
Bimetallic ZnCo-MOF derived porous Ir-doped cobalt oxides for water oxidation with improved activity and stability†
Junliang
Chen
,
Jie
Liu
,
Shaojie
Xu
,
Yi
Wu
,
Yunan
Ye
and
Jinjie
Qian
 *
*
Key Laboratory of Carbon Materials of Zhejiang Province, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, Zhejiang, P. R. China. E-mail: jinjieqian@wzu.edu.cn
First published on 1st July 2024
Abstract
The slow reaction kinetics of the oxygen evolution reaction (OER) significantly impedes the advancement of overall water splitting for practical energy conversion and storage. Transition metal oxides have emerged as competitive alternatives to noble metal-based OER catalysts because of their adaptable composition, tunable electronic structure, and abundant availability. Here, we present a porous bimetallic zinc/cobalt oxide derived from a metal–organic framework (MOF) that is modified by a base-etching process, followed by further iridium loading. The resulting electrocatalyst, Ir-pZCO, exhibited satisfactory OER performance in alkaline conditions, attributed to its high concentration of oxygen vacancies and large specific surface area. Specifically, it demonstrated a small overpotential of 304 mV at 10 mA cm−2 and a Tafel slope of 63.9 mV dec−1, and maintained a decent current retention of 94.8% over 10 hours. This enhanced OER activity stems from the in situ formed oxygen vacancies and Ir introduction, supported by density functional theory calculations. This study offers a novel approach for the rational design and facile fabrication of porous MOF derivatives with abundant oxygen vacancies as well as metal-doping for desired electrochemical properties.
1. Introduction
As the economy continues to evolve, the energy crisis and environmental protection are presenting unprecedented challenges. Currently, the pressing need to address the replacement of unsustainable fossil fuels has become crucial for the future survival and advancement of humanity.1 With its high energy density and non-polluting combustion, green and renewable hydrogen is considered an excellent alternative as a new energy carrier.2,3 In this context, overall water splitting (OWS) is emerging as a promising energy conversion and storage technique.4,5 However, the efficiency of OWS is significantly hindered by the oxygen evolution reaction (OER) at the anode, where the complicated 4-electron transfer occurs, characterized by sluggish reaction kinetics.6–8 While noble metal catalysts, such as iridium, ruthenium, and their derivatives, have demonstrated high OER performance, their low availability and moderate stability limit large-scale applications.9–11 Consequently, enormous efforts have been dedicated to the research on cost-effective, highly active, and stable non-noble metal OER catalysts.Benefiting from flexible compositions, tunable electronic structures, and abundant presence in the Earth's crust, transition metal derivatives,12–15 especially transition metal oxides (TMOs) are suitable candidates to substitute noble metal catalysts.16–18 For example, Co3O4 is a potential TMO for efficient water oxidation which is less toxic, and cost-effective.19–21 Despite their widespread use in OER catalysis, many TMOs still exhibit inferior activity compared to noble metals.22,23 For instance, Nocera et al. reported one OER electrocatalyst formed through the oxidative polarization of an ITO electrode in a neutral aqueous solution comprising phosphate anions and cobalt(II) cations.24 Generally, TMOs are structurally endowed with a low specific surface area and a lack of porous architecture, which hinders the efficient mass transfer during the OER process, adversely affecting the adsorption and desorption of oxygen-related species.25 Moreover, under high applied potentials, the metals in TMOs undergo oxidation to higher valence states and dissolve into the electrolyte, leading to a reduction in catalytic activity.26,27 Therefore, these TMOs also suffer from issues such as low activity and poor stability during the OER.
Recently, porous metal–organic frameworks (MOFs), formed through the self-assembly of inorganic metals and organic ligands, have garnered significant attention as favored precursors for TMOs.28–30 MOF-derived nanomaterials inherently possess advantageous features such as high porosity, large specific surface area, and tunable composition and morphology.31–33 On one hand, the desired porous nanostructures obtained from MOF precursors are beneficial for improving the mass transfer of OER catalysts.34,35 On the other hand, doping with stable metals like Mn and Ir represents an effective strategy to enhance the durability of TMO materials.36,37 According to previous research, the introduced species, typically embedded within the crystal lattice of the material, can modify the electronic structure of TMOs and reinforce chemical bonds, therefore avoiding the dissolution of the active contents. In addition, the dopant can function as a donor under high potentials, thus preventing the active metals from being oxidized into soluble compounds.38 As a result, the facile synthesis of superior and robust OER catalysts from porous MOF precursors, which offer abundant active sites and tunable heterostructures, remains a big challenge.
Herein, we initially synthesized a series of bimetallic ZnCo-based zeolitic imidazolate frameworks (ZIF-ZnCo), and subsequently converted the ZIF-ZnCo precursors into corresponding ZnCo2O4 oxides (ZCO) as outlined in Scheme 1. Upon dissolution of Zn species in a basic solution, the resulting porous ZCO (pZCO) maintained the dodecahedron morphology but displayed a larger specific surface area and more oxygen vacancies. After Ir immersion, the obtained Ir-doped pZCO (Ir-pZCO) exhibited atomically dispersed Ir species, and yielded decent OER activity and stability under alkaline conditions. Specifically, the Ir-pZCO delivered a low overpotential of 304 mV at 10 mA cm−2, a Tafel slope of 63.9 mV dec−1, and a high current retention rate of 94.8% after 10 hours.
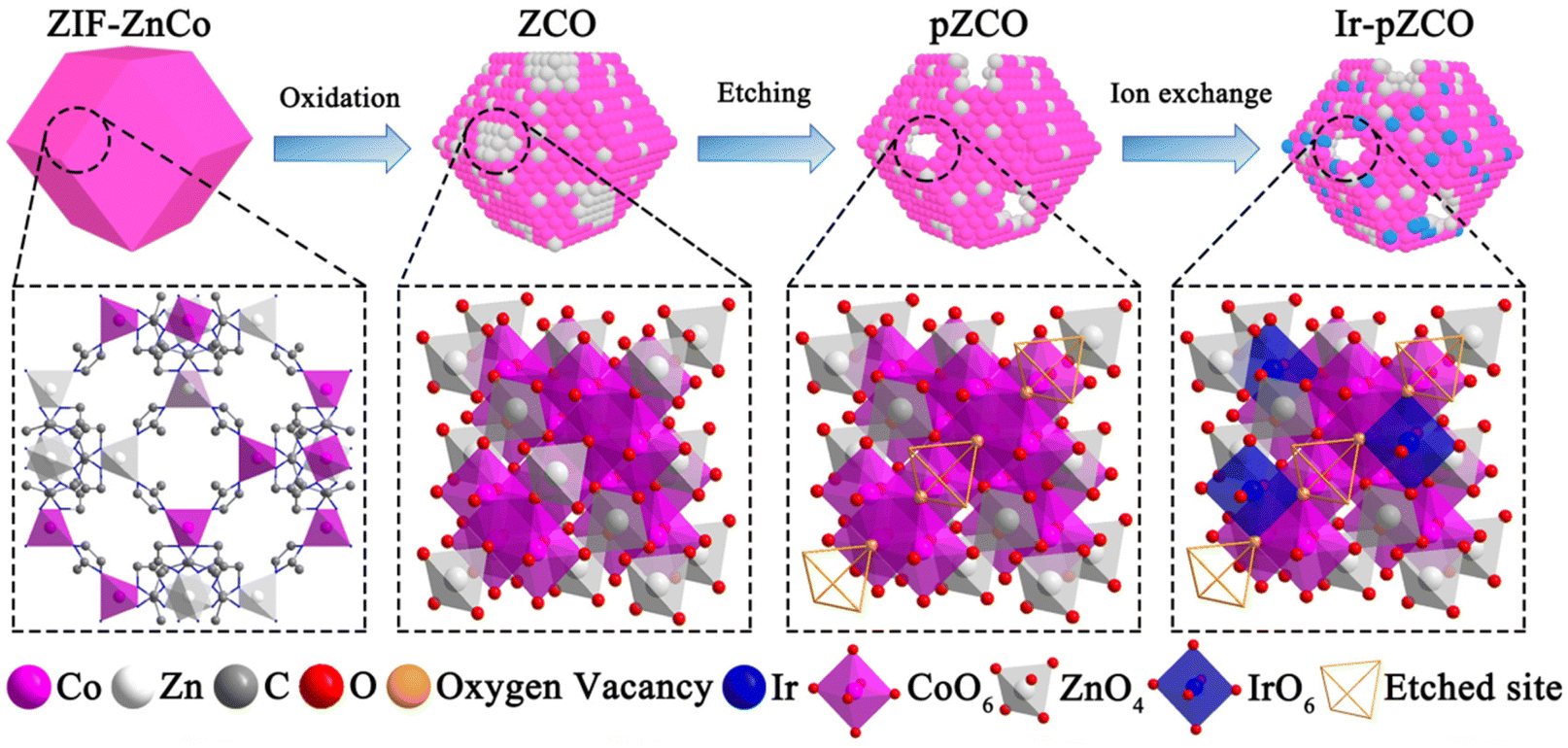 | ||
| Scheme 1 Schematic of the preparation process for base-etched Ir-pZCO with dispersed Ir species. | ||
Furthermore, density functional theory (DFT) calculations further provided additional confirmation of the enhanced activity facilitated by Ir species through the rearrangement of the electronic structure. Our work highlights that the sacrificial MOF template can be reasonably designed and synthesized to adjust both the morphology and electrochemical properties of MOF-derived TMO catalysts for efficient OER applications.
