 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the solvent stability on the cathode surface of Li–O2 batteries by using in situ vibrational spectroscopies
Aimin
Ge†
 a,
Ryuuta
Nagai†
b,
Kota
Nemoto
b,
Bingbing
Li
b,
Koki
Kannari
b,
Ken-ichi
Inoue
b and
Shen
Ye
*b
a,
Ryuuta
Nagai†
b,
Kota
Nemoto
b,
Bingbing
Li
b,
Koki
Kannari
b,
Ken-ichi
Inoue
b and
Shen
Ye
*b
aCenter for Advanced Low-dimension Materials, Donghua University, Shanghai 201620, P. R. China
bDepartment of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: ye.shen@tohoku.ac.jp
First published on 16th October 2023
Abstract
In aprotic lithium–oxygen (Li–O2) batteries, solvent properties are crucial in the charge/discharge processes. Therefore, a thorough understanding of the solvent stability at the cathode surface during the oxygen reduction/evolution reactions (ORR/OER) is essential for the rational design of high-performance electrolytes. In this study, the stability of typical solvents, a series of glyme solvents with different chain lengths, has been investigated during the ORR/OER by in situ vibrational spectroscopy measurements of sum frequency generation (SFG) spectroscopy and infrared reflection absorption spectroscopy (IRRAS). The structural evolution and decomposition mechanism of the solvents during ORR/OER have been discussed based on the observations. Our results demonstrate that superoxide (O2−) generated during the ORR plays a critical role in the stability of the solvents.
Introduction
Among the “beyond lithium-ion battery” technologies, the aprotic rechargeable lithium–oxygen (Li–O2) battery has attracted much attention due to its highest theoretical energy density.1–5 Despite significant efforts, Li–O2 batteries are still far from practical applications due to high charging overpotential and poor cyclability.2,3,6 Many of these problems are related to the properties of aprotic electrolytes. Many efforts have been devoted to developing electrolytes with properties such as wide electrochemical windows, high stability toward the Li anode, and excellent tolerance toward reactive oxygen species (ROS), including superoxide ion (O2−) and singlet oxygen (1O2).2,3 Li–O2 batteries were first employed in organic carbonate-based electrolytes (such as ethylene carbonate and propylene carbonate), which have been widely employed in Li-ion batteries due to their high dielectric constants, good solubilities for lithium salts, low viscosities, and high ionic conductivities. It was recently found that, however, the cyclic carbonate electrolytes can be continuously decomposed by O2− generated during the discharge process and, therefore, are not suitable for Li–O2 batteries.1,7–10The glyme-based electrolytes have been reported to be more stable for the ORR/OER processes than organic carbonate electrolytes.11 McCloskey and co-workers confirmed that dimethoxyethane (DME) is oxidatively decomposed during the charge process.12 Freunberger et al. found that the discharge capacity of porous cathodes decreased mainly after the first ORR/OER cycle in glyme solvents of different chain lengths, which is attributed to the O2−-induced oxidative decomposition of glyme-based electrolytes.11 It was also reported that nucleophilic attack of Li2O2 can result in the oxidative degradation of polyether solvents during ORR.13,14 Recent studies have shown that singlet oxygen (1O2) is a critical factor in the induction of electrolyte degradation in the ORR/OER cycle.15–17 These mechanistic controversies hinder the understanding of the nature of the solvent stability. Therefore, further exploration of the stability mechanism of solvents is crucial for developing high-performance electrolytes.
Previously, we investigated the mechanism of ORR/OER in dimethyl sulfoxide (DMSO), propylene carbonate, ethylene carbonate, and tetraglyme electrolytes using in situ vibrational spectroscopic tools and other methods.9,10,18–22 In particular, our previous results indicate that the potential-dependent behaviors of tetraglyme on the electrode surface highly depend on the types of cations (Li+ or tetrabutylammonium cation (TBA+)).21 It was found that the structural evolution of interfacial tetraglyme exhibits irreversible behaviors in Li+-free solution, which is attributed to the O2−-induced decomposition of tetraglyme. In contrast, tetraglyme is much more stable during ORR in Li+-containing solution, which is understood by the short exposure time of tetraglyme to O2−.21
To gain a deeper understanding of the effect of ether properties on ORR/OER, in this work, we extend the in situ sum frequency generation (SFG) study to a series of ether solvents with regular structural variation (monoglyme (G1 or DME), diglyme (G2), triglyme (G3), and tetraglyme (G4), see Scheme 1). As a second-order nonlinear optical process, infrared-visible SFG is only active in media without centrosymmetry.23,24 Therefore, SFG spectroscopy is an interface-specific spectroscopic method that can detect the molecular structures at the electrode–electrolyte interfaces with high sensitivity.25–28 The present SFG observations demonstrate that the cations can significantly affect the interfacial structures and stabilities of the glyme solvents, possibly due to the different solvation abilities. The glymes become unstable in the ORR in the TBA+-containing solution but improve substantially in the Li+-containing solution. The SFG results show that the structures of glymes on the electrode surface immediately change as soon as the formation of the Li2O2 on the electrode surface occurs during ORR. On the other hand, the structural changes of the glymes are observed from a potential, much more positive than that Li2O2 begins to be oxidized.
 | ||
| Scheme 1 Molecular structures of glymes used in this work. | ||
In addition, in situ IRRAS measurements revealed the decomposition products of G4 molecules during ORR in TBA+-containing and Li+-containing solutions. For the TBA+-containing G4 electrolyte, carbon dioxide (CO2) can be observed when the potential is higher than 2.9 V during the positive-going sweep, which can be attributed to the oxidation of decomposition products formed during ORR. On the other hand, the CO2 band appears when the potential is higher than 3.5 V for the Li+-containing G4 electrolyte, which could be attributed to the oxidation of both decomposition products and G4 molecules. The potential dependences of formation and decomposition of the ORR-induced products correlate well with structural hysteresis observed by the in situ SFG measurement.
