
Injectable smart stimuli-responsive hydrogels: pioneering advancements in biomedical applications
Jiacheng
Liu†
 ,
Chengcheng
Du†
,
Wei
Huang
* and
Yiting
Lei
*
,
Chengcheng
Du†
,
Wei
Huang
* and
Yiting
Lei
*
Department of Orthopedics, Orthopedic Laboratory of Chongqing Medical University, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, China. E-mail: huangw511@163.com; leiyit614@163.com; Tel: +8613883383330 Tel: +8617880267665
First published on 25th October 2023
Abstract
Hydrogels have established their significance as prominent biomaterials within the realm of biomedical research. However, injectable hydrogels have garnered greater attention compared with their conventional counterparts due to their excellent minimally invasive nature and adaptive behavior post-injection. With the rapid advancement of emerging chemistry and deepened understanding of biological processes, contemporary injectable hydrogels have been endowed with an “intelligent” capacity to respond to various endogenous/exogenous stimuli (such as temperature, pH, light and magnetic field). This innovation has spearheaded revolutionary transformations across fields such as tissue engineering repair, controlled drug delivery, disease-responsive therapies, and beyond. In this review, we comprehensively expound upon the raw materials (including natural and synthetic materials) and injectable principles of these advanced hydrogels, concurrently providing a detailed discussion of the prevalent strategies for conferring stimulus responsiveness. Finally, we elucidate the latest applications of these injectable “smart” stimuli-responsive hydrogels in the biomedical domain, offering insights into their prospects.
 Wei Huang | Prof. Wei Huang serves as the Director of the Department of Orthopedics at the First Affiliated Hospital of Chongqing Medical University, where he also holds the position of Director of the Orthopedics Laboratory. Additionally, he holds the position of Vice President of the Orthopedic Physicians Branch of the Chinese Medical Association. Prof. Wei Huang has developed a distinct research focus in areas such as the pathogenesis and treatment of osteoarthritis, biomaterials, stem cell therapy, and the formation and repair of bone and cartilage, and he is dedicated to translating research findings into clinical applications. |
 Yiting Lei | Yiting Lei is currently serving in the orthopedic department of the First Affiliated Hospital of Chongqing Medical University. His research focuses on the design and application of biomaterials for bone and joint diseases. Additionally, he serves as a member of the editorial board for BMC Biomedical Engineering and holds positions as a youth editor for journals such as Exploration, VIEW, Interdisciplinary Medicine, etc. |
1. Introduction
Hydrogels are three-dimensional water-swollen networks with hydrophilic nature crosslinked by various hydrophilic polymers via chemical covalent bonds, physical non-covalent bonds or a combination of both.1–4 Meanwhile, hydrogels could be further classified as natural, synthetic or hybrid based on the source of constituting materials.4–6 Due to their excellent biocompatibility, extracellular matrix (ECM)-like structure and immensely tunable properties (modified through physical/chemical methods) for specific applications, hydrogels are one of the most widely utilized soft biomaterials in tissue engineering (providing mechanical support and scaffold for tissue regeneration, transporting drugs/cells/cytokines/bioactive molecules, etc.).2,7–13 After the first appearance of the term “hydrogel” in the scientific literature in 1894,14 great progress has been made in hydrogels and related fields, especially in the last few decades. To date, the historical development of hydrogels could be divided into three different generations.6 The first generation of hydrogels mainly consisted of water-soluble monomers, synthetic polymers and cellulose, and the polymerization progress was mainly achieved through relatively simple chemical crosslinking techniques.6 The landmark in hydrogel research was reported in 1960 by Wichterle and Lim.15 They synthesized poly(2-hydroxyethyl methacrylate) (pHEMA) hydrogel by free radical polymerization of 2-hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA), and employed it for use in contact lenses with moisture-absorbing ability, which then remained the basis of many contact lenses for five decades thereafter.16,17 Then, in the 1970s, stimuli-responsive properties were introduced into the hydrogel field, endowing the second generation of hydrogels with the capability of responding to specific stimuli (such as temperature and pH) in the application environment.18 This innovation expanded a much larger application range of hydrogels, such as in situ gelling reaction or controlled drug release.19–22 Thereafter, the research focus of hydrogels switched to the preparation of stereocomplexed hydrogels with enhanced thermal, mechanical and degradation properties through more physical interactions (apart from the hydrophobic and ionic interactions in the first generation) in the 1990s.23–26 Nowadays, owing to the increasing knowledge in organic and emerging chemistry, scientists are more intently focused on creating “smart hydrogels” to acquire tailored characteristics, such as in situ-gelling injectable hydrogels with stimulus-reactive and tunable mechanical and/or other physicochemical properties.Although marvelous achievements have been obtained in biomedicine and tissue engineering, such as the emergence of hydrogels, realization of the potential of hydrogels for healthcare and treatment of human diseases is still the tip of the iceberg, and efforts are continuously being made to make this biomaterial play more valuable and beneficial roles in combating various pathophysiological processes. For example, traditional hydrogels are hydrated polymer networks with a characteristic mesh size of nanometer size, allowing the transportation of various biomolecules through the hydrogel network.27 However, the diffusion efficiency for polysaccharides and proteins will be compromised significantly within the nano-sized porous structure in bulk hydrogels larger than 200 μm.28 Therefore, small-size hydrogels (microgels) were then generated to encapsulate bioactive macromolecules, load live cells or for controlled release of drugs, or to achieve higher control over the environmental factors of the ECM for the three-dimensional culture of organoids or cells.27 Additionally, the characteristics of traditional hydrogels are mainly decided by the crosslink density and polymer contents, which actually limit their internal structure and mechanical strength significantly.29 To break these limitations and expand the application areas, numerous methods have been used for the modification of traditional hydrogels. In this context, dynamic-covalent coupling (such as Schiff-base reaction, disulfide exchange and Diels–Alder reaction, etc.) can endow hydrogels with the abilities of self-healing and shear-thinning under shear force, which were then further utilized to generate injectable hydrogels.7,30 Furthermore, microfluidics techniques can be used to prepare hydrogel microspheres (HMs), which can also make hydrogels become injectable, and empower them with the ability to bear lubrication.31,32 In summary, emerging chemistry and new techniques have helped modify the properties of traditional hydrogels and enabled many new applications (such as direct injection then gelling in situ, generating HMs, injectability based on shear-thinning property, and playing a role in 3D printing as bio-ink), then solved many of the remaining obstacles of traditional hydrogels.
Many traditional hydrogels were prefabricated with fixed shapes before use, which cannot accurately fill irregular cavities following solid tumor resection or large and complex bone defects, and thus provided limited effects as therapeutics.33,34 In contrast, injectable hydrogels are more attractive and promising than the conventionally generated hydrogels, as they can adapt to the irregular shape of the targeted tissues/defects through a simple injection.35–37 Moreover, injectable hydrogels can reach deep lesions and fill any cavities easily through injection, as well as release the loaded therapeutic cargo to the surrounding tissue for a prolonged time.38,39 Meanwhile, as a minimally invasive technology through local injection, injectable hydrogels can reduce surgery-induced trauma and blood loss, or even avoid invasive surgery, and overcome the clinical and surgical limitations of traditional hydrogel stenting by tunable sol–gel transition, which has contributed to improved therapeutic efficacy and better patient compliance.40–43 In addition, due to their viscoelastic and diffusive nature, injectable hydrogels can contribute to tissue regeneration in multiple ways, including providing mechanical support, delivering controlled-release cells or therapeutics spatiotemporally, and recruiting and/or modulating host cells.42 As a result, injectable hydrogels have become a research hotspot and are at the forefront of advanced strategies for tissue regeneration.
Besides the mentioned advantages, with the rapid development of emerging chemistry and new technologies, injectable hydrogels have gained “smart” responsiveness to various stimuli, playing a crucial role in diverse biomedical applications. Hence, a timely summary of these advanced injectable hydrogels is necessary to aid researchers in understanding their current status and facilitating future development. However, currently there is no comprehensive summary available for these advanced injectable “smart” hydrogels, such as the selection of raw materials for hydrogel preparation, various crosslinking methods for different raw materials, common strategies for achieving injectability, and how to impart “smart” responsiveness to hydrogels in response to specific stimuli. To address this gap, we first describe the raw materials and the gelation principle of injectable hydrogels. Next, we summarize the common strategies used to endow hydrogels with injectability and responsiveness to specific endogenous/exogenous stimulus. Finally, we focus on the current application of injectable smart stimuli-responsive hydrogels in various diseases and biomedical practices, and give our prospects for their future development.
2. Fabrication of injectable hydrogels
2.1. Biological materials for injectable hydrogels
Biological materials utilized for the preparation of injectable hydrogels are required to meet the following basic requirements, including adequate biocompatibility and appropriate biodegradation rate, low toxicity and no toxic byproducts during gelation, appropriate sol–gel transition ability under physiological conditions, similar structure to the targeted tissue, support for cell adhesion and controlled release of biomolecules.44,45 The raw materials for current injectable hydrogels can be divided into natural and synthetic materials (Fig. 1). Many natural materials exist in the human body, and thus are preferred due to their outstanding advantages (including excellent biocompatibility, biostability, biodegradability, nontoxicity, ecofriendliness, and being economically friendly and readily available) in biomedical research.46–48 Compared with that, synthetic materials are valued for their stronger mechanical strength and readily controlled physical/chemical properties (such as matrix stiffness, pore size, proteolytic degradability and cell adhesion sites), which allow the synthesis of tailored injectable hydrogels for specific circumstances.46 Herein, the most widely used natural and synthetic materials for the preparation of injectable hydrogels are summarized.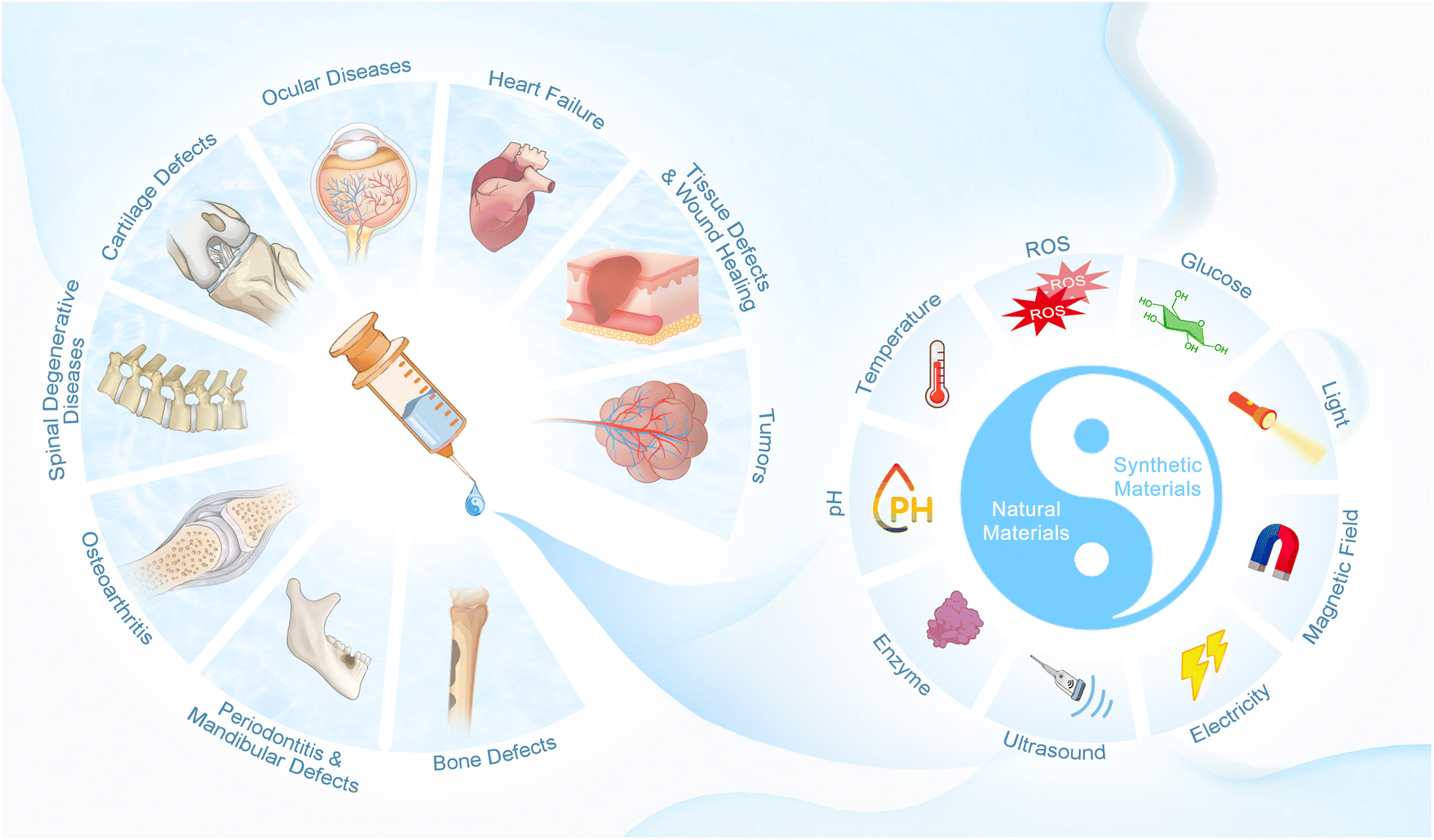 | ||
| Fig. 1 Schematic summary of injectable smart stimuli-responsive hydrogels. | ||
2.1.1.1. Alginate. Alginate is a natural polysaccharide extracted from the cell walls and cytoplasm of algae, and is composed of repeating blocks of polyguluronate and polymannuronate, which is linked by α-1,4 glycosidic bonds.5,47,52,53 Alginate is one of the most commonly used biomaterials in tissue engineering because it can create hydrogel networks rapidly under relatively mild conditions, and it possesses many advantages including low cost and wide sources, minimal toxicity and immunogenicity, high water absorption and biodegradability, and soft protein encapsulation.5,46
As an anionic polysaccharide polymer composed of numerous carboxyl groups, alginate can polymerize with cations in aqueous solution and then transit to gelation rapidly with divalent cations (such as Ca2+ and Ba2+) under relatively mild conditions.5,54 As a result, alginate is widely used as a matrix material for injectable hydrogels. For example, Landa et al. reported an injectable alginate-based hydrogel with sodium alginate and calcium gluconate, which could form gel in situ after injection through calcium crosslinking.55 They found this in situ-forming and biodegradable alginate-based injectable hydrogel could promote cardiac function by preventing adverse cardiac remodeling in myocardial infarctions. Hu and colleagues prepared a special injectable hydrogel dressing with gallic acid-functionalized silver nanoparticles (GA@AgNPs) and sodium alginate molecular chains (SA) by the cross-link via Ca2+.56 This alginate-based hydrogel was able to consistently release Ag+ and inhibit the formation of biofilm, alleviate inflammation and promote angiogenesis, and finally achieve prolonged antimicrobial performance and accelerated healing of bacteria-infected wounds.
