
Electronic modulation by interfacial bridging between Ir nanoparticle and metal–organic framework to enhance hydrogen evolution†
Kaipeng
Cheng
a,
Yinghuan
Xu
a,
Shenmeng
Zhang
a,
Heng
Zhang
 *c,
Yingying
Xue
a,
Xiahui
Huang
a,
Jianbo
Zhao
b,
Pengcheng
Zhang
a,
Shuang
Ma
*d and
Zhiqiang
Hou
*a
*c,
Yingying
Xue
a,
Xiahui
Huang
a,
Jianbo
Zhao
b,
Pengcheng
Zhang
a,
Shuang
Ma
*d and
Zhiqiang
Hou
*a
aSchool of Chemistry and Chemical Engineering, Zhoukou Normal University, Zhoukou 466000, PR China. E-mail: hou_zq@sina.cn
bSchool of Chemistry and Chemical Engineering, Tarim University, Alar 843300, PR China
cScience Island Branch of Graduate School, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: hnzhangheng95@163.com
dSchool of Chemical & Environmental Engineering (Key Lab of Ecological Restoration in Hilly Areas), Pingdingshan University, Pingdingshan, 467000, P.R. China. E-mail: MaShuang@pdsu.edu.cn
First published on 29th April 2024
Abstract
In the quest for amplifying the inherent capabilities of noble metals, crafting precise metal–support interfacial bonds stands as a challenging yet fruitful endeavor. Here, we introduce an approach to synthesize exceptionally small metal nanoparticles (NPs), which are securely anchored onto cobalt-based metal–organic framework (Co-MOF) nanosheets through a wet chemical method. The creation of metal–oxygen linkages between these Ir NPs and Co-MOF not only guarantees structural robustness but also ensures a generous exposure of active sites. Importantly, the distinct interface fine-tunes the electronic configuration of the composites by facilitating charge transfer along the Ir–O–Co pathway, consequently boosting the reaction dynamics. As a result, the hydrogen evolution reaction (HER) performance of our pioneering product, Ir@Co-MOF, in alkaline media, rivals that of the benchmark Pt/C catalyst and surpasses other recent noble metal catalysts. Theoretical analysis reveals that the exceptional HER efficiency of Ir@Co-MOF originates from the optimal adsorption energies for water and hydrogen, a consequence of electron re-allocation owing to its unique interfacial bond.
1. Introduction
In the context of the global transition towards sustainable energy sources, hydrogen energy emerges as a key player due to its clean and efficient characteristics.1–3 Specifically, water electrolysis for the hydrogen evolution reaction (HER) has garnered significant attention for its environmental compatibility and high efficiency.4,5 However, the effectiveness of the HER is highly dependent on the choice of catalyst. Noble metals such as platinum (Pt), palladium (Pd), and ruthenium (Ru) have traditionally served as the foundation for HER catalysts due to their exceptional catalytic activities.6–9 Even so, their extensive application is hindered by high costs, limited availability, and susceptibility to deactivation under specific conditions.In response to these challenges, researchers have explored various strategies for optimizing the use of noble metals in HER. This includes alloying them with less expensive metals, creating bimetallic or multimetallic compounds, and developing core–shell structures where noble metals form a thin layer over a more abundant core.10–15 While these approaches have improved catalytic performance and cost-effectiveness, challenges such as long-term stability and precise control of electronic properties still remain. Metal–Organic Frameworks (MOFs), with their expansive surface area and adjustable pore sizes, have risen as a promising solution to various challenges in the field.16–18 Their distinctive structural properties offer an ideal platform for anchoring noble metal nanoparticles, leveraging these features for enhanced performance in various applications.19,20 This not only allows for a significant reduction in the amount of noble metal used but also enhances the accessibility of active sites, thereby improving catalytic efficiency. For example, Huang et al. successfully synthesized ultra-small Pt nanoparticles (NPs) oriented on MOFs nanosheets by precisely regulating the reduction kinetics of metal ions, which significantly enhanced HER performance.21 Furthermore, the structural versatility of MOFs allows for precise adjustment of the electronic environments surrounding the metal sites, which is essential for achieving optimal catalytic performance.22,23 Chen and colleagues employed MOF-74, known for its structured pore configuration and effective electrical conductivity, as a scaffold to stabilize Pd clusters.24 These clusters were then skillfully transformed into sophisticated HER electrocatalysts (Pd@MOF-74). It is worth noting that the catalysts have a very high precious metal content, with some even reaching 30%. Relative to Pt, Pd and Ru, Ir exhibits superior catalytic performance in HER due to its distinctive chemical and physical attributes, even when used in very small amounts.25–27 Ir exhibits higher corrosion resistance and chemical stability, making it particularly suitable for alkaline environments.28–30 These characteristics make iridium an ideal candidate for enhancing HER efficiency while reducing the overall cost and quantity of catalyst used.
In summary, the integration of Ir NPs with MOF nanosheets holds great promise for enhancing HER performance. Taking inspiration from this, the immobilization of Ir NPs onto the surface of MOF nanosheets is achieved through a wet chemical strategy. The composite material exhibited remarkable catalytic efficiency as anticipated, confirming the effectiveness of our strategy to merge the catalytic capabilities of iridium with the structural advantages of MOFs. To elaborate, this approach boosts the inherent catalytic activity of Co-MOF and lowers the Gibbs free energy of H* intermediates, thus enhancing the electrocatalytic efficiency of Ir@Co-MOF for the HER. This integration not only underscores the potential of Ir as a formidable catalyst in HER but also highlights the effectiveness of Co-MOF as an ideal platform for nanoparticle dispersion and stability, paving the way for a more sustainable and cost-effective approach to hydrogen production.