2. Results and discussion
By subjecting bimetallic ZIF-ZnCo precursors to pyrolysis, we successfully synthesized the ZCO templates and then immersed them in an alkaline solution to obtain the alkali-etched pZCO series (Fig. S1 and 2†). Similarly, the pCO derived from pure ZIF-Co without Zn element was also prepared for comparison. SEM images revealed the preserved morphology of pZCO-1 compared to pCO due to the limited dissolution of Zn species (Fig. 1a and S3–5†). In contrast, the surface of pZCO-2 became rougher with visible folds, while several pores emerged on the surface of pZCO-3, attributed to the higher Zn ratio (Fig. 1b and c).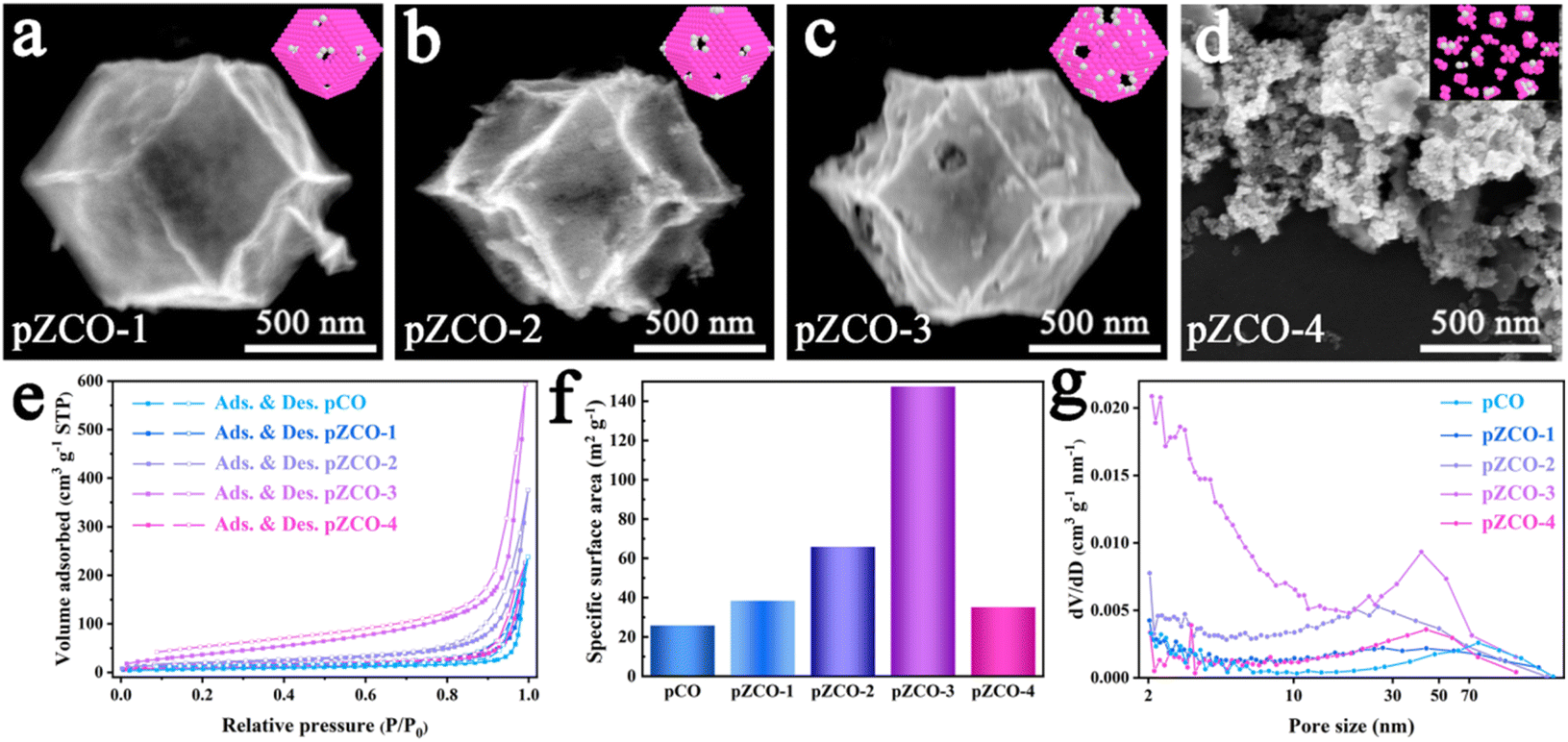 | ||
| Fig. 1 (a–d) SEM images of the pZCO series. (e–g) N2 sorption isotherms, BET specific surface area analysis and the corresponding PSD curves of the pCO and pZCO series. | ||
However, the framework of pZCO-4 collapsed into a chunky structure because of its excessive Zn concentration (Fig. 1d). They exhibited similar type-III N2 isotherms, indicating that this series of MOF-derived metal oxides primarily contained macropores (Fig. 1e). Additionally, the pZCO-3 had the highest surface area of 147.39 m2 g−1, while pCO, pZCO-1, pZCO-2, and pZCO-4 had surface areas of 25.88, 38.43, 65.87, and 35.15 m2 g−1, respectively (Fig. 1f and Table S4†). The calculated pore size distribution (PSD) curves showed a predominance of macropores for all materials, which would enhance the electrochemically active surface area and mass transfer efficiency (Fig. 1g). In this case, the optimal pZCO-3 was rationally chosen for subsequent introduction of Ir species on account of its ample surface area and preserved nanostructure.