Experimental section
Sample preparation
Ethylene glycol dimethyl ether (G1, monoglyme, DME, special grade), diethylene glycol dimethyl ether (G2, diglyme, first grade), triethylene glycol dimethyl ether (G3, triglyme, first grade), tetraethylene glycol dimethyl ether (G4, tetraglyme, TEGDME, >98%) were received from FUJIFILM Wako Pure Chemical Corporation. The molecular structures of these glymes are shown in Scheme 1. Lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, >98%) and tetrabutylammonium bis(trifluoromethanesulfonyl)imide (TBATFSI, 97%) were purchased from Tokyo Chemical Industry. All chemicals were used as received. 0.1 M LiTFSI or TBATFSI in various solvents were prepared and dried with molecular sieves (3 Å) before use. The stock solutions were stored in an argon (Ar)-purged glovebox (Labstar, MBRAUN, O2 concentration <5 ppm, water concentration <1 ppm). Before use, the stock solutions were moved to an O2-purged glovebox (UNICO, water concentration <2 ppm) and purged by an O2 gas stream. The water concentration of the stock solutions was ca. 20 ppm, measured by a Karl Fischer moisture titrator (MKC-710). Single-layer graphene prepared by chemical vapor deposition (CVD) method29 was used as the electrode for in situ SFG measurements. The procedures of CVD and graphene transfer were described in our previous publication.10 In addition, a gold disk electrode (d = 10 mm) was used as the working electrode in the electrochemical IRRAS measurements.21 Before the experiment, the gold electrode surface was mirror-polished by an alumina abrasive with a diameter of 1 μm.Heterodyne-detected (HD) SFG measurements
The HD-SFG measurements at the glyme–air interface were performed using an HD-SFG setup based on a Ti:sapphire regenerative amplifier (Quantronix, Integra-C; average power: 2.5 W, repetition rate: 1 kHz, pulse width: 130 fs, center wavelength: 798 nm). HD-SFG measures the imaginary part of second-order susceptibility (Imχ(2)), which can significantly improve the detection of the molecular orientation and accuracy of peak assignments. The details of this setup have been given elsewhere.30 The HD-SFG measurements were only carried out on the glymes liquid–air interface with the ssp polarization combination (s-SFG, s-visible, and p-infrared). Z-cut quartz was used as the reference. The Imχ(2) spectra are the average of 8 independent measurements.Electrochemical homodyne-detected SFG measurements
The electrochemical SFG measurements were performed using a homodyne-detected setup with a broadband femtosecond laser system based on a 1 kHz Spitfire titanium:sapphire amplifier (Spectra-Physics). The homodyne-detected SFG, which measures the square of χ(2) (|χ(2)|2), was employed in the electrode/solution interface in the study.28 The optical system and spectral processing are relatively simple, but homodyne-detected SFG could suffer from spectral distortion and determination of the absolute orientation. The homodyne-detected SFG will be denoted as SFG unless otherwise specified. The details of this setup have been given elsewhere.10 In the present work, a broadband infrared pulse (centered at 3450 nm, 8 μJ, FWHM ∼200 cm−1, angle of incidence 50°) and narrowband 800 nm pulse (10 μJ, angle of incidence 70°) were spatially and temporally overlapped on the graphene electrode and electrolyte interface surface with an internal reflection mode. All SFG spectra were collected with the ssp polarization combination. The SFG spectra were recorded simultaneously during the cyclic voltammetry (CV) measurements. The integration time for each spectrum is 100 s. Thus, one spectrum represents the averaged SFG signals for a potential range of 100 mV. The raw SFG spectra were smoothed by the 5 points averaging method during the data processing and divided by a ppp-polarized spectrum from the gold film.10 All measurements were performed at room temperature.A homemade spectroelectrochemical cell was used for the SFG measurements. A CaF2/graphene sample was used as the working electrode. Lithium was used as the counter electrode and reference electrode. Electrochemical controls were performed with a potentiostat (Polarization Unit PS-07, Toho Technical Research). The open-circuit potential (OCP) is about 3.1 V (vs. Li+/Li) for solutions used in this work.
Electrochemical IRRAS measurements