2. Results and discussion
The production pathway for Ir@Co-MOF composites is depicted in Fig. 1a. In the initial phase, solvothermal methods are used to grow Co-MOF nanosheet arrays on NF substrates. This is followed by the attachment of Ir NPs onto the MOF nanosheets, utilizing iridium chloride (IrCl3) as the iridium source, achieved through a wet chemical technique. In this process, N,N-dimethylformamide (DMF) and ethanol serve as reducing agents for fabricating Co-MOF nanosheets and converting Ir3+ into Ir NPs, respectively, eliminating the requirement for extra reducing agents or stabilizers. Consequently, this approach enhances the electrocatalytic capabilities of the composite, attributed to the highly accessible Ir active sites, unobstructed by any capping agent. The structure of the samples throughout the fabrication stages was examined using a scanning electron microscope (SEM). Initially, the Co-MOF exhibits a sheet-like form (refer to Fig. S1†). After the introduction of Ir NPs, the Co-MOF nanobelt arrays retain their morphology (as shown in Fig. 1b), offering extensive surface areas and rapid mass transport, vital for effective catalysis. As shown in Fig. 1c, transmission electron microscopy (TEM) also showed similar nanosheet morphology. As seen in Fig. 1d, show a uniform distribution of Ir NPs across the Co-MOF surface, each averaging about 1 nm in diameter (Fig. 1e). The high-resolution TEM (HRTEM) image in Fig. 1f displays a lattice spacing of 0.218 nm, aligning with the (111) plane of Ir.31 Energy-dispersive X-ray spectroscopy (EDX) mappings, depicted in Fig. 1g, confirm the even spread of Co, Ir, and O elements over the entirety of the Co-MOF surfaces. | ||
| Fig. 1 (a) Schematic illustration of the synthesis of Ir@Co-MOF. (b) images of Ir@Co-MOF. (c and d) TEM images at different magnifications of Ir@Co-MOF. (e) The size distribution of Ir nanoparticles. (f) HRTEM and (g) EDS mappings of Ir@Co-MOF. | ||
The X-ray diffraction (XRD) pattern of Ir@Co-MOF, shown in Fig. S2,† closely resembles that of the unmodified Co-MOF, which shares an isostructural relationship with Co-MOF (CCDC no. 985792).32 The absence of distinct Ir NPs peaks in the XRD pattern can likely be attributed to their low concentration and diminutive size, a hypothes is supported by an inductively coupled plasma analysis indicating an Ir content of approximately 6.27 wt%. To delve deeper into the interface interactions between Ir and Co-MOF, X-ray photoelectron spectroscopy (XPS) was employed. Fig. S3† showcases a side-by-side comparison of the XPS spectra for Co-MOF, both in its original form and after Ir integration. In addition to the standard peaks for C, O, and Co, two new, distinct peak range from 300 to 500 eV emerges, signifying the presence of Ir in the resulting composite.33 The high-resolution XPS spectrum of Ir 4f, depicted in Fig. 2a, reveals peaks at 60.8 and 63.8 eV, corresponding to the Ir 4f7/2 and Ir 4f5/2 states of elemental Ir (0), respectively. Moreover, the appearance of peaks at 62.0 and 65.4 eV in the XPS spectrum indicates the formation of Ir–O bonds within the composite.19 The high-resolution Co 2p spectrum of Co-MOF, as shown in Fig. 2b, reveals peaks for Co 2p1/2 and Co 2p3/2 at 797.2 and 781.1 eV, respectively, along with their satellite peaks, confirming the presence of Co2+. Notably, the incorporation of Ir NPs leads to a significant negative shift in these Co 2p1/2 and Co 2p3/2 peaks, which is indicative of a substantial charge transfer from Ir NPs to Co-MOF, further substantiating the interaction between the two components. This data suggests that the electronic structure of the Ir and Co sites is modifiable through interfacial coupling interactions, significantly influencing their catalytic activity in electrochemical reactions. In Fig. S4,† the O 1s spectra of Ir@Co-MOF are meticulously analyzed, showcasing three distinct peaks at energy levels of 531.2, 531.7, and 532.9 eV.25 These peaks are respectively indicative of Co–O, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and adsorbed H2O species. Additionally, a notable peak at 530.7 eV in the Ir@Co-MOF spectrum is observed, signifying the formation of interfacial Ir–O–Co bonds. Moreover, the high-resolution C 1s spectra, illustrated in Fig. S5,† display three distinct peaks at 284.2, 284.6, and 288.0 eV. These are respectively associated with the CC, C–O, and CO bonds.34
O, and adsorbed H2O species. Additionally, a notable peak at 530.7 eV in the Ir@Co-MOF spectrum is observed, signifying the formation of interfacial Ir–O–Co bonds. Moreover, the high-resolution C 1s spectra, illustrated in Fig. S5,† display three distinct peaks at 284.2, 284.6, and 288.0 eV. These are respectively associated with the CC, C–O, and CO bonds.34
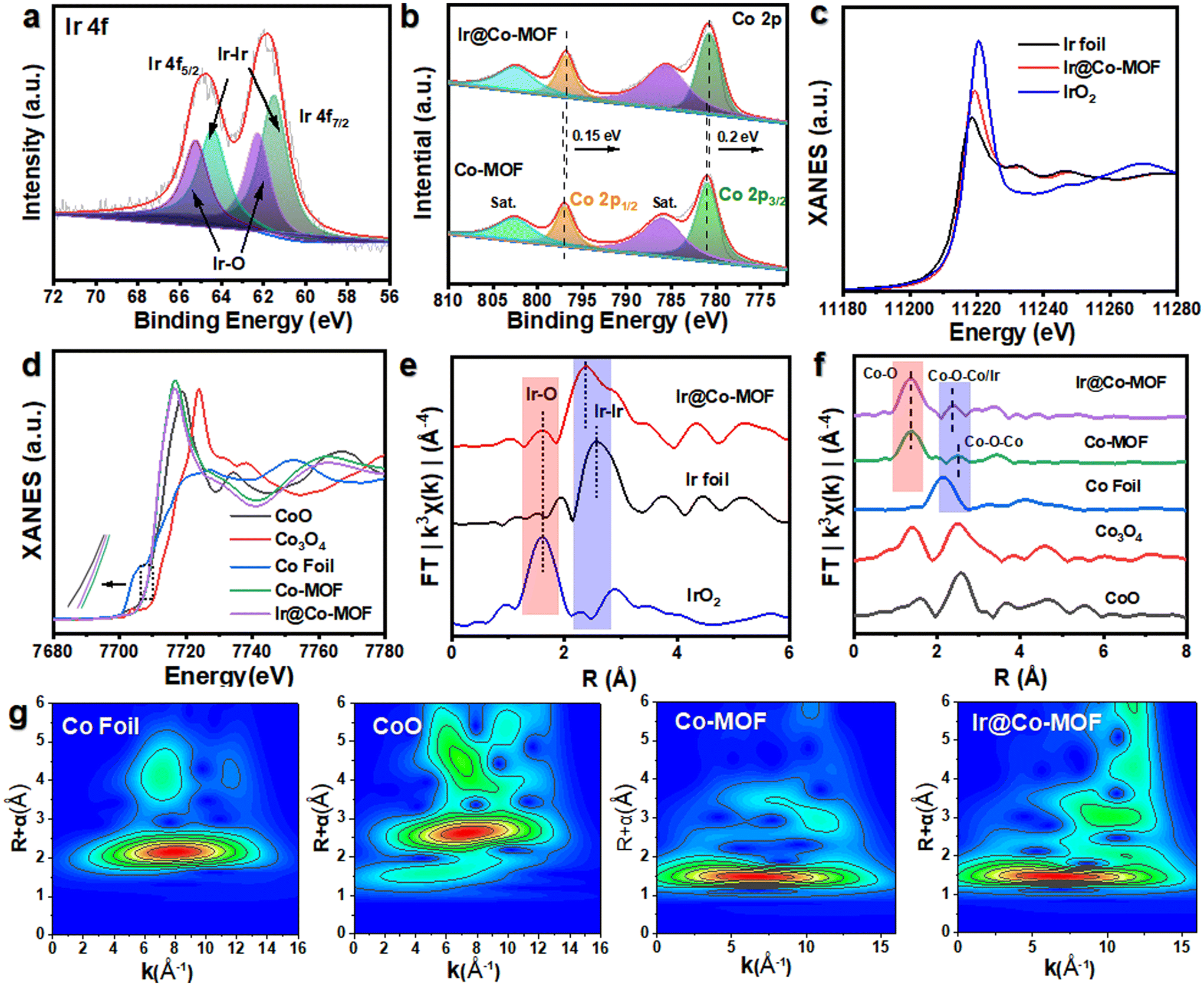 | ||
| Fig. 2 XPS spectra of prepare samples. (a) Ir 4f, (b) Co 2p, of Ir@Co-MOF and Co-MOF. (c and d)The normalized XANES spectra (e and f) FT-EXAFS spectra and (g) Wavelet transform for k3-weighted EXAFS signal of Ir foil, IrO2, Co Foil, CoO, Co-MOF and Ir@Co-MOF, respectively. | ||
