLTO. | ||
The UV-vis spectra of CLTO, BiVO4 and SrTiO3:Rh are compared in Fig. 10a, where the spectra of monochromatic lights are also shown. CLTO absorbs photons with wavelengths up to 600 nm, which is longer than the absorption edge of SrTiO3:Rh (540 nm). To confirm the utilization of long-wavelength photons by CLTO, Z-WS was also examined under irradiation using monochromatic lights with 430 and 560 nm wavelengths (Fig. 10b). Water splitting proceeded under the 430 nm light, and the EQY of the Z-scheme system employing Cr2O3/Ru/CLTO was determined to be 0.5%. No water splitting was induced in the CLTO system when the 560 nm light was solely used. This is because the Z-scheme mechanism cannot be completed without excitation of BiVO4. However, the gas evolution rate was increased when the 560 nm light was added to the basic incident light (430 nm). The additional 560 nm light did not enhance the gas evolution when Ru/SrTiO3:Rh was used as the HEP instead of Cr2O3/Ru/CLTO (Fig. 10c). The difference in the influence of the additional 560 nm light between CLTO and SrTiO3:Rh systems is reasonably explained by the photo absorption properties. The 560 nm light was sufficient to excite CLTO, but insufficient for excitation of SrTiO3:Rh. Thus, it was clearly confirmed that excitation of CLTO by long-wavelength photons contributes to Z-WS.
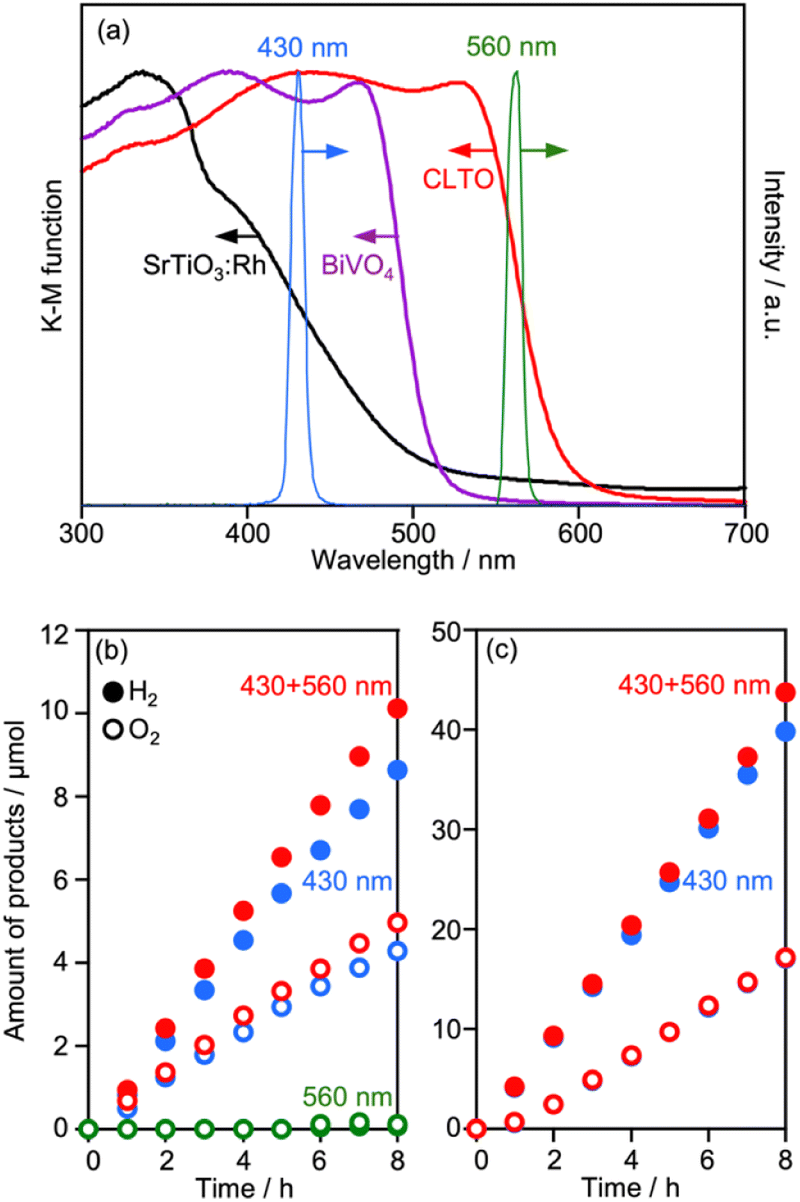 | ||
| Fig. 10 (a) UV-vis spectra of CLTO, BiVO4 and SrTiO3:Rh reduced by H2, and spectra of monochromatic light at 430 and 560 nm. Z-WS in 0.1 mM Co(bpy)3SO4 by (b) Cr2O3/Ru/CLTO-BiVO4 and (c) Ru/SrTiO3:Rh-BiVO4 systems under different irradiation conditions. | ||
Conclusions
Utilization of CLTO in the totally visible responsive Z-scheme system based on the Co(bpy)33+/2+ redox shuttle was demonstrated. Modifying CLTO with the Ru cocatalyst and Cr2O3 was effective to achieve Z-WS. The unusually large Ru cocatalyst was discovered to be an efficient cocatalyst more than general fine particles for CLTO. The advantage of CLTO over SrTiO3:Rh in the response to longer wavelength light was experimentally confirmed, although the EQY of the present Z-scheme system was limited to 0.5% at 430 nm. The Cr2O3 deposition was crucial to suppressing the reduction of Co(bpy)33+ on Ru/CLTO. However, unfavorable backward reactions and reduction of Co(bpy)33+ and O2, still took place. Therefore, the efficiency of the CLTO-BiVO4 system will be improved by establishing advanced surface modification methods to suppress the backward reactions as well as preparation of fine CLTO particles.Author contributions
