
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing the use of a recyclable solid electrolyte for waste minimization in electrosynthesis: preparation of 2-aryl-benzoxazoles under flow conditions†
Francesco
Ferlin
 *,
Federica
Valentini
,
Filippo
Campana
and
Luigi
Vaccaro
*
*,
Federica
Valentini
,
Filippo
Campana
and
Luigi
Vaccaro
*
Laboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123, Perugia, Italy. E-mail: francesco.ferlin@unipg.it; luigi.vaccaro@unipg.it; Web: http://greensoc.chm.unipg.it
First published on 26th April 2024
Abstract
The necessary use of large amounts of a homogeneous electrolyte represents a major issue and challenge for the whole sustainability of electrosynthetic procedures. Herein, we report the use of a solid ammonium salt (e.g. Amberlyst-400-Cl, Amb-400-Cl) as a reusable electrolyte with excellent performance in the representative electrosynthesis of 2-arylbenzoxazoles. Amb-400-Cl works efficiently without adding any additional supporting electrolytes or mediators, and it can be reused without the need for a regeneration procedure. Exploiting this finding, a sustainable electro-promoted protocol has been developed under batch and flow conditions, which proves that the reported chemical and technological innovation leads to significant improvements compared to the literature processes. Extensive green metrics analysis has also been reported to fully quantify the advances in terms of sustainability.
Introduction
The use of electrochemistry in organic synthesis represents a new technological frontier that is proving useful for the development of efficient synthetic methodologies.1 The use of a current of electrons as a substitute for more classic, often toxic, and expensive, “redox” reagents certainly represents a more convenient approach in terms of chemical efficiency2 and overall sustainability.3As proof of this, the “electrification” of organic synthesis has been implemented by chemical industries in order to achieve clean chemical processes.4 The tools that allow the investigation and use of electrochemistry have improved greatly, allowing the definition of procedures for many reactions of general importance, including C–C,5 C–N,6 and C–O7 couplings, protection/deprotection8 strategies, oxidation,9 reduction10 and the synthesis of complex molecular scaffolds.11,12 Notably, very recent efforts have also been devoted to exploiting electrochemistry in the valorization of biomass.13 Despite all these achievements, there are still intriguing challenges in the field, especially if the aim is to define processes featuring the highest sustainability level.
Electrochemical synthesis requires electrolyte additives that dissolve in the reaction medium allowing the transmission of electrons in solution.14 These electrolytes are generally non-recoverable species and used in large amounts. Even when used in catalytic amounts, they and their byproducts formed during the reaction must be separated from the final reaction mixture. Some efforts to solve this issue have been directed towards the use of ionic liquids or their carbon dispersion.15 In both cases, low conductivity could be achieved, limiting one key factor in influencing the efficiency of the process. Therefore, the consequent need for a homogeneous co-mediator is inevitable and the poor solvent compatibility has been highlighted (Figure 1).16
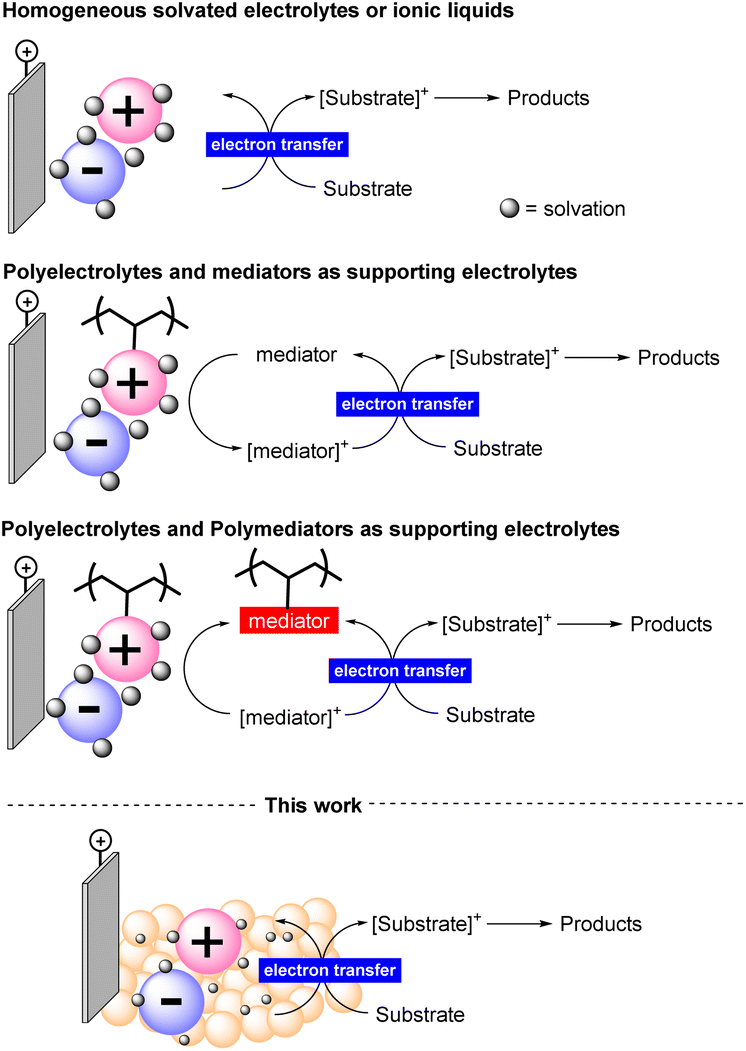 | ||
| Fig. 1 Supporting electrolyte solutions and features of this work. | ||
The amount of starting materials that can be converted in electrosynthesis is related to electron transfer and current density and therefore is directly linked to the quantities of electrolytes used.17,18 Therefore, supporting electrolytes are generally used in equimolar quantities, leading to an inevitable large (at least equimolar) mass of waste. Furthermore, the most commonly used supporting electrolytes are perchlorates or tetraalkylammonium halide salts,19 which require washing with water for their removal. Halogenated aqueous waste (code 070103 of the European Waste Catalogue)20 must eventually be treated by incineration which involves a significant emission of NOx into the environment.21
Recoverable electrolytes are needed to find a solution to this critical sustainability challenge for electrosynthesis. Therefore, we turned our attention to the use of solid polymeric electrolytes under batch conditions with the ultimate goal of defining a tailor-made continuous-flow reactor system that can facilitate their reuse. With this strategy, we intend to propose a new approach to minimize the waste associated with electrosynthesis.
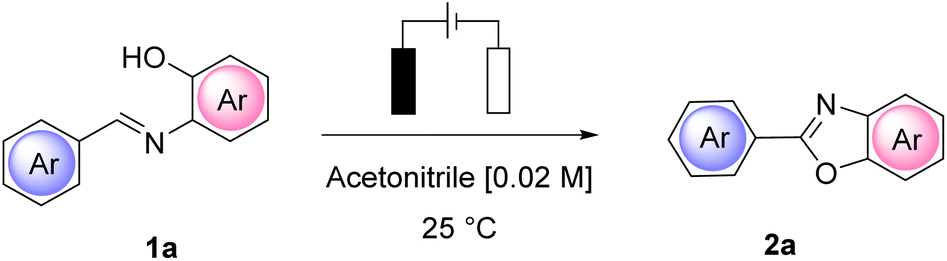
To the best of our knowledge, we describe herein the first utilization of a solid and recyclable electrolyte in an electro-assisted synthetic protocol. We have representatively applied our study to the synthesis of 2-arylbenzoxazoles and to the preparation of a well-known active pharmaceutical ingredient (API), tafamidis.