To enhance understanding of the local electronic state and the emergence of Ir–O–Co bridges, X-ray absorption fine structure (XAFS) analysis was utilized. Regarding the Ir LIII-edge XANES spectra shown in Fig. 2c, the sequence of increasing white line intensity is as follows: Ir foil, Ir@Co-MOF, and then IrO2, which reflects the ascending order of the valence states of the Ir species. The marginally elevated valence state of Ir@Co-MOF compared to Ir foil further substantiates the presence of oxides within the Ir@Co-MOF and corroborates the reduction in electron density at the Ir sites. The Co K-edge XANES spectra, as depicted in Fig. 2d, reveal a shift in the absorption edge of Ir@Co-MOF to a lower energy than that of Co-MOF. This shift suggests an increase in electron density around Co sites due to the immobilization of Ir NPs on Co-MOF.35 Furthermore, the pronounced amplitude variation in the oscillation of the Co K-edge extended X-ray absorption fine structure (EXAFS) function between Ir@Co-MOF and Co-MOF, as shown in Fig. S6,† implies distinct local atomic configurations. Evidently, the XANES findings are entirely consistent with the XPS data, demonstrating that substantial electron transfer occurs from Ir to Co-MOF. Fig. 2e presents the Ir LIII-edge EXAFS spectra, illustrating that, relative to Ir foil, the presence of shorter Ir–Co bonds in the Ir@Co-MOF results in a shorter Ir–Ir bond length and a decreased coordination number for Ir in the Ir–Ir bonds. Furthermore, the coordination number of Ir within the Ir–O bond rises after forming the composite, further confirming the establishment of the Ir–O bond at the heterostructure interface. The EXAFS spectra of Ir@Co-MOF, illustrated in Fig. 2f, indicate that the primary peak corresponds to Co–O single scattering paths, while weaker peaks are related to higher shell interactions involving Co–O–Co/Ir.36–38 Notably, the increased intensity of the Co–O–Co/Ir bond peak in Ir@Co-MOF compared to Co-MOF corroborates the formation of Ir–O–Co bridges.39 This bond's shift suggests robust interaction at the Ir@Co-MOF heterostructure interface, a finding supported by wavelet transform spectroscopic analysis of the Co K-edge EXAFS (Fig. 2g). These combined results point to the formation and electron transfer mechanism in Ir@Co-MOF featuring interfacial Ir–O–Co bridging. Consequently, the joint analysis of XPS and XANES confirms the presence of this bridging in Ir@Co-MOF, which alters the local electronic structure of the catalyst. This alteration potentially improves the adsorption/desorption dynamics of reaction intermediates, thereby enhancing the material's electrocatalytic efficiency.
To evaluate the catalytic prowess of Ir@Co-MOF, we conducted tests on its HER activity within a three-electrode cell framework, using 1 M KOH as the test medium. For a comparison, we also examined Co-MOF, bare NF (nickel foam), and a commercially available Pt/C coated on NF (Pt/C) under identical experimental conditions. The linear sweep voltammetry (LSV) curves reveal a noteworthy performance of the Ir@Co-MOF electrocatalyst in HER. The results, as presented in Fig. 3a, show remarkable efficiency, with the system reaching a current density of 10 mA cm−2 at an exceptionally low overpotential of just 29 mV. This discovery of this is further supported by comparing the overpotentials required at different current densities, specifically at 10 mA cm−2, 50 mA cm−2 and 100 mA cm−2 (Fig. 3b). The Ir@Co-MOF exhibited low overpotentials at these current densities, measuring 29, 71, and 99 mV, respectively. These values are significantly lower than those for Co-MOF, which were 197, 246, and 286 mV, respectively, and are on par with commercial Pt/C catalysts, which showed overpotentials of 24, 35, and 50 mV. The Tafel value for Ir@Co-MOF (87.6 mV dec−1) is lower compared to Co-MOF (123.6 mV dec−1) (Fig. 3c), indicating Ir@Co-MOF demonstrates faster H2 generation kinetic. The electrochemical impedance spectroscopy (EIS) results, including the Nyquist plots of Ir@Co-MOF and Co-MOF, are employed to analyze the efficiency of charge transfer between the Ir nanoclusters and the supporting materials.40Fig. 3d displays these Nyquist plots, where a notably smaller diameter of impedance arc (DIA) for the Ir@Co-MOF modified electrode, compared to commercial Co-MOF, indicates enhanced interfacial charge transport. The impedance spectra were fitted to an equivalent circuit model denoted as R(QR), where Rs symbolizes the electrolyte resistance, Rct denotes the charge-transfer resistance, and Qs represents the constant phase elements (CPE).41 To delve deeper into the intrinsic activity, we assessed the electrochemical active surface area (ECSA) by calculating the double-layer capacitance (Cdl) at various scan rates during cyclic voltammetry (CV) measurements. These measurements were conducted within voltage ranges that excluded redox processes, as shown in Fig. S7.† In comparison to the pristine Co-MOF with a Cdl value of 8.03 mF cm−2, the Ir@Co-MOF hybrids exhibit a substantial increase in Cdl, measuring 145.2 mF cm−2, as illustrated in Fig. 3e. This increase indicates that the introduction of Ir NPs provides a significantly larger number of active sites. To eliminate the influence of ECSA on performance, we have calculated and compared the normalized current density, as depicted in Fig. 3f. Furthermore, the Ir@Co-MOF exhibits the highest ECSA normalized current density for the HER, underscoring its superior intrinsic activity. To further assess the inherent catalytic capabilities of the catalysts, we computed the turnover frequencies (TOF) for each, details of which can be found in the ESI.† The TOF values for the Ir@Co-MOF catalysts were significantly higher than those observed for Co-MOF. Specifically, at an overpotential of 100 mV, the TOF value for each active site on Ir@Co-MOF was measured at 0.07 s−1, which is 5.4 times higher than the TOF of 0.013 s−1 recorded for Co-MOF. Additionally, the amount of hydrogen produced was quantified using the water displacement method (refer to Fig. S8†). The volume of H2 measured experimentally aligns closely with the theoretical predictions. Consequently, we were able to achieve a faradaic efficiency of 99% with the Ir@Co-MOF catalyst.