The loading of IrCl3·xH2O into pZCO-3 was conveniently conducted through a hydrothermal method followed by a secondary annealing. For detailed procedures, please refer to the Experimental section in the ESI.† The morphology of Ir-pZCO largely retained its original form, with some base-etched defects and pores evident, as shown in Fig. S6.† TEM images displayed a distinct contrast between the darker shells and lighter interior cavities of the dodecahedron, indicating the formation of a hollow and porous nanostructure (Fig. 2a). HR-TEM images depicted well-defined characteristic spacings of 0.248 nm, corresponding to the (311) lattice fringe of ZnCo2O4 (Fig. 2b). Moreover, a homogeneous distribution of Ir, Co, Zn, and O elements was clearly observed, with an increased Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn ratio in the EDS pattern confirming Zn species dissolution (Fig. 2c and S7, Table S5†). In Fig. 2d, the collected PXRD patterns revealed four moderate peaks located at 31.2°, 36.8°, 59.2°, and 65.1° indexed to the (220), (311), (511), and (440) planes of ZnCo2O4 (PDF#81-2299), respectively. Notably, the diffraction peaks of pZCO slightly shifted to a higher angle by 0.22° compared to ZCO (Fig. 2d inset), indicating a shorter lattice spacing due to a higher Co ratio and the smaller ionic radius of Co.39 The electron paramagnetic resonance (EPR) spectra in Fig. 2e showed that pZCO exhibited a stronger signal than ZCO, suggesting the creation of more oxygen vacancies. However, the oxygen vacancy concentration of porous Ir-pZCO slightly decreased, possibly due to the utilization of oxygen vacancies after Ir introduction.
:Zn ratio in the EDS pattern confirming Zn species dissolution (Fig. 2c and S7, Table S5†). In Fig. 2d, the collected PXRD patterns revealed four moderate peaks located at 31.2°, 36.8°, 59.2°, and 65.1° indexed to the (220), (311), (511), and (440) planes of ZnCo2O4 (PDF#81-2299), respectively. Notably, the diffraction peaks of pZCO slightly shifted to a higher angle by 0.22° compared to ZCO (Fig. 2d inset), indicating a shorter lattice spacing due to a higher Co ratio and the smaller ionic radius of Co.39 The electron paramagnetic resonance (EPR) spectra in Fig. 2e showed that pZCO exhibited a stronger signal than ZCO, suggesting the creation of more oxygen vacancies. However, the oxygen vacancy concentration of porous Ir-pZCO slightly decreased, possibly due to the utilization of oxygen vacancies after Ir introduction.
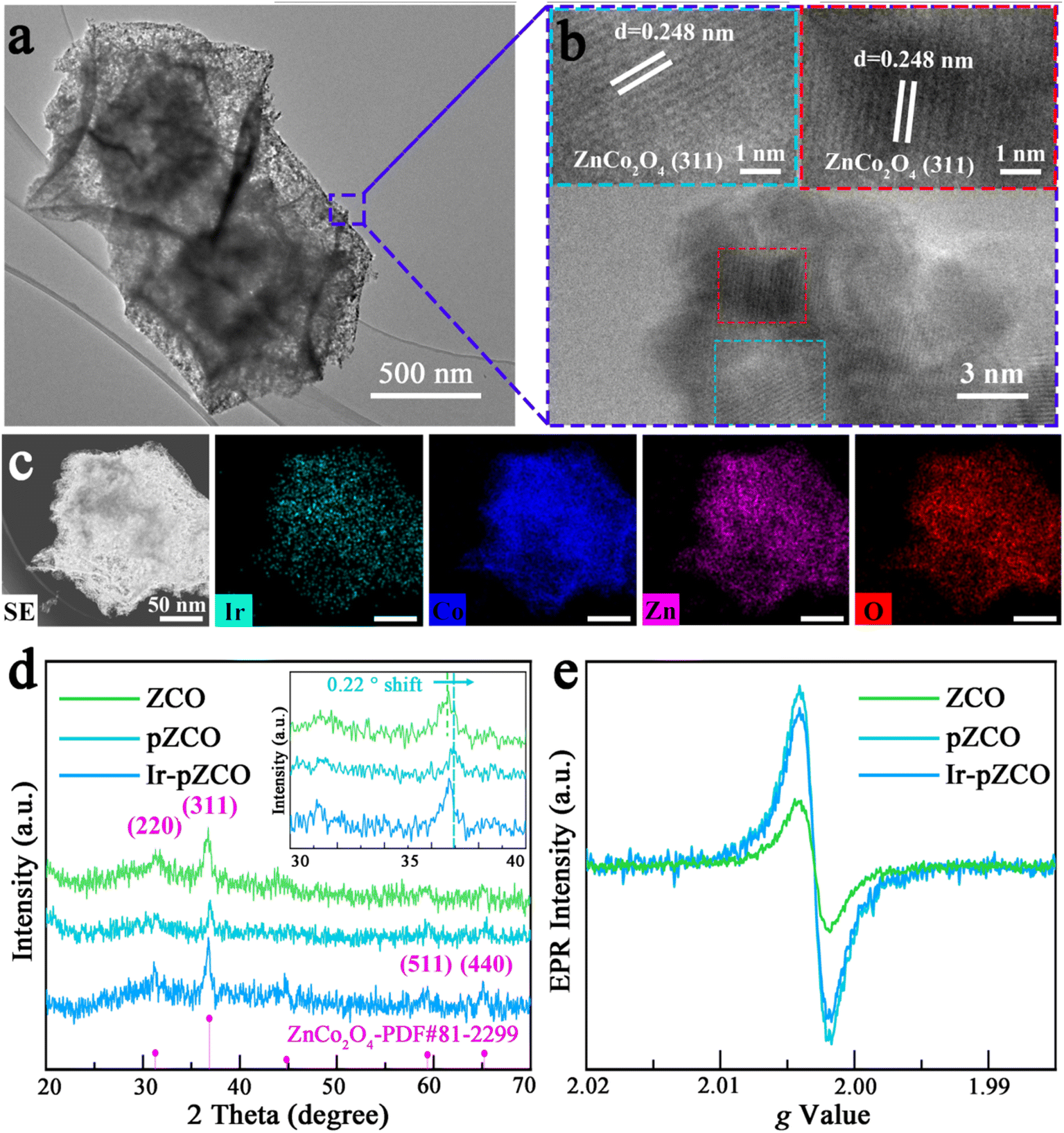 | ||
| Fig. 2 (a) TEM, (b) HR-TEM, and (c) element mapping images of Ir-doped pZCO-3. (d) PXRD patterns and (e) EPR spectra of the as-prepared samples. | ||
To assess the chemical valence states of all elements, X-ray photoelectron spectroscopy (XPS) was further carried out. The full survey XPS spectra of all the as-prepared samples showed distinct signals of C, O, Co, and Zn elements, with only Ir atoms detected in Ir-pZCO (Fig. S8†). The high-resolution O 1s spectra were divided into three subpeaks: lattice oxygen (M–O, 529.4 eV), oxygen vacancy (Ov, 531.6 eV), and adsorbed water (ads. H2O, 533.6 eV). Notably, the M–O peak area in ZCO was conspicuously larger than that of pZCO and Ir-pZCO, which was attributed to the decomposition of Zn–O bonds. Additionally, more Ov sites were detected in pZCO and Ir-pZCO, which corresponded well with the above EPR analysis (Fig. 3a and Table S6†). For the ZCO and pZCO, the deconvoluted peaks located at 779.7/794.7 and 781.6/796.7 eV could be considered as Co3+ and Co2+, respectively. After Ir-doping, the Co3+ and Co2+ peaks of Ir-pZCO presented an obvious positive shift of 0.4 eV as