S. Y. guided the experiments, analyzed the data and wrote the manuscript. T. K., Q. L. and T. Y. performed sample preparation, characterization and photocatalytic experiments. S. N. and H. Ku. performed XAFS measurements and analyses. M. K. performed TEM observation. H. Ka. conceived the idea, supervised the research and wrote the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by KAKENHI from JSPS (Grant Numbers JP17H06438, JP19H02806 and JP20H05085) and the Futaba Foundation. XAFS measurements were performed with the approval of the Photon Factory Program Advisory Committee (Proposal numbers 2020G597 and 2021G597). TEM observation was carried out with the support of the Joint research program of the Institute of Materials and Systems for Sustainability, Nagoya University.References
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 10120–10122 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- Y. Sakata, T. Hayashi, R. Yasunaga, N. Yanaga and H. Imamura, Chem. Commun., 2015, 51, 12935–12938 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- B. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348–1349 CrossRef CAS.
- R. Abe, M. Higashi and K. Domen, ChemSusChem, 2011, 4, 228–237 CrossRef CAS.
- D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS.
- G. Zhao, X. Huang, F. Fina, G. Zhang and J. T. S. Irvine, Catal. Sci. Technol., 2015, 5, 3416–3422 RSC.
- S. Chen, Y. Qi, T. Hisatomi, Q. Ding, T. Asai, Z. Li, S. S. K. Ma, F. Zhang, K. Domen and C. Li, Angew. Chem., Int. Ed., 2015, 54, 8498–8501 CrossRef CAS.
- Q. Wang, T. Hisatomi, Y. Suzuki, Z. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675–1683 CrossRef CAS PubMed.
- A. Nakada, H. Suzuki, J. J. M. Vequizo, K. Ogawa, M. Higashi, A. Saeki, A. Yamakata, H. Kageyama and R. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 45606–45611 CrossRef CAS PubMed.
- S. Nishioka, K. Yanagisawa, D. Lu, J. J. M. Vequizo, A. Yamakata, K. Kimoto, M. Inada and K. Maeda, Sustainable Energy Fuels, 2019, 3, 2337–2346 RSC.
