
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFour new isocoumarins from Cajanus cajan†
Liyuan Yaoabc,
Aikebaier Jumaiab,
Xiaobao Huangae,
Tingting Lina,
Zhengguo Taod and
Sheng-Xiang Qiu *ab
*ab
aState Key Laboratory of Plant Diversity and Specialty Crops & Guangdong Provincial Key Laboratory of Applied Botany, South China Botanical Garden, Chinese Academy of Sciences, Program for Natural Product Chemical Biology, Guangzhou 510650, China. E-mail: sxqiu@scbg.ac.cn; Fax: +86-20-37081190; Tel: +86-13924031914
bSouth China National Botanical Garden, Guangzhou 510650, P. R. China
cGraduate University of Chinese Academy of Sciences, Beijing 100049, P. R. China
dGuangzhou Leader Biotechnology Co., LTD, Guangzhou, 510650, China
eVisiting Student from the Department of Chemistry, University of Wisconsin–Madison, USA
First published on 5th March 2024
Abstract
Four novel new isocoumarins, cajanolactone B, C, D1 and D2 (1–4), were isolated from ethanolic extracts of the leaves of Cajanus cajan. The structural elucidation has been completed mainly depending on extensive spectroscopic analysis including UV, IR, NMR (1D and 2D), HRESIMS and chiral analysis. Notably, all these new isocoumarins were found to exist in racemic forms, among which compounds 3 and 4 share the same planar structure. This finding suggests that at least the biosynthesis of isocoumarin in C. cajan is chiral tolerant. A plausible biogenetic pathway of compounds 1–4 is proposed.
Introduction
Isocoumarins, as isomers of coumarins, feature an inverted lactone ring, namely 1H-2-benzo[c]pyran-1-one, which is a privileged scaffold offering a prodigious diversity of chemical structures that encompass a wide spectrum of pharmacological activities, such as antitumor, anticancer, antiviral, anti-inflammatory and antidiabetic etc.1–5Pigeon pea [Cajanus cajan (L.) Millsp.], belonging to the Leguminosae family, is one of the most valuable legume food crops in the tropical world and a resource for traditional Chinese medicine. Its extracts and active ingredients have received increasing attention in recent years due to their various biological activities,6 among which the main components such as cajaninstilbene acid, cajanonic acid A, longistylin A and pinostrobin have exhibited excellent bioactivities including antioxidant,7 anti-cancer,8 bactericidal,9 antiviral,10 lipid-lowering,11 neuroprotective12 and so on.
Previous studies on the phytochemistry of C. cajan from our and other research groups have focused on its main components, namely, flavonoids and stilbenes. In contrast, there has been limited exploration of smaller-proportion constituents such as isocoumarins. To the best of our knowledge, the only isocoumarin isolated from C. cajan was named cajanolactone A (CLA), which was found to promote osteoblast differentiation in hBMSCs through activating Wnt/LRP5/β-catenin signaling transduction.13 Our ongoing search for more structurally diverse compounds from C. cajan resulted in the isolation of four new isocoumarins (1–4) (Fig. 1), the isolation, structural elucidation of which are reported in the present report.