a result of the electronic interaction between Ir and Co species (Fig. 3b and Table S7†).40 In the Zn 2p spectra, all samples exhibited similar peaks at 1020.7 and 1043.9 eV, which were assigned to Zn 2p3/2 and Zn 2p1/2, respectively. The intensity of the Zn 2p peak in ZCO was apparently stronger than those of pZCO and Ir-pZCO, mainly due to the aforementioned Zn dissolution (Fig. 3c). Furthermore, the obtained Ir-pZCO displayed two peaks at 62.1 and 65.1 eV that indicated the existence of Ir4+, while the signals at 63.3 and 66.1 eV were attributed to Ir3+. The weaker intensity of Ir3+ was probably due to the strong electronic interaction between Co and Ir species. Finally, an overlapped peak at 60.7 eV was attributed to Co 3p, consistent with the previous studies (Fig. 3d).41
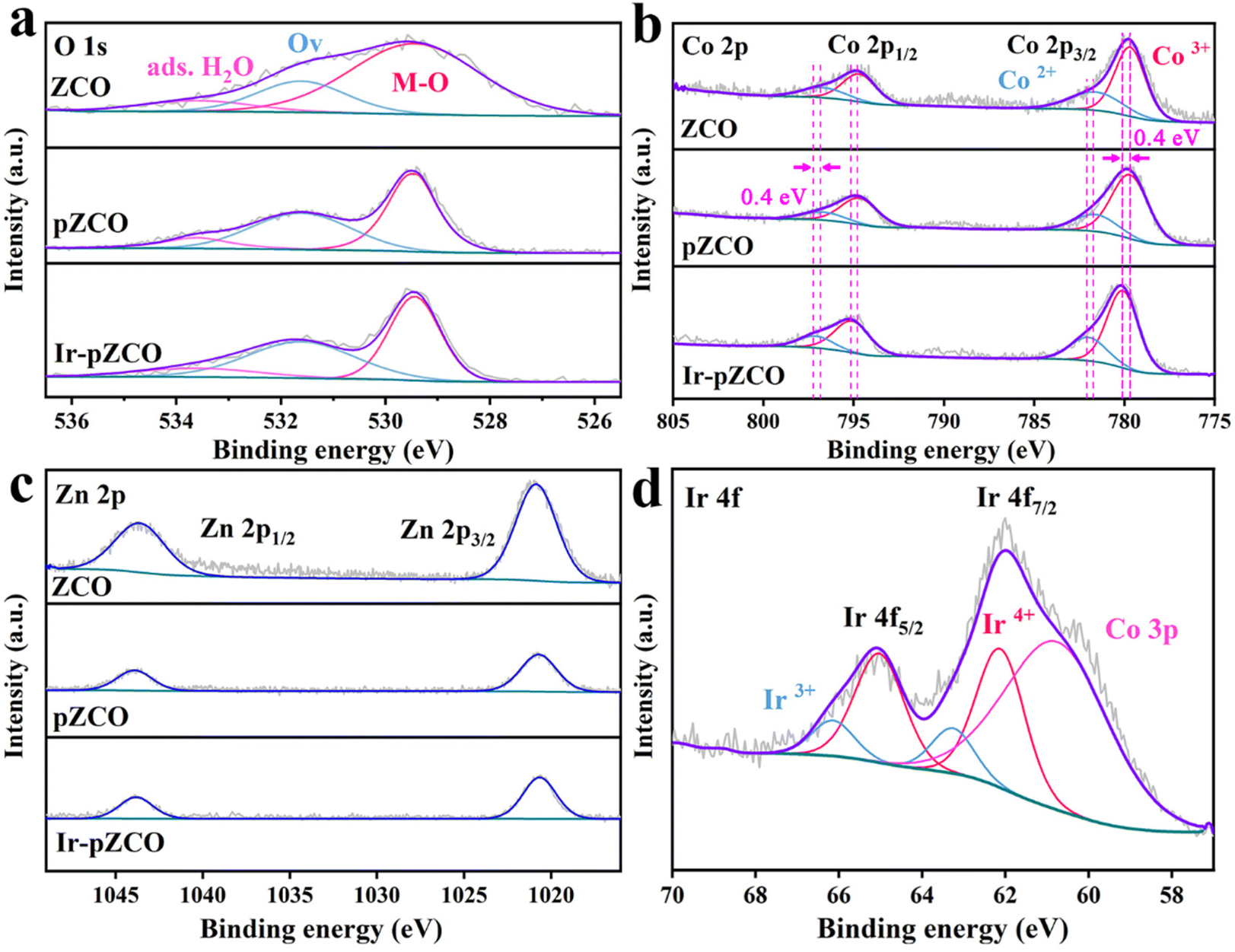 | ||
| Fig. 3 High-resolution XPS spectra of (a) O 1s, (b) Co 2p, and (c) Zn 2p of ZCO, pZCO and Ir-pZCO, and (d) Ir 4f of Ir-pZCO. | ||
The OER activity was assessed using polarization curves without iR-correction, and the applied potentials were calibrated to the reversible hydrogen electrode (RHE), unless otherwise mentioned. The Ir-pZCO exhibited the most favourable OER performance with the lowest overpotential (η10) of 304 mV at 10 mA cm−2, while ZCO, pZCO and commercial RuO2 displayed inferior activity with η10 values of 466, 407, and 346 mV, respectively (Fig. 4a and S9, Table S8†). In Fig. 4b, Ir-pZCO also revealed the lowest Tafel value of 63.9 mV dec−1, indicating superior OER kinetics. Additionally, the electrochemical impedance spectroscopy (EIS) analysis showed that Ir-pZCO exhibited comparatively low charge transfer resistance (Rct, 6.4 Ω) compared to RuO2 (4.4 Ω) and much lower than that of pZCO (15.1 Ω). This improvement can be attributed to the Ir dopant enhancing electrical conductivity and accelerating charge transfer kinetics during the OER process.39 Notably, the Rct value of pZCO was significantly lower than that of ZCO (132.9 Ω), likely due to the higher concentration of oxygen vacancies, which enhance electrical conductivity (Fig. 4c and Table S8†).42,43 The double-layer capacitance (Cdl) analysis is depicted in Fig. 4d, and the Cdl values were calculated to be 1.93, 0.99, and 0.29 mF cm−2 for Ir-pZCO, pZCO, and ZCO, respectively. This implied decent catalytic activity for Ir-pZCO with the maximum active area, matched well with the above pore assessment. Furthermore, the rotating ring-disk electrode (RRDE) analysis in the range of 1.45–1.70 V showed negligible ring current (Iring) compared to disk current (Idisk), indicating a desired quasi-4-electron oxygen evolution process (Fig. 4e and S11†).44 Moreover, the faradaic efficiency (FE) was analyzed through the RRDE technique. For Ir-pZCO, a constant potential of 0.4 V was applied to the ring electrode, and the collected Iring was approximately 39.52 μA while Idisk was controlled at 200 μA, revealing a high FE value of 98.8%. In contrast, the calculated FE values of ZCO and pZCO were 95.2% and 96.5%, respectively, which proved the superior OER efficiency of Ir-pZCO (Fig. S12†). The catalytic stability was analyzed by chronoamperometric examination at a fixed potential of 1.53 V for 10 hours. The collected i–t curve of Ir-pZCO showed no significant attenuation with a compelling current retention of 94.8% (Fig. S13†). Moreover, the LSV curve of Ir-pZCO presented only a slight decay of 8 mV after the long-term durability test, with the PXRD pattern retaining the characteristic peaks of ZnCo2O4 and the XPS spectra exhibiting no obvious change after OER, which further confirmed the improved stability (Fig. 4f and S14, 15†).