In situ IRRAS measurements were performed with an FTIR spectrometer (BioRad FTS 6000) equipped with an MCT detector cooled with liquid nitrogen.21,31 A homemade electrochemical cell was used for the IRRAS measurements. The angle of infrared beam incidence is 60°. A gold disk electrode is pressed against the optical BaF2 window to reduce the infrared absorption from the electrolyte. The IRRAS spectra were recorded simultaneously during the CV (1 mV s−1). Each spectrum accumulated 64 interferograms with a resolution of 4 cm−1, corresponding to a potential window of ca. 7 mV. A spectrum measured at the OCP before the CV was used as the reference spectrum. The IRRAS results are presented in the absorbance change, i.e., −log(Rs/Rr), where Rs and Rr are the reflectances at the sample and the reference potential, respectively. The upward and downward IRRAS bands represent higher and lower absorption than the reference spectrum.Results and discussion
SFG measurements of glyme–air and glyme–graphene interfaces
Fig. 1 shows SFG spectra for the different glymes (G1–G4) at the liquid/air interface in the CH stretch region under (a) heterodyne-detected (HD) mode (Imχ(2)) and (b) homodyne-detected mode (|χ(2)|2). The apparent features of the two spectra for the same solvent are pretty different. While downward and upward peaks can be distinguished in the HD-SFG spectra, only upward peaks are observed in the homodyne-detected SFG spectra. A downward peak at 2817 cm−1 and an upward peak at 2967 cm−1 can be assigned to the symmetric and asymmetric stretches of the OCH3 groups (OCH3ss, OCH3as), respectively.32 Downward peak at 2909 cm−1 is attributable to the Fermi resonance (FR) of the OCH3ss mode with the bending mode (OCH3FR). The band intensity of the OCH3ss and OCH3as modes are not very sensitive to the changes in the chain length of the glyme molecules. In addition, a downward band around 2860 cm−1 can be attributed to the symmetric stretch of the OCH2 group (OCH2ss).32 The intensities of the OCH2ss band increase with the chain length of the glyme molecules, which is consistent with the abundance of the OCH2 groups. The peak direction directly correlates with the direction of the dipole moment of each vibrational mode.33 These downward OCH3ss and OCH2ss peaks indicate that the glymes at the interface orient their hydrogens to the air. In this way, HD-SFG improves the detection of the molecular orientation and accuracy of peak assignments.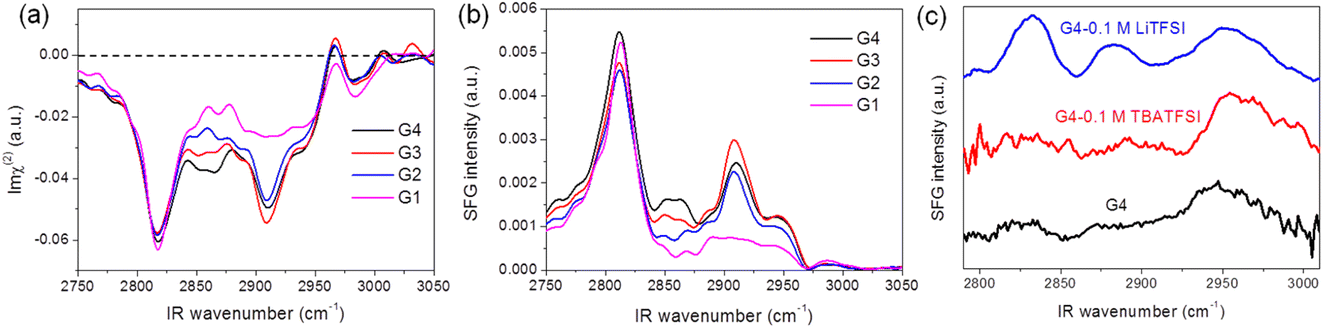 | ||
| Fig. 1 (a) Heterodyne-detected (HD) and (b) homodyne-detected SFG spectra measured at glyme–air interface. (c) Homodyne-detected SFG spectra of G4 electrolyte–graphene interface. | ||
In addition, SFG spectra of the glymes at the liquid/air interface (Fig. 1b) are significantly different from that of the G4/graphene interface (black trace, Fig. 1c), implying some specific interaction between the G4 molecules and the graphene surface. It is also noted that adding 0.1 M TBATFSI salt does not affect the SFG spectrum of G4 much (red trace, Fig. 1c). However, adding 0.1 M LiTFSI salt changes the spectral shape of G4 (blue trace, Fig. 1c). The SFG spectra of the graphene interface with the Li+-contained G4 solution exhibit three bands around 2830, 2880, and 2955 cm−1, which can be tentatively assigned to OCH3ss, OCH3FR, and OCH3as. From these observations, we consider that cations can largely influence the interfacial structure of the glymes on the graphene surface. Since glymes can strongly solvate Li+ in comparison with the large cation of TBA+, the interfacial structure of G4 on the graphene surface can significantly differ between the two electrolytes. Furthermore, we found that the interfacial structures of G4 on the graphene surface in the TBA+-containing solution become significantly irreversible after the ORR, implying the oxidative decomposition of G4 molecules induced by O2−.34 The lower stability of glymes in the Li+-free solution during ORR may also be associated with the low solvation states of the TBA+ cation.
HD-SFG measurements could improve the spectral analysis for the glymes on the graphene electrode and solution interface. However, since the HD-SFG measurements at the graphene–electrolyte interface are still in progress, the interfacial structures of the electrode–electrolyte interfaces will be discussed with homodyne-detected SFG spectra in the Li+-containing glymes in the present study.
Structural evolution at the electrode–electrolyte interface