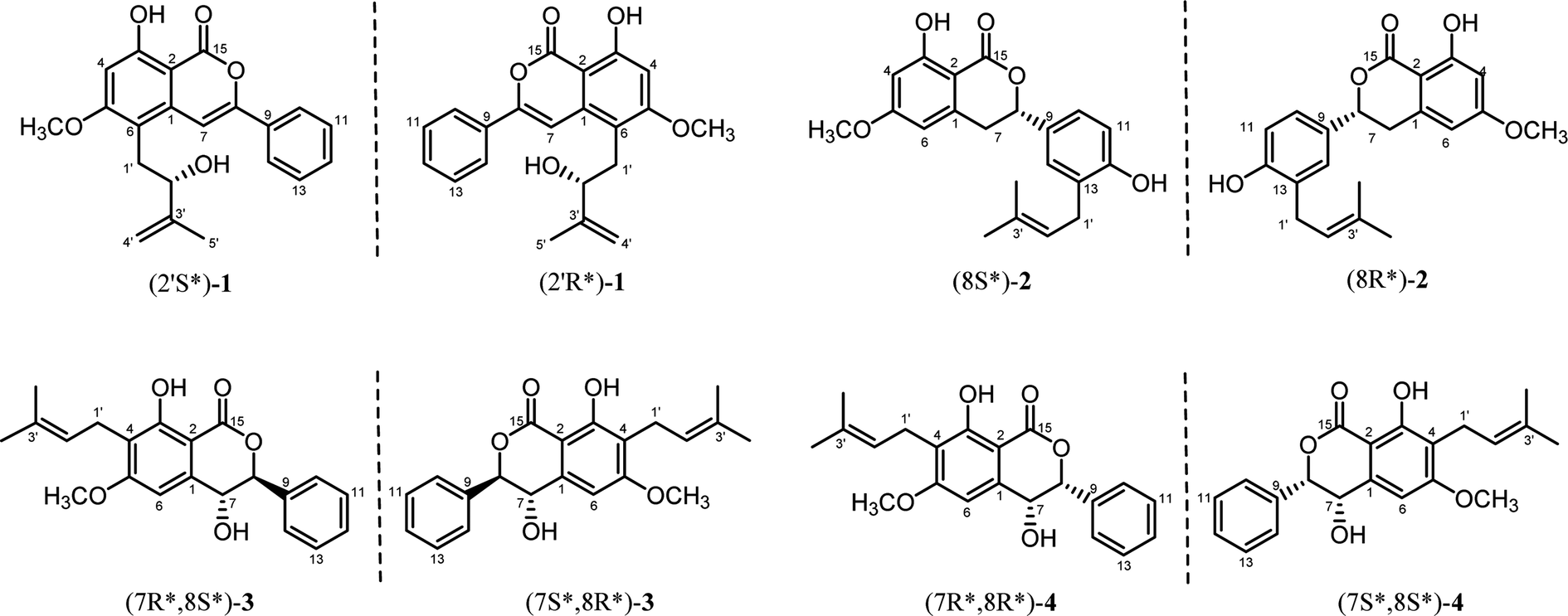 | ||
| Fig. 1 Chemical structures (relative configurations) of compounds 1–4. | ||
Results and discussion
Structure elucidation and identification
Compound 1 was purified as a colorless powder. Its molecular formula was determined to be C21H20O5 by HRESIMS from the ion peak at m/z 353.1382 [M + H]+ (calcd 353.1384) with twelve degrees of unsaturation. The typical IR resonance absorptions at 3374 and 1668 cm−1 suggested the existence of hydroxyl and carbonyl functionalities. Inspection of the 1D NMR data (Table 1) revealed a series of characteristic resonances as follow: a penta-substituted benzene ring [δH 6.56 (1H, s)], a monosubstituted benzene ring [δH 7.42–7.49 (3H, m), δH 7.84–7.86 (2H, m)], a methoxy (δC 56.2), as well as a chelated hydroxyl group [δH 11.42 (1H, s)]. Signals at 2.95 (1H, dd, J = 14.2, 8.5 Hz), 3.09 (1H, dd, J = 14.3, 4.2 Hz), 4.26 (1H, dd, J = 8.3, 4.0 Hz), 4.98 (1H, s), 4.87 (1H, s), and 3.92 (3H, s), coupling with carbon resonances at δC 31.6, 76.1, 147.6, 110.8, 18.4 ppm denoted the presence of an isopentyl group. Furthermore, on the basis of twelve indices of hydrogen deficiency, in conjunction with the high value of the corresponding carbonyl resonance at δC 166.6 ppm, a lactone ring was proposed.| 1 | 2 | ||||
|---|---|---|---|---|---|
| No. | δH (J in Hz) | δC, type | No. | δH (J in Hz) | δC, type |
| 1 | 137.4, C | 1 | 141.2, C | ||
| 2 | 100.0, C | 2 | 102.0, C | ||
| 3 | 163.2, C | 3 | 164.9, C | ||
| 4 | 6.56, s | 98.4, CH | 4 | 6.40, d (2.2) | 99.7, CH |
| 5 | 165.1, C | 5 | 166.1, C | ||
| 6 | 113.4, C | 6 | 6.28–6.29, m | 106.4, CH | |
| 7 | 7.19, s | 100.5, CH | 7 | 3.26, dd (12.2) | 35.5, CH2 |
| 3.01, dd (3.1) | |||||
| 8 | 152.9, C | 8 | 5.45, dd (3.1) | 80.5, CH | |
| 9 | 132.1, C | 9 | 130.4, C | ||
| 10/14 | 7.84–7.86, m | 125.5, CH | 10 | 7.16–7.17, m | 125.8, CH |
| 11/13 | 7.42–7.49, m | 129.1, CH | 11 | 6.83, d (8.1) | 116.0, CH |
| 12 | 7.42–7.49, m | 130.2, CH | 12 | 155.0, C | |
| 15 | 166.6, C | 13 | 127.4, C | ||
| 1′ | 3.09, dd (14.3, 4.2), 2.95, dd (14.2, 8.5) | 31.6, CH2 | 14 | 7.18–7.19, m | 128.4, CH |
| 2′ | 4.26, dd (8.3, 4.0) | 76.1, CH | 15 | 170.0, C | |
| 3′ | 147.6, C | 1′ | 3.37, d (7.1) | 30.0, CH2 | |
| 4′ | 4.98, s, 4.87, s | 110.8, CH2 | 2′ | 5.31, t (7.2) | 121.5, CH |
| 5′ | 1.88, s | 18.4, CH3 | 3′ | 135.5, C | |
| 5-OCH3 | 3.92, s | 56.2, CH3 | 4′ | 1.78, s | 25.9, CH3 |
| 3-OH | 11.42, s | 5′ | 1.78, s | 18.1, CH3 | |
| 5-OCH3 | 3.84, s | 55.8, CH3 | |||
| 3-OH | 11.23, s | ||||
| 12-OH | 5.26, s |
The identification of the planar structure of 1 was accomplished by a comprehensive interpretation of its 2D NMR spectra (Fig. 2). The HMBC contour maps allowed determination of the position of the following fragments: the conclusive correlations between H-7 to C-2, C-6 and C-8, H-10/14 to C-8, H2 -1'to C-1, C-5 and C-6, together with 5-OCH3 to C-5, strongly suggested that 1 has a structure very similar to that of CLA. The only difference between 1 and CLA is that 1 has an extra isopentenyl group that can rationally attributed at the C-6.