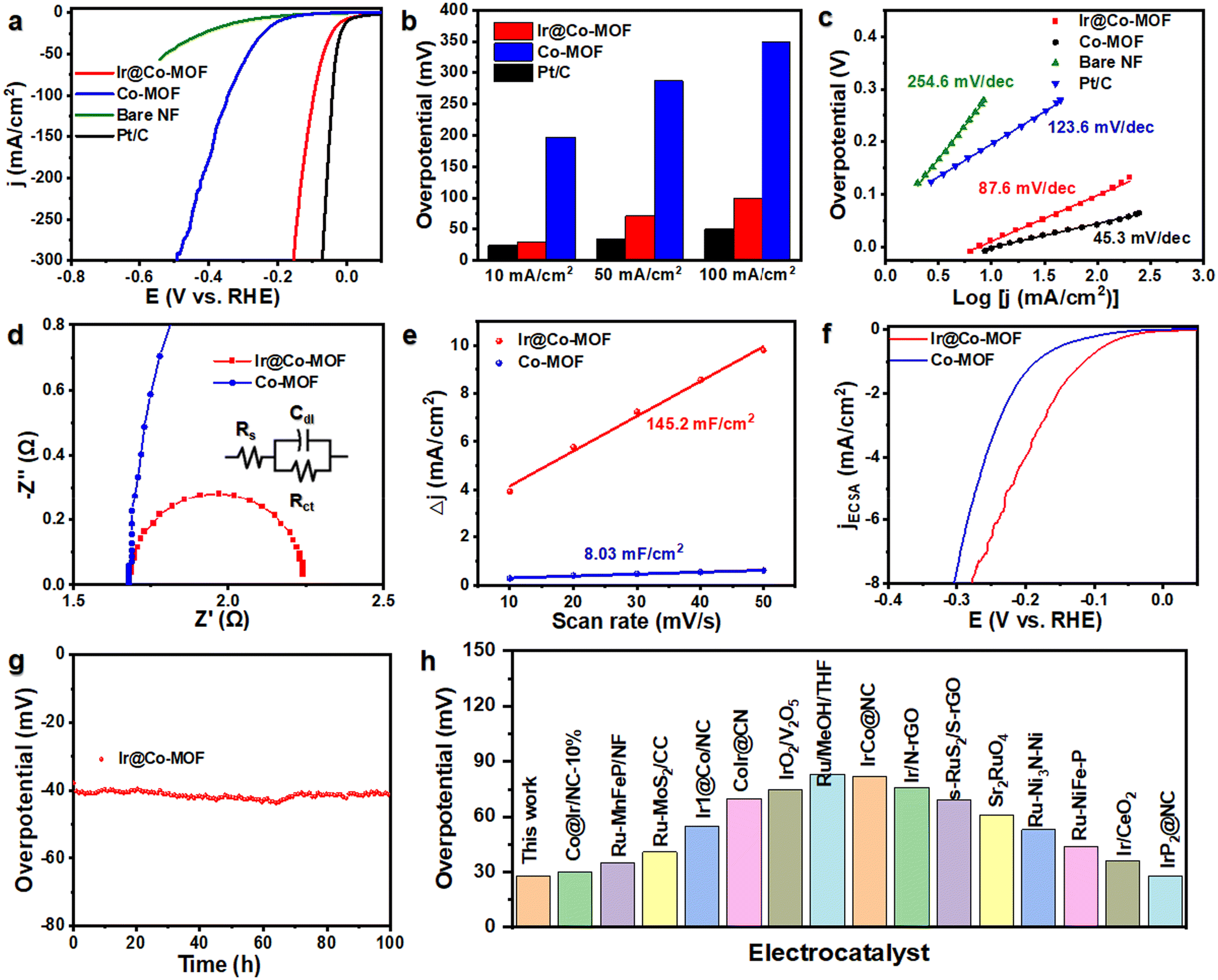 | ||
| Fig. 3 (a) HER polarization curves for Ir@Co-MOF, Co-MOF, Pt/C, and Bare NF. (b and c) Corresponding overpotentials at 10 mA cm−2, 50 mA cm−2, 100 mA cm−2 and Tafel plots. (d) Nyquist plots of different samples. (e) Double-layer capacity Cdl of Ir@CoMOF and Co-MOF. (f) LSV curve after ECSA normalization. (g) Chronoamperometry curves. (h) HER activity comparison graph of the overpotentials at 10 mA cm−2 for the reported Pt-based. | ||
In addition to its electrocatalytic activity, the Ir@Co-MOF demonstrates excellent durability, with no significant decay observed during a chronoamperometry measurement conducted over 100 hours at 10 mA cm−2, as depicted in Fig. 3g. The exceptional durability of the Ir@Co-MOF composite can be attributed to its unique heterostructure. In this structure, the Co-MOF nanosheets play a crucial role in preventing the aggregation of Ir NPs, thanks to the strong interface interaction between them. Furthermore, it's worth noting that Ir@Co-MOF exhibits outstanding performance in alkaline HER with a low overpotential, surpassing the performance of most noble-metal-based electrocatalysts that have been reported to date (Fig. 3h and Table S1†). The SEM and XRD characterization of Ir@Co-MOF after HER reveals that their nanosheet morphologies remain almost unchanged (Fig. S9 and S10†). Furthermore, XPS analyses of the Ir@Co-MOF catalyst, as shown in Fig. S11,† confirm the stability of the Ir–O bond, which remains intact even after the electrochemical testing. These results collectively highlight the remarkable electrochemical performance and endurance of the Ir@Co-MOF catalyst in HER applications.