Fig. 2a shows the CV (3.1 V → 1.7 V → 4.5 V → 3.1 V) of ORR/OER on a graphene electrode in an O2-saturated 0.1 M LiTFSI/G1 solution at a scan rate of 1 mV s−1. During the negative-going sweep from OCP (3.1 V), a reduction peak appears at 2.0 V, which can be attributed to ORR in which Li2O2 is formed on the electrode surface. During the reversed positive-going sweep, a broad oxidation wave starts at 2.9 V. This oxidation wave could be attributed to the oxidation of Li2O2.35,36 The oxidation current significantly increases when the potential is higher than 4.3 V, possibly due to Li2O2 oxidation together with the oxidative decomposition of G1 solvent.37 Generally, the CV curve of ORR/OER in G1 shows similar features to G4 except for the large oxidation current in the positive potential region.21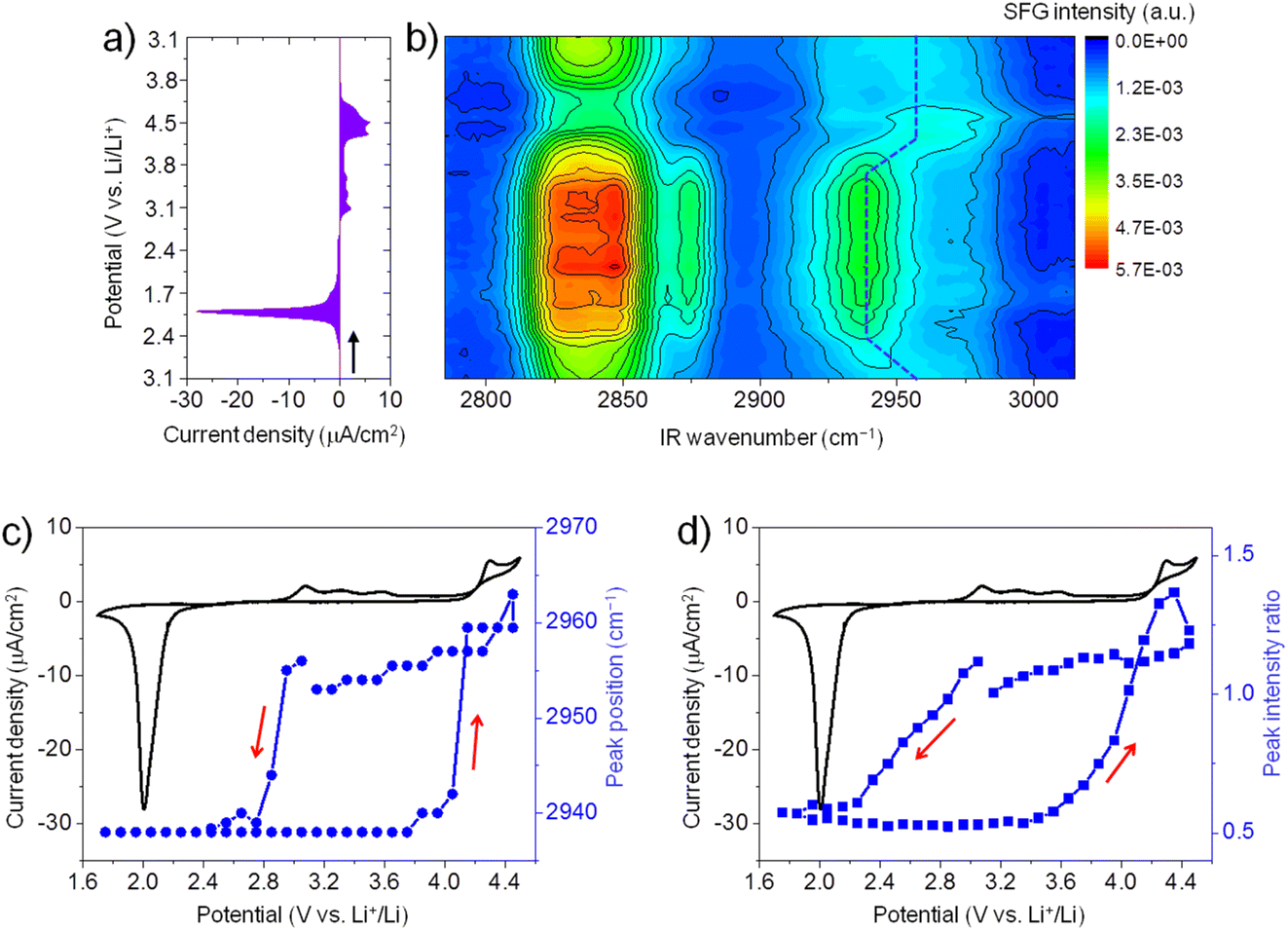 | ||
| Fig. 2 (a) CV curve of ORR/OER on a graphene electrode in O2-saturated 0.1 M LiTFSI/G1 solution (3.1 V → 1.7 V → 4.5 V → 3.1 V, scan rate 1 mV s−1). (b) Contour plot of SFG spectra measured during ORR/OER. The Y-axis is the potential of the graphene electrode, corresponded to that in (a). Dashed blue lines indicate the shifts of the C–H stretch for OCH3as. (c) Potential dependence of peak position of the SFG band around 2955 cm−1 during ORR/OER. (d) SFG intensity ratio (2955 cm−1/2938 cm−1) versus potential. CV curves are given in parts c and d for comparison. | ||
Fig. 2b illustrates in situ SFG spectra in the C–H stretching region of G1 molecules recorded simultaneously during the potential sweep (3.1 V → 1.7 V → 4.5 V → 3.1 V). The SFG signals change significantly upon ORR/OER. Before ORR at OCP (3.1 V), the SFG spectrum is dominated by two bands around 2830 cm−1 and 2955 cm−1. As discussed above in Fig. 1, these two peaks can be tentatively assigned to the symmetric stretch of OCH3 (OCH3ss) and the asymmetric stretch of OCH3 (OCH3as).32 In addition, a weaker band around 2875 cm−1 is assigned to OCH3FR. These C–H stretches can be attributed to the G1 molecules adsorbed at the electrode–electrolyte interface. The peak positions differ from those observed on the G1/air interface by HD-SFG, which may be associated with the different adsorption structures of G1 molecules on the graphene electrode surface.
During ORR/OER processes, significant changes in SFG intensity and peak position can be observed. Here, we focus on the SFG band around 2955 cm−1. To analyze the trend in the spectra semi-quantitatively, we show the potential dependence of the peak position of the SFG band around 2955 cm−1 in Fig. 2c. During the negative-going sweep, it can be seen that when the potential becomes more negative than 3.0 V, the peak frequency sharply decreases to 2938 cm−1 and then keeps almost constant. However, during the anodic potential sweep, the peak frequency quickly increases to 2960 cm−1 when the potential becomes more positive than than 3.7 V. These spectral changes in the peak positions indicate that the interfacial structures of G1 molecules change as ORR starts. Such changes can be attributed to the influence of ORR products (Li2O2, LiO2, and products of solvent decomposition) deposited on the electrode surface. Solvent–product interaction may change the conformation or orientation of the interfacial solvent molecules. During the anodic sweep from 1.7 V to 3.7 V, no noticeable spectral changes are observed even if the oxidation current starts at 2.9 V. It indicates the interfacial structures of G1 molecules remain stable in the potential range of 2.9 V to 3.7 V during the OER. When the potential becomes more positive than 3.7 V, the structures of the interfacial G1 molecules should return to the original state on the graphene electrode surface. We also plot the SFG intensity ratio (2955 cm−1/2938 cm−1) versus potential in Fig. 2d, which shows a similar trend as that in Fig. 2c.
Similarly, we have also conducted in situ SFG measurements for ORR/OER in O2-saturated G2, G3, and G4 electrolytes. Generally, for all ether solvents used in this work, the peak position of the SFG band around 2955 cm−1 shows a similar trend during ORR/OER (blue symbols, Fig. 3a–c). It is also noted that such changes in the peak position are not observed for the Ar-saturated G4 electrolyte where no ORR/OER occurs (blue symbols, Fig. 3d). It indicates that such spectral changes observed cannot be attributed to the electric field-induced structural changes of the solvent at the electrode surface.
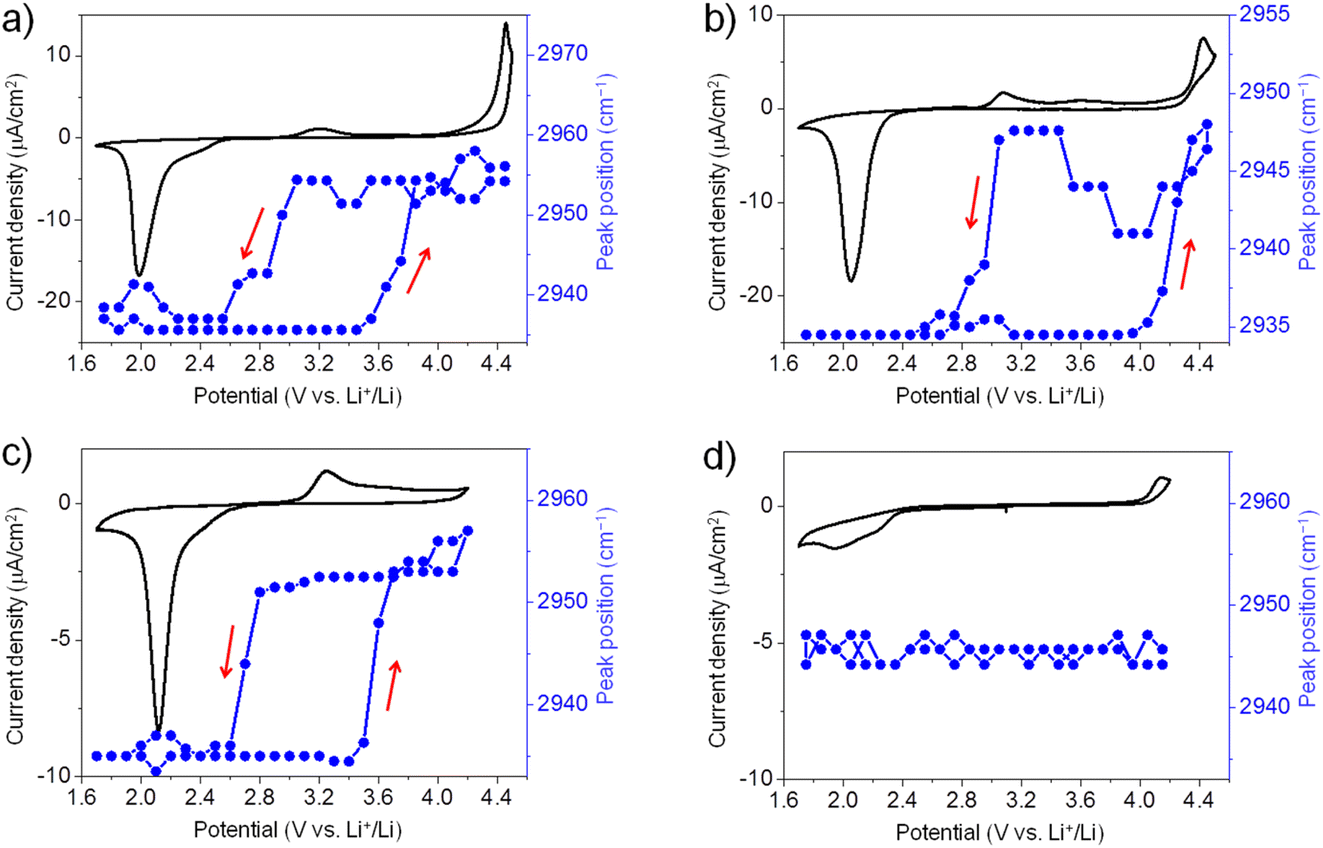 | ||