Nevertheless, the cell adhesion property and mechanical strength of alginate are quite limited, and thus limit the application of the pure alginate-based hydrogel in biomedicine.53 Hence, many other polymers and/or particles are mixed with alginate to break its inherent limitations in practice. Based on the good cell recruitment physical properties of silica, Ghanbari et al. added silica nanoparticles into injectable hydrogels consisting of oxidized alginate and gelatin, and found the hydrogel was endowed with increased cell recruitment activity.57 Similarly, the team of Balakrishnan prepared a self-assembled gel by the Schiff-base reaction between periodic acid-oxidized alginate and gelatin in the presence of borax, and found it could recruit cells and ECM.54 On the other hand, Wang et al. used polyglutamic acid and sodium alginate to fabricate an injectable dynamic crosslinked hydrogel system via Schiff-base bonds, and added microcrystalline cellulose to prolong the degradation time and reduce the swelling rate.58 The addition of microcrystalline cellulose was demonstrated be an effective way to increase the mechanical strength of the alginate-based hydrogel system, which was achieved by forming concentration-dependent multiple hydrogen bonds and reducing the degradation of the dynamic crosslinking networks effectively. Moreover, the team of Growney Kalaf et al. assessed the different gelation characteristics of 1% sodium alginate-based injectable hydrogels with various molar concentrations of CaCO3 and glucono-δ-lactone (GDL), while the ratio of CaCO3 and GDL was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.59 After a series of experiments, they finally found that the optimal concentrations of GDL were 30 mM and 60 mM to synthesize a mechanically stronger injectable alginate hydrogel with prolonged crosslinking time, better water retention property and long-term mechanical stability.
:2.59 After a series of experiments, they finally found that the optimal concentrations of GDL were 30 mM and 60 mM to synthesize a mechanically stronger injectable alginate hydrogel with prolonged crosslinking time, better water retention property and long-term mechanical stability.
In addition, owing to the lack of angiogenic and osteogenic ability, alginate is greatly restricted in tissue engineering such as bone/cartilage regeneration.60 However, due to the polycation behavior of alginate in solution, it can crosslink with many cations with biological effects and form three-dimensional injectable hydrogel networks rapidly, which provides opportunities for alginate-based hydrogels in the regeneration process of irregular tissue defects.5,61 Zhang and colleagues prepared a novel injectable hydrogel system (composed of sodium alginate, akermanite and glutamic acid) for irregular bone repair.60 The akermanite of this hydrogel can release Si4+, Ca2+ and Mg2+ in aqueous solution, which enables this system to form gel as well as affect cell behaviors (Si4+ and Ca2+ contribute to the mineralized deposition of mesenchymal stem cells, and Mg2+ enhances cell adhesion and proliferation).60,62–64 Compared with the alginate control group, this system was then proved to promote osteogenic differentiation and almost double the migration ability of human bone marrow stromal cells, and finally contributed to bone regeneration. Furthermore, Zhu et al. designed an injectable hydrogel system based on alginate to promote osteochondral regeneration.65 This novel injectable system possessed stratified structures with different functionally biomimetic constructs, which were designed to enhance subchondral bone regeneration and articular cartilage regeneration, respectively. Bioglass (BG), which has been well demonstrated to induce bone marrow stem cells’ (BMSCs) osteogenic differentiation and bone formation, was added to this composite hydrogel.66,67 Meanwhile, the added BG could release bivalent cations to enable the system to gel with alginate. Finally, this composite injectable hydrogel with stratified structures was proved to contribute to both the subchondral bone and hyaline cartilage regeneration, and improve the integration between the host tissues and the new tissues.
2.1.1.2. Chitosan. Chitosan is a naturally derived linear polysaccharide by partial deacetylation of chitin, one of the major polysaccharides found in the shells of insects, lobsters and crabs.68 It is mainly composed of glucosamine and N-acetyl glucosamine units, and its structure is similar to the glycosaminoglycan of human ECM.5,46 Chitosan is valued as an important source of injectable hydrogels for many advantages, including low cost, high bioactivity and biocompatibility, good antibacterial quality and tunable biodegradability through the change of the deacetylation level.69
However, the chitosan hydrogel is crosslinked by hydrogen bonding, which results in limited compressive strength and elastic modulus.5 Furthermore, the application of pure chitosan is significantly restricted as it degrades easily in the presence of lysozyme, and it is fragile with weak mechanical property.46,61 Fortunately, the combination of chitosan with gelatin and/or chemical polymers can empower the hydrogel with stronger mechanical properties and stability.70 Ghorbani et al. enhanced the mechanical strength of chitosan by adding fibroin silk, gelatin, collagen II and chondroitin sulfate, and found an optimal proportion of components to acquire the best energy storage modulus.71 Ghavami and colleagues found the addition of calcium phosphate into the cellulose nanocrystal/chitosan hydrogel endowed it with significant osteogenic induction ability as well as improved compressive strength.72
As a cationic polymer with numerous amino groups on the backbone, chitosan is an ideal choice for the preparation of injectable hydrogels based on imine reactions (like the Schiff-base reaction).73 For example, Deng et al. synthesized an injectable hydrogel with rapid self-healing characteristic through the Schiff-base reaction between hydroxypropyltrimethyl ammonium chloride chitosan (HACC) and dialdehyde-modified bacterial cellulose (DABC).74 The conversion from chitosan to HACC added the water solubility of chitosan, and the Schiff-base bond between the amine groups of HACC and the aldehyde groups of DABC enabled the hydrogel's injectability and self-healing ability.
Moreover, since chitosan is easily affected by pH and temperature, it is well suited for the preparation of pH-responsive and temperature-responsive injectable hydrogels.46,75 The team of Guedes utilized chitosan to create a double dynamic network (imine bond and electrostatic interactions) containing doxorubicin (DOX) and {Mo154} (a polyoxometalate) for synergistic chemotherapy and photothermal therapy.76 According to the release profile, this chitosan-based hydrogel performed a pH-responsive release behavior, which enabled it to play a role as a controlled-release drug carrier in the acidic environment of tumors. Meanwhile, Kim et al. found the mixed injectable hydrogel of oxidized succinoglycan (OSG) with chitosan performed a pH-controlled release, and the release rate of loaded 5-fluorouracil was increased from 60% to 90% when the pH was changed from 7.4 to 2.0.77 On the other hand, Ghorbani and colleagues assessed the viscoelastic properties of a chitosan-based injectable hydrogel, and they found this hydrogel could remain as solution at 4 °C and would transit into gelation at 37 °C, under which the storage modulus of the hydrogel could be constant over a wide strain range.71 Similarly, Zhu et al. prepared a thermosensitive injectable self-assembled hydrogel (TISH) using chitosan which could transition from liquid phase to gelation phase upon incubation at 37 °C.78 This hydrogel was loaded with resveratrol and granulocyte-macrophage colony-stimulating factor (GM-CSF) to release the bioactive molecules to induce tolerogenic dendritic cells and regulatory T-cells, and finally attenuate diabetic periodontitis.
2.1.1.3. Chondroitin sulfate. Chondroitin sulfate (CS) is one of the main components of the chondrocyte ECM, and the most abundant glycosaminoglycan in the human body, which is composed of alternating units of glucuronic acid and N-acetylglucosamine.53,61 Generally, the CSs can be divided into 4 different types according to the sulfated sites, including GlcA-GalNAc-4-SO4 (CS-A), GlcA-GalNAc-6-SO4 (CS-C), GlcA-2-SO4-GalNAc-6-SO4 (CS-D) and GlcA-GalNAc4,6-diSO4 (CS-E).79 Meanwhile, the CSs in the human body are linked to various core proteins, producing proteoglycans.79 In the practice of biomedicine, CS is a commonly used natural biomaterial with prominent advantages, such as cell adhesion and ECM aggregation, supporting the signaling and intercellular communication of chondrocytes, promoting chondrogenesis and providing resistance to stress, inhibiting cartilage degradation and reducing inflammatory reaction.36,61,80
As one of the main components of cartilage, CS is rich in sulfate groups and possesses large negative charge, which is crucial for ionic interactions, steric hindrance, mechanical strength and resistance of cartilage to compression.61,79 This obvious negative-charge property makes CS-based hydrogel a promising delivery system for cationic drugs. Ornell and colleagues prepared a blended injectable CS-based hydrogel with CS-methacrylate (CSMA) and poly(vinyl alcohol)-methacrylate (PVAMA).81 According to the reported results, the release of DOX and sunitinib (two cationic oncology therapeutics) was prolonged to up to 6 weeks, and thus this electrostatic coupling strategy of cationic drugs with CS-based hydrogels was promising for contributing to the sustained release of cationic drugs in oncology therapeutics. Similarly, according to the reported results from the team of Keutgen, the injectable CS-based hydrogel loaded with sunitinib via electrostatic interaction could be degraded by endogenous hyaluronidases after injection into the tumor, release the sunitinib it contained in a sustained manner, and finally suppress the growth of pancreatic neuroendocrine tumors (PanNETs) without inducing significant systemic side effects.82
In addition, due to the antioxidant activity of CS,83 some CS-based injectable hydrogels are loaded with other antioxidants to achieve an enhanced curative effect of some oxidative stress-related diseases. He et al. utilized CS hydrogels modified with methacryloyl groups (ChsMA) to develop novel injectable microspheres, which were then endowed with dual antioxidant capacity by anchoring with liquiritin-loaded liposomes.84 According to the results of the in vivo experiments, the CS monomers from the degradation of ChsMA by endogenous enzymes and the released liquiritin (an antioxidant drug) could play a synergistic role in eliminating the reactive oxygen species (ROS) in the osteoarthritis (OA) model of rats. Meanwhile, this hybrid system also inhibited the interleukin-1β (IL-1β)-induced ECM degradation, M1 macrophage polarization and the activation of the inflammasome, and finally retarded the progress of OA.
However, although CS can effectively simulate the micro-environment of native cartilage, the application of pure CS-based hydrogels in cartilage regeneration is significantly limited due to the rapid degradation rate. Consequently, several inert synthetic molecules are utilized to regulate the degradational and mechanical properties of CS-based hydrogels. For example, Li et al. fabricated a functionalized injectable hydrogel (CS-SH/HB-PEG) with thiol-functionalized CS (CS-SH) and hyperbranched multifunctional PEG copolymer (HB-PEG), which was crosslinked via thiol–ene reaction.85 Compared with the pure CS-SH hydrogel, this novel hybrid CS-based injectable hydrogel was characterized by a significantly prolonged degradation time, rapid gelation, outstanding mechanical property and suitable porosity for cell loading, providing a favorable micro-environment for both the loaded mesenchymal stem cells (MSCs) and host cartilage cells, and finally contributed to chondrogenesis.
2.1.1.4. Hyaluronic acid. Hyaluronic Acid (HA) is a water-soluble natural linear anionic polysaccharide, and comprises the primary component of the ECM in epithelial, connective and neural tissues (including cartilage, skin, synovial joint fluid and the vitreous humor of the eyes).79,86–89 It consists of repeated alternate disaccharide units of D-glucuronic acid and N-acetylglucosamine, linked via β-1,4 and β-1,3 glycosidic bonds.90,91 As a negatively charged non-branched polymer consisting of non-sulfated glycosaminoglycans, HA has various unique characteristics, including outstanding hydrophilicity and water-binding capacity, satisfactory biocompatibility and biodegradability, low immunoreactivity and high bioactivity (such as modulating cell function as a signaling molecule).87,92,93 Simply put, HA exhibits various unexpected complex biological functions, because it can interact with cells through the molecular interactions with specific receptors on the cell membrane, such as CD44, toll-like receptor-4 (TLR-4), lymphatic vessel endothelial receptor-1 (LYVE-1) and receptor for hyaluronan-mediated motility (RHAMM), and thus contributes to the regulation of the activity and function of cells.86,94 As great progress has been made in the extraction of HA and hydrogel preparation technology, more functional HA-based injectable hydrogels with stronger mechanical properties and more stable structure are being developed for various specific biomedical applications, including tissue engineering, drug delivery and medical cosmetology.87
Generally, the molecular weight of HA found within the ECM is extremely high (>10000 kDa), and plays a crucial role in sequestering the cell-produced proteins in vivo.30,95 The common range of HA molecular weight is between 5 kDa to 200 kDa, and HA with different chain lengths exhibits quite different biological functions in tissues.87,92 HA with high molecular weight contributes to various biological processes (such as maintaining cell communication and integrity, tissue hydration and integrity) in vivo, and is found to play an important role in the inhibition of cell proliferation (like angiogenesis) and inflammation.96,97 However, when it comes to a condition of inflammation or injury, the HA with high molecular weight is degraded to low molecular weight (<100 kDa) by the upregulated hyaluronidase enzyme in vivo, and then stimulates wound healing by promoting inflammation, immunoreaction and angiogenesis.30 Although it is mainly degraded by ubiquitous hyaluronidase enzyme and oxidative processes in vivo, HA with high molecular weight can also be hydrolyzed into smaller fragments with low molecular weight under certain acidic or basic conditions in vitro.30,95 Hence, based on the size-dependent effect, HA can be used for the synthesis of tailored injectable hydrogel with defined functions for specific biological applications. Jung and colleagues designed a thermo-sensitive injectable hydrogel based on a high molecular weight HA (∼1000 kDa), which was physically mixed with Pluronic F-127, to achieve a sustained delivery of Piroxicam (PX) to the joint.98 According to the reported results, the high molecular weight HA-based injectable hydrogel not only exhibited significantly improved mechanical strength under physiological conditions, but also achieved a sustained release of the loaded drug. This was presumed to be the result of the inter-micellar packing in the inner structure of the hydrogel, which was provided by the high molecular weight HA. Similarly, Chen et al. prepared an in situ-forming and injectable hybrid hydrogel (oxi-HAG-ADH) based on oxidized high molecular weight HA (1900 kDa), gelatin and adipic acid dihydrazide (ADH).99 Compared with the hydrogel developed from 320 kDa HA in their earlier work, this oxi-HAG-ADH hydrogel was stiffer and exhibited stronger mechanical strength. This may be a result of the increased chain length of HA, which contributed to the steric hindrance between oxi-HA and ADH during the gelation process. Meanwhile, this hybrid injectable hydrogel was found to increase the expression of ECM-related genes (including AGN, COL2A1, HIF-1A and SOX-9) in nucleus pulposus (NP) cells.