- X. Guan, L. Tian, Y. Zhang, J. Shi and S. Shen, Nano Res., 2023, 16, 4568–4573 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef CAS.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CrossRef CAS.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS.
- Q. Jia, A. Iwase and A. Kudo, Chem. Sci., 2014, 5, 1513–1519 RSC.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS.
- M. Higashi, R. Abe, T. Takata and K. Domen, Chem. Mater., 2009, 21, 1543–1549 CrossRef CAS.
- M. Tabata, K. Maeda, M. Higashi, D. Lu, T. Takata, R. Abe and K. Domen, Langmuir, 2010, 26, 9161–9165 CrossRef CAS PubMed.
- W. Wang, J. Chen, C. Li and W. Tian, Nat. Commun., 2014, 5, 4647 CrossRef CAS PubMed.
- W. Zhao, K. Maeda, F. Zhang, T. Hisatomi and K. Domen, Phys. Chem. Chem. Phys., 2014, 16, 12051–12056 RSC.
- K. Iwashina, A. Iwase, S. Nozawa, S. Adachi and A. Kudo, Chem. Mater., 2016, 28, 4677–4685 CrossRef CAS.
- R. Kobayashi, T. Takashima, S. Tanigawa, S. Takeuchi, B. Ohtani and H. Irie, Phys. Chem. Chem. Phys., 2016, 18, 27754–27760 RSC.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS.
- Z. Pan, T. Hisatomi, Q. Wang, S. Chen, A. Iwase, M. Nakabayashi, N. Shibata, T. Takata, M. Katayama, T. Minegishi, A. Kudo and K. Domen, Adv. Funct. Mater., 2016, 26, 7011–7019 CrossRef CAS.
- A. Iwase and A. Kudo, Chem. Commun., 2017, 53, 6156–6159 RSC.
- T. Hisatomi, T. Yamamoto, Q. Wang, T. Nakanishi, T. Higashi, M. Katayama, T. Minegishi and K. Domen, Catal. Sci. Technol., 2018, 8, 3918–3925 RSC.
- S. Sun, T. Hisatomi, Q. Wang, S. Chen, G. Ma, J. Liu, S. Nandy, T. Minegishi, M. Katayama and K. Domen, ACS Catal., 2018, 8, 1690–1696 CrossRef CAS.
- A. Kudo, S. Yoshino, T. Tsuchiya, Y. Udagawa, Y. Takahashi, M. Yamaguchi, I. Ogasawara, H. Matsumoto and A. Iwase, Faraday Discuss., 2019, 215, 313–328 RSC.
- H. Kumagai, R. Aoyagi, K. Kato, A. Yamakata, M. Kakihana and H. Kato, ACS Appl. Energy Mater., 2021, 4, 2056–2060 CrossRef CAS.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS.
- A. Iwase, R. Sakamoto and H. Misono, Chem. Commun., 2022, 58, 12951–12954 RSC.
- H. Kato, T. Fujisawa, M. Kobayashi and M. Kakihana, Chem. Lett., 2015, 44, 973–975 CrossRef CAS.
- R. Abe, K. Sayama and H. Arakawa, Chem. Phys. Lett., 2003, 371, 360–364 CrossRef CAS.
- H. Kato, Y. Sasaki, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 2457–2464 CrossRef CAS.
- Y. Qi, S. Chen, J. Cui, Z. Wang, F. Zhang and C. Li, Appl. Catal., B, 2018, 224, 579–585 CrossRef CAS.
- Y. Miseki and K. Sayama, Chem. Commun., 2018, 54, 2670–2673 RSC.
- H. P. Duong, T. Mashiyama, M. Kobayashi, A. Iwase, A. Kudo, Y. Asakura, S. Yin, M. Kakihana and H. Kato, Appl. Catal., B, 2019, 252, 222–229 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133–137 CrossRef CAS.
- H. Suzuki, S. Nitta, O. Tomita, M. Higashi and R. Abe, ACS Catal., 2017, 7, 4336–4343 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01622f |
| This journal is © The Royal Society of Chemistry 2024 |