| Fig. 3 Potential dependence of peak position of the SFG band around 2955 cm−1 (blue symbols) during ORR/OER. (a) O2-saturated 0.1 M LiTFSI/G2; (b) O2-saturated 0.1 M LiTFSI/G3; (c) O2-saturated 0.1 M LiTFSI/G4; (d) Ar-saturated 0.1 M LiTFSI/G4. CV curves are given for comparison (solid trace). | ||
The large potential hysteresis in the reorganization of the interfacial solvent structure during the OER process has been observed for four glymes as shown in Fig. 2c and 3a–c. It indicates that the graphene surface is blocked by the ORR products at the initial stage of OER. Taking G4 as an example (Fig. 3c), the oxidation current starts at 3.0 V, which can be attributed to the Li2O2 oxidation. However, the SFG signals of the interfacial G4 molecules do not change significantly until the potential is higher than 3.5 V. Obviously, the decomposition of the Li2O2 layer cannot explain this potential hysteresis phenomenon well. It is noted that such potential hysteresis phenomenon is not observed for the DMSO-based electrolyte solutions.20 Combined with the IRRAS results that will be discussed below, we believe that this may be due to the presence of a layer of G4 decomposition products formed during ORR between the electrode surface and the Li2O2 layer. The ORR-induced decomposition product layer can exist stably on the electrode surface below 3.5 V. As a result, the G4 molecules attached to the electrode surface cannot undergo significant structural changes. When the potential is higher than 3.5 V, the layer of ORR-induced products undergoes oxidative decomposition. Therefore, the structure of the G4 molecules on the electrode surface return to their original state. Although the exact reaction mechanism is still unknown, the in situ SFG spectroscopy has been demonstrated as a unique tool to investigate the ORR/OER mechanism in glyme-based electrolytes by tracking the structural evolution of interfacial glyme molecules.
Moreover, although there is potential hysteresis for reorganizing interfacial glyme molecules, the interfacial structures generally return to the original state after one ORR/OER cycle. It indicates that the glyme molecules are relatively stable upon ORR/OER in the present electrolytes. In contrast, as pointed out in our previous work, the interfacial structures of G4 exhibit irreversible changes during ORR/OER in TBA+-containing electrolyte due to the superoxide-induced decomposition of G4.21 As shown in Fig. 1c, the interfacial structures of G4 in TBA+-containing and Li+-containing electrolytes are significantly different. The solvation effect of Li+ may play a crucial role in the stability of G4 molecules at the electrode–electrolyte interface.
Probing decomposition products of G4 solvents by IRRAS measurements
Furthermore, in situ IRRAS measurements were applied to investigate the stability of G4-based electrolyte solutions without (Fig. 4a and b) and with Li+ (Fig. 4c and d). In comparison with SFG, IRRAS has lower surface selectivity and sensitivity. In contrast, IRRAS can evaluate the amounts of the reaction products and intermediates accumulated in the thin electrolyte layer between the electrode surface and the optical window. Furthermore, IRRAS measurements can obtain the infrared spectrum in a wide frequency region by using FTIR.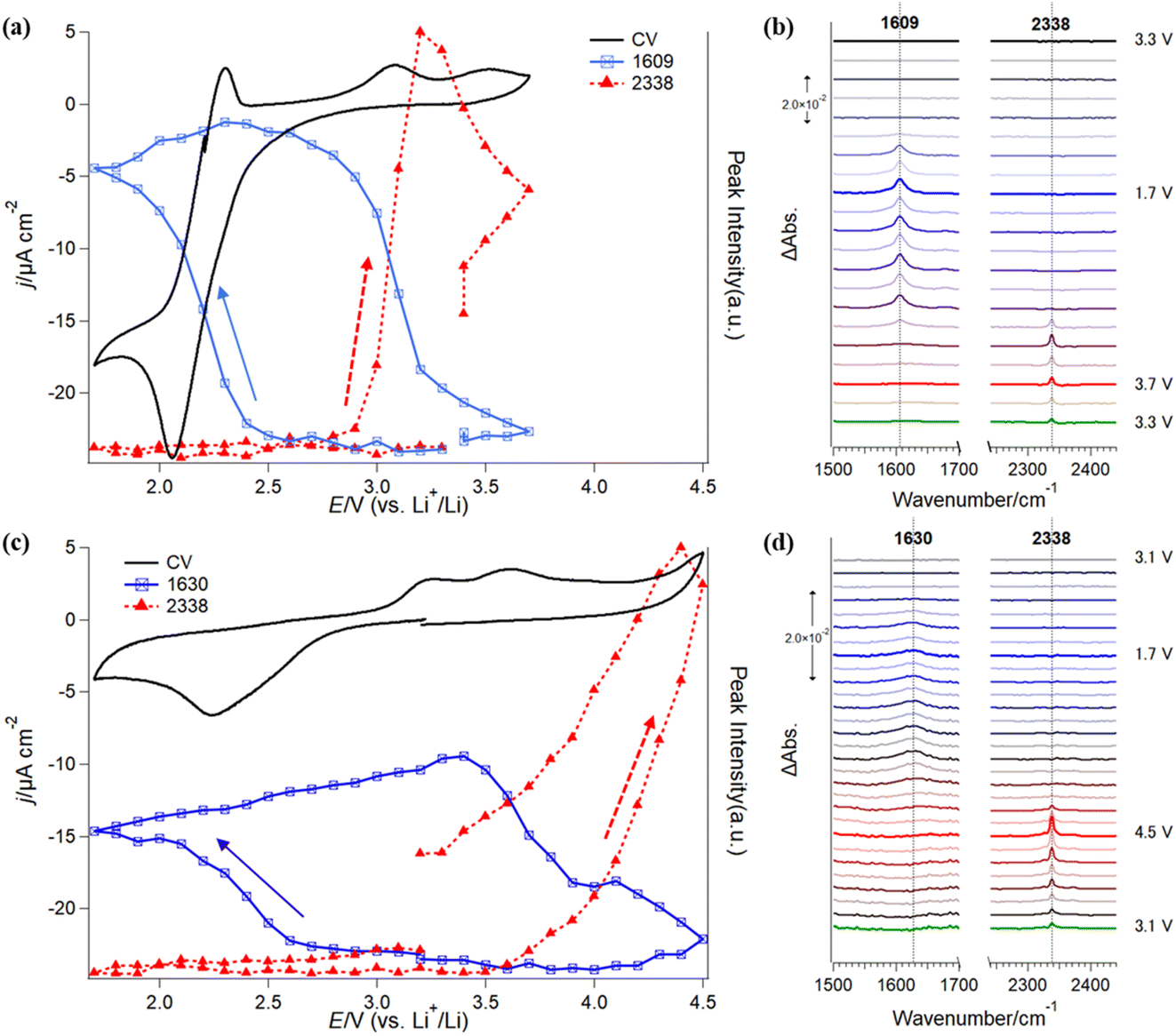 | ||
| Fig. 4 CVs and in situ IRRAS spectra obtained in (a and b) O2-saturated 0.1 M TBATFSI/G4 and (c and d) O2-saturated 0.1 M LiTFSI/G4 electrolytes. The IRRAS spectra are shown every 0.2 V and are offset on the y-axis for clarity. The spectral region between 1800 cm−1 and 2240 cm−1 is omitted for clarity. | ||
Fig. 4a (black trace) shows a CV of ORR/OER in an O2-saturated TBATFSI/G4 electrolyte at a scan rate of 1 mV s−1 on a gold electrode. The potential was first swept from OCP (ca. 3.3 V) in the negative direction in a potential region between 1.7 V and 3.7 V. In the negative-going sweep, a cathodic current started to flow from 2.8 V with a large reduction at 2.1 V. An oxidation peak was observed at 2.3 V in the subsequent positive-going sweep. The cathodic and anodic peak pair should be attributed to the redox reaction between O2 and its one-electron reduction product, superoxide (O2−) ion, in an aprotic electrolyte solution.38,39 Interestingly, the cathodic charge passed is much higher than the anodic one, showing fewer O2− species are oxidized to O2 in the reversed potential sweep. As discussed previously, this irreversible redox behavior implies that some of