HA is an non-branched polymer chain which cannot generate hydrogel networks by itself without forming cross-linkages between the chains through physical/chemical/combined reactions.27 Nevertheless, of note, there are many active groups on the chain of natural HA (such as amino, hydroxyl and carboxyl groups, etc.), which provide numerous possibilities to modify HA and generate injectable hydrogels through a variety of chemical methods (including carbodiimide crosslinking, enzymatic crosslinking, protein crosslinking, photocrosslinking, Schiff base crosslinking, Michael addition crosslinking and click-chemistry crosslinking).5,79 As a result, many chemical crosslinking agents are utilized to graft crosslinkable functional groups to HA, and the most commonly grafted sites on the HA chain are primary alcohol and carboxylic acid.27 Lei et al. inducted methacrylic anhydride into the chain of HA to generate methacrylated HA (HAMA), which was thus empowered with a photocrosslinking ability.32 Then, they utilized HAMA to synthesize injectable microspheres by photopolymerization processes and microfluidics technology, which carried liposomes encapsulating rapamycin, to alleviate osteoarthritis. However, due to the toxicity of the added chemical initiators, catalysis or crosslinking agents, these modified injectable HA hydrogels are reported to have a potential negative impact on cell vitality and activity and adverse reactions in vivo.30,100 Therefore, new methods to form HA-based hydrogels without the use of chemical crosslinking agents have also been explored a lot. In the research reported by Kim et al., sodium periodate was used to generate aldehyde groups in the backbone of HA, and this modification gave the HA-based hydrogel the ability to form gels with glycol chitosan via Schiff base reaction under mild conditions, which avoided the use of chemical crosslinking agents.101 This Schiff base bonds-based injectable HA hydrogel was regarded as a promising carrier of cells in the practice of tissue engineering (such as cartilage regeneration) due to its excellent biocompatibility and prolonged durability.
However, like other naturally derived biomaterials, the application of HA hydrogels is troubled by poor mechanical properties and relatively rapid degradation by endogenous enzymes.5,46,87 These disadvantages result in the vulnerability of HA-based injectable hydrogels under mechanical stress and short half-life period in vivo, and thus have significantly limited its application in tissue engineering requiring load-bearing.46,87,102,103 Therefore, appropriate modification is required to improve these properties of HA before expecting it to exhibit better therapeutic effects. According to the published reports, some novel strategies (such as double-crosslinking methods and low-temperature free-radical polymerization) have been explored to improve the mechanical properties of HA-based hydrogels.104,105 However, HA hydrogels modified through these methods are not the final answer, because there still remain some problems including the surgical trauma of implantation, failure in filling irregular-shaped defects completely and instability of structures under frequent stress.7,106 Hence, smart HA-based hydrogels with more advanced functions (such as injectability, the properties of shear-thinning or self-healing) have emerged as the times require in recent years.61 One of the most widely used methods to endow HA-based hydrogel with the above advanced functions is dynamic covalent coupling (DCC) chemistry.107 DCC is a series of dynamic reactions (including esterification, disulfide exchange and Diels–Alder reactions, the formation of imine, oxime, hydrazone and boronic ester) which results in the adaptable and reversible poly networks of hydrogels, and then endows the hydrogel with stimuli-responsive properties like shearing thinning and self-healing.108 Lei et al. prepared a shear-responsive injectable HA hydrogel, which carried liposomes encapsulating celecoxib inside, via the Schiff base reaction between the adipic dihydrazide-modified HA (HA-ADH) and aldehyde-modified HA (HA-CHO).109 This smart hydrogel could provide sustained boundary lubrication based on the structural rearrangement under shearing stress, while alleviating osteoarthritis by delivering celecoxib. On the other hand, the addition of other biomaterials and/or crosslinking with specific molecules are also effective methods to enhance the mechanical strength and prolong the degradation time of HA-based hydrogel. Zhu et al. added chitosan and glycerol phosphate to HA and formed a hybrid injectable hydrogel loaded with kartogenin (KGN), the Young's modulus of which was found to be enhanced to that of the intervertebral disc.110 Meanwhile, this hybrid hydrogel could release KGN sustainedly after injection, which then promoted the proliferation of adipose-derived stem cells and the differentiation of NP, and finally contributed to the repair process of the degenerative NP tissue. In addition, Su and colleagues synthesized an injectable HA-based hydrogel with oxidized HA and dihydrazine adipate, which could form hydrogel under physiological conditions.111 According to the results, this hydrogel could maintain a stable morphological structure for 5 weeks, and the degradation rate was only 40%.
2.1.1.5. Collagen. Collagen is one of the major components of the ECM, and is important in cell migration, adhesion, proliferation and differentiation in vivo.112 It is essentially a structural protein consisting of RGD sequences, rich in glycine, aspartate and arginine sequences, and plays a vital role in the anti-tensile and anti-shear property of tissues.5,113 Natural collagen is mainly derived from some connective tissues, including cartilage, disc, skin and tendon, and is widely used in tissue engineering for its outstanding biocompatibility, low immunogenicity, robust cellular activity, heat reversibility, hemostasis and biodegradability.5,10,46,114
According to a previously published report, the majority of the ECM of myocardium consists of 12% type III and 70% type I collagen.115 As the dominant extra-cellular protein, collagen is of vital importance in transmitting the cardiomyocyte-generated force and providing the mechanical strength in the myocardium.10 Based on this background, Dai et al. injected 100 μl of saline or collagen hydrogel randomly into the scars of 24 Fischer rats one week after myocardial infarction (MI).116 They found the collagen hydrogel contributed to a better preservation of cardiac function after MI when compared with the control group, as it could thicken the infarct scar, and increase the ejection fraction and left ventricular (LV) stroke volume. Similarly, Blackburn and colleagues attempted to treat infarcted mice with injectable collagen-based hydrogel, and compared the efficacy of this therapy at 3 different time points after MI.117 This injectable collagen-based hydrogel was found to promote angiogenesis, reduce cell death, alleviate fibrosis, positively regulate the repair processes of myocardial tissues and finally stabilize cardiac function for a prolonged time up to 3 months, and the optimal therapeutic effect was achieved when the hydrogel was injected 3 hours after MI.
Nevertheless, pure collagen hydrogel is also limited due to the low physical strength and mechanical properties. Hence, hybrid injectable hydrogels consisting of collagen and various biomaterials have been developed to broaden the application sphere. Wong et al. created an injectable hydrogel with composite alginate–collagen (CAC) for local drug delivery in ocular diseases through a minimal invasive intravitreal injection.118 When compared with pure collagen hydrogels, the elongation at break and tensile strength of this composite injectable hydrogel was better, and exhibited satisfactory ocular drug encapsulation ability, mechanical stability and cell compatibility. Sarker and colleagues incorporated tannic acid (TA) microparticles into injectable collagen-based hydrogels, and found the yield stress and elastic modulus of hydrogel were enhanced.119 This change of mechanical properties may be a result of the numerous non-covalent interactions (such as hydrogen bonds) between the different functional groups of collagen and TA particles (including carboxylic and phenolic/hydroxyl functional groups).120 Meanwhile, the yield stress of the hydrogel exhibited a positive correlation with the concentration of the added TA microparticles, which may be the result of a greater contribution from their material properties to the hydrogel as well as more surfaces for interaction. On the other hand, several chemical molecules have been used to improve the mechanical strength and stability of collagen hydrogels. Poly(ethylene glycol) ether tetrasuccinimidyl glutarate (4S-StarPEG) is a typical representative, which was previously proved to crosslink type I collagen without significant cell toxicity and thus has been advocated in the modification of hydrogels.121 The team of Collin investigated a new injectable hydrogel composed of type II collagen and HA, which was crosslinked by 4S-StarPEG, for intervertebral disc regeneration.122 Similarly, the stability and mechanical strength of this composite system was significantly improved due to the cross-linking of the amine groups of type II collagen and the N-hydroxysuccinimidyl terminal groups of 4S-StarPEG.
Despite the above advantages, there are still several shortcomings of animal-derived collagens, including insolubility in water, limited sources, time-consuming and difficult extraction, and the potential risks of carrying viruses and inducing the immune reaction.123 In recent years, biosynthesis technology has developed rapidly, and has contributed to the preparation of reliable, low-immunogenicity, highly pure collagens with defined chemical structures, named recombinant humanized collagen.10 During the classic biosynthesis progress of recombinant human collagen, the cDNA fragment of the targeted human collagen is determined first, then it is cloned into the vector and translated into expression cells, and finally, the targeted product is obtained after purification.124 Recombinant humanized collagen not only possesses the same basic characteristics of the general animal-derived collagen (including good biocompatibility and positive effects on cell activity), but also can be endowed with specific modified properties under certain conditions, such as improved water solubility, stronger processability and lower immunogenicity.10,125 Yang et al. developed an ECM-mimetic coating with recombinant human type III collagen (rhCOLIII) and HA for thrombo-protective cardiovascular stents.126 Due to the absence of binding sites for platelets while reserving the affinity for vascular endothelial cells, this rhCOLIII-based coating contributed to prominent thrombo-protection and enhanced endothelialization in the in vivo experiments of rabbits. Moreover, in a recent report from Hu et al., tailored rhCOLIII and anti-inflammatory nanoparticles were encapsulated in a MI-responsive injectable hydrogel, which could respond to the pathological micro-environment of MI and then release rhCOLIII and curcumin for myocardial repair.127 Based on the reported results, the addition of the tailored rhCOLIII was demonstrated to contribute to the improved properties of this injectable hydrogel, including enhanced cell migration, adhesion, proliferation and angiogenesis. In addition, Guo et al. designed a new injectable hydrogel based on a tailored recombinant human collagen (rhCol) composed of partial fragments from human type I and type III collagens, which could crosslink with transglutaminase (TG) and then carry basic fibroblast growth factor (bFGF) to promote bone repair.128 The rhCol-based injectable hydrogel had a porous structure with satisfactory mechanical properties, and contributed to cell migration, adhesion and proliferation.
2.1.1.6. Gelatin. Gelatin is a water-soluble protein derivative formed by breaking the natural triple helix of collagen into single-strand molecules, and is usually found in corneal and scleral stroma in vivo.5,47,79 It is also one of the main components of the ECM, characterized by excellent biocompatibility, biodegradability, water absorption and expansibility.5,79,129 Besides, the rich RGD sequence within its structure contributes to its outstanding biomedical properties (including the promotion of cell migration, adhesion, growth, proliferation and differentiation), and thus it has numerous biomedical applications.90,130
As a natural polymer derived from the thermal denaturization or physical and/or chemical degradation of collagen, gelatin eliminates the concerns about the potential risk of immune response and transmission of pathogens related to collagen.130 However, on the other hand, owing to the destruction of the triple helix structure of the collagen, the mechanical strength of gelatin is significantly decreased compared with that of its precursor.131 Meanwhile, the application of gelatin is further limited due to its fast degradation rate in vivo, as the crosslinked network of gelatin can be hydrolyzed by proteinase K within 1 h.130,132 Hence, several crosslinking agents, such as genipin and glutaraldehyde, have been used to mitigate these deficiencies by stabilizing its structure.130 Nevertheless, as agents like glutaraldehyde have potential cytotoxic effects, other more advanced crosslinkers like methacrylic anhydride (MA) have attracted increasing attention due to their inherent non-cytotoxicity, biocompatibility, biodegradability.133 The introduced MA groups endow the GelMA hydrogel with the ability to be photocrosslinked with photointiator under illumination and mild conditions via the irreversible covalent bond.129,134 Consequently, GelMA is currently one of the most widely used gelatin-based hydrogels in biomedical research. However, the ability of the pure GelMA to promote tissue regeneration without other bioactive materials was significantly limited.130 As reported by Sergi et al., the addition of bioactive glass particles of nano-/micro-size was an effective solution to improve the mechanical strength and enhance the bioactivity of gelatin-based hydrogels.130 In addition, the combination of organic gelatin and inorganic particles is also an effective way to overcome the limited bioactive and mechanical properties of gelatin-based hydrogels. Haghniaz et al. added silicate nanoplatelets (SNs) and zinc ferrite (ZF) nanoparticles into GelMA hydrogel to develop an injectable hydrogel sealant.135 This novel hydrogel containing functional inorganic particles had a significantly improved stretch property and sealing capacity, and was found to reduce the viability of bacteria by ∼90% and decrease blood loss in a rat bleeding model by ∼50%. Similarly, the team of Wang et al. added hydroxyapatite microspheres and Ag+ to GelMA hydrogel to form a multi-functional injectable hydrogel.136 Owing to the added hydroxyapatite, this hybrid hydrogel exhibited improved microstructural stability and bending resistance property, increased viscosity and lower swelling rate. Moreover, synthesizing composite hydrogels with two or multiple organic biomaterials is another promising method to further enhance the mechanical property of gelatin-based hydrogel. For instance, Fu and colleagues developed an injectable hydrogel with methacrylated silk fibroin, GelMA and Pluronic F127 diacrylate, which was crosslinked via covalent and non-covalent bond interactions.137 The novel hydrogel could fill arbitrarily shaped defects perfectly and transform into gel to provide stable bio-adhesive and sealing properties within 90 s after injection. It was characterized by significantly enhanced mechanical properties (including toughness, stretchability and viscoelasticity), as it provided stable support for bladder during large stress–strain encounters and cyclic stress changes.
One of the most prominent characteristics of gelatin is thermal responsiveness. The reversible sol–gel transition occurs when the temperature is cooled to 25–35 °C (the critical solution temperature), and thus paves the way for gelatin in synthesizing injectable thermal-responsive hydrogels in tissue engineering.53 Kim et al. prepared an injectable biological ink consisting of gelatin and sodium alginate to 3D print micro/macropore-forming hydrogels.138 They first 3D printed the hybrid hydrogel with macropores, and then crosslinked the hydrogel by 3% CaCl2. Thereafter, the hydrogel was incubated with natural killer (NK) cells under 37 °C, and interconnected micropores were generated as the gelatin was removed due to the its thermal sensitivity. This hydrogel with micro/macropores contributed to the increased cell viability, adhesion, aggregation and release of cytokine, and finally achieved the targeted antitumor effect. Besides, Liang and colleagues designed a novel injectable hydrogel adhesive with gelatin, sodium alginate, protocatechualdehyde and ferric ions.139 Most notably, this composite hydrogel was characterized by not only outstanding injectability, shape adaptability, biocompatibility and antibacterial activity, but also by temperature-dependent adhesive and self-healing capacity, which provided extra fault-tolerant chances for repeated adhesion by adjusting its adhesive strength through the change of temperature.