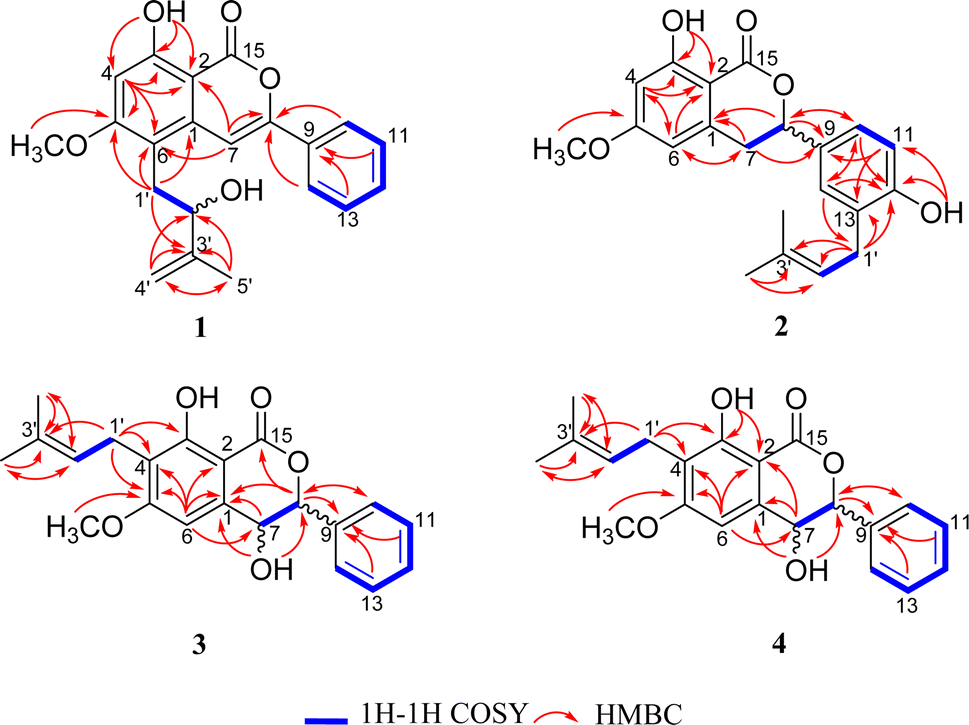 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of compounds 1–4. | ||
Additionally, the optical rotation of 1 was measured as being close to zero, suggesting it might be a pair of racemates with almost equal abundance. As shown in Fig. 4A, this deduction was further confirmed by chiral analysis of 1, wherein two well-resolved peaks were observed in the chromatogram. Thus, the structure was finally identified and named as racemic cajanolactone B.
Compound 2 was obtained as a yellow oil. The HRESIMS data showed a protonated-molecular ion at m/z 355.1539 [M + H]+ (calcd 355.1540), corresponding to a molecular formula of C21H22O5 with eleven degrees of unsaturation. The combination of 1D NMR (Table 1) and 2D NMR correlation signals (Fig. 2) showed the presence of a tetra-substituted benzene ring [δH 6.28–6.29 (1H, m), 6.40 (1H, d, J = 2.2 Hz)], a tri-substituted benzene ring [δH 6.83 (1H, d, J = 8.1 Hz), 7.16–7.17 (1H, m), 7.18–7.19 (1H, m)], two linked aliphatic carbons (δC 35.5, δC 80.5), a typical isoprenyl moiety [δC 30.0, 121.5, 135.5, 25.9, 18.1], a chelated hydroxyl group [δH 11.23 (1H, s)] and a methoxy (δC 55.8).
Further close comparison of the NMR data of 2 with those of 1 suggested that 2 shared a similar isocoumarin skeleton. It was also found that the differences between them mainly revolved on the location of the free hydroxyl group and the isoprenyl moiety, as well as the absence of hydrogenation of the double bond at C-7/C-8 in 2. This conclusion could be further strengthened by the HMBC correlations from 12-OH to C-12, and H2 -1'to C-12, C-13.
Since the optical rotation of compound 2 was found being close to zero, it is speculated to be a pair of racemates and was then subjected to chiral analysis, in which two well-resolved peaks could be observed with almost identical abundance (Fig. 4B). Accordingly, the structure of compound 2 was established and named as racematic cajanolactone C.
Compound 3 was isolated as a yellowish oil. Its HRESIMS spectrum with the protonated molecular ion measured at m/z 355.1541 [M + H]+ (calcd 355.1540) suggested a molecular formula of C21H22O5, corresponding to eleven indices of hydrogen deficiency. Its IR showed characteristic resonance absorptions at 3379 and 1614 cm−1, accounting for the existence of hydroxyl and carboxyl functionalities, respectively.
The 1H-NMR and 13C-NMR spectra (Table 2) indicated the presence of a penta-substituted benzene ring [δH 6.71 (1H, s)], a monosubstituted benzene ring [δH 7.42–7.49 (5H, m)], a methoxy (δC 55.9), an isopentyl group [δH 3.37 (2H, d, J = 7.1 Hz), 5.20 (1H, t, J = 7.1 Hz), 1.79 (3H, s), 1.68 (3H, s)], a chelated hydroxyl group [δH 11.44 (1H, s)], together with a free hydroxyl group [δH 2.09 (1H, s)]. Analyses of the 1D and 2D NMR data of 3 and 2 revealed their close resemblance except for the positions of the free hydroxyl group and the isoprenyl moiety in 3, which was further assigned by the observed HMBC cross-peaks of 7-OH to C-1 and C-8, and H2-1′′ to C-3 and C-4, respectively. Detailed inspection of 2D NMR (HSQC, 1H–1H COSY, HMBC) spectra established the planar structure of 3 as depicted in Fig. 1.