To investigate the impact of the synthesis method on performance, we incorporated IrCl3 during the MOF synthesis. The results indicated that while the direct integration of Ir ions in a one-step process preserves the crystallinity of the MOF, it significantly compromises the structural integrity of the MOF nanosheets, adversely affecting their catalytic performance (Fig. S12†). Moreover, IrCl3 content was varied to produce Ir1.32 wt%@Co-MOF and Ir13.53 wt%@Co-MOF materials with Ir contents of 1.32 wt% and 13.53 wt%, respectively. The XRD pattern, as depicted in Fig. S13,† indicates that the incorporation of Ir into Co-MOF has a negligible impact on its crystalline structure. Despite this, the presence of Ir markedly enhances the electrocatalytic performance. The polarization curves in Fig. S14† clearly illustrate that samples containing 6.27 wt% of Ir display the most effective HER performance. Specifically, the Ir6.27 wt%@Co-MOF sample distinguishes itself by exhibiting the lowest charge transfer resistance and the most advantageous Tafel slope among all tested samples, as evidenced in Fig. S15.† Additional electrochemical tests, detailed in Fig. S16,† further confirm the exceptional HER activity of Ir@Co-MOF. This enhanced activity is attributed to its minimal charge transfer resistance and maximal ECSA. Collectively, these findings indicate that a 6.27 wt% concentration of Ir optimally improves the structural stability, catalytic activity, and interfacial synergistic effects in Co-MOF. In pursuit of further validating the universality of our synthetic methodology, we extended our investigations to include additional metal–organic frameworks (MOFs), those derived from biphenyldicarboxylate (BPDC) and benzene-1,3,5-tricarboxylate (BTC). XRD analysis confirmed the successful synthesis of these MOFs and their similar nanosheet structures (Fig. S17†). Subsequent evaluations focused on the HER performance of these synthesized MOFs to assess their catalytic efficacy. Among the various MOFs tested, the framework synthesized using benzene-1,4-dicarboxylate (BDC) exhibited superior performance (Fig. S18†). This result not only emphasizes the versatility of our synthetic approach but also highlights the potential for optimizing electrocatalytic performance through strategic selection of MOF precursors.
To elucidate the mechanism behind the high activity and outstanding kinetics of Ir@Co-MOF, DFT calculations were performed. In Fig. 4a and b, you can observe the structure model of Ir@Co-MOF. As depicted in Fig. 4c, it is evident that there is a rearrangement of electrons at the interface, leading to a loss of electrons at the Ir site and an increase of electrons at the Co site. This observation aligns with the XPS and XANES analysis results and provides further evidence of the strong electronic interaction between the Ir NPs and the Co-MOF substrate. The analysis of the total density of states (DOS) for Ir@Co-MOF indicates an increased electron density near the Fermi level, especially when contrasted with that of Co-MOF. The incorporation of Ir NPs into the structure, as depicted in Fig. 4d, implies an enhancement in electrical conductivity and electron transfer efficiency during the HER. Additionally, this electron transfer adeptly modulates the d-band center of Ir. As illustrated in Fig. 4e, this modulation involves shifting the d-band center towards a lower energy level at the interface, which in turn facilitates the adsorption of hydrogen onto the surface.42,43 Such a shift is instrumental in boosting the catalytic performance of the system. The calculated adsorption energies of H2O and H* on the surfaces of Ir@Co-MOF and Co-MOF were further analyzed. Comprehending the adsorption process of H2O molecules on the catalyst surface is essential for facilitating water dissociation in alkaline environments during the HER.44–46 The analysis of binding energies for H2O on both pure Co-MOF and the enhanced Ir@Co-MOF reveals that the adsorption of H2O is more thermodynamically favorable on Ir@Co-MOF, as depicted in Fig. 4f. This observation implies that H2O molecules can be effectively adsorbed and activated on Ir@Co-MOF, thereby enhancing the HER process. Moreover, the adsorption free energy of hydrogen atoms (H*) denoted as ΔGH* is pivotal in determining the efficiency of HER.47,48 Values of ΔGH* nearing zero suggest an optimal balance between hydrogen adsorption and H2 desorption. Illustrated in Fig. 4g, the ΔGH* for Ir@Co-MOF is −0.11 eV, significantly closer to the ideal thermoneutral value than that of pure Co-MOF (−0.98 eV). This indicates reduced barriers in the adsorption–desorption cycle, leading to an elevated HER performance for Ir@Co-MOF. These results demonstrate that the electronic structure modification, achieved through the formation of Ir–O–Co bonds, synergistically optimizes the free energies associated with hydrogen and water adsorption, contributing to the superior HER activity. This optimization facilitates the HER process in alkaline solutions, leading to the exceptional performance of Ir@Co-MOF as an electrocatalyst.
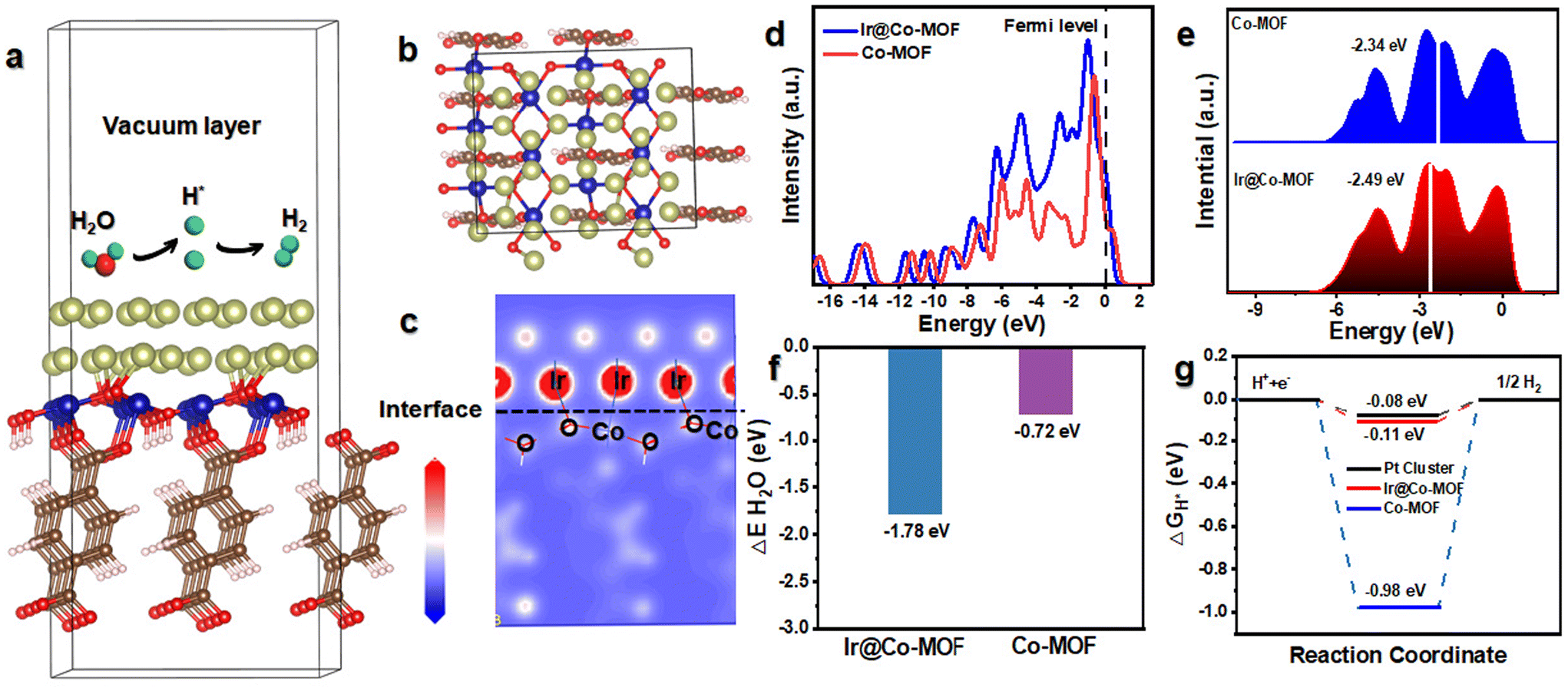 | ||
| Fig. 4 (a and b) HER reaction mechanism on the Ir@Co-MOF model (green, light blue, red, and dark blue represent hydrogen, cobalt, oxygen, and iridium atoms, respectively). (c) Differential charge density of the Ir@Co-MOF model. (d) DOS of Ir@Co-MOF and Co-MOF, where the red line represents the Fermi level. (e) Partial electronic density of states of the Co d orbital in Ir@Co-MOF and Co-MOF. (f) H2O adsorption energy for the three systems. (g) Calculated free-energy diagram of H adsorption. | ||