2.1.1.7. Silk fibroin. Silk fibroin is the majority of Bombyx mori silk, which is a natural biomaterial derived from lepidopteran insects and arthropods (especially silkworms).47 It contains 18 amino acids identical to those in the human body, and mainly consists of numerous inter-linked light-chain and heavy-chain polypeptides.140,141 Besides, it can achieve self-assembly via the formation of β-sheets between the adjacent light and heavy chains.61 Notably, as a natural biomaterial, silk fibroin has drawn increasing attention in the design of advanced multifunctional hydrogel due not only to its good hydrophilicity, biocompatibility, biodegradability and low immunogenicity, but also its rare favorable mechanical and physical properties.47
For example, Bhunia et al. utilized two types of silk fibroin (from Bombyx mori and Antheraea assamensis) to develop an injectable hydrogel which could achieve self-assembly and gelation in situ by simple blending.142 This injectable hydrogel composed of pure silk fibroin has been proved to be competent in load-bearing applications like disc degeneration therapy, and its mechanical properties can be adjusted by changing the ratios of silk fibroin. Similarly, Hu et al. formed an injectable hydrogel based on silk fibroin and liquid polyurethane, which could achieve in situ gelation under mild physiological conditions after injection.143 This silk fibroin-based hydrogel was presumed to be a promising alternative for replacement of the nucleus pulposus of the intervertebral disc, because it could fill the irregular defects after the ejection of nucleus completely due to its injectability, and it could maintain its diameter and height within one million cycles of the fatigue tests due to its astonishing stress resistance and self-renewable capacity. This remarkable mechanophysical characteristic of silk fibroin is mainly determined by the heavy-chains, as the heavy-chains can develop β-sheet crystallites while the light-chains with smaller size contribute little.130 Nevertheless, despite the rare strong mechanical property it possesses as a natural material, silk fibroin is not the final answer for the preparation of ideal hydrogels, as its inherent bioactivity is quite poor.144–146 Therefore, silk fibroin is usually added into other natural biomaterials whose bioactivity is good but mechanical strength is poor, and plays a role as a mechanical reinforcement block in the hybrid hydrogel.61 Ziadlou and colleagues prepared a novel injectable hydrogel with silk fibroin and hyaluronic acid-tyramine to deliver drugs and contribute to the repair process of cartilage defects.145 This hybrid hydrogel was demonstrated to have excellent bioactivity as it increased the expression level of cartilage matrix protein and promoted the production of ECM. Meanwhile, its compressive modulus was significantly improved due to the addition of silk fibroin, which in return also contributed to the deposition of ECM.
In addition, the gelation process of silk fibroin-based hydrogel is slow, but can be accelerated by several external stimulations (such as pH, shear and ultrasound).61 Consequently, silk fibroin is also one the most widely used raw materials for the preparation of injectable smart stimuli-responsive hydrogels with tunable gelation time. Lv et al. designed an injectable smart hydrogel consisting of silk fibroin, chitosan, platelet-derived growth factor-BB (PDGF-BB) and MgFe-layered double hydroxide (LDH) functionalized by bone morphogenetic protein 2 (BMP-2), which was endowed with thermo-responsive property and controlled release of PDGF-BB and BMP-2 to enhance bone regeneration.147 The addition of LDH significantly improved the thermo-responsive property, reduced the sol–gel transition temperature, shortened the gelation time and enabled the composite hydrogel gel rapidly under 37 °C. Meanwhile, owing to the sustained release of growth factors and bioactive ions contained in the hydrogel, this multifunctional hydrogel exhibited excellent abilities in promoting osteogenesis and angiogenesis. Besides, Wang and colleagues reported an interesting and valuable strategy to prevent the recurrence and metastasis of breast cancer based on a smart photoresponsive injectable hydrogel.148 It was a composite hydrogel that consisted of silk fibroin and polydopamine crosslinked collagen, which could achieve nutrition deprivation of breast cancer by clogging the tumor-related blood vessels and inhibiting angiogenesis under near-infrared (NIR) light. On exposure to NIR light, the thrombin loaded within the composite hydrogel could be released from the hydrogel into the residual vessels near the targeted tissues, promote blood coagulation, and finally interrupt the current nutrient supply of the tumor. Meanwhile, the photothermal effect brought about by exposure to NIR light decreased the secretion level of vascular endothelial growth factor (VEGF) in the adjacent tissues and thus restricted the future nutrient supply of tumor.
2.1.1.8. Extracellular matrix. Extracellular matrix (ECM) is a naturally occurring complex bio-polymer in the intercellular space, which mainly consists of water, polysaccharides, glycoproteins and proteins.149,150 The cell-produced ECM develops complex networks between the local cells, and then provides them with solid structure, and various biophysical and biochemical support.30,151 Since the components of ECM are released by the local cells, the ECM compositions of different tissues are unique and match the different needs of specific tissues.151 The numerous biomolecules contained in the ECM play an essential role in tissue maintenance and regeneration, and have significant impacts on cell behaviors (including cell migration, growth, differentiation and production of neo-ECM).152 Of note, the decellularization technique (including physical, chemical and biological methods) can remove the cells and immunogenic molecules within the targeted tissue, while preserving most of the structural proteins and macromolecules.151,153 Therefore, decellularized ECM (dECM) is regarded as a promising biomaterial for tissue engineering and regenerative medicine, as it can maximize the retention of bioactive molecules and provide a favorable adhesion surface and biological activity for cells at the same time.53,152
Ungerleider et al. utilized skeletal muscle obtained from farm pigs to prepare an injectable decellularized porcine skeletal muscle (SKM) hydrogel.154 In the rat model of hindlimb ischemia, the injected SKM hydrogel was demonstrated to improve the perfusion and related kinetics of ischemic tissues by stimulating arteriogenesis, enhancing the recruitment of skeletal muscle progenitors, inducing a shift of the inflammatory response and reducing cell death. Moreover, the team of Traverse et al. conducted the first clinical trial assessing the efficacy and safety of a dECM-based injectable hydrogel in treating myocardial infarction among human patients.155 The used hydrogel (VentriGel) was derived from porcine cardiac ECM, and could achieve gelation and form a porous fibrous structure rapidly after intra-cardiac injection, which provided favorable conditions for endogenous cell infiltration and cardiac regeneration. According to the reported results, this dECM-based hydrogel exhibited the ability to recruit stem cells and induced the differentiation of the recruited cells towards heart, and no VentriGel-related adverse events were reported. In addition, other injectable dECM hydrogels based on various tissues have also been developed in a variety of biological applications and disease treatments, including cartilage and bone repair, functional nerve regeneration, wound healing, liver tissue engineering, ulcerative colitis, human islet culture and others.156–162
However, it is worth noting that the process of decellularization will significantly diminish the mechanical strength and structural stability of the original tissues, which may result in reduced matching of the dECM and targeted tissues, suboptimal interactions between dECM and targeted cells, and finally exhibiting lower effects than expected.152 To enhance the mechanical property of dECM-based hydrogel, Basiri et al. added silk fibroin into the injectable hydrogel made from the decellularized extract from Wharton's jelly (DEWJ).161 The DEWJ-based hydrogel was proved to provide a favorable microenvironment for the targeted cells, as its components were similar to the ECM of articular cartilage, and it could release various cytokines at the same time which promoted cellular proliferation and differentiation continuously. Combined with the improved mechanical property, this hydrogel was regarded to have great potential in cartilage tissue engineering. Hence, the appropriate addition of a mechanical reinforcement block seems to be an easy but effective solution to overcome the low inherent mechanical property of dECM hydrogel. Apart from directly filling the defective tissues with the whole dECM material, researchers have recently explored a new method to fill irregular defects completely as well as match mechanical properties perfectly, which is the addition of micronized dECM particles into injectable hydrogels based on other biomaterials. For instance, Almeida and colleagues first prepared dECM microparticles by low-temperature milling and freeze-drying porcine articular cartilage, and then the dECM microparticles were incorporated into injectable fibrin hydrogel at a ratio of 2% (w/v).163 This dECM-containing hybrid hydrogel was then found to deliver transforming growth factor (TGF)-β3 to infrapatellar fat pad (IFP)-derived stem cells, promote the accumulation of collagen and sulphated glycosaminoglycan (sGAG), and finally induce cartilage regeneration. Similarly, Wang et al. formed an injectable dECM microparticle formulation derived from cardiac tissue and applied it into the infarcted mouse heart; this was then proved to protect cardiac function by inhibiting left ventricular remodeling and promoting vessel density.164 Besides, when compared with the traditional ECM hydrogel (the precursor of dECM microparticles), this dECM microparticle formulation was found to have higher stiffness, longer retention and slower release of proteins.
In addition, as a naturally derived material with multiple bioactivities, dECM has also been used to prepare smart stimuli-responsive injectable hydrogels in recent years. Zhu et al. utilized mannitol particles, the dECM from urinary bladder and poly(N-isopropylacrylamide-co-N-vinylpyrrolidone-co-methacrylate-polylactide) (NIPAAm-co-VP-co-MAPLA) to prepare an injectable thermo-responsive hydrogel.165 Due to thermal responsiveness, this smart hydrogel could gel in situ and form multiple porous structures with appropriate pore sizes after injection. Similarly, the team of Varshosaz prepared another injectable thermo-responsive hydrogel with dECM (derived from human adipose tissue) and aminated guaran (AGG), and it was loaded with gold nanoparticles and atorvastatin lipid nano-capsules (LNCs).166 This hybrid thermo-responsive dECM-based hydrogel exhibited good syringeability, conductivity and tensile strength, and the contained atorvastatin could be released sustainedly for over 30 days.
2.1.2.1. Polyethylene glycol. Polyethylene glycol (PEG) is synthetic polyether which is polymerized from ethylene oxide and polyethylene, and the length of its molecular chain can be effectively controlled by adjusting the ratio of chemical reactants.5,169 As a result of its non-toxicity, low immunogenicity, good biocompatibility and biodegradation, PEG is one of the most widely used synthetic materials in the preparation of injectable hydrogels.5,170
PEG is a hydrophilic polymer which has been approved by the Food and Drug Administration (FDA), and PEG-based injectable hydrogels demonstrate good performance as a drug/bioactive molecule carrier. Meng et al. prepared an injectable PEG hydrogel which was loaded with an anthracycline anticancer drug (epirubicin).171 Through injection, this drug-loaded hydrogel could coat the tumor completely before its gelation and continuously release of epirubicin to achieve a long-term tumor-suppression effect. The team of Lao et al. created a PEG-based injectable hydrogel, which was immobilized by recombinant human bone morphogenic protein-2 (rhBMP-2) and loaded with rat bone marrow mesenchymal stem cells (rBMSCs).172 This hydrogel exhibited potent regenerative capacity in the repair of bone defect due to the synergetic effect of the spatiotemporally controlled release of rhBMP-2 and rBMSCs.
However, as a synthetic material, PEG has an inherent lack of adequate bioactive activity. To endow it with specific functions and expand its application, various natural materials with high bioactivity and multiple functions have been added into PEG-based hydrogel in recent biomedical practices. Ju and colleagues used the dECM derived from porcine cartilage to crosslink with PEG and prepared an injectable suspension, which could form hydrogel scaffolds in situ after injection.173 This hydrogel was proved to be a safe implantation and was endowed with better bio-activity, as it exhibited improved host-cell infiltration and prolonged retention time in vivo. According to the report of Li et al., the addition of hydroxypropyltrimethyl ammonium chloride chitosan (HACC) into benzaldehyde-terminated PEG gave this hydrogel the ability to significantly accelerate wound healing by inhibiting bacteria, and promoting host cell migration and proliferation.174 This is consistent with the similar acquired antibacterial property of PEG-based hydrogel reported by Balitaan et al., which was found to inhibit S. aureus, P. aeruginosa and E. coli without any antibiotics.175 Moreover, Ha et al. developed an in situ-gelation injectable hydrogel consisting of methoxy polyethylene glycol-b-polycaprolactone (MPEG-PCL) and collagen-mimetic peptide GFOGER-conjugated PEG-PCL (GFOGER-PEG-PCL).176 Owing to the added GFOGER, it significantly increased the expression of integrins and FAK, and induced downstream signaling of p38 and ERK, which finally exhibited remarkable promotion of osteochondral regeneration.
The physical, chemical or biological properties of the tissue micro-environment may change significantly during the pathophysiological process of diseases. Therefore, smart hydrogels and/or the therapeutic agents they are loaded with, which can respond to these changes, make it possible to achieve more precise treatment for the targeted tissues and minimize related side effects. Recently, several smart injectable hydrogels based on PEG have been reported to be endowed with special responsiveness to certain stimuli under pathological conditions. Chen et al. developed a novel smart injectable hydrogel (MPGC4) based on PEG, which could respond to the significantly overexpressed matrix metalloproteinase (MMP) 2/9 in myocardial infarction.177 This hydrogel consisted of composite gene nanocarrier (CTL4) and tetra-poly (ethylene glycol), and was proved to automatically release CTL4 on demand in the rat model of myocardial infarction to inhibit ECM degradation, promote angiogenesis and regulate the immune microenvironment. Fang and colleagues combined two simple components, dibenzaldehyde-terminated poly(ethylene glycol) (DB-PEG2000) and atechol-functionalized quaternized chitosan (CQCS), and prepared a multifunctional injectable hydrogel for hemostasis and infected wound healing.178 This smart hydrogel system could be responsive to the local acidic microenvironment induced by lactic acid produced during bacterial metabolism, and was characterized by many favorable properties (such as self-healing, tissue adhesiveness, antioxidant and antibacterial). In addition, as tumor is a glutathione (GSH)-rich tissue, Liu et al. created a novel smart injectable hydrogel with PEG and poly (thioctic acid) (PTA), which would break up and release the loaded drugs via the thiol exchange reaction between its disulfide bonds and the local high concentrations of GSH.179 According to the experimental results, this system achieved on-demand release of drugs, as the loaded anticarcinogen (DOX) could only be released in the presence of GSH. This kind of smart hydrogel is now regarded as a potential intelligent drug delivery system that can maximize the therapeutic effects while minimizing the side effects of anticarcinogens.
2.1.2.2. Polyvinyl alcohol. Polyvinyl alcohol (PVA) is a common long-chain synthetic material, which is derived from the polymerization and hydrolysis of vinyl acetate.5 Like PEG, PVA is also widely used to form injectable hydrogel due to its advantages, such as non-toxicity, water-solubility, good histocompatibility and mechanical properties.