| 3a | 4b | ||||
|---|---|---|---|---|---|
| No. | δH (J in Hz) | δC, type | No. | δH (J in Hz) | δC, type |
| a Recorded in CDCl3.b Recorded in acetone-d6. | |||||
| 1 | 140.5, C | 1 | 141.0, C | ||
| 2 | 100.2, C | 2 | 100.6, C | ||
| 3 | 161.0, C | 3 | 159.9, C | ||
| 4 | 117.0, C | 4 | 116.1, C | ||
| 5 | 164.0, C | 5 | 162.7, C | ||
| 6 | 6.71, s | 98.9, CH | 6 | 6.76, s | 101.8, CH |
| 7 | 4.91, d (9.5) | 69.3, CH | 7 | 4.88, dd (6.3, 1.6) | 67.3, CH |
| 8 | 5.22, d (9.5) | 84.4, CH | 8 | 5.74–5.75, m | 82.2, CH |
| 9 | 135.4, C | 9 | 136.1, C | ||
| 10/14 | 7.42–7.49, m | 127.4, CH | 10/14 | 7.60, d (7.5) | 126.6, CH |
| 11/13 | 7.42–7.49, m | 129.5, CH | 11/13 | 7.36–7.45, m | 127.5, CH |
| 12 | 7.42–7.49, m | 129.0, CH | 12 | 7.36–7.45, m | 127.5, CH |
| 15 | 168.8, C | 15 | 169.2, C | ||
| 1′ | 3.37, d (7.1) | 21.8, CH2 | 1′ | 3.35, d (7.3) | 21.0, CH2 |
| 2′ | 5.20, t (7.2) | 121.6, CH | 2′ | 5.21, t (7.3) | 121.4, C |
| 3′ | 132.2, C | 3′ | 130.6, C | ||
| 4′ | 1.79, s | 17.8, CH3 | 4′ | 1.77, s | 16.4, CH3 |
| 5′ | 1.68, s | 25.8, CH3 | 5′ | 1.65, s | 24.5, CH3 |
| 5-OCH3 | 3.92, s | 55.9, CH3 | 5-OCH3 | 3.96, s | 55.1, CH3 |
| 3-OH | 11.44, s | 3-OH | 11.44, s | ||
| 7-OH | 2.09, s | 7-OH | 4.66, d (6.3) | ||
Similar to compound 2, the optical rotation of 3 was measured to be close to zero, implying its racematic character. Further chiral analysis evidenced that compound 3 is a mixture of two racemates with almost equal abundance (Fig. 4C). Moreover, the NOE correlations between H-7 and H-10/14 (Fig. 3), coupled with characteristic coupling constant JH-7/H-8 = 9.5 Hz, which led to a trans configuration according to Karplus equation,14 confirmed that 3 is a mixture of (7R*, 8S*) and (7S*, 8R*) racemates. Based on the above analysis, the structure of 3 was determined and named as racematic cajanolactone D1.