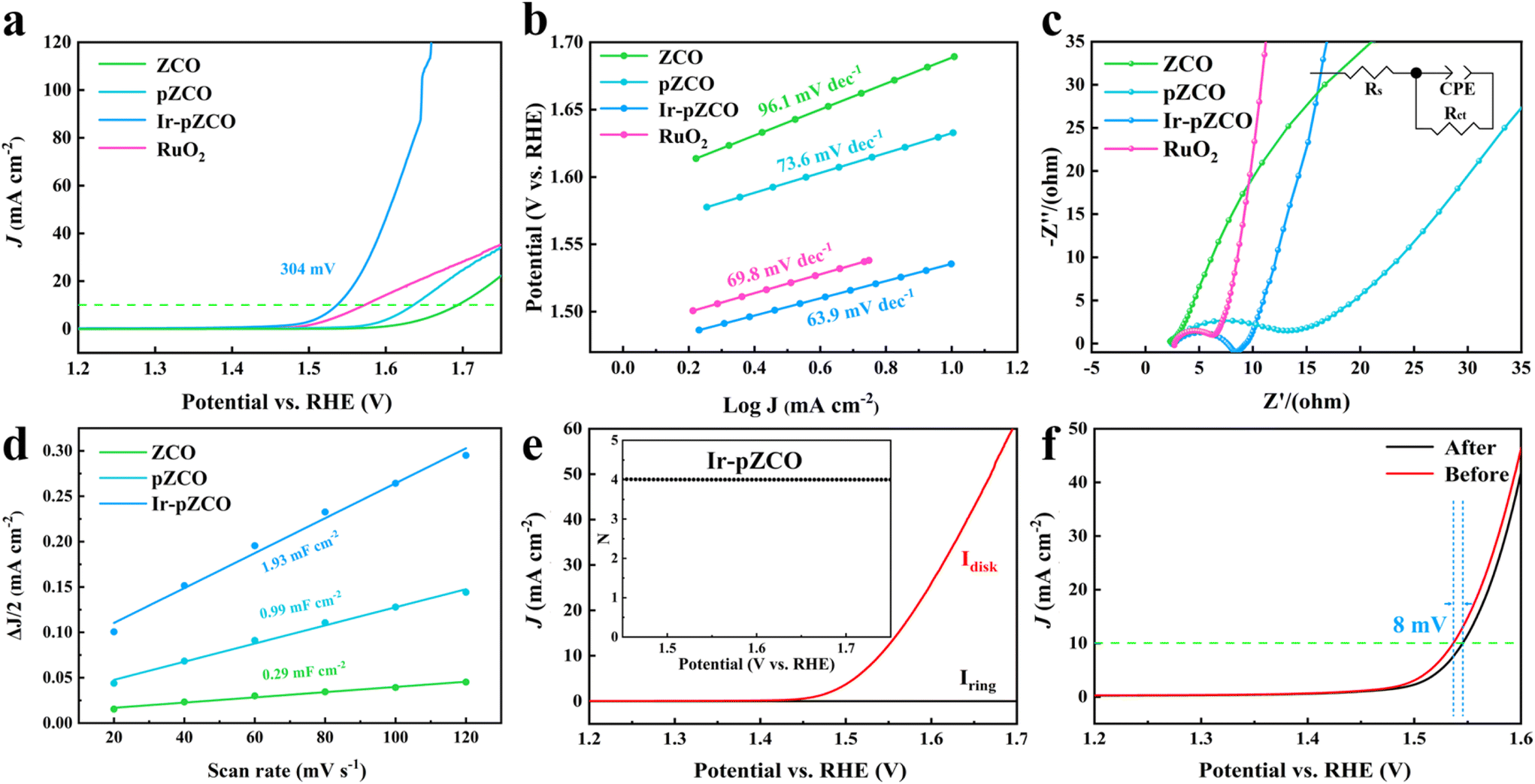 | ||
| Fig. 4 (a) LSV curves, (b) Tafel plots, (c) EIS analysis, and (d) Cdl values of all samples. (e) RRDE voltammogram and the calculated N for Ir-pZCO. (f) LSV curves of Ir-pZCO before and after a 10-hour OER. | ||
To further estimate the electrocatalytic OER activity, we conducted an analysis of temperature-dependent performance. Subsequently, the activation energy (Ea) can be determined to provide evidence of the improved intrinsic OER properties of these materials. As shown in Fig. S17,† all three samples displayed the same trend of increasing current densities and decreasing overpotentials with rising temperature, which indicated the temperature dependence of the OER. The corresponding Arrhenius plots are depicted in Fig. 5a–c, and a summary of the Ea values for the catalysts at different overpotentials (η = 300, 350, 400, 450 mV) is presented in Fig. 5d and Table S9.† Firstly, the calculated Ea values of pZCO at varying overpotentials were much lower than those of ZCO, suggesting that the base-etching process inducing Ov sites was beneficial for enhancing catalytic activity. Secondly, the optimized Ir-pZCO illustrated the lowest Ea value compared to ZCO and pZCO, likely attributable to the interaction between Ir and Co species and the abundance of Ov sites, confirming the favourable reaction thermodynamics.