3. Conclusion
In summary, a simple wet chemical strategy has been successfully employed to synthesize Ir NPs anchored on Co-MOF nanosheet arrays (Ir@Co-MOF), resulting in high-performance electrocatalysts for the HER in alkaline solutions. The Co-MOF nanosheets act as a durable support for the catalyst, effectively preventing the agglomeration of Ir NPs. This stability ensures a vast exposure of active surface sites and facilitates efficient electron transfer across interfaces via the Ir–O–Co bonds. As a result, the finely tuned Ir@Co-MOF electrocatalyst showcases exceptional HER activity. It achieves a notable current density of 10 mA cm−2 at a remarkably low overpotential of just 29 mV in an alkaline setting. Additionally, this catalyst maintains its impressive performance with remarkable long-term stability. This promising electrocatalyst holds significant potential for various applications in hydrogen generation and energy conversion technologies. Theoretical calculations have provided valuable insights into this work, revealing that the charge relocation induced by interfacial bonds can enhance the kinetics of H2O adsorption and improve the kinetics of HER by reducing the barriers for hydrogen adsorption. This research not only improves the catalytic performance of precious metals through electronic structure modulation but also introduces a novel approach to the synthesis of efficient HER catalysts utilizing MOFs as a platform.Author contributions
Kaipeng Cheng: conceptualization, methodology, writing – original draft. Yinghuan Xu: methodology. Shenmeng Zhang: data curation. Heng Zhang: investigation, software, formal analysis. Yingying Xue: formal analysis. Xiahui Huang: validation. Jianbo Zhao: data curation. Pengcheng Zhang: investigation, Shuang Ma: formal analysis, data curation. Zhiqiang Hou: writing – review & editing, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Data availability Data will be made available on request.Acknowledgements
K. P. Cheng thanks the funding support from PhD Research Startup Foundation (ZKNUC2021017), Henan Province Science Foundation for Youth (242300420580), Key Scientific Research Projects of Colleges and Universities in Henan Province (24A530011). Y. Y. Xue and J. B. Zhao thank the funding support from National Natural Science Foundation of China (No. 22108320, 22268038).References
- L. Q. Wang, Y. X. Hao, L. L. M. Deng, F. Hu, S. Zhao, L. L. Li and S. J. Peng, Rapid complete reconfiguration induced actual active species for industrial hydrogen evolution reaction, Nat. Commun., 2022, 13, 5785 CrossRef CAS PubMed.
- M. A. Ahsan, A. R. Puente Santiago, Y. Hong, N. Zhang, M. Cano, R.-C. Enrique, L. Echegoyen, T. S. Sreeprasad and C. N. Juan, Tuning of trifunctional NiCu bimetallic nanoparticles confined in a porous carbon network with surface composition and local structural distortions for the electrocatalytic oxygen reduction, oxygen and hydrogen evolution reactions, J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed.
- S. Zhao, F. Hu, L. J. Yin, L. L. Li and S. J. Peng, Manipulating electron redistribution induced by asymmetric coordination for electrocatalytic water oxidation at a high current density, Sci. Bull., 2023, 68, 1389–1398 CrossRef CAS PubMed.
- S. Ma, P. Y. Yang, J. L. Chen, Z. H. Wu, X. Q. Li and H. Zhang, NiCu alloys anchored Co3O4 nanowire arrays as efficient hydrogen evolution electrocatalysts in alkaline and neutral media, J. Colloid Interface Sci., 2023, 642, 604–611 CrossRef CAS PubMed.
- B. W. Jiang, J. W. Zhu, Z. Z. Xia, J. H. Lyu, X. C. Li, L. R. Zheng, C. Chen, S. Chaemchuen, T. Bu, F. Verpoort, S. C. Mu, J. Wang and Z. K. Kou, Correlating single–atomic ruthenium interdistance with long–range interaction boosts hydrogen evolution reaction kinetics, Adv. Mater., 2024, 36, 2310699 CrossRef CAS PubMed.
- L. Zhang, J. W. Zhu, X. Li, S. Mu, F. Verpoort, J. M. Xue, Z. K. Kou and J. Wang, Nurturing the marriages of single atoms with atomic clusters and nanoparticles for better heterogeneous electrocatalysis, Interdiscip. Mater., 2022, 1, 51–87 CrossRef.
- J. Li, N. T. Wu, J. Zhang, H.-H. Wu, K. M. Pan, Y. X. Wang, G. L. Liu, X. M. Liu, Z. P. Yao and Q. B. Zhang, Machine learning-assisted low-dimensional electrocatalysts design for hydrogen evolution reaction, Nano-Micro Lett., 2023, 15, 227 CrossRef CAS PubMed.
- L. L. Li, D. S. Yu, P. Li, H. J. Huang, D. Y. Xie, C.-C. Lin, F. Hu, H.-Y. Chen and S. J. Peng, Interfacial electronic coupling of ultrathin transition-metal hydroxide nanosheets with layered MXenes as a new prototype for platinum-like hydrogen evolution, Energy Environ. Sci., 2021, 14, 6419–6427 RSC.
- X. L. Li, Y. Q. Huang, Z. Y. Chen, S. Q. Hu, J. L. Zhu, P. Tsiakaras and P. K. Shen, Novel PtNi nanoflowers regulated by a third element (Rh, Ru, Pd) as efficient multifunctional electrocatalysts for ORR, MOR and HER, Chem. Eng. J., 2023, 454, 140131 CrossRef CAS.
- C. T. Zhang, Q. Liu, P. Y. Wang, J. W. Zhu, D. Chen, Y. Yang, Y. F. Zhao, Z. H. Pu and S. C. Mu, Molybdenum carbide–PtCu nanoalloy heterostructures on MOF–derived carbon toward efficient hydrogen evolution, Small, 2021, 17, 2104241 CrossRef CAS PubMed.