the highly reactive O2− species are consumed by an oxidative decomposition reaction with G4 solvent.21 Thus, O2− concentration near the electrode surface significantly decreases and induces more reduction of O2 to match the polarization potential. The redox peaks are similar to that previously observed on the graphene electrode surface.21 Slight differences in the peak position and anodic currents in the positive potential region should be related to the cell configuration of the two experiments, especially the influence of the thin electrolyte layer geometry used in the IRRAS measurements. In addition, a small anodic current was also observed in the potential region more positive than 2.6 V, with two small oxidation peaks at 3.0 V and 3.5 V.
Fig. 4b shows the in situ IRRAS spectra in selected frequency regions simultaneously recorded with the CV (Fig. 4a) in O2-saturated 0.1 M TBATFSI/G4 electrolyte. Since the complicated IR bands from the potential-induced migration of the TFSI− anion in the thin layer during the ORR/OER are significantly involved in the low-frequency region of the spectra, we will focus only on major spectral features in the IRRAS spectra between 1500 cm−1 and 2500 cm−1 in the paper. First, as the potential was swept to a potential more negative than 2.5 V, an upward peak appeared at 1609 cm−1 and rapidly increased with decreasing potential. Even though the sweep direction was changed to positive, the band kept growing slightly. Then, the band intensity started to decline from 2.9 V and became almost ignorable around 3.7 V. Second, a new upward band appeared at 2338 cm−1 in the reversed positive-going sweep at 3.0 V and immediately increased with the potential increase.
As mentioned in the Experimental section, the upward IR band indicates a formation or an accumulation of the species on the electrode surface in the thin layer. The present observations show that at least two new species are generated during the ORR/OER process in the Li+-free electrolyte.
The IR peak at 1609 cm−1 can be assigned to the asymmetric stretching mode of the carboxylic groups (COO−) of carboxylic species, such as acetate and formate anions.40 The appearance of the 1609 cm−1 band indicates that the G4 solvent becomes unstable during the ORR process in the Li+-free electrolyte solution. On the other hand, the IR peak at 2338 cm−1 can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric stretching mode of carbon dioxide (CO2),40,41 a specific oxidation final product of the carbon species, and demonstrates further oxidation of the G4 solvent in the OER region.
O asymmetric stretching mode of carbon dioxide (CO2),40,41 a specific oxidation final product of the carbon species, and demonstrates further oxidation of the G4 solvent in the OER region.
The blue and red symbols in Fig. 4a show potential dependences of the IR peak intensities of 1609 cm−1 and 2338 cm−1, respectively, together with the CV (black trace) observed in the Li+-free G4 solution. One can see a good correlation between the electrochemical behaviors and IR peak evolutions. IR band intensity at 1609 cm−1 increases with the reduction current of O2 in the negative-going sweep but does not decrease with the appearance of the corresponding oxidation peak at 2.3 V in the positive-going sweep. This indicates that the IR peak should be associated with the novel species generated during the ORR, but not O2 and O2− species. From the IR peak assignments described above, this band is an important indicator for the decomposition products or intermediates of G4 molecules with the reactive oxygen species (O2−). On the other hand, it is interesting to note that this IR peak intensity decreases simultaneously with the additional oxidation current and the evolution of CO2 species in the positive-going sweep (Fig. 4a). The band intensity for the CO2 reached a maximum at 3.2 V and then continually decreased even though the potential became more positive. These behaviors imply that the CO2 observed in Fig. 4b should be mainly contributed by the further oxidation of carboxylic species formed in the preceding ORR stage. As soon as the ORR-induced species was oxidized, the CO2 peak intensity started to decrease. The CO2 observed here is not newly formed but is that remaining in the thin layer after the slow diffusion process. These results indicate that the ORR process promotes the decomposition of the G4-based electrolyte solution in the Li+-free G4 electrolyte.