Since the physicochemical characteristics of PVA hydrogel are very similar to those of the intervertebral disc, PVA-based hydrogels have attracted much attention in the treatment of the diseased nucleus pulposus. Jia and colleagues synthesized an injectable hydrogel based on the crosslinking of PVA chains and glycerol which possessed a comparable compressive and storage modulus to that of nucleus pulposus, and exhibited protective effects against the pathological mechanical loading on the nucleus pulposus cells.180 Leone et al. prepared an injectable hydrogel based on PVA, whose core contained hydrophilic poly-vinyl pyrrolidone (PVP), and it could preform a 3D network as well as maintain injectability.181 They found that when the molar ratio of PVA and PVP was 1:1, this hydrogel reached the optimal balance of the mechanical property and easy injectability, and it was regarded as a promising alternative in the replacement of nucleus pulposus. Moreover, the team of Allen et al. even completed a preclinical evaluation of the injectable hydrogel made of PVA in the baboon discectomy defects model, and found this implant exhibited good toleration over 24 months without significant evidence of adverse events or toxicity.182
The same as PEG, PVA-based hydrogel also has a lack of favorable bioactivity, but can be improved by crosslinking with other molecules or loading of therapeutic agents. Zhang et al. developed an injectable hydrogel with self-healing property via the dynamic boronic ester bond between PVA and boronic acid-grafted carboxyethyl cellulose (CMC-BA).183 This PVA-based hydrogel exhibited rapid hemostasis and wound-repairing effects, and preserved the tumor inhibition performance while reducing the acute in vivo toxicity of DOX through controlled release. In the injectable wound dressing hydrogel prepared by Xiang and colleagues, the combination of GelMA enhanced the tissue affinity of PVA-based hydrogel, and the addition of zwitterionic silver nanoparticles significantly increased its antibacterial efficiency.184 In addition, the team of Shi et al. synthesized a novel injectable PVA-based hydrogel via the dynamic boronic ester covalent bond between PVA and the phenylboronic acid-modified hyaluronic acid (HA-PBA), and assessed its potential in various biomedical applications, such as drug delivery, cell carrier, in vitro 3D culture of cells and 3D bioprinting.185 It was demonstrated to provide a good micro-environment for cell adhesion and proliferation, while exhibiting a favorable anti-oxidative property that protected the loaded cells from ROS-stress and related death.
However, although great progress has been made, there remain some issues that current PVA-based hydrogel with the above improved properties cannot solve. For example, most of the current modified PVA-based hydrogels can achieve a sustained release of loaded drugs, but it is actually a non-targeted treatment with prolonged duration. They fail to deliver the therapeutic agents to the cells/tissues needing treatment on demand, which would truly achieve long-term effective therapy with minimal side effects, especially for those hypertoxic agents. In recent years, smart injectable hydrogels based on PVA have emerged as the times require. Gong et al. prepared a smart hydrogel system containing anti-programmed cell death protein ligand 1 antibodies (aPDL1) via the covalent bonds between PVA and phenylboronic acid-modified 7-ethyl-10-hydroxycamptothecin (SN38-SA-BA).186 After injection, this drug-loaded hydrogel could respond to endogenous ROS and break up the networks, then achieve the on-demand release of the loaded SN38 and aPDL1 to inhibit tumors. Similarly, Kuddushi and colleagues developed another smart hydrogel system by adding multiple biocompatible components into the PVA-based hydrogel, which endowed it with various biomedical functions, such as good injectable and self-healable properties, favorable cell adhesion and proliferation and, most importantly, the dual-responsiveness to both temperature and pH.187 These additives enabled the injected hydrogel to release the encapsulated DOX only when it was exposed to a specific temperature or the acidic micro-environment produced by the cancerous tissues, which achieved the targeted release of DOX, improved drug uptake and further decreased DOX-related cytotoxicity. Therefore, smart stimuli-responsive hydrogels may represent a novel drug delivery strategy with high precision and delivery effectiveness, as well as minimal undesirable off-target toxicities.
2.1.2.3. Poly lactic-co-glycolic acid. Poly lactic-co-glycolic acid (PLGA) is a synthetic polymer with good biosafety and biodegradability, and has been approved by the FDA for clinical use, such as in artificial catheters, as a sustained-release drug/functional nano-particle carrier and as tissue engineering scaffold material.53,188 It is free from immunogenicity and antigenicity, and the abundant carboxyl groups contained in the side chains provide great potential for various chemical modifications.53 Currently, PLGA is mainly used in particulate form and plays a role in targeted delivery as a transport platform for drugs, proteins and adherent cells.189–191 Besides, the hydrogel made from PLGA is characterized with favorable liquid fluidity, and has also been well developed to apply in biomedical practices over the past decades.
Owing to the hydrophobic chains within its structure, the application of PLGA-based hydrogel is limited due to poor hydrophilicity. Endres and colleagues combined PLGA with HA to improve its hydrophilicity, cell migration and adhesive properties, and enhanced cell migration and disc repair were found after the application of this hybrid hydrogel.192 Li et al. designed a novel injectable hydrogel with self-healing ability based on the Schiff base reaction between benzaldehyde-terminated PEG (PEG-CHO) and adipic dihydrazide-modified PLGA (PLGA-ADH), which exhibited favorable support towards the proliferation of BMSCs and the deposition of GAGs in a rat cartilage defect model.193 Recently, interest has also been raised in designing smart PLGA-based hydrogels, which can respond to specific endogenous and/or exogenous stimuli, to further develop targeted therapy and precision medicine. A thermo-responsive PLGA-PEG-PLGA hydrogel encapsulating interleukin-36 receptor antagonist (IL-36Ra) was prepared by Yi et al., which could gel rapidly after intra-articular injection, release IL-36Ra slowly over a prolonged period, up-regulate the expression of collagens and GAGs while down-regulating that of MMP-13 and ADAMTS-5.194 In the combined treatment of cancer chemo- and immunotherapy, Wu and colleagues developed an injectable nano-composite hydrogel based on the framework of PLGA-PEG-PLGA, which was then grafted with R837-loaded CaCO3 nano-particles and paclitaxel (Pac)-loaded mesoporous silica nanoparticles (MSNs), to release the highly toxic agents on demand only in cancer tissues and achieve targeted treatment.195 Upon injection, this hybrid hydrogel could gel in situ due to its thermo-responsiveness and exhibited a GSH-dependent release of the loaded antitumor agents over time, which suggested a controlled spatiotemporal release of therapeutics.
In addition to the above synthetic polymers, researchers have explored many other synthetic materials, such as poly(urethane) (PU), poly(alanine) (PA), poly(amide) (PAM), poly(organophosphazenes) (PNP), polyoxyethylene (PEO), poly(propylene oxide) (PPO), poly(galacturonic acid) (PGA), poly(caprolactone) (PCL) and poly(caprolactone-co-lactide) (PCLA), to meet various requirements of advanced hydrogels.61,196,197 Although there are numerous kinds of synthetic materials, their synthetic processes and characteristics are similar, and here they are not described in detail repetitively. Of note, since the advantages and drawbacks of synthetic polymers are opposite to those of natural materials (similar to the yin-yang in Chinese traditional culture, Fig. 1), it seems to be a reasonable strategy to generate multi-functional injectable hydrogels with comprehensive properties (such as good biodegradability with favorable mechanical strength) by combining both the synthetic and natural materials.
2.2. Injectable strategies
Compared with the traditional hydrogel systems, injectable hydrogels are preferred and come with great expectations. They can deliver therapeutic drugs/cell factors/bio-functional molecules deep into the targeted tissues by a simple injection (a minimally invasive administration), avoid unexpected spreading to healthy tissues and achieve sustained release of loaded therapeutics, which is of vital importance in avoiding the pain and injury of traditional treatments (such as surgery and repeated injections) and further reducing undesirable treatment-related effects. Over the last decades, a variety of advanced methods and techniques have been explored and developed to endow conventional hydrogels with injectability. Herein, in order to help researchers rapidly understand the existing synthesis techniques and facilitate their selection of appropriate methods for certain conditions in future research, we have classified the most widely used strategies into four main types, and described their principles and synthetic methods, respectively.2.2.1.1. Photoinitiated gelation. The technique turning liquid monomers into gel polymers through the exposure of light is called light curing, and the main mechanisms of light curing consist of two representative chemical reactions, photoinduced radical polymerization and photoinduced click crosslinking.
Due to the mild reaction conditions (such as neutral pH and room temperature) and the spatial/temporal controllability of reactions, photoinduced radical polymerization is the most widely accepted pathway for researchers in the synthesis of in situ-forming hydrogels.198 It is based on the polymerization of monomers initiated by the reactive species (including cations, anions and radicals) generated by photoirradiation. Methacryloyl (MA) is the most widely used substituent to enable the grafted molecules to polymerize via photoinduced radical polymerization under the exposure of ultraviolet (UV), and it provides a spatiotemporal controllable polymerization owing to its chain-growth mechanism (Fig. 2A).198 Among the biomedical applications of photoinduced radical polymerization, gelatin, HA and PEG are the most common polymers utilized to prepare photoinitiated in situ-forming hydrogels.199 Due to the intact preservation of RGD sequences during the grafting process, GelMA preserves the original superior bioactive functions of gelatin (such as cell migration, adhesion, proliferation and differentiation), and is endowed with favorable photocrosslinking ability under the exposure of UV. For example, Shen et al. prepared an injectable in situ-forming hydrogel with GelMA to achieve a sustained release of triamcinolone acetonide in the vitreous cavity.200 After intravitreal injection, this hydrogel could form gel in situ at the injection site after a 60-second exposure to visible light at a wavelength of 405 nm, and provided a safe, biocompatible and controlled drug-release platform in a posterior vitreous location for ophthalmic applications. Similarly, HA can also be modified to HAMA through the same chemical modification of the active groups (such as hydroxy and carboxyl groups) contained in the backbone, and acquire stronger crosslinking and mechanical properties. In the study of Chen et al., the solution of HAMA was injected into the cartilage defect (1.5 mm in depth and 1.5 mm in diameter) first, and then formed a completely fitted hydrogel in situ after exposure to UV (365 nm, 5 minutes), which enhanced cartilage regeneration by promoting the integration between native and neo-cartilage.201 With respect to PEG, dimethacrylated PEG (PEGDA) is usually used to prepare hydrogels based on photoinitiated radical polymerization, and the chain polymerization occurs as the propagation of the radicals (generated from the photocleavage of initiator molecules propagate) on the unsaturated vinyl bonds on PEGDA.198 As reported by the team of Meng et al., the precursor solution of PEGDA could be transformed to hydrogel under the 660 nm light-emitting diode (LED) light irradiation through the polymerization induced by the ROS generated by the photoinitiator (Chlorin e6).202 This in situ-forming hydrogel resulted in long-term retention and even distribution of therapeutic agents in tumor tissues, which achieved multi-round photodynamic therapy (PDT) and enhanced immune responses.
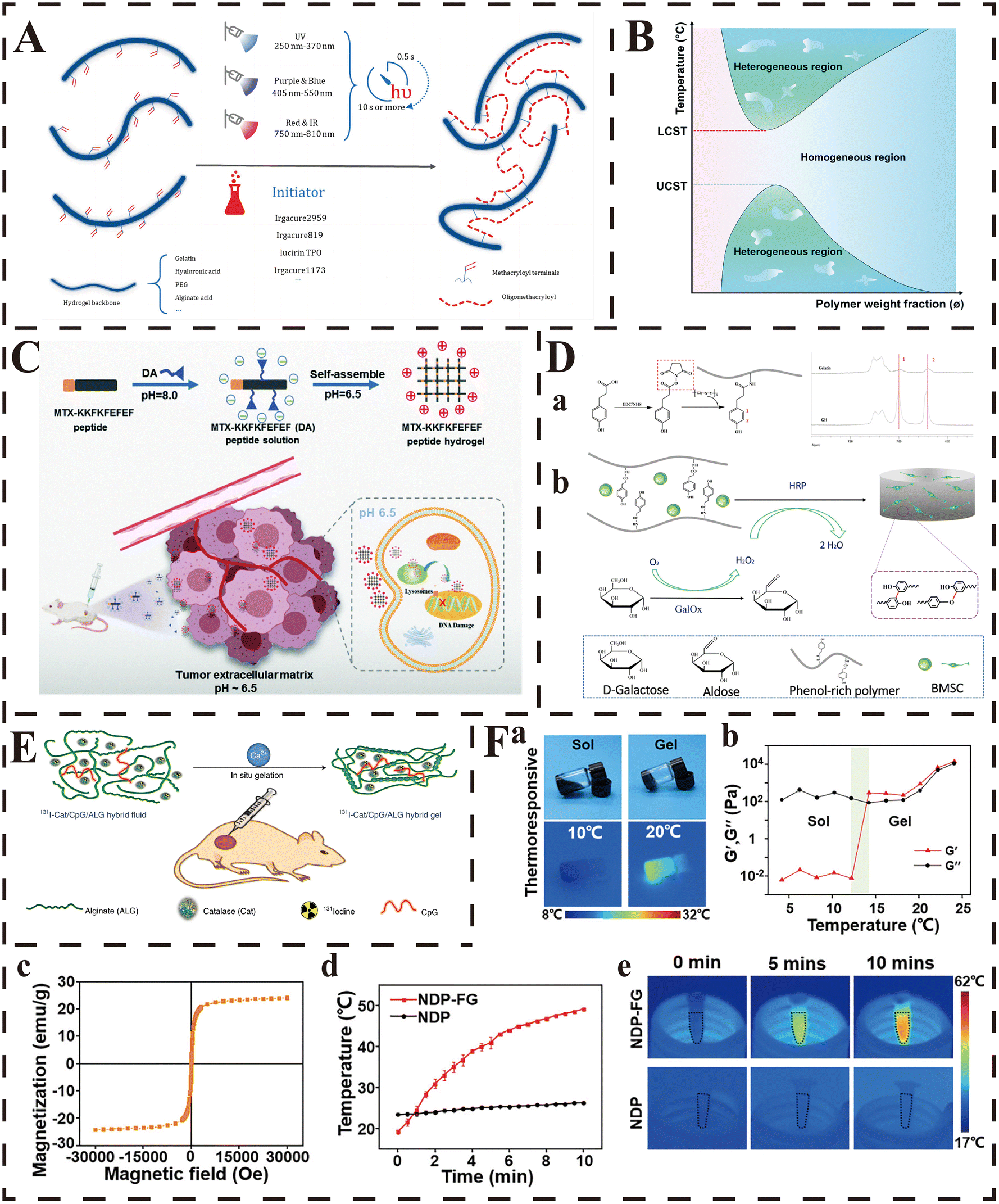 | ||
| Fig. 2 Strategies for in situ gelation. (A) Upon generation of a free radical (by different types light exposure that depend on the type of photoinitiator), the methacryloyl terminals on hydrogel backbone chains, such those of as GelMA, hyaluronic acid MA and PEGDA, polymerize to generate a more connected network through the formation of short oligomethacryloyl chains. From ref. 198 licensed under Creative Commons Attribution 4.0 (CC BY 4.0). (B) Phase diagrams of LCST- and UCST-type polymer aqueous solutions (temperature versus polymer weight fraction). Reproduced from ref. 207 with permission from the Royal Society of Chemistry. (C) Schematic illustration of the formation of the pH-responsive peptide hydrogel and the anti-tumor mechanism of the pH-responsive peptide hydrogel at the tumor site. Reproduced from ref. 214 with permission from the Royal Society of Chemistry. (D) (a) Synthetic scheme of gelatin–hydroxyphenyl conjugate and the 1H NMR spectra of the GH conjugate. (b) Synthetic scheme of BMSC-loaded GH hydrogel dual-enzymatically cross-linked by HRP and GalOx. Reproduced with permission from ref. 225, copyright 2019, American Chemical Society. (E) Scheme illustrating in situ gelation of the 131I-Cat/ALG hybrid fluid after local injection into tumours. Adapted with permission from ref. 227. Copyright 2018, Springer Nature. (F) Characterization of the NDP-FG hybrid hydrogels. (a) Digital images and infrared thermal images of NDP-FG aqueous dispersion under the heating process. (b) G′/G′′ measurements of the NDP-FG aqueous dispersion. (c) Hysteresis loops of FG at 300 K. (d) Time-dependent temperature curves and (e) infrared thermal images of NDP and NDP-FG under AMF. Reproduced with permission from ref. 231, copyright 2022, American Chemical Society. | ||
In addition, photoinduced click chemistry is also an ideal choice to prepare photoinduced in situ-forming hydrogels with multiple components, as it provides a spatiotemporally controllable strategy and produces highly uniform polymer networks.198 As understanding of the mechanisms about photoinitiator and photoinduced polymerization has deepened over the last decades, more adaptive photoinitiated click reactions (including thiol–ene and thiol–yne photoclick reactions, hydrazone reaction, Diels–Alder reaction, etc.) have been explored to broaden the synthesis strategies of photoinduced in situ-forming hydrogels. Photoinitiated click reactions exhibit great adaptivity of the spatiotemporal regulation during the polymerization process. Thiol–ene and thiol–yne click reactions comprise the main type of photoclick chemistry, which improve the overall compatibility and simplicity of the synthetic process, and maintain the classic benefits of photoinduced click reactions at the same time.203 This hybrid polymerization can achieve highly precise polymerization at specific times and locations, minimize extra stress and shrinkage, and optimize the kinetics.198 Based on the thiol–ene photoclick reactions mediated by visible light (wavelength: 400–700 nm), Shih and their group generated a step-growth hydrogel with multiple-layer structures.204 Since the loaded eosin-Y (a non-cleavage type photoinitiator) could diffuse into the adjacent layers and was demonstrated to retain the ability to re-initiate the thiol–ene photoclick reaction, this multi-layer hydrogel could be acquired just through sequential light exposure without additional new initiator. Besides, the thickness of this hydrogel could be adjusted easily by changing the reaction parameters, such as the exposure time of light, the initial concentration of eosin-Y and macromers. Hence, this method represents a significantly simplified strategy compared with conventional photoinduced polymerization. Additionally, Hao et al. generated a fully biodegradable hydrogel platform based on the light-induced thiol–yne click reaction between HA and poly (butynyl phospholane)-random-poly (ethylethylene phosphate) (PBYP-r-PEEP), which was initiated by irgacure 2959 (a photoinitiator) under UV irradiation.205 It provides a simple yet highly effective strategy for the preparation of multi-functional and fully degradable hydrogels under physiological conditions.