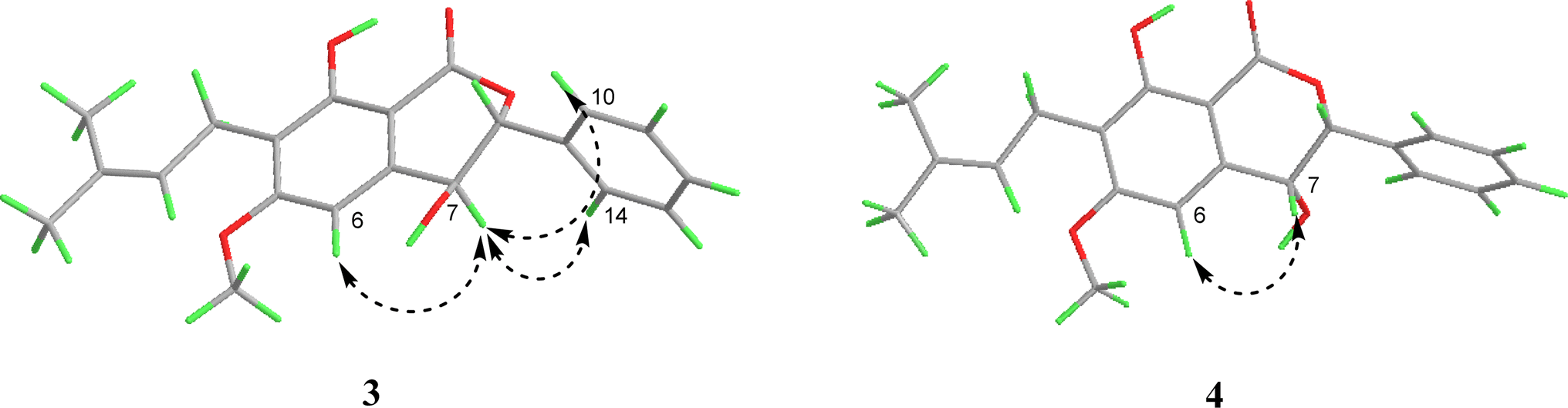 | ||
| Fig. 3 Key NOESY correlations of compounds 3–4. | ||
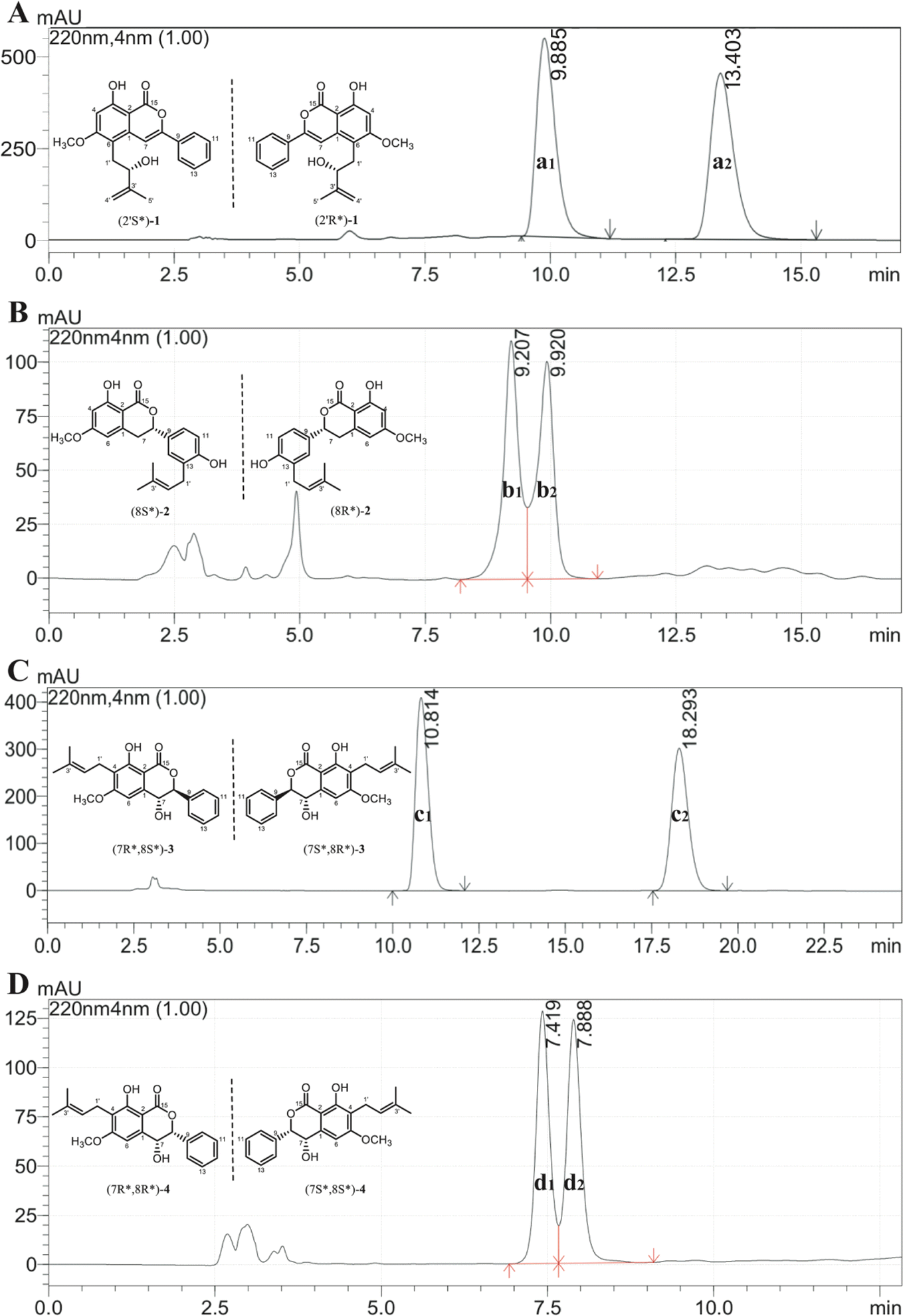 | ||
Fig. 4 Chiral analysis of compounds 1–4. (A) Relative peak area ratio of compound 1 was calculated as a1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :a2 = 49.4:50.6; (B) relative peak area ratio of compound 2 was calculated as b1:b2 = 51.3:48.7; (C) relative peak area ratio of compound 3 was calculated as c1:c2 = 49.2:50.8; (D) relative peak area ratio of compound 4 was calculated as d1:d2 = 50.5:49.5. :a2 = 49.4:50.6; (B) relative peak area ratio of compound 2 was calculated as b1:b2 = 51.3:48.7; (C) relative peak area ratio of compound 3 was calculated as c1:c2 = 49.2:50.8; (D) relative peak area ratio of compound 4 was calculated as d1:d2 = 50.5:49.5. | ||
Compound 4 was also isolated as a yellowish oil, which exhibited an ion peak at m/z 355.1531 [M + H]+ (calcd 355.1540) in HRESIMS spectrum with a positive mode, giving the molecular formula of C21H22O5 with eleven degrees of unsaturation.
The planar structure of 4 was determined to be the same as 3 based on a comparison of 1D and 2D NMR data. Similarly, compound 4 was considered as possible racemates owing to its optical rotation being close to zero, which was also verified via chiral analysis (Fig. 4D). However, there's a decent chance that compound 4 was a mixture of (7R*, 8R*) and (7S*, 8S*) racematic isomers since 3 was determined as a mixture of (7R*, 8S*) and (7S*, 8R*) racemates. The absent NOESY correlation of H-7 to H-10/14 (Fig. 3), along with the coupling constant of H-7 and H-8 (JH-7/H-8 = 1.6 Hz) arising from the small spatic dihedral angle, were consistent with a cis configuration, namely a (7R*, 8R*) or (7S*, 8S*) relative configuration assignment. Collectively, compound 4 was elucidated and given a trivial name as racematic cajanolactone D2.