- K. Guo, D. P. Fan, J. C. Bao, Y. F. Li and D. D. Xu, Atomic–level phosphorus–doped ultrathin Pt nanodendrites as efficient electrocatalysts, Adv. Funct. Mater., 2022, 32, 2208057 CrossRef CAS.
- Y. Hao, S. F. Hung, W.-J. Zeng, Y. Wang, C. C. Zhang, C.-H. Kuo, L. Q. Wang, S. Zhao, H.-Y. Chen and S. J. Peng, Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics, J. Am. Chem. Soc., 2023, 145, 23659–23669 CrossRef CAS PubMed.
- R. Z. Jiang, D. T. Tran, J. T. Li and D. Chu, Ru@ RuO2 core–shell nanorods: a highly active and stable bifunctional catalyst for oxygen evolution and hydrogen evolution reactions, Energy Environ. Mater., 2019, 2, 201–208 CrossRef CAS.
- H. Z. Yu, S. Q. Zhu, Y. X. Hao, Y.-M. Chang, L. L. Li, J. Ma, H.-Y. Chen, M. H. Shao and S. J. Peng, Modulating Local Interfacial Bonding Environment of Heterostructures for Energy-Saving Hydrogen Production at High Current Densities, Adv. Funct. Mater., 2023, 33, 2212811 CrossRef CAS.
- P. P. Su, W. Pei, X. W. Wang, Y. F. Ma, Q. K. Jiang, J. Liang, S. Zhou, J. J. Zhao, J. Liu and G. Q. Lu, Exceptional electrochemical HER performance with enhanced electron transfer between Ru nanoparticles and single atoms dispersed on a carbon substrate, Angew. Chem., 2021, 133, 16180–16186 CrossRef.
- A. Radwan, H. H. Jin, D. P. He and S. C. Mu, Design engineering, synthesis protocols, and energy applications of MOF-derived electrocatalysts, Nano-Micro Lett., 2021, 13, 1–32 CrossRef PubMed.
- C.-F. Li, T.-Y. Shuai, L.-R. Zheng, H.-B. Tang, J.-W. Zhao and G.-R. Li, The key role of carboxylate ligands in Ru@Ni-MOFs/NF in promoting water dissociation kinetics for effective hydrogen evolution in alkaline media, Chem. Eng. J., 2023, 451, 138618 CrossRef CAS.
- R.-F. Li, H. Zhang, M.-Z. Hong, J.-G. Shi, X.-F. Liu and X. Feng, Two Co (ii)/Ni (ii) complexes based on nitrogenous heterocyclic ligands as high-performance electrocatalysts for the hydrogen evolution reaction, Dalton Trans., 2022, 51, 3970–3976 RSC.
- J. Yang, Y. Shen, Y. M. Sun, J. H. Xian, Y. J. Long and G. Q. Li, Ir nanoparticles anchored on metal–organic frameworks for efficient overall water splitting under pH–universal conditions, Angew. Chem., 2023, 135, e202302220 CrossRef.
- F. L. Yang, Y. T. Chen, G. Z. Cheng, S. L. Chen and W. Luo, Ultrathin nitrogen-doped carbon coated with CoP for efficient hydrogen evolution, ACS Catal., 2017, 7, 3824–3831 CrossRef CAS.
- J. Li, H. L. Huang, Y. Li, Y. Z. Tang, D. H. Mei and C. L. Zhong, Stable and size-controllable ultrafine Pt nanoparticles derived from a MOF-based single metal ion trap for efficient electrocatalytic hydrogen evolution, J. Mater. Chem. A, 2019, 7, 20239–20246 RSC.
- Y. J. Long, Y. Shen, P. P. Jiang, H. Su, J. H. Xian, Y. M. Sun, J. Yang, H. L. Song, Q. H. Liu and G. Q. Li, Ultrafine Ru nanoparticles stabilized by V8C7/C for enhanced hydrogen evolution reaction at all pH, Sci. Bull., 2024, 69, 763–771 CrossRef CAS PubMed.
- Y. M. Sun, Z. Q. Xue, Q. L. Liu, Y. L. Jia, Y. L. Li, K. Liu, Y. Y. Lin, M. Liu, G. Q. Li and C.-Y. Su, Modulating electronic structure of metal-organic frameworks by introducing atomically dispersed Ru for efficient hydrogen evolution, Nat. Commun., 2021, 12, 1369 CrossRef CAS PubMed.
- F. Q. Zheng, C. M. Zhang, X. H. Gao, C. Du, Z. H. Zhuang and W. Chen, Immobilizing Pd nanoclusters into electronically conductive metal-organic frameworks as bi-functional electrocatalysts for hydrogen evolution and oxygen reduction reactions, Electrochim. Acta, 2019, 306, 627–634 CrossRef CAS.
- R. F. Li, X. Y. Chen, H. Zhang, Y. R. Wang, Y. Lv, H. P. Jiang, B. W. Guo and X. Feng, Ultrafine iridium nanoparticles anchored on Co-based metal–organic framework nanosheets for robust hydrogen evolution in alkaline media, Inorg. Chem., 2024, 63, 2282–2288 CrossRef CAS PubMed.
- L. N. Wang, R. F. Du, X. Liang, Y. C. Zou, X. Zhao, H. Chen and X. X. Zou, Optimizing Edge Active Sites via Intrinsic In-Plane Iridium Deficiency in Layered Iridium Oxides for Oxygen Evolution Electrocatalysis, Adv. Mater., 2024, 36, 2312608 CrossRef CAS PubMed.
- P. F. Liu, X. Zhang, J. W. Fei, Y. Shi, J. W. Zhu, D. Zhang, L. Zhao, L. Wang and J. P. Lai, Frank Partial Dislocations in Coplanar Ir/C UltrathinNanosheets Boost Hydrogen Evolution Reaction, Adv. Mater., 2024, 36, 2310591 CrossRef CAS PubMed.
- J. Kwon, S. Sun, S. Choi, K. Lee, S. Jo, K. Park, Y. K. Kim, H. B. Park, H. Y. Park, J. H. Jang, H. Han, U. Paik and T. Song, Tailored electronic structure of Ir in high entropy alloy for highly active and durable Bi–functional electrocatalyst for water splitting under acidic environment, Adv. Mater., 2023, 35, 2300091 CrossRef CAS PubMed.
- W. H. Lee, Y.-J. Ko, J.-H. Kim, C. H. Choi, K. H. Chae, H. Kim, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, High crystallinity design of Ir-based catalysts drives catalytic reversibility for water electrolysis and fuel cells, Nat. Commun., 2021, 12, 4271 CrossRef CAS PubMed.
- Y. H. Xie, X. X. Yu, X. W. Li, X. Long, C. F. Chang and Z. H. Yang, Stable and high-performance Ir electrocatalyst with boosted utilization efficiency in acidic overall water splitting, Chem. Eng. J., 2021, 424, 130337 CrossRef CAS.