2.2.1.2. Temperature-initiated gelation. Recently, several physiologically relevant stimuli (such as physiological temperature, pH, enzymes) have attracted increasing interest owing to their easily controlled gelation process without any chemical agents. The hydrogel, whose sol–gel transition occurs with the increase/decrease of temperature, is called thermo-sensitive hydrogel, and provides feasibility of injection of the pre-gel as an aqueous solution into the body, followed by in situ gelation driven by in vivo temperature.206 One of the main common characteristics of thermo-sensitive hydrogels is the co-existence of hydrophilic and hydrophobic groups, and there is a critical solution temperature (CST) which controls the crosslinking of this kind of hydrogel (Fig. 2B).207 More specifically, CST can be further divided into two different types, the upper CST (UCST) and the lower CST (LCST): the gelation occurs when the temperature is greater than the LCST for hydrogels with a LCST, and occurs when the temperature is lower than UCST for those with a UCST.199,208 For example, the enthalpy change plays a dominant role when the temperature is lower than LCST, and the pre-gel is dissolved in water due to the hydrogen bonds between the polar groups contained in the polymer chain and water molecules; while the entropy change will replace the enthalpy change to play a dominant role when the temperature is over LCST, and the polymer molecules will precipitate and form a hydrogel network due to the dehydration of the hydrophobic isopropyl groups during the sol–gel transition.209,210
Several natural biomaterials (including carrageenan, gelatin, collagen, elastin, fibrin, cellulose and agarose) are appropriate candidates for the preparation of temperature-induced gelation hydrogel due to their inherent thermo-sensitivity, as they can change from liquid to solid by adjusting the temperature.199 For example, Yeh et al. developed a series of injectable thermo-sensitive hydrogels with different gel strengths and stiffness by adjusting the hydrophobic–hydrophilic balance with different types and contents of gelatin and Pluronic F127 (an amphiphilic copolymer).211 This composite hydrogel was proved to form gel at the injection site under 37 °C rapidly, and exhibited a thermo-reversible property in the cycle between 25 °C and 37 °C. In addition, due to the inherent thermo-sensitivity of collagen, a composite collagen-based hydrogel was synthesized by Sarker et al. to provide an in situ-forming hydrogel platform in tissue engineering.119 This new hydrogel system remained in the liquid state before and during injection (at 4 °C), but could gel promptly and cover the targeted tissue defects completely at 37 °C (body temperature). Similarly, the team of Jia et al. designed a thermo-initiated gelation hydrogel based on the “Double H-bond” of hydrazone bond and hydrogen bond, which could self-gel rapidly in situ at the injection site upon the exposure of mild conditions (37 °C, pH = 7.4) in vivo and then release the loaded gelatin microsphere as well as metformin to promote the repair of diabetic wound.212
Another common strategy to prepare temperature-induced gelation hydrogels is introducing thermo-sensitive chains, such as poly(N-isopropylacrylamide) (PNIPAM), poly (ethylene glycol) (PEG)-based block copolymers and poly (ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO), into the polymers via physical and/or covalent crosslinking reaction. For example, Pourjavadi and colleagues produced an injectable in situ gelation hydrogel with Au nanoparticle-modified chitosan, κ-carrageenan PNIPAM, which was enabled with the ability to transform into hydrogel from homogeneous solution within 5 min at 37 °C due to the physical interaction of PNIPAM with the other components in the hydrogel.213 In the study of Yi et al., the authors synthesized an injectable thermo-sensitive hydrogel based on PLGA-PEG-PLGA (a typical PEG-based block copolymer) to alleviate osteoarthritis through in situ gelation and the sustained release of the loaded IL-36Ra.194 According to the experimental results, this hydrogel-system was in the liquid form at 25 °C (room temperature) and could transform into gel at the injection site automatically at 37 °C within 60 s. That was actually the result of the temperature-induced change of the hydrophobic–hydrophilic balance (the hydrophilicity of PEG vs. the hydrophobicity of PLGA) of this system at the molecular level: the hydrogen bonds between the PEG segments and water molecules dominate the aqueous solution at room temperature and kept this system in a liquid state; however, the hydrogen bonds would get weaker while the hydrophobic forces between the PLGA segments would become stronger as the temperature rose, and finally the system changed from solution to gel.
2.2.1.3. pH-Initiated gelation. Another type of hydrogel whose gelation is initiated by the acidic/basic tissue microenvironment is called pH-sensitive hydrogel. With changes from physiological pH to disease-induced microenvironmental pH, the structure of this kind of hydrogel can shrink or expand accordingly, which then provides another strategy to prepare injectable in situ-gelation hydrogel. Generally, the phase transition ability of pH-sensitive hydrogel is determined by the weak acidic or basic groups (such as amines, imines, carboxylic acids) contained in the hydrogel matrix, which can accept or donate protons with the change of the surrounding pH, changing the solubility and volume of polymers, and finally resulting in the formation or collapse of hydrogel networks. For example, one of the most prominent characteristics of solid tumors is their acidic microenvironment owing to the overproduction of lactate by tumor cells. After injection into this acidic tissue microenvironment, the polymer chains containing acidic groups (like carboxylic acids) will shrink and form compact hydrogel networks in situ due to the electrostatic interaction. In contrast, the basic groups (such as amines) will be positively charged by accepting protons, and with the increase of the electrostatic repulsion between charges, the hydrogel networks will be enlarged or even collapse due to the expansion of the polymer chains. Consequently, hydrogels with the latter characteristic are also called pH-responsive hydrogels, and have been investigated in local anti-tumor therapy for the release of cytotoxic drugs exclusively in tumor tissues to minimize systematic toxicity. More details about smart pH-responsive injectable hydrogels will be described later in this review.
In the study of Zhang et al., the authors prepared a novel injectable hydrogel (MTX-KKFKFEFEF(DA)) by grafting methotrexate (MTX, a small-molecule drug with carboxyl groups) and 2,3-dimethylmaleic anhydride (DA, a pH-responsive linker) onto the KKFKFEFEF peptide through amidation reactions, and MTX-KKFKFEFEF(DA) was charged negatively in the neutral condition (pH = 7.4).214 After intra-tumoral injection, this hybrid hydrogel system was activated to be positive, and achieved an efficient sol–gel transition upon exposure to the tumor-related acidic microenvironment (pH = 6.5) (Fig. 2C). According to the subsequent results of both in vitro and in vivo experiments, this hydrogel achieved a rapid in situ gelation and long retention time after injection, and provided a high inhibition rate of tumor with negligible adverse effects in the mouse model of breast cancer. In addition, another commonly used method to prepare pH-sensitive hydrogels is forming polyelectrolyte complexes between anionic and cationic polymers, and chitosan and its derivative are the most widely used biomaterials in this regard. According to the report of Chiu et al., the investigators developed a pH-triggered injectable hydrogel based on a hydrophobically modified chitosan (N-palmitoyl chitosan, NPCS).215 Palmitoyl groups (a kind of hydrophobic group) were grafted onto the free amine groups of chitosan to obtain the comb-like associating polyelectrolyte (NPCS), which could achieve in situ gelation triggered simply by the surrounding environmental pH through an appropriate balance between the hydrophobic interaction and charge repulsion. In the following experiments, this hydrogel system was proved to form massive hydrogel in situ rapidly after subcutaneous injection in a rat model, and could be degraded gradually within 6 weeks without significant toxicity.
2.2.1.4. Enzyme-initiated gelation. Enzymes are catalytically active substances that are essential to ensure the proper functioning of various stages in the life process. Since the type, quantity and activity of enzymes are different in distinct tissues/organs under physiological/pathological conditions, the synthesis of in situ gelation hydrogel triggered by enzymes is also a reasonable and promising strategy that has been proposed and investigated for years.216,217 In general, there is usually at least one chemical moiety within the enzyme-initiated gelation hydrogel matrix which is a specific substrate (or its mimic) of the targeted enzyme that can access the active center of the enzyme. After injection into the local tissues, with the occurrence of the specific enzymatic reaction (enzyme-mediated crosslinking or cleavage), the physicochemical properties of the hydrogel matrix change (sol–gel or polymerization–collapse transition) subsequently.218 Based on this, smart injectable hydrogels that can be responsive to specific enzymes (called enzyme-responsive hydrogels) have also been explored and applied in biomedical practices for controlled delivery of therapeutic agents, which will be discussed in the next section. Due to host-derived enzymes and the inherent high efficiency of enzymatic reactions, the enzyme-initiated in situ gelation is characterized by several outstanding advantages, including high substrate specificity with regio- and stereo-selectivity, highly efficient reaction rate under mild body conditions, no need for extra chemical agents, avoiding the potential toxicity of both substrates and products during the whole reaction process, tunable reaction rate by adjusting the concentrations of the enzymes and/or substrates, and favorable and stable mechanical properties of the acquired hydrogels.199,219–223
For example, Li et al. designed a novel injectable enzyme-initiated gelation hydrogel (GGA@Lipo@PTH (1–34), GLP) that could gel in situ rapidly in the presence of transglutaminase (TG) after intra-articular injection, regulate cartilage metabolism through the sustained release of teriparatide (PTH (1–34)) and finally alleviate osteoarthritis.224 They first obtained a gelatin derivative (GGA) which was grafted with gallic acid via the crosslinking of the carboxyl groups in gallic acid and the amino groups in gelatin. Then, liposomes encapsulating PTH (1–34) in the core were incorporated into the GGA solution to obtain the pre-gel of GLP. Finally, due to the existence of TG in vivo, the injected pre-gel of GLP achieved sol–gel transition in the joint cavity within 5 min, and through the sustained release of PTH (1–34), this hybrid hydrogel system promoted the proliferation of ATDC5 cells, increased the production of GAG and decreased the degradation of cartilage ECM. Similarly, to accelerate wound healing, the team of Yao et al. developed a gelatin-hydroxyphenyl hydrogel, which could gel in situ at the injection site, based on the dual enzyme crosslinking of galactose oxidase (GalOx) and horseradish peroxidase (HRP) (Fig. 2D).225 After injection, the GalOx contained in the pre-gel oxidized the galactose in vivo and produced H2O2 indirectly, which was an essential substance in the crosslinking reaction mediated by HRP, and finally resulted in the formation of the three-dimensional networks of hydrogel. This enzyme-initiated in situ hydrogel exhibited great application potential in wound repair as it formed completely fitted hydrogel in situ, provided a 3D biomimetic micro-environment for the loaded BMSCs and improved their survival, finally accelerating the process of wound closure.
2.2.1.5. Ion-initiated gelation. Several ionizable and zwitterionic polymers (including alginate, pectin, polyacrylic acid and carboxymethyl dextran) can be crosslinked with oppositely charged polymers/materials (such as Na+, K+, Ca2+, Ba2+, etc.), and then the changes of their conformations at molecular level result in the sol–gel transition, which is called ion-initiated gelation.222,226 Alginate is the typical and most widely used biomaterial in the preparation of this kind of hydrogel, and the abundant hydroxyl and carboxyl groups contained in its molecule enable it to crosslink with cations (mostly Ca2+ and Ba2+) in vivo and form gel rapidly in situ. This in situ gelation system has attracted increasing attention in recent years due to the simple and controllable fabrication process, mild reaction conditions, no addition of cytotoxic chemical agents, and rapid formation of biocompatible and biodegradable hydrogels in situ.222 For example, Chao and colleagues utilized several purely biocompatible components (alginate, catalase and CpG oligonucleotide) to develop an injectable hydrogel, which could achieve in situ gelation after an intra-tumoral injection in the presence of endogenous Ca2+via the changes of the conformations at molecular level (Fig. 2E).227 This in situ-forming hydrogel provided a highly effective strategy for local tumor treatments in a three-pronged way: the sustained release of the evenly distributed immunostimulatory CpG oligonucleotide and catalase contributed to the stronger systemic anti-tumor immune response and improvement of the tumor oxygenation status, respectively, while the 131I contained in the hydrogel was fixed within the tumors to provide local radiotherapy continuously for a long time. Based on the same principle, their team designed a novel injectable hydrogel in 2020 with a “cocktail” formulation for chemoimmunotherapy, which was composed of alginate, imiquimod (R837, an immune adjuvant) and immunogenic cell death (ICD)-inducing chemotherapeutics (DOX or oxaliplatin (OXA)).228 This hybrid system could form hydrogel at the injection site of tumors rapidly due to the crosslinking between the contained alginate and the Ca2+ within the tumor. Owing to the in situ-forming principle, the loaded therapeutic agents were distributed evenly within the tumor tissues, and exhibited sustained release of therapeutic agents to amplify immune responses, trigger ICD and enhance anti-tumor immunity over a long period. In addition to the endogenous Ca2+in vivo, CaCO3 has also been reported to play a role in the preparation of ion-initiated hydrogel as an exogenous ionic cross-linker in recent years. The team of Yan et al. synthesized an alginate-based injectable hydrogel with hydroxyapatite (HAp), gelatin microspheres (GMs), CaCO3 and glucono-D-lactone (GDL).229 This system was proved to achieve in situ gelation with the slow release of Ca2+ from the added CaCO3, and the added Hap and GMs successfully increased the mechanical strengths of hydrogel, while the loaded tetracycline hydrochloride contributed to the improved osteogenesis.