Fig. 4c and d show in situ IRRAS results in selected frequency regions in O2-saturated LiTFSI/G4 electrolyte (3.1 V → 1.7 V → 4.5 V → 3.1 V). The positive limit of the measurement (4.5 V) is higher than the measurement mentioned above in the Li+-free solution (3.7 V) due to the high OER overpotential in the Li+-containing electrolyte.42 In the first negative-going ORR sweep (3.1 V → 1.7 V), the cathodic current appeared from 2.8 V with a reduction peak at 2.2 V. In the positive-going sweep, a large anodic current flowed from 3.0 V to 4.5 V with two oxidation peaks at 3.2 V and 3.6 V. However, the large cathodic current observed in the Li+-free solution (Fig. 4a) was not found in the Li+-containing solution (Fig. 4c). A sequential two-electron ORR occurs when Li+ is included in the solution, forming lithium superoxide (LiO2) and lithium peroxide (Li2O2).43 Li2O2 is known to be less reactive in comparison with LiO2 or O2−, but the reversibility for ORR and OER becomes worse than that in the Li+-free solution.21
The in situ IRRAS spectra (Fig. 4d, 1500 cm−1 to 2500 cm−1) recorded in the Li+-containing solution show a small and broad upward peak at 1630 cm−1 in the first negative-going ORR sweep at a potential more negative than 2.5 V. In the following positive-going sweep, an upward peak at 2338 cm−1 was observed from 3.2 V.
The upward peak at 1630 cm−1 was not found in our previous study21 but could be reproducibly observed in recent measurements in the O2-saturated Li+-containing G4 electrolyte. However, this peak was not observed in the Ar-saturated solution, so it should be attributed to the species generated in the ORR in the Li+-containing G4 electrolyte. The peak position (1630 cm−1) differs from that observed in Li+-free solution (1609 cm−1, Fig. 4b). This peak at 1630 cm−1 is tentatively assigned to the CC stretching mode of vinyl hydrocarbon species generated during ORR. After the hydrogen is abstracted from G4 molecules by LiO2 or O2−, vinyl hydrocarbon species and alcohol, such as (2-methoxyethyl) vinyl ether and 2-methoxyethanol, can be formed by C–O cleavage reactions.44–46 Similar to that observed in Li+-free G4 electrolytes, the IR peak at 2338 cm−1 is assigned to CO2. Assuming the absorbance coefficients for the species are comparable, the amount of the ORR-induced species in the Li+-containing solution is much less than that in the Li-free solution, indicating the higher reactivity of O2− compared to Li2O2.
The blue and red symbols in Fig. 4c show the potential dependences of the IR peak intensities at 1630 cm−1 and 2338 cm−1, respectively, together with the CV (black trace) observed in the Li+-containing G4 solution. IR peak intensity at 1630 cm−1 increases in the negative-going ORR sweep and the positive-going sweep until 3.4 V, indicating that the ORR-induced products (such as vinyl ether) continually accumulate until this potential. When the potential becomes more positive than 3.4 V, the IR band intensity at 1630 cm−1 gradually decreases with an increase in the IR band at 2338 cm−1.
In comparison to the Li+-free G4 electrolyte solution, the CO2 band appears at a more positive potential. The re-oxidative decomposition of the ORR-induced species occurs with difficulty since the electrode surface was covered by the Li2O2 species under the conditions. The decomposition of the ORR-induced species, as well as the CO2 formation, gradually increased the free electrode surface. The peak intensity for CO2 continuously increased until 4.5 V with the decrease in the ORR-induced species (such as vinyl hydrocarbon).
On the other hand, the oxidation current became higher when the potential was higher than 4.0 V. Suppose we first sweep the potential in the positive direction (i.e., OER before ORR). In that case, we can also see the formation of the CO2 species by the in situ IRRAS around 4.0 V. These features indicate that an additional oxidation reaction occurs at the positive potential region, probably the electrochemical oxidation of the G4 solvent. Therefore, when the potential becomes very positive, the contribution from the direct electrochemical oxidation of the G4 solvent will be more significant.
It is interesting to note that the onset potentials for formation (ca. 2.7 V) and decomposition (ca. 3.4 V) for the ORR-induced species (such as vinyl hydrocarbon) coincide well with the low and high limits of the potential hysteresis obtained by the above SFG measurements for G4 (Fig. 3c, blue symbols). We speculate that a layer of solvent decomposition products is present on the graphene electrode surface during the ORR, which significantly changes the interaction between G4 and graphene and thus induces a large redshift in the frequency. In the OER process, the frequency returns to the original value as soon as the oxidation of the layer occurs. Therefore, the adsorption layer of the ORR-induced decomposition products of G4 is expected to be one of the possible reasons for the potential hysteresis observed by SFG. More detailed IRRAS studies on the other glymes are in progress. It should be mentioned that such ORR-induced decomposition was not observed in the DMSO-based elctrolyte solutions, indicating that DMSO solvent is stable to LiO2 and O2− species. This is considered as a origin for the absence of potential hysteresis phenomenon in the DMSO-based solutions.
Conclusions
In summary, we have employed in situ vibrational SFG and IRRAS spectroscopies to investigate the mechanisms of ORR/OER with glyme-based electrolytes. Our SFG study revealed that the cations significantly affect the interfacial structures and stabilities of the glyme solvents due to different solvation abilities. The SFG results show that the adsorption states of glymes on the electrode surface change with the ORR/OER process, showing a large potential hysteresis. Furthermore, in situ IRRAS measurements revealed the decomposition products of G4 molecules during ORR in TBA+-containing and Li+-containing solutions. For the TBA+-containing G4 electrolyte, carbon dioxide (CO2) can be observed when the potential is higher than 2.9 V during the positive-going sweep, which can be attributed to the oxidation of decomposition products formed during ORR. On the other hand, the CO2 band appears when the potential is higher than 3.5 V for the Li+-containing G4 electrolyte, which could be attributed to the oxidation of both decomposition products and G4 molecules. Formation and decomposition of the ORR-induced decomposition products in the Li+-containing electrolyte solution may be one of the origins of the potential hysteresis observed by SFG measurements.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
SY thanks the funding from the Advanced Low Carbon Technology Research and Development Program (ALCA), specially promoted research for innovative next-generation batteries (SPRING), from the Japan Science and Technology Agency (JST). RN thanks a fellowship from the Graduate Program for Green and Digital Innovation (GreDi) of Tohoku University, Japan.References
- K. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS.