We have focused on the use of cheap and easily accessible polymeric ammonium salts (Amberlyst IRA-400-Cl, Amb-400-Cl) as solid electrolytes for the indirect synthesis of 2-arylbenzoxazoles using electricity as an oxidative cyclization promoter and acetonitrile as a reaction medium.
The indirect synthesis of 2-arylbenzoxazoles from imines is a well-known and synthetically useful 2e− oxidative process that has been realized with the use of many different conditions.22 We considered it of general interest to explore the contribution of the solid electrolyte to the overall conductivity. In agreement with our research,23 we have here also designed and built a customized electrochemical flow reactor to better highlight the utility of using a reusable solid electrolyte to enhance the productivity and scalability of the process. Importantly, the flow electrochemical cell allowed us to also improve the usage of the solid electrolyte in terms of current efficiency and electricity consumption.
Results and discussion
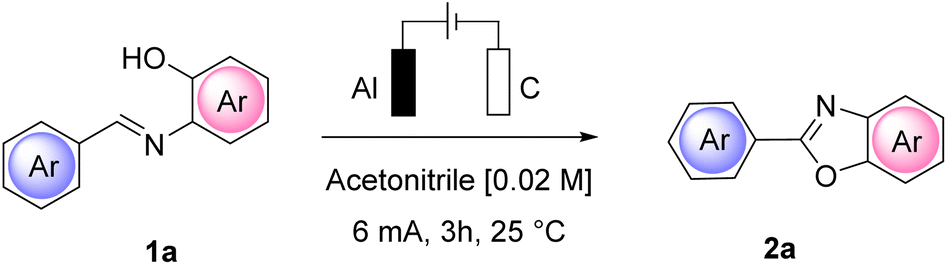
We began our investigation by performing a batch experiment set to preliminarily evaluate the best anode/cathode couples for the conversion of representative arylimine 1a to 2-arylbenzoxazoles 2a. In an undivided cell, at a constant current mode, (Table 1) we screened different materials and current intensities, keeping constant the use of chloride24 as a benchmark electrolyte. The addition of water was expected to be beneficial for achieving a stable constant current during the experiments, due to the increased solubility of ammonium chloride in the reaction medium.|
|
|||
|---|---|---|---|
| Entry | Anode/cathode | CCE | 2a (%)b |
| a Reaction conditions: undivided cell, 1a (0.1 mmol), 5 mL of acetonitrile, NH4Cl (0.5 mmol), 100 μL of water, at a constant current, 3 h at 25 °C. b Isolated yield. c Reaction run without current at 24 h. d Reaction run without NH4Cl was not conductive and led to partial decomposition of 1a. | |||
| 1 | Al/Al | 8 mA | 54 |
| 2 | C/Al | 8 mA | 89 |
| 3 | Al/C | 8 mA | 14 |
| 4 | C/C | 8 mA | 68 |
| 5 | C/SS | 8 mA | 37 |
| 6 | GC/Pt | 10 mA | 90 |
| 7 | Pt/Pt | 8 mA | 86 |
| 8 | C/Pt | 8 mA | 88 |
| 9 | C/Cu | 8 mA | 64 |
| 10 | C/Al | 6 mA | 90 |
| 11 | C/Al | 4 mA | 86 |
| 12c | C/Al | — | 6 |
| 13d | C/Al | 2 mA | — |
At a low current intensity of 6 mA using easily accessible and convenient electrodes, graphite as the anode and aluminum as the cathode, the reaction proceeded smoothly, affording 90% yield of 2a in 3 h with 6.7 F mol−1 of charge passed (Table 1, entry 10) and a faradaic efficiency of 30%. Importantly, the reaction performed in the absence of current led to a completely unsuccessful reaction even after 24 h (Table 1, entry 13).
With the preliminary conditions set, we evaluated the use of a solid electrolyte, namely Amb-400-Cl. As commonly performed for homogeneous electrolytes, we correlated the masses of different commercially available solid electrolytes with both the equivalents of 1a and the volume of reaction medium used (see the ESI† for the complete details of the procedure). Interestingly, we found that a direct correlation between the current intensity and the resulting cell voltage can be established considering the mass ratio of Amberlyst/volume of medium (gAmb/Vmed) rather than the mass ratio of Amberlyst/mmol of the substrate (gAmb/mmol 1a) (Scheme 1).
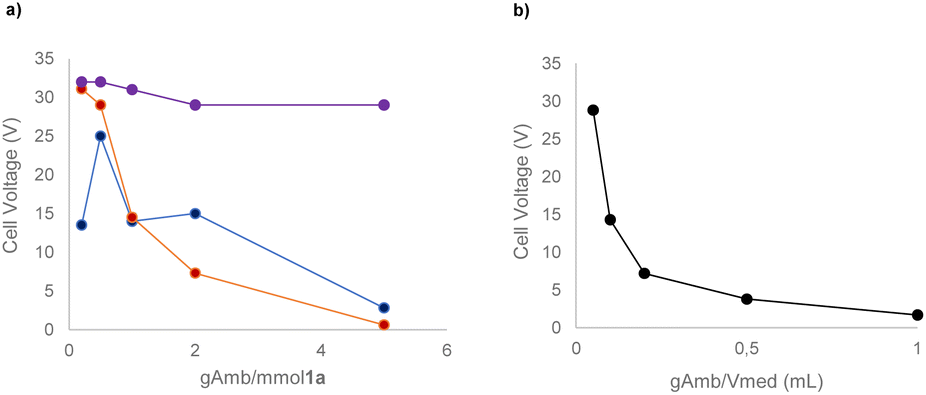 | ||
| Scheme 1 Mass/volume and mass/mmol plot to identify the suitability of the solid electrolyte at 6 mA constant current using a C anode and an Al cathode. (a) Grams of Amb-400-Cl/mmol of the 1a ratio over the cell voltage potential plot; blue line: in 2 mL of acetonitrile; red line: in 5 mL of acetonitrile; purple line: in 10 mL of acetonitrile. (b) Grams of Amb-400-Cl/mL of the acetonitrile ratio over the cell voltage potential plot. | ||
With these experiments, it was also possible to determine that 0.2 g of Amb-400-Cl per mL of acetonitrile allowed good electrical conduction of the cell with a reasonable voltage (7.2 V) of the power supply instrument. By determining and evaluating the resistance of the solution (see the ESI†) it is possible to see that, in all the cases tested, an exponential decay of the solution resistance occurred by increasing the gAmb/Vmed ratio with a consequent increase in the ionic concentration of the reaction medium.
Intriguingly, with this unique behavior, the possibility emerges of decoupling the mass of starting materials from the mass of electrolytes required, allowing for a deviation from the conventional intimate correlation between these parameters in steering the process. In addition, this information is also very useful in view of transferring this protocol under flow conditions. In fact, the increase of the surface/volume ratio of the flow reactor could represent an additional advantage in terms of the conductivity and overall stability of the system.