Biogenetically, isocoumarins are usually polyketide in origin and arise from the acetate–malonate pathway via cyclization reactions at the end(s) of a polyketide chain.15 However, compounds 1–4 are more likely to originate biogenetically from the same bio-precursor named 2-hydroxy-4-methoxy-6-(2-phenylcinyl) -benzoic acid (5), which is a stilbenoid produced by three malonyl-CoAs and one p-coumaroyl-CoA through stilbene synthase (STS).16 The methylation and divergent prenylation of 5 lead to different intermediates, followed by critical oxidation and cyclization to form the lactone nucleus, which set the stage for generating 1–4. The possible biosynthetic reaction was proposed as outlined in Scheme 1. Notably, bio-precursors 6–7 were also found as natural products from the leaves of C. cajan.17 Compounds 1–4 represent the first members of a novel biogenetic pathway for the isocoumarins, which could be of great significance by providing a new viewpoint on isocoumarin metabolism in the plant kingdom.
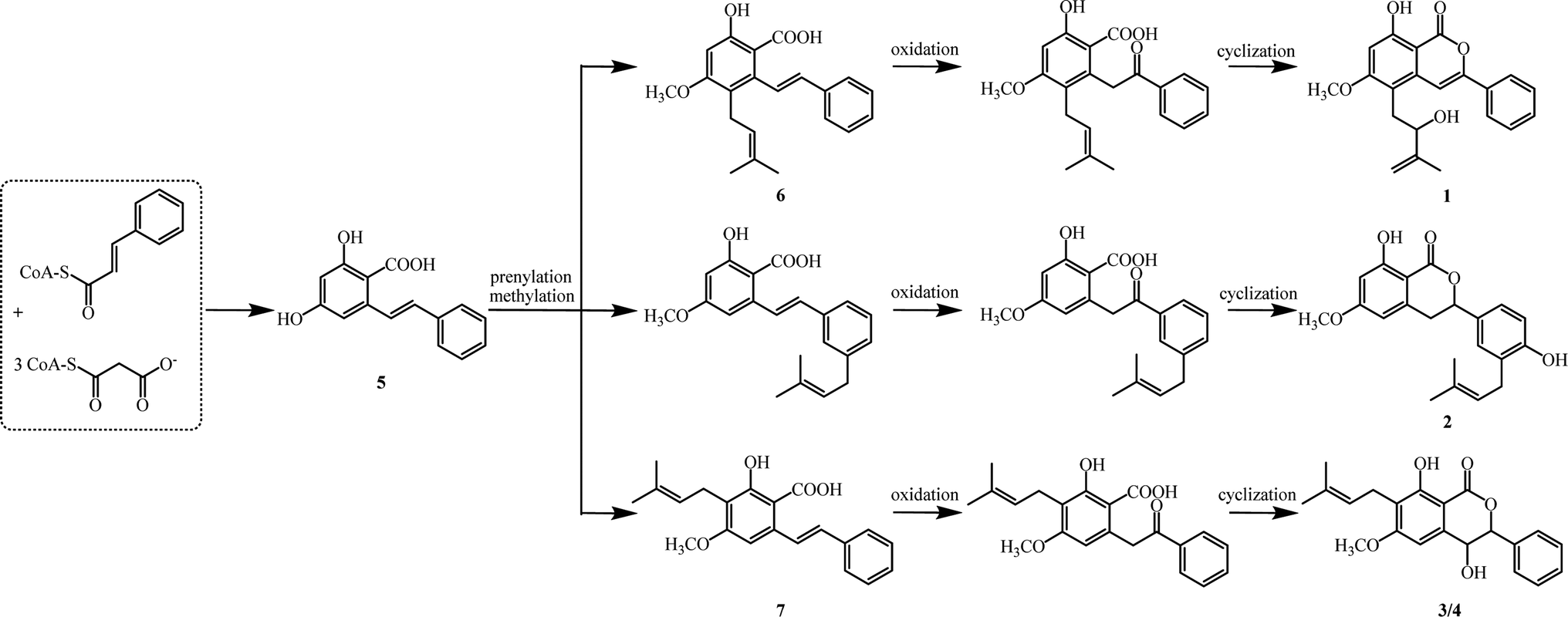 | ||
| Scheme 1 Possible biogenetic pathway to 1–4. | ||
Biological evaluation
The bioassay was performed to evaluate the cytotoxicity for new compounds 1–4, which were found to be devoid of detectable cytotoxicity against HRT-18, Huh-7, and A549 cell lines even at high concentrations of 100 μM.Experimental section
General experimental procedures
The values of optical rotations were recorded using an MCP-500 spectropolarimeter (Anton Paar, Graz, Austria). The Applied Photophysis Chirascan were used to determine the ECD spectra of tested compounds. UV and IR data were acquired by a Shimadzu UV-2600 spectrophotometer (Shimadzu, Kyoto, Japan) and a Shimadzu IR Affinity-1 spectrometer (Shimadzu, Kyoto, Japan), respectively. HRESIMS spectra were obtained through a Thermo MAT95XP high resolution mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The measurement of 1D and 2D NMR data were collected on a Bruker Avance-500 spectrometer with aids of TMS as internal standard (Bruker, Fällanden, Switzerland). The 200–300 mesh silica gel (Qingdao Marine Chemical Inc., Qingdao, China) was used for column chromatography. TLC analysis was carried out on silica gel plates (Merck KGaA, Darmstadt, Germany). A Hitachi Primaide [Hitachi Instruments (Dalian) Co., Ltd.] equipped with a diode array detector (DAD) using a preparative YMC ODS C18 reverse column (20 × 250 mm, 5 μm) was used for semi-preparative HPLC separation. Organic solvents used were of analytical grade (Guangzhou Chemical Regents Company, Ltd., Guangzhou, China).Plant material
Leaves of C. cajan were collected from Honghe, Yunnan, P. R. China in August 2009 and authenticated by Professor Yunfei Deng of the South China Botanical Garden. A voucher specimen (SCIB 20210521) was deposited at the key laboratory of Natural Product Medicinal Chemistry, South China Botanical Garden, Chinese Academy of Sciences.Extraction and isolation
The mixture of air-dried, milled leaves of C. cajan (30.0 kg) were extracted with 95% EtOH (130 L) at room temperature for 72 h to provide a residue (5.0 kg), which was partitioned in n-hexane and EtOAc to obtain sequential extraction. The crude EtOAc extract was then subjected to a column chromatography over silica gel CC (100–200 mesh) and eluted with n-hexane/EtOAc (v/v 100:0 → 0:1) to yield fractions F1–11.