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- T. Liu, J. P. Vivek, E. W. Zhao, J. Lei, N. Garcia-Araez and C. P. Grey, Chem. Rev., 2020, 120, 6558–6625 CrossRef CAS.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- J.-H. Kang, J. Lee, J.-W. Jung, J. Park, T. Jang, H.-S. Kim, J.-S. Nam, H. Lim, K. R. Yoon, W.-H. Ryu, I.-D. Kim and H. R. Byon, ACS Nano, 2020, 14, 14549–14578 CrossRef CAS PubMed.
- W. Xu, K. Xu, V. V. Viswanathan, S. A. Towne, J. S. Hardy, J. Xiao, Z. Nie, D. Hu, D. Wang and J.-G. Zhang, J. Power Sources, 2011, 196, 9631–9639 CrossRef CAS.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- Q. Peng, Y. Qiao, K. Kannari, A. Ge, K. Inoue and S. Ye, J. Phys. Chem. C, 2020, 124, 15781–15792 CrossRef CAS.
- A. Ge, D. Zhou, K. Inoue, Y. Chen and S. Ye, J. Phys. Chem. C, 2020, 124, 17538–17547 CrossRef CAS.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, G. Girishkumar and A. C. Luntz, J. Phys. Chem. Lett., 2011, 2, 1161–1166 CrossRef CAS PubMed.
- D. Sharon, V. Etacheri, A. Garsuch, M. Afri, A. A. Frimer and D. Aurbach, J. Phys. Chem. Lett., 2013, 4, 127–131 CrossRef CAS PubMed.
- D. Sharon, D. Hirshberg, M. Afri, A. Garsuch, A. A. Frimer and D. Aurbach, Isr. J. Chem., 2015, 55, 508–520 CrossRef CAS.
- J. Wandt, P. Jakes, J. Granwehr, H. A. Gasteiger and R. A. Eichel, Angew. Chem., 2016, 128, 7006–7009 CrossRef.
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine and D. Kramer, Nat. Energy, 2017, 2, 17036 CrossRef CAS.
- E. Mourad, Y. K. Petit, R. Spezia, A. Samojlov, F. F. Summa, C. Prehal, C. Leypold, N. Mahne, C. Slugovc, O. Fontaine, S. Brutti and S. A. Freunberger, Energy Environ. Sci., 2019, 12, 2559–2568 RSC.
- Q. Yu and S. Ye, J. Phys. Chem. C, 2015, 119, 12236–12250 CrossRef CAS.
- C. Liu and S. Ye, J. Phys. Chem. C, 2016, 120, 25246–25255 CrossRef CAS.
- Q. Peng, J. Chen, H. Ji, A. Morita and S. Ye, J. Am. Chem. Soc., 2018, 140, 15568–15571 CrossRef CAS PubMed.
- A. Ge, R. Nagai, C. Xu, K. Kannari, B. Peng, K. Inoue, A. Morita and S. Ye, J. Phys. Chem. C, 2022, 126, 2980–2989 CrossRef CAS.
- C. Xu, A. Ge, K. Kannari, B. Peng, M. Xue, B. Ding, K. Inoue, X. Zhang and S. Ye, ACS Energy Lett., 2023, 8, 1289–1299 CrossRef CAS.
- Y. Shen, Fundamentals of Sum-Frequency Spectroscopy, Cambridge University Press, 2016 Search PubMed.
- H. F. Wang, W. Gan, R. Lu, Y. Rao and B. H. Wu, Int. Rev. Phys. Chem., 2005, 24, 191–256 Search PubMed.
- A. J. Cowan and L. J. Hardwick, Annu. Rev. Anal. Chem., 2019, 12, 323–346 Search PubMed.
- S. Baldelli, Acc. Chem. Res., 2008, 41, 421–431 CrossRef CAS.
- N. G. Rey and D. D. Dlott, J. Electroanal. Chem., 2017, 800, 114–125 CrossRef CAS.
- A. Ge, K. Inoue and S. Ye, J. Chem. Phys., 2020, 153, 170902 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung and E. Tutuc, Science, 2009, 324, 1312–1314 CrossRef CAS.
- K. Inoue, C. Takada, L. Wang, A. Morita and S. Ye, J. Phys. Chem. B, 2020, 124, 5246–5250 CrossRef CAS PubMed.
- S. Ye, A. Yashiro, Y. Sato and K. Uosaki, J. Chem. Soc., Faraday Trans., 1996, 92, 3813–3821 RSC.
- Z. Chen, R. Ward, Y. Tian, S. Baldelli, A. Opdahl, Y.-R. Shen and G. A. Somorjai, J. Am. Chem. Soc., 2000, 122, 10615–10620 CrossRef CAS.
- S. Nihonyanagi, J. A. Mondal, S. Yamaguchi and T. Tahara, Annu. Rev. Phys. Chem., 2013, 64, 579–603 CrossRef CAS PubMed.
- C. Fu, L. Xu, F. W. Aquino, A. v. Cresce, M. Gobet, S. G. Greenbaum, K. Xu, B. M. Wong and J. Guo, J. Phys. Chem. Lett., 2018, 9, 1739–1745 CrossRef CAS.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2010, 114, 9178–9186 CrossRef CAS.
- C. O. Laoire, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, J. Electrochem. Soc., 2011, 158, A302 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS.
- D. Vasudevan and H. Wendt, J. Electroanal. Chem., 1995, 392, 69–74 CrossRef.
- M. E. Peover and B. S. White, Electrochim. Acta, 1966, 11, 1061–1067 CrossRef CAS.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, in Introduction to Infrared and Raman Spectroscopy, Academic Press, San Diego, 3rd edn, 1990 Search PubMed.
- G. Horwitz, E. J. Calvo, L. P. Méndez De Leo and E. de la Llave, Phys. Chem. Chem. Phys., 2020, 22, 16615–16623 RSC.
- B. D. McCloskey, R. Scheffler, A. Speidel, G. Girishkumar and A. C. Luntz, J. Phys. Chem. C, 2012, 116, 23897–23905 CrossRef CAS.
- C. Laoire, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, J. Electrochem. Soc., 2011, 158, A302 CrossRef CAS.
- M. Ue, H. Asahina, S. Matsuda and K. Uosaki, RSC Adv., 2020, 10, 42971–42982 RSC.
- Y. Gao, H. Noguchi and K. Uosaki, RSC Adv., 2023, 13, 5467–5472 RSC.
- Y. Gao, H. Noguchi and K. Uosaki, ACS Energy Lett., 2023, 8, 1811–1817 CrossRef CAS.
Footnote |
| † Equal contributions. |
| This journal is © The Royal Society of Chemistry 2024 |