We screened different types of polymeric ammonium salts of the Amberlyst series. As solid electrolytes, we also evaluated their recyclability under optimized conditions for the electro-assisted synthesis of 2a (Table 2).
|
|
|||
|---|---|---|---|
| Entry | Solid electrolyte | Recyclabilityb | 2a (%)c |
| a Reaction conditions: 1a (0.1 mmol), 5 mL of acetonitrile, 1 g of solid electrolyte, 100 μL of water, 6 mA CCE using a C anode and an Al cathode, 3 h at 25 °C. b Recyclability was tested over two consecutive reactions run under the same reaction conditions. c Conversion determined by GC analysis. The remaining material is unreacted 1a; isolated yield is given in parentheses. | |||
| 1 | Amberlyst 400 (Cl) | ✓ | >99 (92) |
| 2 | Amberlyst A26 (OH) | × | 70 |
| 3 | Amberlyst 900 (Cl) | ✓ | >99 (87) |
| 4 | Amberlyst 958 (Cl) | ✓ | >99 (90) |
| 5 | NH4Cl | × | >99 (90) |
Amberlyst-400 (chloride form) (Amb-400-Cl) easily enables a 6 mA constant current, leading to an excellent isolated yield of 2-arylbenzoxazole 2a. It was also preliminarily found that Amb-400-Cl can be reused. In contrast, Amberlyst A26 (OH− form) was not efficient for the reaction itself and in terms of recyclability, due to the reasonably lower stability of the OH− form compared to the Cl− form. Other solid chloride forms (Amberlyst 900 and 958) were also reusable, but they did not lead to sufficiently satisfactory results compared to Amb-400-Cl. It is worth highlighting that the different Amberlyst-Cl materials reported in Table 2 share the same quaternary ammonium cation and the same chloride anions while they differ in the polymeric form. Amberlyst-400 is a gel-type resin while Amberlyst-900 and -958 are macroreticular polymers. The different pore sizes can cause the slight differences observed due to possible fouling of the polymer making more or less efficient the recovery and stability of the organic materials.
We continued our study to fully confirm the compatibility of the reaction conditions (electrode materials and current intensity) and the use of the solid electrolyte selected (see the ESI† for further details).
We prepared and compared different Amberlyst 400 polymers by exchanging the anion to introduce different halides and different poor nucleophilic anions such as BF4−, PF6−, and ClO4−. We tested these materials in terms of efficiency and reusability (Table 3).
|
|
||||
|---|---|---|---|---|
| Entry | Solid electrolyte | Recyclabilityb | 2a (%) | 2a yield % in 2nd run |
| a Reaction conditions: 1a (0.1 mmol), 5 mL of acetonitrile, 1 g of solid electrolyte, 100 μL of water, 6 mA CCE using a C anode and an Al cathode, 3 h at 25 °C. b Recyclability was tested over two consecutive reactions run under the same reaction conditions. c Conversion determined by GC analysis. The remaining material is unreacted 1a; isolated yield is given in parentheses. d Reaction run at 4 mA CCE and 6 h. e No stable constant current operative. | ||||
| 1 | Amberlyst 400-Cl | ✓ | >99 (92) | 92 |
| 2 | Amberlyst 400-Br | × | 80 (72) | 7 |
| 3 | Amberlyst 400-I | × | 68 (55) | 12 |
| 4 | Amberlyst 400-F | — | —e | — |
| 5d | Amberlyst 400-Cl | ✓ | >99 (92) | 92 |
| 6d | Amberlyst 400-Br | × | 64 (55) | 10 |
| 7d | Amberlyst 400-I | × | 43 (35) | 14 |
| 8d | Amberlyst 400-F | ✓ | 44 (32) | 28 |
| 9 | Amberlyst 400-PF6 | × | 28 (19) | —e |
| 10 | Amberlyst 400-BF4 | × | 20 (13) | —e |
| 11 | Amberlyst 400-ClO4 | ✓ | 41 (29) | 21 |
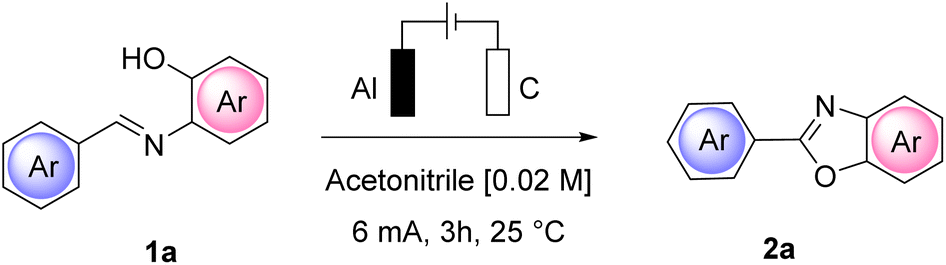
We observed that Amberlyst-Br allowed a 72% isolated yield of 2a while the remaining material (28%) consisted of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of benzaldehyde and starting 1a. Amberlyst-I gave a 55% yield of 2a, while Amberlyst-F did not allow a constant current to be reached and no product 2a was formed. Notably, Amberlyst-Br and -I were also not reusable in a second run. Performing the same experiments at a current intensity of 4 mA but for a prolonged time (6 h, 9 F mol−1 of current), similar results could be achieved when using Amb-400-Cl, -Br, and -I. Amb-400-F at 4 mA led to a 32% yield of product 2a and also a 28% yield of 2a in a second reaction run.
:1 mixture of benzaldehyde and starting 1a. Amberlyst-I gave a 55% yield of 2a, while Amberlyst-F did not allow a constant current to be reached and no product 2a was formed. Notably, Amberlyst-Br and -I were also not reusable in a second run. Performing the same experiments at a current intensity of 4 mA but for a prolonged time (6 h, 9 F mol−1 of current), similar results could be achieved when using Amb-400-Cl, -Br, and -I. Amb-400-F at 4 mA led to a 32% yield of product 2a and also a 28% yield of 2a in a second reaction run.
When Amberlyst-PF6 and -BF4 were used, after only 20 minutes, the reaction mixture became black. Product 2a was formed in a low yield with partial decomposition (45% and 52% for Amberlyst-PF6 and -BF4, respectively) to benzaldehyde. This behaviour can be ascribed to a degradation process that occurs in Amberlyst-PF6 and -BF4 under our reaction conditions.24 In addition, Amberlyst-PF6 and -BF4 could not be reused at all for a second run, leading to an unstable current and no product formation. Amberlyst-ClO4 was found to be reusable for at least a second reaction run but product 2a was obtained in poor yield.
To have a better insight into the behaviour of the system upon changing the anion of the Amberlyst polymers and to acquire information regarding the mechanism, we performed a cyclic voltammetry study. We used NH4Cl as a benchmark and then we tested different Amberlyst 400 polymers with and without 1a (Scheme 2).