Fraction F6 (20.0 g) was resolved into eight sub-fractions (F6-1 to F6-8) by silica gel RP C18 using MeOH/H2O (70 → 100%) as the eluent. F6-4 (2.8 g) was applied to Sephadex LH-20 column chromatography, eluting with CHCl3/MeOH (v/v 1:3) to obtain further fractions (F6-4-1 to F6-4-5). F6-4-5 (1.5 g) was applied to silica gel CC (200–300 mesh) eluting with n-hexane/EtOAc (v/v 5:1) and then purified by RP-HPLC (YMC-Pack C18) with 60% acetonitrile in H2O to afford 3 (2.8 mg, tR = 32.0 min) and 4 (5.9 mg, tR = 35.0 min), respectively.
Fraction F10 (39.8 g) was divided into nine further fractions (F10-1 to F10-9) by column chromatography using silica gel RP C18 under gradient conditions, using MeOH/H2O (40 → 100%) followed by MeOH/acetone (v/v 20:1 → 0:1) as mobile phase. F10-5 (9.3 g) was isolated through Sephadex LH-20 column chromatography eluting with MeOH to acquire a number of sub-fractions (F10-5-1 to F10-5-6), and F10-5-3 was subjected to silica gel CC (200–300 mesh) to afford 2 (2.7 mg), eluting with n-hexane/EtOAc (v/v 5:1).
F10-6 (2.2 g) was separated over a Sephadex LH-20 column chromatography eluting with CHCl3/MeOH (v/v 1:1) to acquire sub-fractions F10-6-1 to F10-6-5. F10-6-2 (246.0 mg) was subjected Sephadex LH-20 column chromatography repeatedly and then purified with RP-HPLC (YMC-Pack C18) with 75% acetonitrile in H2O to yield 1 (4.7 mg, tR = 16.5 min).
Cajanolactone B (1) White powder; UV (MeOH) λmax/nm (log ε): 265 (3.09); IR νmax: 3374, 2920, 2851, 1668, 1622, 1576, 1472, 1456, 1242, 1204, 1148, 1103, 997, 764, 689 cm−1; 1H (500 MHz) and 13C (125 MHz) NMR data, see Table 1; ESIMS m/z 353 [M + H]+; HRESIMS m/z 353.1382 [M + H]+ (for C21H21O5+, calcd 353.1384).
Cajanolactone C (2) Yellow oil; UV (MeOH) λmax/nm (logε): 267 (2.84), 302 (2.53); IR νmax: 3368, 2932, 1717, 1653, 1489, 1356, 1242, 1203, 1155, 1107, 1024, 949, 908, 795, 698, 567 cm−1; 1H (500 MHz) and 13C (125 MHz) NMR data, see Table 1; ESIMS m/z 355 [M + H]+; HRESIMS m/z 355.1539 [M + H]+ (for C21H23O5+, calcd 355.1540).
Cajanolactone D1 (3) Yellow oil; UV (MeOH) λmax/nm (logε): 222 (3.46), 260 (2.95), 295 (2.50); IR νmax: 3379, 2940, 2847, 1728, 1614, 1466, 1456, 1339, 1242, 1196, 1126, 1090, 1016, 764, 719, 700, 631, 554 cm−1; 1H (500 MHz) and 13C (125 MHz) NMR data, see Table 2; ESIMS m/z 355 [M + H]+; HRESIMS m/z 355.1541[M + H]+ (for C21H23O5+, calcd 355.1540).
Cajanolactone D2 (4) Yellow oil; UV (MeOH) λmax/nm (logε): 222 (3.41), 260 (2.96), 295 (2.55); IR νmax 3385, 2939, 2843, 1728, 1612, 1466, 1454, 1435, 1337, 1236, 1090, 1020, 764, 721, 700, 600, 575 cm−1; 1H (500 MHz) and 13C (125 MHz) NMR data, see Table 2; ESIMS m/z 355 [M + H]+; HRESIMS m/z 355.1531 [M + H]+ (for C21H23O5+, calcd 355.1540).
Chiral analysis
Chiral analysis of compounds 1 and 4 was performed on a Shimadzu LC-15C HPLC with an EnantioPak® Y1 column (250 × 4.6 mm, 5 μm) using n-hexane/ethanol (v/v 85:15) and n-hexane/ethanol (v/v 90:10) as the eluent, respectively. Compound 2 was subjected to EnantioPak® R-C column (250 × 4.6 mm, 5 μm) eluting with n-hexane/ethanol (v/v 80:20), while 3 was analyzed using EnantioPak® Y3 column (250 × 4.6 mm, 5 μm) with n-hexane/ethanol (v/v 80:20) as the eluent. All these chiral columns were purchased from Guangzhou Research & Creativity Biotechnology Co., Ltd, China.