2.2.1.6. Magnetic field-initiated gelation. Magnetic field is also one of the trigger factors of in situ gelation hydrogels, whose structure and physical properties can change with the change of external magnetic field. These kinds of hydrogels are called magnetic field-initiated gelation hydrogels, and are usually obtained by inserting exogenous ferromagnetic or paramagnetic compounds (such as Fe2O3, Fe3O4 and CoFe2O4) within the polymeric matrix.230 These hydrogels are composed of three-dimensional polymeric networks and magnetic nanoparticles, and the most widely used compound is iron oxide nanoparticles with paramagnetic properties. The inserted magnetic nanoparticles are usually vibrated under the exposure of an external magnetic field, resulting in a dramatic increase of the local temperature. Since the increased temperature induced by the magnetic field can promote the sol–gel transition and the controlled encapsulation/release of drugs, the system of magnetic field-sensitive hydrogels generally combines magnetic field-sensitivity with thermo-sensitivity at the same time. For example, in the study of Yan et al., the authors developed a novel injectable hydrogel (NDP-FG) based on a biocompatible triblock co-polymer of poly-((N-isopropylacrylamide-co-dopamine)-b-poly-(ethylene-glycol)-b-poly-(N-isopropylacrylamide-co-dopamine)) (poly-(NIPAM-co-DOPA)-PEG-poly-(NIPAM-co-DOPA), NDP) by adding reduced graphene oxide nanosheets decorated with iron oxide nanoparticles (Fe3O4@rGO, FG).231 Within 10 min of exposure under an alternating magnetic field (AMF), the temperature of the hybrid pre-gel system increased rapidly from 20 to 49 °C due to the vibration of the inserted magnetic nanoparticles, and the aqueous dispersion of NDP-FG was then transformed into hydrogel in situ (Fig. 2F). According to the reported results, this magnetic field-initiated hydrogel was indicated to contribute to the controllable in situ gelation, lead to effective intra-operative hemostasis, enhance the magnetic hyperthermia efficacy and prevent tumor recurrence.
It is worth mentioning that due to the inherent property that magnetic fields can make magnetic substances arrange in a regular form, magnetic-sensitive hydrogels have also been investigated to provide ordered 3D structures for in situ arrangement of cells, so that the cells can grow in the right direction and differentiate adequately.232 In addition, as a result of the intrinsic properties, magnetic nanoparticles have been found to be able to regulate the behaviors of cells loaded in the hydrogels under the exposure of external magnetic fields, and magnetic materials have enabled hydrogels to possess the anisotropy required for various uses in tissue engineering. For example, the magnetic forces at the interface between inserted composites and cells have been found to activate sensitive receptors on the cell surface, enhance cell activity, contribute to the bone formation process and promote the integration of scaffolds into the host bone.233–235 Besides, magnetic nanoparticles contribute to the controlled design of anisotropic magnetic field-sensitive hydrogels as they can be used to provide the hydrogel matrix with a visually anisotropic hierarchical architecture. However, although magnetic field-sensitive hydrogels have attracted special attention due to their minimal invasiveness and excellent permeability to deep tissues, because of the potential cytotoxicity of magnetic materials and the low reproducibility of in vivo outcomes, it remains a challenge to design an ideal magnetic field-sensitive hydrogel and apply it in biomedical processes.236
 | ||
| Fig. 3 Strategies for shear-thinning hydrogels. (A) Schematic behavior of a self-healing injectable hydrogel with (i) gel-like properties at rest, (ii) fluidization under shear due to reversible chemistry and/or alignment in the flow field, and (iii) self-healing of the original structure and mechanical properties after flow. From ref. 42 licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). (B) Schematic illustrations of the fabrication of CLX@Lipo@HA-gel. The CLX@Lipo@HA-gels were constructed by incorporating CLX-loaded HSPC liposomes within Schiff base bond-based HA hydrogels. From ref. 109 licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0). (C) Synthesis of hyaluronic acid (HA)-based biopolymer binders. Bisphosphonate (BP) groups or BP and acrylamide (Am) groups were chemically linked to backbone of HA macromolecules using carbodiimide coupling and thiol-disulfide exchange reactions. Linkers 1, 2, and 3 were used for HA modification. Adapted with permission from ref. 251. Copyright 2017, John Wiley and Sons. (D) Illustration of the orbital interactions in normal (a) and inverse (b) electron-demand Diels–Alder reactions and the most common examples of these reactions for normal (a′) and inverse (b′) electron-demand Diels–Alder reactions. From ref. 255 licensed under Creative Commons Attribution 4.0 (CC BY 4.0). (E) Thiol-collagen synthesis scheme. Reproduced with permission from ref. 272, copyright 2019, American Chemical Society. (F) Self-assembly. From ref. 199 licensed under Creative Commons Attribution 4.0 (CC BY 4.0). | ||
2.2.2.1. Schiff-base reaction. Schiff-base reaction is a chemical reaction that forms dynamic covalent crosslinks between the amine and carbonyl groups (with the formation of imine bonds, hydrazone bonds or oxime bonds).238 As a result of the reversible groups exchange reaction, the reversibility of this chemical reaction is usually utilized to empower hydrogels with injectability, shear-thinning and self-healing property.30 Besides, since the Schiff-base reaction can occur under mild conditions, and water is the only by-product and the reaction products are compatible with water, this reaction is appropriate for various biomedical applications. In summary, the Schiff-base reaction has been widely used in the preparation of injectable hydrogels due to its advantages of mild reaction conditions, simple reaction principles, reversible process, pH sensitivity and good biocompatibility.92 Lei and colleagues prepared a dynamic covalent bond-based hydrogel (CLX@Lipo@HA-gel) by the Schiff base reaction between the adipic dihydrazide-modified HA (HA-ADH) and aldehyde-modified HA (HA-CHO) (Fig. 3B).109 This hydrogel was demonstrated to possess favorable injectability as it could be easily injected through a 0.6 mm needle, and the shear-thinning property was further assessed quantitatively by rheometer, which revealed that its viscosity decreased with the increase of shear rate. Moreover, the incorporated celecoxib (CLX)-loaded liposomes were found to provide effective shear-responsive boundary lubrication and release CLX to maintain the anabolic–catabolic balance of chondrocytes, and finally contributed to the alleviation of osteoarthritis. Similarly, in a recent report, Fang et al. synthesized an injectable, adhesive and self-healing hydrogel based on the Schiff-base bonds between the oxidized hyaluronic acid (OHA) and carbohydrazide-modified gelatin (Gel-CDH), which further encapsulated mesenchymal stem cell-secreted small extracellular vesicles (MSC-sEV) and polydopamine nanoparticles (PDA-NPs).239 This hybrid hydrogel was enabled with favorable injectability as it could be injected continuously and used to write any letters smoothly through a syringe. Furthermore, in the results, this hydrogel promoted the healing rate of radiation combined with skin wounds (RCSWs) by relieving radiation-induced tissue inflammation, reducing oxidation impairment and enhancing cell vitality and angiogenesis.
The hydrazone bond is a dynamic covalent linkage generated by the condensation of hydrazine and carbonyl groups, and the formation and dissociation rates are mainly dependent on the structures of hydrazine and aldehyde.240,241 The dynamic nature makes the hydrazone bond suitable for the synthesis of viscoelastic and self-healing hydrogels. However, as a result of the slow exchange kinetics, hydrazone bond-based hydrogels have the obvious disadvantage of poor injectability. Besides, there is a dilemma in designing dynamic crosslinking hydrogels like hydrazone bond-based hydrogels: the rapid exchange rate of crosslinks contributes to easier injection using less force, but lacks long-term polymer stability; conversely, a slow exchange rate improves the stability of networks, but compromises injectability.240 Notably, the addition of appropriate catalysts has been proposed as a potential strategy to have it both ways in recent years. For example, under the catalysis of sulfonated amino-benzimidazole (a non-toxic cyto-compatible catalyst), Lou and co-workers synthesized a novel shear-thinning hydrogel with both high injectability and high stability, which was crosslinked through dynamic hydrazone bonds between the hydrazine-modified HA and aldehyde-modified HA.240 Compared with the hydrogel without catalyst, the gelation time of the novel catalyst-containing hydrogel was significantly decreased due to the significantly accelerated formation and exchange rates of the dynamic covalent hydrazone bonds in the hydrogel matrix. This also contributed to the smooth injection of the hydrogel through a 28 G needle without clogging and the improved cell protection during injection. Moreover, according to the results from the erosion kinetics and oscillatory frequency sweep tests, the addition of catalyst did not alter the network structure and storage modulus of the hydrogel, but rather slowed the degradation rate while improving the long-term stability owing to the rapid diffusion of catalyst out of the hydrogel after injection. In addition, the ketoester-type acylhydrazone bond is regarded as another promising method to prepare injectable hydrazone-bond based hydrogel, as its rapid formation and exchange kinetics enable fast hydrazone crosslinking and rapid reconstruction of the dynamic networks under physiological conditions. Based on this, Jiang et al. developed a novel cellulose-based hydrogel (CPK) via the ketoester-type acylhydrazone bond between the adipic dihydrazide-grafted carboxyethyl cellulose (CEC-ADH) and ketoester-grafted PVA (PVA-Ket).242 Benefitting from the rapid kinetic rates and favorable reversibility of the ketoester-type acylhydrazone bonds, the gelation time was dramatically shortened from 600 s to 3 s, and the CPK hydrogel exhibited excellent shear-thinning behavior as well as good injectability, rapid self-healing and strong mechanical properties.
The oxime bond is formed through the condensation of hydroxylamine with aldehyde/ketone, which is a bio-orthogonal chemo-specific “click” reaction in which the two specific substrates can react with each other efficiently and specifically in the presence of various other functional groups.243–245 Compared with imines and hydrazones, oximes exhibit better hydrolytic stability with the equilibrium lying far to the oxime.246 The kinetics of the formation of oxime bonds can be easily adjusted through changing pH, adding salts or catalytic amine buffers, replacing aldehyde with ketone groups.247–249 Due to the inherent reversibility, the oxime reaction has also been applied to prepare injectable and self-healing hydrogels. For example, Grover et al. fabricated an injectable hydrogel based on the oxime crosslinking of the two PEG derivatives.243 They first synthesized 4-armed ketone-PEG (ket-PEG) through the carbodiimide coupling with levulinic acid, and then prepared 4-armed aminooxy-PEG (AO-PEG) via the Mitsunobu reaction between N-hydroxyphthalimide and the terminal PEG-alcohols and the following deprotection with hydrazine. The oxime bond-based hydrogels were developed by mixing the above two PEG-derivatives at pH 5.0. According to the results, this hydrogel was capable of fast gelation and allowed multiple injections through a catheter at 37 °C. In another study of Baker et al., the same principle was utilized to prepare an injectable HA-based hydrogel which was fabricated to act as a vitreous substitute and was found effective in filling the vitreous cavity through injection, resorbing over time, and maintaining normal ocular pressure and a healthy functional retina.250
2.2.2.2. Disulfide exchange reaction. The nucleophilic substitution reaction between a thiolate ion and an active disulfide that leads to the liberation of another thiolate ion is called the disulfide exchange reaction. Thiolate is the core intermediate which drives forward the reaction, and thus the disulfide exchange reaction is usually performed under basic conditions (such as pH > 9) to ensure enough thiolate.30 The thiol-disulfide exchange reaction, during which the sulfur atom of disulfide is attacked by the thiolate ion (an effective nucleophile), is one of the most representative types that have been applied to prepare injectable hydrogels with self-healing properties. The shear-thinning (then injectability) and self-healing properties of hydrogels are acquired due to the reversible crosslinks, which are formed based on the dynamic behavior of disulfide bonds in the presence of nucleophilic thiols under alkaline or neutral conditions.42 For example, Shi et al. prepared a robust dually crosslinked silk fibroin-based hydrogel via the disulfide exchange reaction and photoinduced polymerization, which was proved to be a promising injectable biomaterial to fill irregularly shaped bone defects (Fig. 3C).251 As verified by the quantitative rheology test, the reversible properties of these interactions enabled the hybrid hydrogel with good shear-thinning and autonomous self-healing characteristics, and the dual-crosslink contributed to the ten-fold improved storage modulus of hydrogel. In addition, since there are abundant free thiol groups and disulfide bonds within extracted keratin, Chen et al. proposed a novel strategy to prepare keratin-based hydrogels with dynamic crosslink networks.252 They provided an alkali condition (the pH value was adjusted to 9.5–10.0) for the keratin solution, under which the free thiol groups and the disulfide bonds of keratin could form dynamic covalent bonds via the thiol-disulfide exchange reaction. According to the reported results, this hydrogel could be easily injected out through a narrow needle to form the pre-set shape, and the results from steady shear measurements further demonstrated its favorable injectability and shear-thinning property. This reversible behavior was mainly the result of the disulfide bonds generated through the oxidation of free thiol groups within the type II keratin, which then promoted the conformation transformation from α-helix to β-sheets. Additionally, this principle has also been investigated and demonstrated to be effective in many other biomedical processes, such as hemostasis, wound healing, liver injury and fabrication of artificial nucleus pulposus.253,254 Notably, disulfide bonds can cleave rapidly in the presence of glutathione (GSH), and thus injectable GSH-responsive hydrogels have been developed for biomedical applications. For example, the team of Liu et al. prepared an injectable hydrogel loaded with DOX using poly (thioctic acid) (PTA) and PEG, which achieved GSH-responsive “smart” drug release in the tumor microenvironment.179 Due to the abundant disulfide bonds contained in PTA, the crosslinked network of the hydrogel rapidly degraded upon the injection into the tumor tissue, where the GSH concentration was elevated abnormally. This resulted in the substantial release of the loaded DOX and induced tumor cell death. On the other hand, due to the lower GSH concentration in normal tissues, the hydrogel exhibited low leakage in the normal physiological environment. Thus, the GSH-responsive hydrogel greatly enhanced the therapeutic efficacy of DOX while significantly reducing off-target and related toxic side effects in normal tissues.