- Y. X. Zhao, Q. Y. Sun, X. Y. Zhou, Z. Y. Duan, C. F. Zhang, G.-R. Xu, D. X. Ju and L. Wang, Scalable Synthesis of Ir Cluster Anchored on Porous Hollow Carbon Nanobowls for Enhancing pH-Universal Hydrogen Evolution, Small, 2023, 19, 2305343 CrossRef CAS PubMed.
- B. B. Liu, X. Y. Wang, Y. Y. Zhai, Z. L. Zhang, H. Q. Liu, L. Li and H. R. Wen, Facile preparation of well conductive 2D MOF for nonenzymatic detection of hydrogen peroxide: Relationship between electrocatalysis and metal center, J. Electroanal. Chem., 2020, 858, 113804 CrossRef CAS.
- H. F. Zhang, P. D. Shi, X. Ma, C. Q. Ma, S. M. Han, C. H. He, H. M. Wu, L. J. Zhu, B. W. Zhang, Y. Z. Lu, W. B. Cao, H. Q. Yin, X. M. Meng, J. Xia, J. W. Zhang, A.-L. Wang and Q. P. Lu, Construction of ordered atomic donor–acceptor architectures in bcc IrGa intermetallic compounds toward highly electroactive and stable overall water splitting, Adv. Energy Mater., 2023, 13, 2202703 CrossRef CAS.
- C. F. Yang, R. Zhao, H. Xiang, J. Wu, W. D. Zhong, W. L. Li, Q. Zhang, N. J. Yang and X. K. Li, Ni–activated transition metal carbides for efficient hydrogen evolution in acidic and alkaline solutions, Adv. Energy Mater., 2020, 10, 2002260 CrossRef CAS.
- H. J. Huang, D. S. Yu, F. Hu, S.-C. Huang, J. Song, H.-Y. Chen, L. L. Li and S. J. Peng, Clusters induced electron redistribution to tune oxygen reduction activity of transition metal single-atom for metal–air batteries, Angew. Chem., Int. Ed., 2022, 61, 202116068 CrossRef PubMed.
- S. L. Zhao, Y. Wang, J. C. Dong, C.-T. He, H. J. Yin, P. F. An, K. Zhao, X. F. Zhang, C. Gao, L. J. Zhang, J. W. Lv, J. X. Wang, J. Q. Zhang, A. M. Khattak, N. A. Khan, Z. X. Wei, J. Zhang, S. Q. Liu, H. J. Zhao and Z. Y. Tang, Ultrathin metal–organic framework nanosheets for electrocatalytic oxygen evolution, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- G. K. Li, H. Jang, S. G. Liu, Z. J. Li, M. G. Kim, Q. Qin, X. Liu and J. Cho, The synergistic effect of Hf-O-Ru bonds and oxygen vacancies in Ru/HfO2 for enhanced hydrogen evolution, Nat. Commun., 2022, 13, 1270 CrossRef CAS PubMed.
- L. Q. Wang, L. Song, Z. Y. Yang, Y.-M. Chang, F. Hu, L. Li, L. L. Li, H.-Y. Chen and S. J. Peng, Electronic modulation of metal–organic frameworks by interfacial bridging for efficient pH-universal hydrogen evolution, Adv. Funct. Mater., 2022, 33, 2210322 CrossRef.
- F. He, Y. J. Zhao, X. X. Yang, S. X. Zheng, B. Yang, Z. J. Li, Y. B. Kuang, Q. H. Zhang, L. C. Lei, M. Qiu, L. M. Dai and Y. Hou, Metal–organic frameworks with assembled bifunctional microreactor for charge modulation and strain generation toward enhanced oxygen electrocatalysis, ACS Nano, 2022, 16, 9523 CrossRef CAS PubMed.
- G. L. Liu, T. Zhang, X. J. Li, J. Li, N. T. Wu, A. Cao, W. W. Yuan, K. M. Pan, D. L. Guo and X. M. Liu, MoS2@C with S vacancies vertically anchored on V2C-MXene for efficient lithium and sodium storage, Inorg. Chem. Front., 2023, 10, 1587–1602 RSC.
- K. A. K. Raju and A. Biswas, Corrosion behavior of self-organized TiO2 nanotubular arrays grown on Ti6Al4V for biomedical applications, Mater. Chem. Phys., 2023, 305, 128011 CrossRef.
- W. Q. Li, H. Zhang, K. Zhang, Z. Z. Cheng, H. P. Chen, G. Tan, X. Feng, L. Y. Wang and S. C. Mu, Altered electronic structure of trimetallic FeNiCo-MOF nanosheets for efficient oxygen evolution, Chem. Commun., 2023, 59(32), 4750–4753 RSC.
- R. F. Li, H. Zhang, J. L. Chen, K. Zhang, W. Q. Li, X. Feng, H. J. Zhang, I. D. Abdoulkader, X. Zhang and T. X. Zhang, Interfacial engineering of FeWO4/Fe2O3 homometallic heterojunctions for synergistic electrocatalytic water splitting, Inorg. Chem. Front., 2023, 10, 3675–3685 RSC.
- W. Q. Li, B. W. Guo, K. Zhang, H. Zhang, K. Q. Bu, H. P. Chen and X. Feng, A co-axial structure composed of RuO2 on defective N-doped carbon nanotubes as a highly efficient electrocatalyst for overall water splitting, Inorg. Chem. Front., 2024, 11, 745–755 RSC.
- T. T. Wang, P. Y. Wang, W. J. Zang, X. Li, D. Chen, Z. K. Kou, S. C. Mu and J. Wang, Nanoframes of Co3O4−Mo2N heterointerfaces enable high–performance bifunctionality toward both electrocatalytic HER and OER, Adv. Funct. Mater., 2022, 32, 2107382 CrossRef CAS.
- J. W. Zhu, F. J. Xiao, Y. Guo, R. H. Lu, L. Gong, D. Chen, P. Y. Wang, L. Chen, J. Yu, J. S. Wu and S. C. Mu, Electron accumulation effect over osmium/erlichmanite heterointerfaces for intensified pH-universal hydrogen evolution, ACS Catal., 2022, 12, 13312–13320 CrossRef CAS.
- T. T. Wang, J. C. Huang, W. Sang, C. Zhou, B. H. Zhang, W. Zhu, K. Du, Z. K. Kou and S. X. Wang, Correlative Mn-Co catalyst excels Pt in oxygen reduction reaction of quasi-solid-state zinc-air batteries, Nano Res., 2023, 1–7 Search PubMed.
- Y. P. Zhu, K. Fan, C. S. Hsu, G. Chen, C. S. Chen, T. C. Liu, Z. Z. Lin, S. X. She, L. Q. Li, H. M. Zhou, Y. Zhu, H. M. Chen and H. T. Huang, Supported ruthenium single–atom and clustered catalysts outperform benchmark Pt for alkaline hydrogen evolution, Adv. Mater., 2023, 35, 2301133 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi00515e |
| This journal is © the Partner Organisations 2024 |