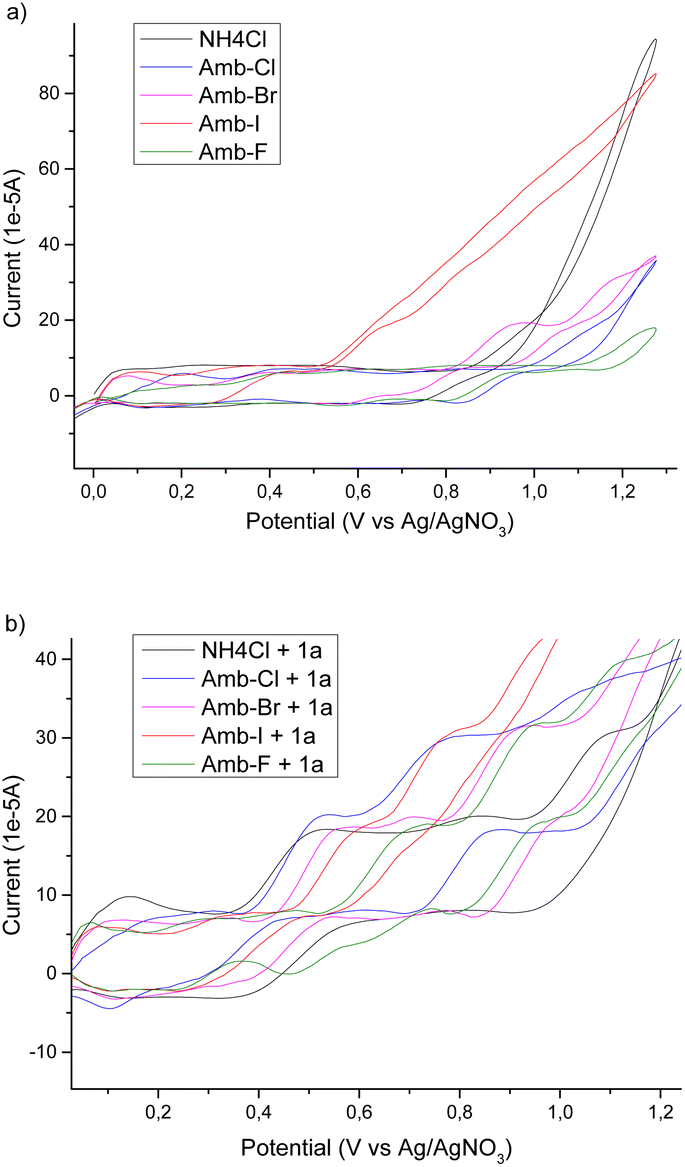 | ||
| Scheme 2 Cyclic voltammetry experiments: (a) solid electrolytes without 1a; (b) solid electrolytes with 1a. CV data are reported in IUPAC conventions; a glassy carbon working electrode and a platinum plated counter electrode were used with an Ag/AgNO3 reference electrode filled with a 0.1 M solution of AgNO3 in acetonitrile. Measurements were made at 25 °C at an initial potential of 0 V in the oxidative direction with a sweep of 100 mV s−1. | ||
From the cyclovoltammetry data illustrated in Scheme 1a, it is possible to obtain information regarding the potential range in which the tested Amberlyst resins can be used as electrolytes without interfering with the anodic oxidation of substrate 1a. Amberlyst-Cl exhibits an oxidation peak associated with the chloride anion at potentials exceeding 1 V, similar to the benchmark NH4Cl, and the same applies to Amberlyst-400-F. Amberlyst-Br presents an oxidation peak at lower values than Amberlyst-Cl at a value of 0.95 V, while Amberlyst-I already begins to oxidize at potential values of around 0.5 V. After recording the CV curve of 1a in NH4Cl solution as a reference (two oxidation peaks were observed at 0.53 V and 0.84 V at values of 18.4 μA and 20.0 μA, respectively), we then added 1a to each of the Amberlyst-400-X solutions tested. The general trend observed is that moving from the -Cl anion to -Br, -I, and -F anions, the first oxidation peak shifted progressively at higher potentials (0.54 V, 0.58 V, 0.61 V, and 0.73 V) with a decrease of current intensity. The second oxidation peak potential shifted nonlinearly with an increase in the current. Considering this, Amberlyst-Cl, -Br, and -F can be used under optimized experimental conditions without major interferences due to parasitic reactions triggered by the oxidation of anion halides. Amberlyst-I, on the other hand, given its low oxidation voltage, can interfere with the progress of the reaction (Scheme 3).
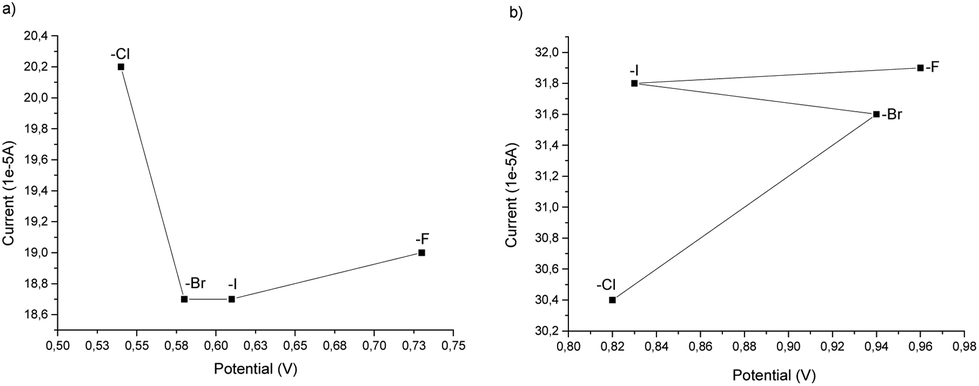 | ||
| Scheme 3 Anodic oxidation peak shift: (a) first oxidation peak; (b) second oxidation peak. | ||
Based on the cyclic voltammetry experiments, we could confirm that the use of a solid electrolyte did not change the general mechanism25 (Scheme 4) of the reaction itself while it only affected the potentials and current intensities at which anodic oxidation can occur.
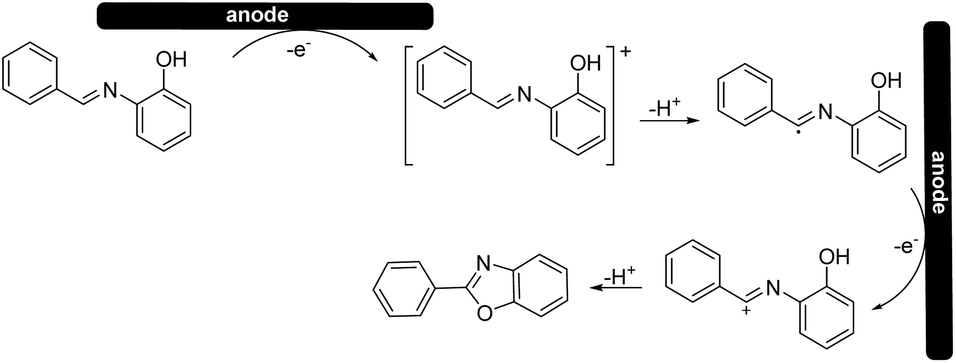 | ||
| Scheme 4 Plausible mechanism. | ||
During our experimentation, we noted an increase in the conductivity when the electrodes were in contact with the polymer surface. Therefore, we hypothesized that the solid Amberlyst-400 (Cl) presumably works with an ion-hopping, transporting-type mechanism, where when current is applied, the movement of ions on the polymer surfaces allows the entire solution to be conductive.26 The nature of the ionic interaction of the surface and the maintenance of conductivity for cycles of successive reactions are still being studied in our laboratories.
With the optimized conditions and system at our disposal, we tested the reusability of the Amb-400-Cl solid electrolyte by performing several consecutive reactions. Gratifyingly, we found that the same Amb-400-Cl can be reused up to 10 times before observing a rise in the cell voltage at a constant current intensity of 6 mA (Scheme 5) with the chemical efficiency remaining unchanged after 15 runs; under these conditions, we obtained a constant yield of 92% for product 2a with a stable faradaic efficiency of 30% over consecutive runs.