Cytotoxicity assay
Compounds 1–4 were evaluated for their cytotoxic activity against HRT-18, Huh-7, and A549 cell lines using Cell Counting Kit-8 (CCK-8, Beyotime Biotechnology) according to the manufacturer's protocol. Doxorubicin was used as a positive control with IC50 values of 1.4, 0.8, 1.2 μM for HRT-18, Huh-7, and A549 cell lines, respectively.Conclusion
Four new isocoumarins (1–4) were discovered during the investigation of C. cajan. Their structures were determined to be mixtures of racemic pairs, evident from chiral analysis, among which 3 and 4 share the same planer structure, which enriched the chemical diversity of the plant C. cajan. A plausible biogenetic pathway was proposed to account for the biosynthesis of compounds 1–4. However, further biological evaluation of these constituents is ongoing with an aim to explore the chemistry and biology of this plant.Conflicts of interest
The authors declare no conflict of interest that could have appeared to influence the work reported in this paper.Acknowledgements
We're thankful to the National Natural Science Foundation of China (NSFC) (No. 82073733), the Science and Technology Service Network (STS) Initiative of the Chinese Academy of Sciences (CAS) (KFJSTS-QYZD-2021-03-002), the Innovation Leading Team Project of Guangzhou City (No. 202009020004) and Academician (Expert) Workstation of Yunnan Province Program (No. 202005AF150039).References
- A. Saeed, Eur. J. Med. Chem., 2016, 116, 290–317, DOI:10.1016/j.ejmech.2016.03.025.
- G. Shabir, A. Saeed and H. R. El-Seedi, Phytochemistry, 2021, 181, 112568, DOI:10.1016/j.phytochem.2020.112568.
- M. A. Tammam, M. I. Gamal El-Din, A. Abood and A. El-Demerdash, RSC Adv., 2023, 13, 8049–8089, 10.1039/D2RA08245D.
- P. Wang, H. Wang, J. Yang, L. Yang, C. Cai, J. Yuan, F. Wu, C. Gai, W. Mei and H. Dai, Mar. Drugs, 2023, 21, 150, DOI:10.3390/md21030150.
- E. R. Sukandar, S. Kaennakam, P. Raab, X. Nöst, K. Rassamee, R. Bauer, P. Siripong, T. Ersam, S. Tip-Pyang and W. Chavasiri, Molecules, 2021, 26, 6626, DOI:10.3390/molecules26216626.
- B. Gargi, P. Semwal, S. B. Jameel Pasha, P. Singh, S. Painuli, A. Thapliyal and N. Cruz-Martins, Molecules, 2022, 27, 6877, DOI:10.3390/molecules27206877.
- N. Wu, Y. Kong, Y. Fu, Y. Zu, Z. Yang, M. Yang, X. Peng and T. Efferth, J. Agric. Food Chem., 2011, 59, 437–443, DOI:10.1021/jf103970b.
- Y. Fu, O. Kadioglu, B. Wiench, Z. Wei, C. Gao, M. Luo, C. Gu, Y. Zu and T. Efferth, Phytomedicine, 2015, 22, 462–468, DOI:10.1016/j.phymed.2015.02.005.
- J. Wu, B. Li, W. Xiao, J. Hu, J. Xie, J. Yuan and L. Wang, Int. J. Antimicrob. Agents, 2020, 55, 105821, DOI:10.1016/j.ijantimicag.2019.10.002.
- L. Zhao, L. Yao, R. Chen, J. He, T. Lin, S. Qiu, G. Chen, H. Chen and S. X. Qiu, Antiviral Res., 2023, 212, 105570, DOI:10.1016/j.antiviral.2023.105570.
- R. Yang, L. Wang, J. Xie, X. Li, S. Liu, S. Qiu, Y. Hu and X. Shen, Int. J. Mol. Med., 2018, 42, 2329–2342, DOI:10.3892/ijmm.2018.3836.
- Y. Liu, S. Shen, Z. Li, Y. Jiang, J. Si, Q. Chang, X. Liu and R. Pan, Neurochem. Int., 2014, 78, 43–52, DOI:10.1016/j.neuint.2014.08.007.
- S. Liu, Z. H. Luo, G. M. Ji, W. Guo, J. Z. Cai, L. C. Fu, J. Zhou, Y. J. Hu and X. L. Shen, Molecules, 2019, 24, 271, DOI:10.3390/molecules24020271.
- M. Karplus, J. Chem. Phys., 1959, 30, 11–15, DOI:10.1063/1.1729860.
- A. A. Magid, L. Voutquenne-Nazabadioko, G. Moroy, C. Moretti and C. Lavaud, Phytochemistry, 2007, 68, 2439–2443, DOI:10.1016/j.phytochem.2007.05.011.
- J. Fliegmann, G. Schroder, S. Schanz, L. Britsch and J. Schroder, Plant Mol. Biol., 1992, 18, 489–503, DOI:10.1007/BF00040665.
- C. J. Cooksey, J. S. Dahiya, P. J. Garratt and R. N. Strange, Phytochemistry, 1980, 21, 2935–2938, DOI:10.1016/0031-9422(80)85072-2.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08149d |
| This journal is © The Royal Society of Chemistry 2024 |