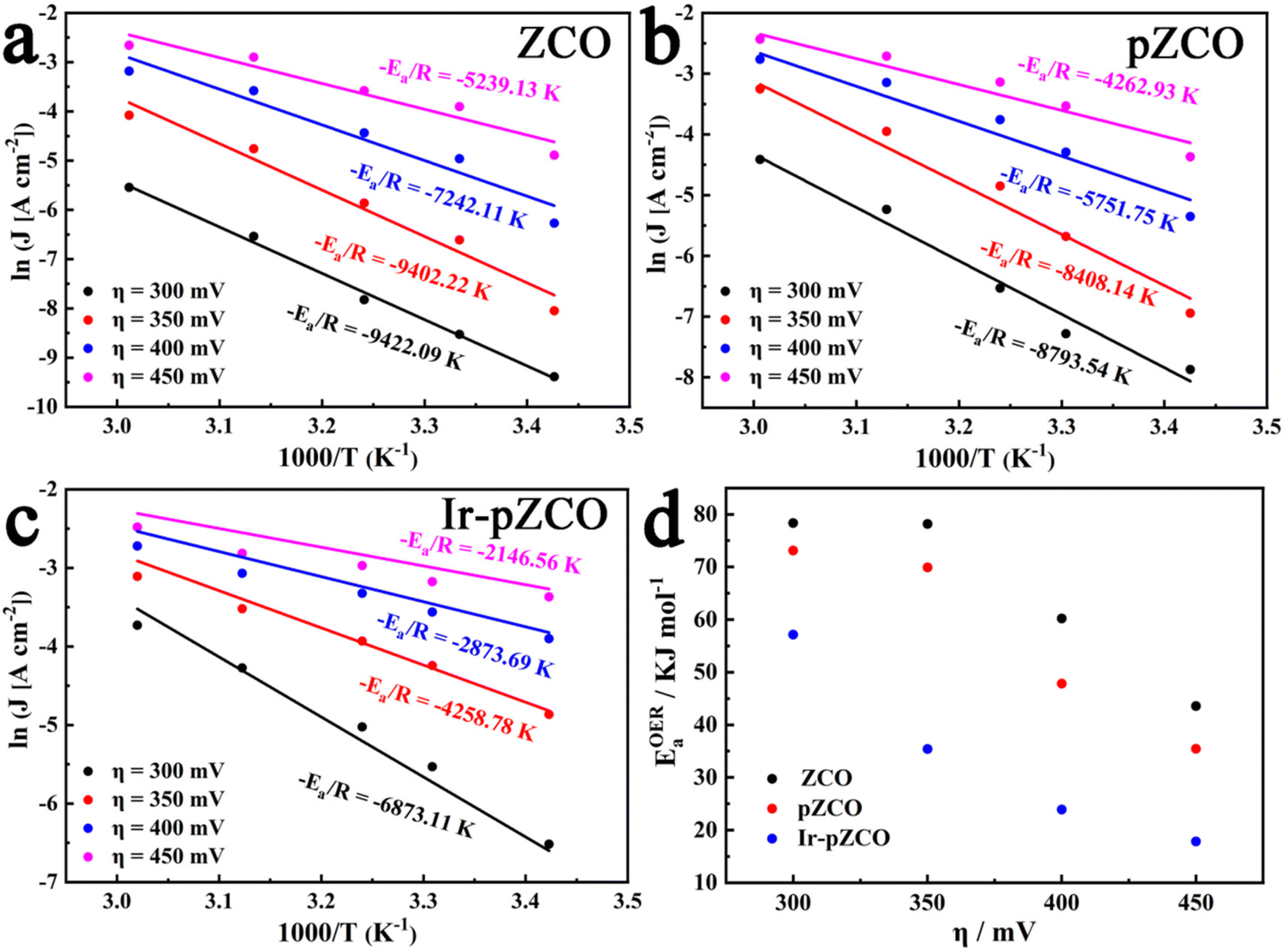 | ||
| Fig. 5 (a–c) Arrhenius plots of ZCO, pZCO, and Ir-pZCO at different overpotentials, respectively. (d) The corresponding activation energies of all three samples. | ||
Previous studies have indicated that the operative active species during the oxygen evolution reaction (OER) process for Co3O4 materials is CoOOH with a high oxidation state. Co3O4 possessing oxygen defects tends to transform more readily into CoOOH during the OER.45 Hence, in this study, we employed Ir-doped CoOOH as the computational model to explore the source of the enhanced OER activity. Fig. 6a depicts the lateral and top views of the crystal structure of Ir-CoOOH, showing the binding of Ir dopants within the lattice of Ir-CoOOH. Fig. 6b illustrates the alteration in charge density in Ir-CoOOH, validating that the incorporation of Ir effectively modulated charge redistribution.
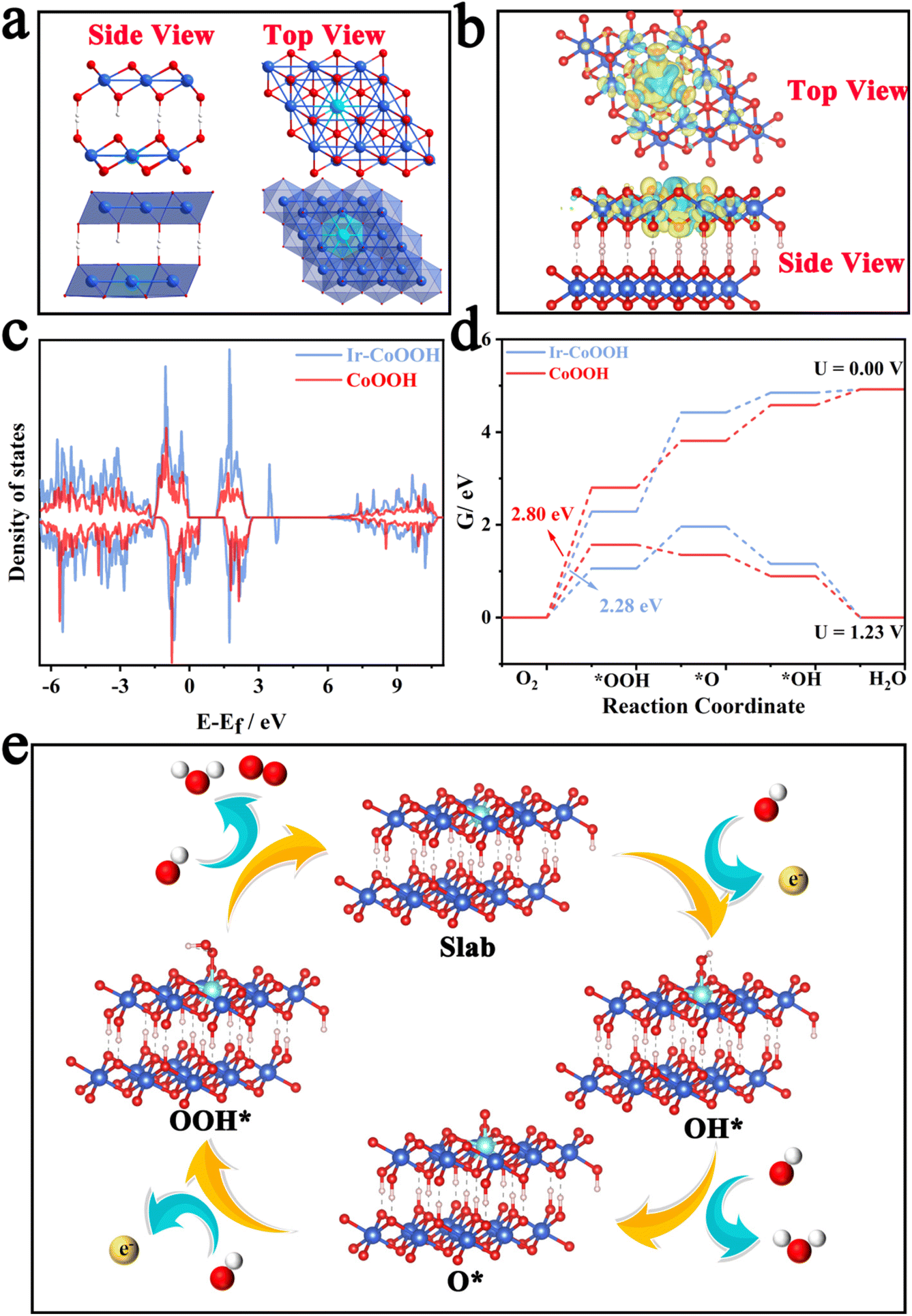 | ||
| Fig. 6 (a) Top and side view crystal structures of Ir-CoOOH, (b) charge density difference over Ir-CoOOH, (c) DOS of Ir-CoOOH and CoOOH (Fermi level is set as 0 eV), (d) Gibbs free energy diagrams of the 4-electron OER process for the Ir-CoOOH and CoOOH catalysts at an applied overpotential of 1.23/0.00 V vs. RHE, (e) the proposed 4-electron OER mechanism for the Ir-CoOOH catalyst. | ||
This observation underscored a significant electron interaction between the doped Ir site and the CoOOH matrix, inducing Ir into an electron-deficient state and a higher valence state, thereby enhancing OER performance.46Fig. 6c presents the total density of states DOS plot of Ir-CoOOH and CoOOH. Clearly, the introduction of Ir atoms induced changes in the electronic state of Ir-CoOOH, leading to an increased density of states (DOS) near the Fermi level. The discerned alterations in the electronic structure of Ir-CoOOH contribute to its superior electron conductivity relative to pure CoOOH.47 This heightened electron conductivity, in turn, facilitated charge transfer between the active site and the surface-adsorbed intermediates, thereby bolstering the OER performance of Ir-CoOOH. Additionally, Fig. 6d delineated the Gibbs free energy changes for OER intermediates, such as OH*, O*, and OOH*. Notably, the transition from H2O to OH− signified the rate-determining step (RDS) of Ir-CoOOH and CoOOH in the OER process, as evidenced by the substantial alteration in Gibbs free energy (ΔG). Particularly, the ΔG value of Ir-CoOOH was 2.28 eV, markedly lower than the ΔG value of CoOOH (2.80 eV). Fig. 6e elucidates the four-electron OER reaction process of Ir-CoOOH, commencing with the adsorption of OH− from the electrolyte at the active surface site to form OH* via electron loss. This OH* species then interacted with another OH−, transitioning to O* by releasing water molecules and electrons. Subsequently, the coupling of the O* species with another OH− gave rise to OOH* via electron loss. The OER process culminated in the reaction of the OOH* species with OH− to yield O2, while releasing water molecules and electrons. Hence, the theoretical computational results demonstrated that Ir-CoOOH formed by Ir-doping of ZnCo2O4 serves as the genuine active species. And the electron interaction between the doped Ir and the CoOOH substrate augmented the valence state of Ir, thereby enhancing the OER performance.