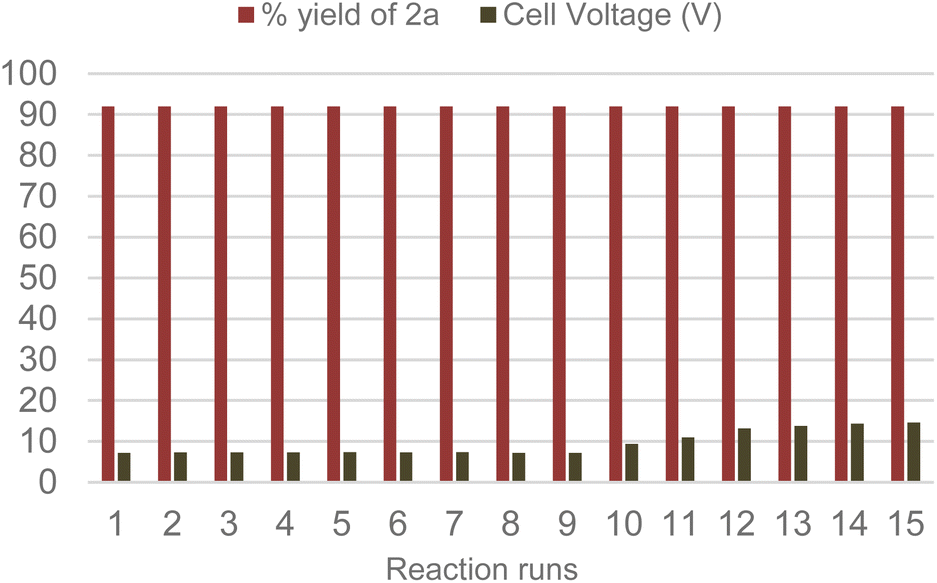 | ||
| Scheme 5 Reusability of Amberlyst 400-Cl as an electrolyte. | ||
It is worth noticing that the inherent clean profile of the output reaction mixture after electrolysis makes almost unnecessary any further purification. The final acetonitrile solution after filtration of the solid electrolyte can been distilled and recovered (in 80% w/w), furnishing almost pure 2a in 92% yield.
MP-AES analysis of crude 2a was also performed to check whether aluminum particles could contaminate the product. A value of 11 ppm was found which is lower than the PDI of 25 ppm established by the FDA,27 confirming that with no further purification, the purity achieved is very good. In order to test the utility of the conditions we defined, we extended our protocol to different substrates using a variety of substituted aryl imines (Scheme 6).
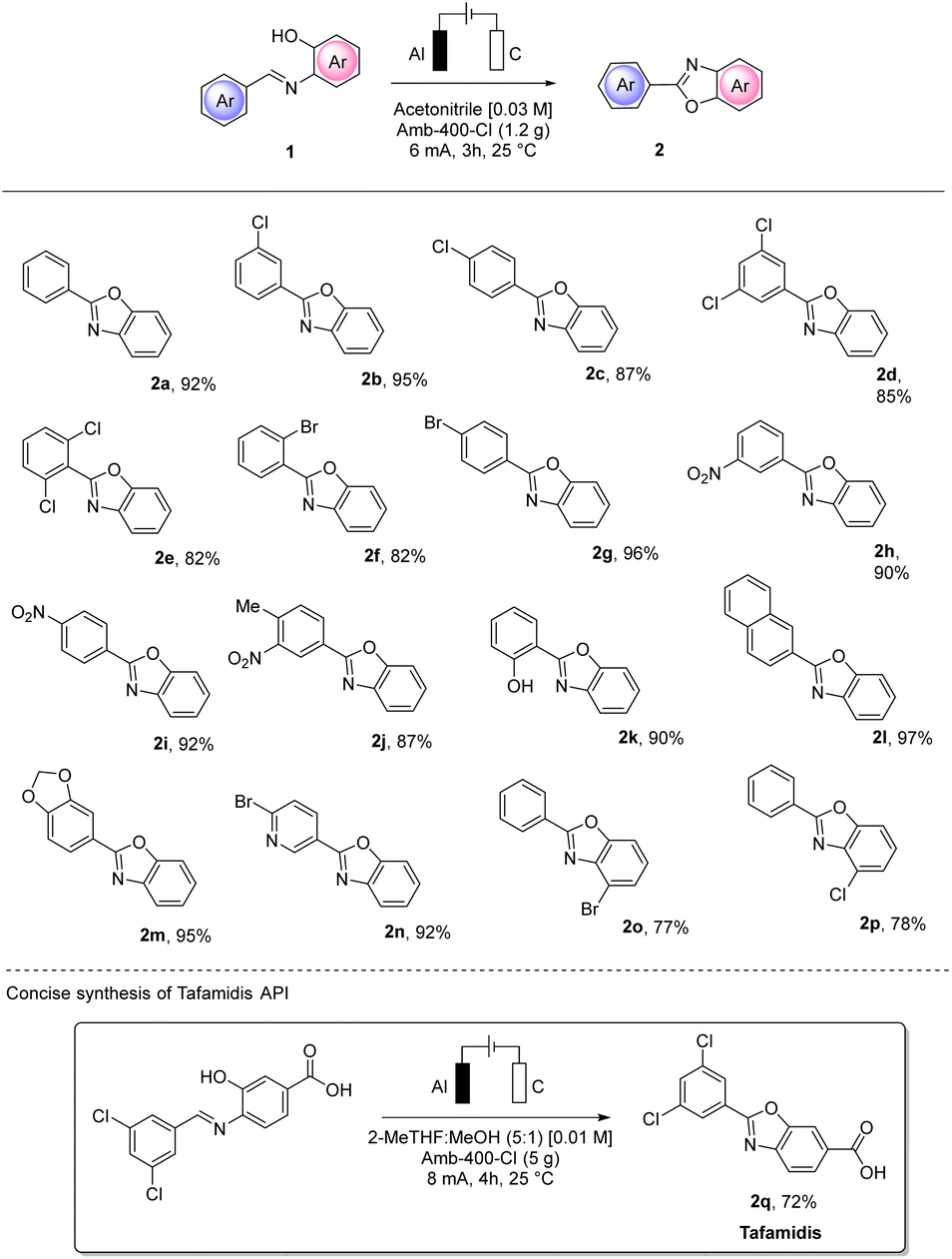 | ||
| Scheme 6 Scope for using the solid electrolyte in the synthesis of 2-arylbenzoxazoles 2a–s. | ||
The electro-assisted oxidative cyclization of aryl imines 1a–q occurred smoothly for almost all the substrates tested. Notably, halogen substitutions were well tolerated in different positions (2b–g and 2n–p), opening up an interesting orthogonal functionalization. Notably, with such substrates, no cathodic dehalogenation byproduct was observed.
The efficiency and the yield of the process were not influenced by electron-donating or electron-withdrawing substituents and good tolerance to multiple substitution was also observed (2d, 2e and 2j). Naphthyl (2l) and piperonyl (2m) moieties resulted in excellent yields (97% and 95%, respectively) of the corresponding benzoxazoles. Interestingly, the salicyl-derived compound (1k) bearing a hydroxy substitution and the 4-bromo-nicotin aldehyde derivative (1n) also gave excellent isolated yields of the corresponding 2k and 2n (90% and 92%, respectively). To prove the synthetic utility of our procedure, we also successfully performed the synthesis of the first-in-class medication tafamidis API 2q in 72% yield.
As anticipated, to improve the reusability of the solid electrolyte, we also designed and realized a new electrochemical flow reactor composed of an aluminum plate interfaced by a customized 2 mm thick PTFE plate. The flow bed was carved into the PTFE plate which was filled with 1 g of Amb-400 (Cl). The anodic compartment was composed of a graphite foil and the whole system was properly tightened to avoid leakage (see the ESI† for further details).
We next screened the appropriate residence time required to optimize the access of 2a under flow conditions (Table 4).
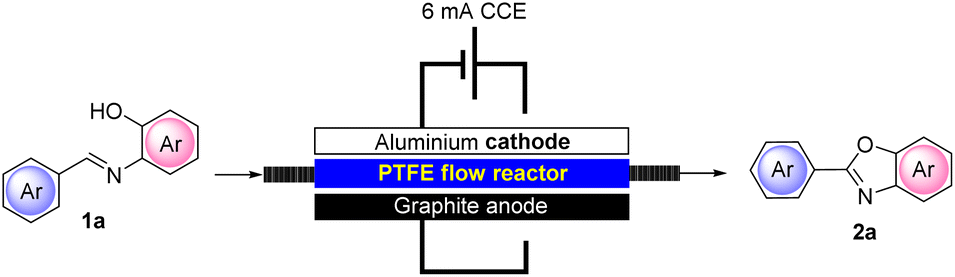
Due to the proximity of the electrodes that can be realized in our newly defined flow reactor and also due to the packed-bed reactor containing the solid electrolyte, we observed that at an operating constant current intensity of 6 mA, the flow cell voltage was strongly reduced compared to that under batch conditions (6.5–7.2 under batch conditions vs. 1.1–1.8 under flow conditions). This can be ascribed to a more efficient conductivity that can be achieved in our flow reactor compared to that under batch conditions. In addition, the miniaturization of the device also resulted in an expected improved utilization of the current allowing a better yield (95%) of representative 2a to be achieved and in only 30 min of residence time. The flow conditions also enabled the adoption of a more concentrated solution of 1a (0.05 M) compared to batch conditions (0.02–0.03 M) with consequent optimization in terms of solvent utilization. Interestingly, we also found that, with the adoption of our flow reactor, the addition of water to the reaction mixture is not necessary, further lowering the input material required for the process.
Intrigued by these results and with the intention of pushing the reusability limits of the solid electrolyte, we performed a continuous synthesis of 2a with a productivity of 77 mg h−1 by running the flow electrochemical cell for up to 12 h, observing no loss in efficiency. The resulting mixture at the outlet of the electrochemical flow reactor was evaporated, allowing us to recover 90% w/w of the acetonitrile used and furnishing 2a in 0.900 g yield. We also replicated the substrate scope under flow conditions always obtaining comparable results under better conductivity conditions.
To quantify the benefits and the advances in terms of the sustainability of our procedure, we compared some green metrics (E-factor, RME and MRP) calculated for our and other electrochemical procedures.28
A worthy focus for this type of work could be comparison of the percentage of the kernel derived from the use of an homogeneous electrolyte and that of our recoverable supporting electrolyte: we refer to the Environmental electrolyte factor (Ee-factor).
From the results summarized in Fig. 2, it is evident how significant the minimization of waste resulting from the use of a polymeric recoverable electrolyte is. The E-factor distribution (for clarity in this part presented with the exclusion of the work-up materials) shows that, besides the expected relevant contribution of the reaction medium, the mass of the electrolyte constitutes 25–68% of the entire kernel mass. An exception can be noted (Fig. 2, example f) for a flow protocol where a further reduction of the electrolyte was achieved. However, in this case, there is a large overall E-factor value anyway due to the reactive iodine(III) species involved in the process.
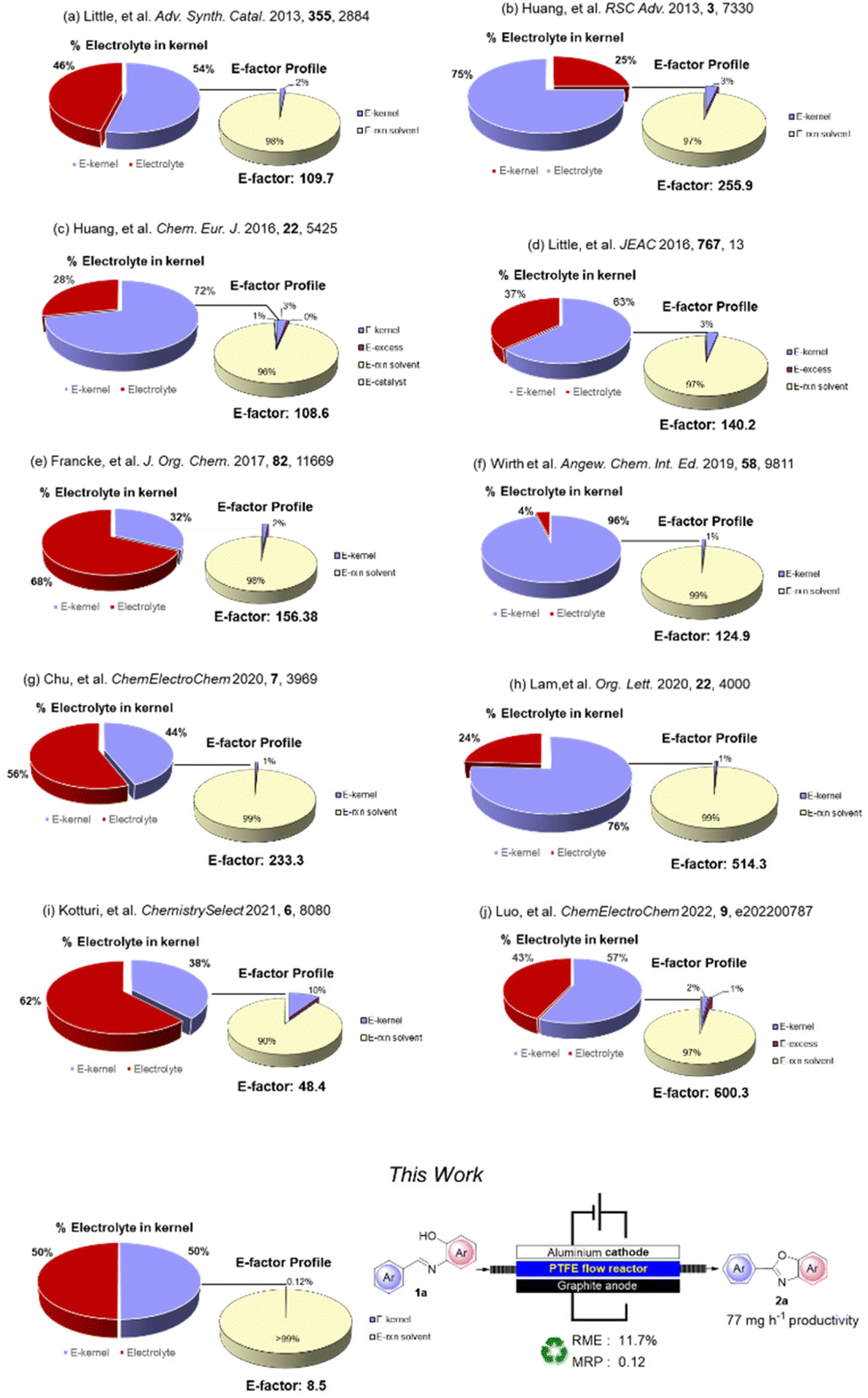 | ||
| Fig. 2 Sustainability features and comparison of the electrochemical procedure for the synthesis of 2a. | ||
In general, E-factor values are at least one order of magnitude larger than the E-factor value associated with our procedure under flow conditions using a solid and recyclable electrolyte.
It is also worth noting that our protocol led to a kernel percentage as low as 0.12%. This result confirms that this approach can significantly reduce the actual contribution of the electrolyte and the kernel to the waste produced during the process.