3. Conclusion
In summary, a porous zinc/cobalt oxide with Ir loading was successfully fabricated by a base-etching process followed by immersion. Initially, a range of bimetallic ZCO oxides with different Zn ratios were first reacted with 2.0 M NaOH solution, revealing that the pZCO-3 showed the largest specific surface area while maintaining its dodecahedron morphology. Subsequently, the presence of in situ formed pores and oxygen vacancies was confirmed through electron microscope images, EPR, and XPS characterization. Upon introducing Ir species, the Ir-pZCO showcased markedly enhanced OER activity with a high current retention rate. This improvement can be reasonably attributed to the large specific surface area facilitating mass transfer, the abundance of oxygen vacancies, and the introduction of Ir species modifying the electronic structure to enhance the activity and durability, which was further supported by DFT calculations. Overall, this work presents a facile way to develop porous, vacancy-rich, and high-performance TMO-based catalysts for practical OWS applications.Author contributions
Junliang Chen: methodology, data curation, formal analysis, investigation, validation, writing – original draft. Jie Liu: methodology, data curation, formal analysis, validation, theoretical calculation, writing – original draft. Shaojie Xu: data curation, formal analysis. Yi Wu: data curation, formal analysis. Yunan Ye: data curation, formal analysis. Jinjie Qian: conceptualization, formal analysis, funding acquisition, investigation, project administration, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21601137), Natural Science Foundation of Zhejiang Province (LQ16B010003), Basic Science and Technology Research Project of Wenzhou, Zhejiang Province (H20220001), and the Special Basic Cooperative Research Programs of Yunnan Provincial Undergraduate Universities Association (202101BA070001-042).References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- D. E. McCoy, T. Feo, T. A. Harvey and R. O. Prum, Structural absorption by barbule microstructures of super black bird of paradise feathers, Nat. Commun., 2018, 9, 1 CrossRef CAS PubMed.
- R. Abbasi, B. P. Setzler, S. Lin, J. Wang, Y. Zhao, H. Xu, B. Pivovar, B. Tian, X. Chen, G. Wu and Y. Yan, A roadmap to low-cost hydrogen with hydroxide exchange membrane electrolyzers, Adv. Mater., 2019, 31, 1805876 CrossRef PubMed.
- K. Jiang, M. Luo, M. Peng, Y. Yu, Y.-R. Lu, T.-S. Chan, P. Liu, F. M. F. de Groot and Y. Tan, Dynamic active-site generation of atomic iridium stabilized on nanoporous metal phosphides for water oxidation, Nat. Commun., 2020, 11, 2701 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Metallic nanostructures with low dimensionality for electrochemical water splitting, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Homogeneously dispersed multimetal oxygen-evolving catalysts, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- H. B. Wu and X. W. Lou, Metal-organic frameworks and their derived materials for electrochemical energy storage and conversion: Promises and challenges, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Recent advances on water-splitting electrocatalysis mediated by noble-metal-based nanostructured materials, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- L. Dai, Z.-N. Chen, L. Li, P. Yin, Z. Liu and H. Zhang, Ultrathin Ni(0)-Embedded Ni(OH)2 heterostructured nanosheets with enhanced electrochemical overall water splitting, Adv. Mater., 2020, 32, 1906915 CrossRef CAS PubMed.
- P. Bhanja, Y. Kim, B. Paul, Y. V. Kaneti, A. A. Alothman, A. Bhaumik and Y. Yamauchi, Microporous nickel phosphonate derived heteroatom doped nickel oxide and nickel phosphide: Efficient electrocatalysts for oxygen evolution reaction, Chem. Eng. J., 2021, 405, 126803 CrossRef CAS.
- Y. Guo, C. Zhang, J. Zhang, K. Dastafkan, K. Wang, C. Zhao and Z. Shi, Metal–organic framework-derived bimetallic NiFe selenide electrocatalysts with multiple phases for efficient oxygen evolution reaction, ACS Sustainable Chem. Eng., 2021, 9, 2047–2056 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, Y. Guo, Y. Ide, B. Yuliarto, X. Jiang, Nugraha, H. K. Dipojono, D. Golberg and Y. Yamauchi, Tailorable nanoarchitecturing of bimetallic nickel–cobalt hydrogen phosphate via the self-weaving of nanotubes for efficient oxygen evolution, J. Mater. Chem. A, 2020, 8, 3035–3047 RSC.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, K. Kani, A. E. Allah, B. Yuliarto, Nugraha, H. K. Dipojono, Z. A. Alothman, D. Golberg and Y. Yamauchi, Self-assembly of two-dimensional bimetallic nickel–cobalt phosphate nanoplates into one-dimensional porous chainlike architecture for efficient oxygen evolution reaction, Chem. Mater., 2020, 32, 7005–7018 CrossRef CAS.
- T. X. Nguyen, Y.-C. Liao, C.-C. Lin, Y.-H. Su and J.-M. Ting, Advanced high entropy perovskite oxide electrocatalyst for oxygen evolution reaction, Adv. Funct. Mater., 2021, 31, 2101632 CrossRef CAS.
- F. Hu, D. Yu, M. Ye, H. Wang, Y. Hao, L. Wang, L. Li, X. Han and S. Peng, Lattice-matching formed mesoporous transition metal oxide heterostructures advance water splitting by active Fe–O–Cu bridges, Adv. Energy Mater., 2022, 12, 2200067 CrossRef CAS.
- R. R. Chen, Y. Sun, S. J. H. Ong, S. Xi, Y. Du, C. Liu, O. Lev and Z. J. Xu, Antiferromagnetic inverse spinel oxide LiCoVO4 with spin-polarized channels for water oxidation, Adv. Mater., 2020, 32, 1907976 CrossRef CAS PubMed.
- X. Zou, J. Su, R. Silva, A. Goswami, B. R. Sathe and T. Asefa, Efficient oxygen evolution reaction catalyzed by low-density Ni-doped Co3O4 nanomaterials derived from metal-embedded graphitic C3N4, Chem. Commun., 2013, 49, 7522–7524 RSC.