Further analysis (Table 5) of the other green metrics and the complete E-factor (including work-up materials) was performed to give an overall quantification of the benefits derived from the utilization of solid and recyclable electrolytes.
| As Fig. 2 | RME (%) | MRP | E-factor | eE-factor |
|---|---|---|---|---|
| a | 0.9 | 0.012 | 489.1 | 1.23 |
| b | 0.4 | 0.004 | 255.9 | 2.48 |
| c | 0.9 | 0.032 | 1148.5 | 1.36 |
| d | 0.7 | 0.012 | 1139.2 | 2.09 |
| e | 0.6 | 0.007 | 858.5 | 0.15 |
| f | 0.8 | 0.008 | 813.0 | 0.08 |
| g | 0.4 | 0.005 | 2623.8 | 1.59 |
| h | 0.2 | 0.007 | 514.3 | 1.21 |
| i | 2 | 0.044 | 48.3 | 3.50 |
| j | 0.2 | 0.018 | 8603.2 | 7.88 |
| This work | 10.5 | 0.117 | 8.5 | 0.00 |
As can be seen, the complete E-factor values clearly show that when a homogeneous supporting electrolyte is used, the corresponding final work-up is significantly wasteful as the removal of the electrolyte or its byproducts from the reaction mixture is necessary before performing the purification procedure necessary to isolate the pure product. By calculating the eE-factor (electrolyte Environmental factor) simply as the mass ratio of supporting electrolyte to that of the product isolated, we can truly confirm how more efficient the use of a recyclable electrolyte is in terms of sustainability with an eE-factor value of zero compared to 1.21–7.88. In addition, very low values of eE-factor can be obtained using recoverable homogeneous electrolytes (protocol e in Table 5) or by using flow conditions for the generation of hypervalent iodine species (protocol f in Table 5). Anyway, in these two cases, the large amounts of solvent (Et2O) used to recover the electrolyte lead to very large values of complete E-factor.
Conclusions
In conclusions, we have proved for the first time that the use of a solid and recyclable electrolyte (Amberlyst 400-Cl) is possible, and it can be exploited for the electro-assisted synthesis of 2-arylbenzoxazoles 2. The novel and interesting ability of the polymer-supported electrolyte allowed the synthesis of 17 different benzoxazoles in good to excellent yields. Cyclic voltammetry (CV) experiments were performed to determine the intensity of electrolytic effects in comparison with the benchmark homogeneous electrolyte (NH4Cl). Thus, solid Amberlyst 400-Cl was efficiently reused for several reaction runs with very consistent promising and solid results. A tailor-made continuous flow reactor was realized and a flow methodology was also presented as further proof that it is possible to improve the usage of a solid electrolyte and to optimize the current utilization and the overall sustainability of the process. Simple, inexpensive, and readily available materials were employed as electrodes (graphite and aluminium). The reaction medium used could also be largely recovered, allowing us to minimize the waste production at a very low level. Ultimately, the final products were found to be almost free from metal contaminants. With a complete green metrics analysis, it can also be quantified how the use of solid electrolytes in electrosynthesis is a very promising and a valuable approach to minimize the quantity and quality of the waste produced in this valuable synthetic strategy.Efforts are now being directed towards a deep comprehension of the efficient electron-transfer nature that we have found in the use of a solid electrolyte. The development of other polymer-based ionic tags which can be used as solid electrolytes is ongoing in our laboratories together with the implementation of their synthetic utility.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript. F. F.: conceptualization, project administration, investigation, methodology, data analysis, and manuscript preparation – review/editing; F. V., data analysis and manuscript editing; F. C. data analysis and manuscript editing; L. V.: conceptualization, project administration, and manuscript review/editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality. MUR is also thanked for the PRIN-PNRR 2022 project “P2022XKWH7 – CircularWaste. The National PhD program in Catalysis coordinated by the University of Perugia is also thanked.References
- (a) C. Yan, X. Jiang, J. Yu, Z. Ding, L. Ma, T. Su, Y. Wang, C. Wang, G. Huang and S. Xu, Green Chem., 2023, 25, 3816–3846 RSC; (b) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS; (c) R. D. Little and K. D. Moeller, Chem. Rev., 2018, 118, 4483–4448 CrossRef CAS PubMed; (d) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 21, 13230–13319 CrossRef PubMed; (b) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- (a) O. Y. Gutierrez, K. Grubel, J. Kothandaraman, J. A. Lopez-Ruiz, K. P. Brooks, M. E. Bowden and T. Autrey, Green Chem., 2023, 25, 4222–4233 RSC; (b) C. Tang, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed; (c) Q. Zhao, X. Lu, Y. Wang, S. Zhu, Y. Liu, F. Xiao, S.-X. Dou, W.-H.- Lai and M. Shao, Angew. Chem., Int. Ed., 2023, 62, e202307123 CrossRef CAS PubMed; (d) Y. Yuan and A. Lei, Nat. Commun., 2020, 802 CrossRef CAS PubMed; (e) R. Francke, Curr. Opin. Electrochem., 2022, 36, 1011 Search PubMed; (f) S. R. Waldvogel, A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo and M. Zirbes, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef PubMed; (g) B. A. Frontana-uribe, R. D. Little, J. G. Ibanez and R. Vasquez-medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- (a) J. Xu, F. Liu, L. Sun, M. Huang, J. Jiang, K. Wang, D. Ouyang, L. Lu and A. Lei, Green Chem., 2022, 24, 7350–7354 RSC; (b) J.-Y. He, W.-F. Qian, Y.-Z. Wang, C. Yao, N. Wang, H. Liu, B. Zhong, C. Zhu and H. Xu, Green Chem., 2022, 24, 2483–2491 RSC; (c) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC; (d) S.-H. Shi, Y.-J. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed.
- (a) N. Kurig and R. Palkovitz, Green. Chem., 2023, 25, 7508–7517 RSC; (b) M. D. Karkas, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- P. Villo, A. Shatskiy, M. D. Karkas and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed.
- (a) M. C. Leech and K. Lam, Acc. Chem. Res., 2020, 53, 121–134 CrossRef CAS PubMed; (b) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- (a) F. Sprang, J. D. Herszman and S. R. Waldvogel, Green Chem., 2022, 24, 5116–5124 RSC; (b) F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53(3), 561–574 CrossRef CAS PubMed.
- M. Klein and S. R. Waldvogel, Angew. Chem., Int. Ed., 2022, 61, e202204140 CrossRef CAS PubMed.
- (a) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS PubMed; (b) S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122–7123 CrossRef CAS PubMed.
- (a) C. Kingston, M. D. Palkovitz, Y. Takaira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53(1), 72–83 CrossRef CAS PubMed; (b) C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willians, Green Chem., 2020, 22, 3358–3375 RSC.
- (a) S. Sun, X. Qiu, S. Hao, S. Ravichandran, J. Song and W. Zhang, Green Chem., 2023, 25, 3127–3136 RSC; (b) M. C. Morejon, A. Franz, R. Karande and F. Harnisch, Green Chem., 2023, 25, 4662–4666 RSC; (c) X. Yang, Y. Zhang, M. Ye, Y. Tang, Z. Wen, X. Liu and C. C. Li, Green Chem., 2023, 25, 4154–4179 RSC; (d) J. Kümper, J. Meyers, R. Sebers, N. Kurig and R. Palkovits, Green Chem., 2023, 25, 6231–6237 RSC; (e) T. Lenk, V. Rueß, J. Gresch and U. Schröder, Green Chem., 2023, 25, 3077–3085 RSC.
- (a) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (b) M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef PubMed; (c) F. Dorchies and A. Grimaud, Chem. Sci., 2023, 14, 7103–7113 RSC.
- (a) W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC; (b) B. Schille, N. O. Giltzau and R. Francke, Angew. Chem., Int. Ed., 2018, 57, 422–426 CrossRef CAS PubMed; (c) S. J. Yoo, L.-J. Li, C.-C. Zeng and R. D. Little, Angew. Chem., Int. Ed., 2015, 54, 3744–3747 CrossRef CAS PubMed.
- (a) T. Tajima and T. Fuchigami, J. Am. Chem. Soc., 2005, 127, 2848 CrossRef CAS PubMed; (b) T. Tajima, H. Kurihara and T. Fuchigami, J. Am. Chem. Soc., 2007, 129, 6680 CrossRef CAS PubMed; (c) T. Tajima and T. Uchiyama, Angew. Chem., Int. Ed., 2005, 44, 4760 CrossRef CAS PubMed.
- (a) O. Hammerich and B. Speiser, Organic Electrochemistry: Revised and Expanded, CRC Press, 2015 CrossRef; (b) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- (a) K. Izutsu, Electrochemistry in Nonaqueous Solutions, Wiley, 2011 Search PubMed; (b) H. Tanaka, M. Kuroboshi and S. Torii, in Organic Electrochemistry, CRC Press, 5th edn, 2015, pp. 1267–1307 Search PubMed.
- (a) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Khampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed; (b) C. Stang and F. Harnisch, ChemSusChem, 2016, 9, 50–60 CrossRef CAS PubMed; (c) F. Bures, Top. Curr. Chem., 2019, 377, 14–35 CrossRef PubMed; (d) E. Nouri-Nigjeh, M. P. de Vries, A. P. Bruins, R. Bischoff and H. P. Permentier, Electrochem. Commun., 2012, 21, 54–57 CrossRef CAS.
- https://ec.europa.eu/eurostat/documents/342366/351806/Guidance-on-EWCStat-categories-2010.pdf/0e7cd3fc-c05c-47a7-818f-1c2421e55604 .
- (a) P. I. Hora, S. G. Pati, P. J. McNamara and W. A. Arnold, Environ. Sci. Technol. Lett., 2020, 7, 622–631 CrossRef CAS PubMed And references cited herein; (b) J. Gyorgy, WO2007107804A2, 2006.
- For recent examples summarizing the diverse methods for the synthesis of 2-arylbenzoxazoles: (a) S. R. Patel, R. V. Patil, J. U. Chavan and A. g. Beldar, ChemistrySelect, 2023, 8, e20230197 Search PubMed; (b) S. Soni, N. Sahiba, S. Teli, P. Teli, L. K. Agarwal and S. Agarwal, RSC Adv., 2023, 13, 24093–24111 RSC; (c) K. Kant, C. K. Patel, S. Banerjee, P. Naik, A. K. Atta, A. K. Kabi and C. C. Malakar, ChemistrySelect, 2023, 8, e20230398 CrossRef; (d) J. Sharma, P. Mishra and J. Bhadoria, Results Chem., 2022, 4, 100670 CrossRef CAS; (e) S. Sunny, S. E. John and N. Shankaraiah, Asian J. Org. Chem., 2021, 10, 1986–2009 CrossRef CAS; (f) M. C. Henry, V. M. Abbinante and A. Sutherland, Eur. J. Org. Chem., 2020, 2819–2826 CrossRef CAS; (g) Y. Zhang and M. Ji, Eur. J. Org. Chem., 2019, 7506–7510 CrossRef CAS; (h) A. Ibrar, I. Khan, N. Abbas, U. Farooq and A. Khan, RSC Adv., 2016, 6, 93016–93047 RSC.
- For recent examples of our research line in customized flow reactors: (a) F. Ferlin, I. Anastasiou, L. Carpisassi and L. Vaccaro, Green Chem., 2021, 23, 6576–6582 RSC; (b) F. Ferlin, D. Sciosci, F. Valentini, J. Menzio, G. Cravotto, K. Martina and L. Vaccaro, Green Chem., 2021, 23, 7210–7218 RSC; (c) N. Salameh, F. Ferlin, F. Valentini, I. Anastasiou and L. Vaccaro, ACS Sustainable Chem. Eng., 2022, 10, 3766–3776 CrossRef CAS; (d) G. Brufani, F. Valentini, G. Rossini, L. Carpisassi, D. Lanari and L. Vaccaro, Green Chem., 2023, 25, 2438–2445 RSC.
- (a) M. G. Freire, C. M. S. S. Neves, I. M. Marrucho, J. O. A. P. Coutinho and A. M. Fernandes, J. Phys. Chem. A, 2009, 114, 3744–3749 CrossRef PubMed; (b) J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- Q. An, C. He, X. Fan, C. Hou, J. Zhao, Y. Liu, H. Liu, J. Ma, Z. Sun and W. Chu, ChemElectroChem, 2020, 7, 3969–3974 CrossRef CAS.
- H. Yang and N. Wu, Energy Sci. Eng., 2022, 10, 1643–1671 CrossRef CAS.
- Small Volume Parenteral Drug Products and Pharmacy Bulk Packages for Parenteral Nutrition: Aluminum Content and Labeling Recommendations. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/small-volume-parenteral-drug-products-and-pharmacy-bulk-packages-parenteral-nutrition-aluminum. Document identifier: FDA-2022-D-2301.
- (a) W.-C. Li, C.-C. Zang, L.-M. Hu, H.-Y. Tian and R. D. Little, Adv. Synth. Catal., 2013, 355, 2884–2890 CrossRef CAS; (b) Y. Shih, C. Ke, C. Pan and Y. Huang, RSC Adv., 2013, 3, 7330–7336 RSC; (c) Y.-L. Lai, J.-S. Ye and J.-M. Huang, Chem. – Eur. J., 2016, 22, 5425–5429 CrossRef CAS PubMed; (d) L.-S. Kang, H.-l. Xiao, C.-C. Zeng, L.-M. Hu and R. D. Little, J. Electroanal. Chem., 2016, 767, 13–17 CrossRef CAS; (e) O. Koleda, T. Broese, J. Noetzel, M. Roemelt, E. Suna and R. Francke, J. Org. Chem., 2017, 82, 11669–11681 CrossRef CAS PubMed; (f) M. Elsherbini, B. Winterson, H. Alharbi, A. A. Folgueiras-Amador, C. Génot and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 9811–9815 CrossRef CAS PubMed; (g) Q. An, C. He, X. Fan, C. Hou, J. Zhao, Y. Liu, H. Liu, J. Ma, Z. Sun and W. Chu, ChemElectroChem, 2020, 7, 3969–3974 CrossRef CAS; (h) A. D. Garcia, M. C. Leech, A. Petti, C. Denis, I. C. A. Goodall, A. P. Dobbs and K. Lam, Org. Lett., 2020, 22, 4000 CrossRef CAS PubMed; (i) T. Ghoshal, T. M. Patel and S. Kotturi, ChemistrySelect, 2021, 6, 8080–8084 CrossRef CAS; (j) Y.-L. Lai, X. Yang, S.-C. Wu, X.-Y. Li, Y.-X. Gong, S.-L. Zhang, R.-M. Zhong, J.-H. Liao and J.-M. Luo, ChemElectroChem, 2022, 9, e202200787 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, full characterization of the synthesized compounds and copies of 1H, 13C and 19F NMR spectra. See DOI: https://doi.org/10.1039/d4gc00930d |
| This journal is © The Royal Society of Chemistry 2024 |