- A. V. Munde, B. B. Mulik, R. P. Dighole and B. R. Sathe, Cobalt oxide nanoparticle-decorated reduced graphene oxide (Co3O4−rGO): active and sustainable nanoelectrodes for water oxidation reaction, New J. Chem., 2020, 44, 15776–15784 RSC.
- P. K. Bhoj, G. P. Kamble, J. B. Yadav, T. D. Dongale, B. R. Sathe and A. V. Ghule, Economic and binder-free synthesis of NiCo2O4 nanosheets on a flexible stainless steel mesh as a bifunctional electrode for water splitting, Appl. Surf. Sci., 2024, 648, 159083 CrossRef CAS.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Self-supported transition-metal-based electrocatalysts for hydrogen and oxygen evolution, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Dynamic stability of active sites in hydr(oxy)oxides for the oxygen evolution reaction, Nat. Energy, 2020, 5, 222–230 CrossRef.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, Mechanistic studies of the oxygen evolution reaction by a cobalt-phosphate catalyst at neutral pH, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- A. H. Al-Naggar, N. M. Shinde, J.-S. Kim and R. S. Mane, Water splitting performance of metal and non-metal-doped transition metal oxide electrocatalysts, Coord. Chem. Rev., 2023, 474, 214864 CrossRef CAS.
- N. Zhang and Y. Chai, Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- H. Wang, T. Zhai, Y. Wu, T. Zhou, B. Zhou, C. Shang and Z. Guo, High-valence oxides for high performance oxygen evolution electrocatalysis, Adv. Sci., 2023, 10, 2301706 CrossRef CAS PubMed.
- Y. Liu, X. Xu, Z. Shao and S. P. Jiang, Metal-organic frameworks derived porous carbon, metal oxides and metal sulfides-based compounds for supercapacitors application, Energy Storage Mater., 2020, 26, 1–22 CrossRef.
- X. Wang, L. Yu, B. Y. Guan, S. Song and X. W. Lou, Metal–organic framework hybrid-assisted formation of Co3O4/Co-Fe oxide double-shelled nanoboxes for enhanced oxygen evolution, Adv. Mater., 2018, 30, 1801211 CrossRef PubMed.
- Q. Qian, Y. Li, Y. Liu and G. Zhang, General anion-exchange reaction derived amorphous mixed-metal oxides hollow nanoprisms for highly efficient water oxidation electrocatalysis, Appl. Catal., B, 2020, 266, 118642 CrossRef CAS.
- L. Chai, Z. Hu, X. Wang, Y. Xu, L. Zhang, T.-T. Li, Y. Hu, J. Qian and S. Huang, Stringing bimetallic metal–organic framework-derived cobalt phosphide composite for high-efficiency overall water splitting, Adv. Sci., 2020, 7, 1903195 CrossRef CAS PubMed.
- Z. Liang, T. Qiu, S. Gao, R. Zhong and R. Zou, Multi-scale design of metal–organic framework-derived materials for energy electrocatalysis, Adv. Energy Mater., 2022, 12, 2003410 CrossRef CAS.
- X. Wang, Z. Ma, L. Chai, L. Xu, Z. Zhu, Y. Hu, J. Qian and S. Huang, MOF derived N-doped carbon coated CoP particle/carbon nanotube composite for efficient oxygen evolution reaction, Carbon, 2019, 141, 643–651 CrossRef CAS.
- G. Song, Y. Shi, S. Jiang and H. Pang, Recent progress in MOF-derived porous materials as electrodes for high-performance lithium-ion batteries, Adv. Funct. Mater., 2023, 33, 2303121 CrossRef CAS.
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. Lou, Interfacing manganese oxide and cobalt in porous graphitic carbon polyhedrons boosts oxygen electrocatalysis for Zn–Air batteries, Adv. Mater., 2019, 31, 1902339 CrossRef PubMed.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid, Nat. Catal., 2022, 5, 109–118 CrossRef CAS.
- S. Hong, K. Ham, J. Hwang, S. Kang, M. H. Seo, Y.-W. Choi, B. Han, J. Lee and K. Cho, Active motif change of Ni-Fe spinel oxide by Ir doping for highly durable and facile oxygen evolution reaction, Adv. Funct. Mater., 2023, 33, 2209543 CrossRef CAS.
- D. Chen, X. Ji, X. Zhou, Q. Sun, S. Xu, L. Mao, Z. Guo, J. Guan, T.-T. Li and J. Qian, MOF-on-MOF-derived hollow FeNi3/N-doped carbon nanorods for efficient oxygen evolution, Chem. Eng. J., 2023, 470, 144418 CrossRef CAS.
- A. Wang, W. Wang, J. Xu, A. Zhu, C. Zhao, M. Yu, G. Shi, J. Yan, S. Sun and W. Wang, Enhancing oxygen evolution reaction by simultaneously triggering metal and lattice oxygen redox pair in iridium loading on Ni-doped Co3O4, Adv. Energy Mater., 2023, 13, 2302537 CrossRef CAS.
- S. Jung, R. A. Senthil, C. J. Moon, N. Tarasenka, A. Min, S. J. Lee, N. Tarasenko and M. Y. Choi, Mechanistic insights into ZIF-67-derived Ir-doped Co3O4@N-doped carbon hybrids as efficient electrocatalysts for overall water splitting using in situ Raman spectroscopy, Chem. Eng. J., 2023, 468, 143717 CrossRef CAS.
- Y. Zhu, J. Wang, T. Koketsu, M. Kroschel, J.-M. Chen, S.-Y. Hsu, G. Henkelman, Z. Hu, P. Strasser and J. Ma, Iridium single atoms incorporated in Co3O4 efficiently catalyze the oxygen evolution in acidic conditions, Nat. Commun., 2022, 13, 7754 CrossRef CAS PubMed.
- Z. Wang, R. Lin, Y. Huo, H. Li and L. Wang, Formation, detection, and function of oxygen vacancy in metal oxides for solar energy conversion, Adv. Funct. Mater., 2022, 32, 2109503 CrossRef CAS.
- K. Zhu, F. Shi, X. Zhu and W. Yang, The roles of oxygen vacancies in electrocatalytic oxygen evolution reaction, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- D. Chen, Q. Sun, C. Han, Y. Guo, Q. Huang, W. A. Goddard and J. Qian, Enhanced oxygen evolution catalyzed by in situ formed Fe-doped Ni oxyhydroxides in carbon nanotubes, J. Mater. Chem. A, 2022, 10, 16007–16015 RSC.
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, Operando identification of the dynamic behavior of oxygen vacancy-rich Co3O4 for oxygen evolution reaction, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- B. Yang, M. Li, Z. Zhang, S. Chen, M. Wang, L. Sheng, L. Deng, R. Si, M. Fan and H. Chen, Spatial and electronic effects synergistically enhanced electrocatalytic oxygen evolution using atomic iridium-anchored cobalt oxyhydroxide nanosheets, Appl. Catal., B, 2024, 340, 123227 CrossRef CAS.
- X. Ji, Y. Lin, J. Zeng, Z. Ren, Z. Lin, Y. Mu, Y. Qiu and J. Yu, Graphene/MoS2/FeCoNi(OH)x and Graphene/MoS2/FeCoNiPx multilayer-stacked vertical nanosheets on carbon fibers for highly efficient overall water splitting, Nat. Commun., 2021, 12, 1380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi01022a |
| This journal is © the Partner Organisations 2024 |