2.2.2.3. Diels–Alder reaction. The diene components with a conjugated system of double bonds and the dienophilic components with double/triple bonds can generate 6-membered cycles through a “2 + 4” cyclo-addition reaction, which is also called the Diels–Alder (DA) reaction.255 Since it is a thermal reversible chemical reaction, the 6-membered cycle generated from DA reaction can be decomposed back into dienes and dienophiles under high temperature. As a member of the “click chemistry” family, the DA reaction possesses various advantages, such as high regioselectivity, partial stereoselectivity and stereospecificity, and can be proceeded efficiently under mild conditions, without the need for catalysts and with the absence of any by-products.42 Besides, the DA reaction has excellent bio-orthogonality, meaning that the reaction can occur inside living systems without influencing the normal biochemical processes in vivo, and the formed covalent bonds can significantly increase the mechanical strength of the crosslinked networks.255 As a result, the DA reaction is regarded as a promising tool in the preparation of hydrogels with favorable biocompatibility. Meanwhile, since the DA reaction-induced gelation time (varies from tens of seconds to several minutes) can be easily tailored by adjusting the temperature parameter and crosslinking density, hydrogels are endowed with good injectability through injection within the “gelation time window”.255,256
According to the nature (electron donor or acceptor) of the substituents of diene and dienophile, the DA reaction is further divided into two types (normal and inverse electron-demand type) (Fig. 3D). Generally, the normal electron-demand DA reaction is proceeded through the reaction between the electron-rich donating diene groups (EDG) and the electro-poor withdrawing dienophilic groups (EWG).257 In modern polymer chemistry, the most commonly used diene–dienophile pair for the normal electron-demand DA reaction is the furan-maleimide coupling. For instance, Bai et al. prepared an injectable chondroitin sulfate-based hydrogel through the DA reaction between the maleimido-terminated Pluronic F127 and the furfurylamine-grafted chondroitin sulfate.258 This hybrid hydrogel could gel in tens of seconds, and injection during the “gelation time window” contributed to the in situ gelation of the hydrogel and sustained release of the loaded bone morphogenetic protein-4 (BMP-4), and finally promoted the bone regeneration of cranial bone defects.
Compared with the above type, the inverse electron-demand DA reaction involves the opposite groups of diene and dienophile, which corresponds to the interactions between dienophiles with EDG and dienes with EWG, respectively.255 One of the most outstanding advantages of this inverse electron-demand DA reaction is that it can greatly shorten the gelation time and thus reduce the hydrogel swelling when compared with the normal electron-demand DA reaction. The most used diene–dienophile functional pair for the inverse electron-demand DA reaction is norbornene–tetrazine coupling. For example, Vu and colleagues synthesized an injectable hydrogel, which was crosslinked through the inverse electron-demand DA reaction between the norbornene groups of the chitosan derivatives and the tetrazine groups of the disulfide-based crosslinker, to deliver DOX in a controlled manner.259 The results showed the gelation time varied from 90 to 500 s, which reserved sufficient time for the injection of the pre-gel solution into the body and thus achieved injectability. As the N2 gas produced by the click reaction contributed to the porosity-creating process during gelation, this hydrogel exhibited high drug-loading capability. In another study from the same team, the newly designed hydrogel, which was developed based on the inverse electron-demand DA reaction between the alginate-norbornene and the PEG based disulfide crosslinker containing two terminal tetrazine groups, was also found to be easily injected through a needle within the several-minute-long gelation time window.260
However, as an exothermic reaction, the products of the DA reaction are stable, and thus it requires large energy (such as >100 °C) to proceed the retro DA reaction, which obviously limits its application in mild biological systems.261,262 Of note, when the reaction substrates are electron-rich dienes and electron-poor dienophiles (such as anthracene and tetracyanoethylene), the retro DA reaction can be proceeded at room temperature, and the equilibrium is not sensitive to water.263,264 Hence, this reversible DA reaction under mild conditions provides a new method to develop injectable hydrogels with shear-thinning and self-healing properties. In the report of Wei et al., the investigators designed a novel injectable hydrogel based on a reversible DA reaction, in which the fulvene-modified hydrophilic dextran (Dex-FE) acted as dienes and the dichloromaleic-acid-modified PEG (PEG-DiCMA) acted as dienophiles.261 According to the results from the rheological recovery test, the reversible changes of G′ and G′′ values in the strain cycle (from 1.0% to 1000%) demonstrated the good injectability, and favorable shear-thinning and self-healing properties of this hydrogel. Meanwhile, its self-healing ability was further proved through the macro- and microscopic assessments. All the above excellent performances of this hydrogel were owing to the reversible linkages based on DA bonds, which further contributed to the effective dynamical reconstruction and self-healing following mechanical disruption.
2.2.2.4. Michael addition reaction. Michael addition reaction is a conjugate addition of enolate-type nucleophiles with α, β unsaturated carbonyl groups, which is usually catalyzed by bases or nucleophiles without the use of toxic chemical agents.265 The nucleophile is called the Michael donor while the α, β unsaturated compound is termed the Michael acceptor, and the reaction product is named the Michael adduct.266 Highly specific polymer products with crosslinked architectures can be easily synthesized under mild conditions via Michael addition reaction, and due to the facile and orthogonal nature, this reaction is considered beneficial for biological applications.265–267 According to the formed bonds, Michael addition reactions can be further divided into several different types, including thiol-, aza-, oxa- and carbon-Michael addition reactions.265,267–269 Of note, thiol-Michael addition reaction is a typical type, which is a nucleophilic addition of thiols (the Michael donor) on olefines conjugated with EWG (the Michael acceptor).270 This type of reaction is heavily utilized for materials synthesis (including hydrogels) for its mild reaction conditions, high efficiency and yields, specific selectivity and tunable reaction parameters.271 For example, Pupkaite et al. fabricated a novel injectable hydrogel for cell encapsulation and targeted delivery, which was crosslinked through the thiol-Michael addition reaction between the thiol-grafted collagen and the 8-armed PEG-maleimide (Fig. 3E).272 The rheological results revealed the excellent self-healing and shear-thinning properties, which contributed to its outstanding injectability; this hydrogel could be injected smoothly through a 27-gauge needle even 5 and 16 h after preparation. Besides, owing to the good shear-thinning property, the encapsulated cells maintained high viability even at the 4th day following injection. Similarly, the team of Fan et al. fabricated an injectable hydrogel via the covalent bonds generated through the double Michael addition of thiols to alkynones on dextran-derived polymers, and this hydrogel was demonstrated to be a promising injectable depot for the sustained and prolonged delivery of vaccines.273
2.2.2.5. Self-assembly. In addition to the above chemical strategies, there are so called self-assembly hydrogels that can gel spontaneously under mild conditions only via physical crosslinking without dependence on chemical agents/initiators and related chemical reactions, which then contributes to their high bio-compatibility. Self-assembly is a physical crosslinking process that is based on non-covalent interactions and weak forces (including hydrogen bonds, electrostatic repulsion and attraction, hydrophilic and hydrophobic interactions, π-stacking and van der Waals forces), and maintains suitable stability and balance between the various countering forces (Fig. 3F).199,236,274,275 Due to the reversible linkages within the networks, the crosslinked hydrogels exhibit low viscosity under shear stress and re-gel rapidly with the removal of stress, and thus these hydrogels are endowed with favorable self-healing and shear-thinning properties.199 As a result, the dynamic crosslinking maintains the integrity of the hydrogels, and also mechanically protects the encapsulated cells from shear damage during injection.276 Self-assembling peptide (SAP) is one of the most widely used types of this system, which is characterized by a simple synthesis method with high yield, a reproducible synthesis process, flexibility for modification and enzymatic degradability in vivo.236,277 As a synthetic short poly-peptide consisting of user-specified amino acids, SAP not only has low immunogenicity and can degrade in vivo slowly, but also provides great convenience for the network assembly control of hydrogels.46,278 Depending on the acquired secondary structures, SAP can be further divided into nature-mimicking structures (such as α-helices, coiled coils, β-sheets and β-hairpins) and newly designed derivatives (amphiphilic and short aromatic peptides).279 The team of Yan et al. investigated the mechanism of shear-thinning and rapid self-healing of injectable hydrogels based on β-hairpin peptides in detail.280 Specifically, they provided comprehensive reports (including rheological behavior and mechanism, mechanical changes and the impact on loaded cells) of the two representative β-hairpin peptides, VKVKVKVK–VDPPT-KVKVKVKV-NH2 (MAX1) and VKVKVKVK–VDPPT-KVEVKVKV-NH2 (MAX8), upon various experiments such as rheometer tests, shear recovery experiments, rheo-small-angle neutron scattering (SANS) and small-angle X-ray scattering (SAXS). The two β-hairpin peptide hydrogels exhibited shear-thinning behaviors and recovered to solids right after shearing stress, obtaining varying immediate stiffness depending on the different shear rates and duration. The results from SANS and SAXS indicated that the network and physical crosslinking was broken due to the applied shear force, but the fibril network structure of hydrogels remained unchanged after the shearing tests with different shear rates. Moreover, the shear-thinning hydrogel was proved to be an excellent candidate as live-cell delivery vehicle due to the small impact on cell viability and spatial distribution, which was demonstrated by 3D confocal microscope imaging.
The self-assembly mechanisms are phase-separation and amphiphilic, and these hydrogels can be divided into two groups according to the different interaction types, i.e., host–guest interactions and complementary binding.223 Host–guest interactions are usually proceeded by cyclodextrins or cucurbituril. For example, Feng and co-workers prepared a highly resilient gelatin hydrogel based on the efficient host–guest interaction between the free diffusing photocrosslinkable acrylated β-cyclodextrin (β-CD) monomers and the aromatic residues of gelatin.281 The excellent shear-thinning property enabled the injection of the hydrogel in gelation state though a needle and filling of the targeted geometries completely. Besides, since this hydrogel was crosslinked only via the weak host–guest interactions, it not only sustained high tensile/compressive strain and self-healing rapidly after mechanical disruption, but also provided mechanical protection for the encapsulated cells while facilitating the migration and infiltration of host cells into the hydrogels. On the other hand, complementary bindings (including base-pairing interactions, antigen–antibody pairs and ligand–receptor pairs) are also widely used to fabricate injectable shear-thinning hydrogels.282–284 Due to the extremely high binding affinity and strong tendency toward each other of ligand–receptor pairs, the complex between the ligand and receptor is utilized to drive gelation.285 One of the typical examples is the streptavidin-biotin pair, which has been used to develop an injectable cell-laden hydrogel that could retain the original shape within minutes following injection into a cavity.286 In addition, several self-assembly hydrogels based on deoxyribonucleic acid (DNA) and/or ribonucleic acid (RNA) have been well developed in recent years, and also play a role in various biomedical applications.287–289
Nevertheless, as a result of the weak linkages within the networks, the stability and mechanical properties of these self-assembly hydrogels are quite poor.199,236 To overcome these disadvantages, some researchers have combined (dynamic) covalent bonds with the weak non-covalent bonds within the self-assembly hydrogels to improve the inherent poor physical properties through the formation of double or multiple crosslinked networks, and the physical crosslinking was regarded as a pre-crosslinking form prior to the chemical ones in these cases.199,290,291
2.2.3.1. Microfluidics. Microfluidics is a commonly employed detection technique in the field of biopharmaceutical analysis, playing an important role in applications such as single-cell multi-omics analysis, hemostasis monitoring, thrombus diagnosis, and liquid biopsy of tumor-derived exosomes.293–295 With its downscaled “chip laboratory” on a micrometer scale, microfluidics systems enable precise manipulation of minuscule volumes of fluids, resulting in a substantial reduction in both testing expenses and sample consumption. Concurrently, microfluidics has emerged as a novel technique to develop spherical droplets with uniform size of the aqueous hydrogel precursor within a continuous oil phase, and plays a vital role in fabricating HMs.296 Generally, the size of the droplets developed by microfluidics ranges from 5 to 1000 μm, which can be easily adjusted to a specific size by changing the device channel geometry, the flow ratios of the oil and aqueous phase flow, and the interfacial tension between the immiscible fluids.28,297 One of the most excellent advantages of microfluidics is the extremely high uniformity and mono-dispersity of the formed microspheres, whose diameter variation was reported to be as low as 1 to 2%.298
The poly(dimethylsiloxane) (PDMS) microfluidics device is a classical tool to develop HMs, which is treated with on-chip plasma-assisted deposition of PVA to modify the hydrophobicity of the micro-channels. Samandari et al. developed a simple but rapid method to prepare mono-disperse HMs, which was based on the double emulsion system on a chip-microfluidics device.299 According to the experimental results, the formed hydrogels not only exhibited uniform size with narrow size distribution (C.V. = 2.4%), which could be easily adjusted through changing the rate of the oil phase flow and aqueous phases flow, but also achieved the efficient encapsulation of cells with high viability and desired concentration.
In addition to the classical devices like PDMS chips, there are many other methods to achieve microfluidics due to its inherent easy feasibility. Researchers can make a microfluidics device by themselves with existing equipment and materials, such as syringe needles, rubber hoses and plastic catheters. The team of Lei et al. constructed a simple microfluidics device with a glass slide, two syringe needles and three plastic catheters (one for the dispersed aqueous phase, one for the continuous oil phase and one for the collection of microspheres) (Fig. 4A).32 According to the light microscopy images, the microspheres developed through this self-designed microfluidics device exhibited good dispersion, and the diameters were 208.36 ± 7.37 μm with a narrow size distribution.
 | ||
| Fig. 4 Strategies for the preparation of microgels. (A) The principle and fabrication of RAPA@Lipo@HMs. (a) The fabrication of RAPA@Lipos, photocrosslinkable HAMA matrix, and microfluidic RAPA@Lipo@HMs. (b) The design of RAPA@Lipo@HMs for treating osteoarthritis based on combining hydration lubrication and ball-bearing lubrication and maintaining cellular homeostasis. From ref. 32 licensed under Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC). (B) Scheme of the fabrication of CMS via a combination of an emulsion technique with gradient-cooling cryogelation. Adapted with permission from ref. 301. Copyright 2021, John Wiley and Sons. (C) Graphical abstract: (a) Bulk hydrogel is mechanically extruded through a grid to deconstruct it into microstrands. In this process, microstrands randomly entangle within each other and form entangled microstrands—a stable material with properties relevant for tissue engineering: moldability, stability in aqueous solutions, porosity, printability, and alignment of microstrands by extrusion. (b) A bioink can be created by embedding cells in bulk hydrogel before sizing that results in a spatial deposition of cells inside the gel phase. Alternatively, cells can be mixed in between already prepared entangled microstrands, so cells occupy the space outside the gel phase. From ref. 303 licensed under Attribution 4.0 International (CC BY 4.0). | ||
Recently, there have been some interesting reports that have developed microspheres with non-spherical shapes through the combination of microfluidics devices with different sizes. Cai and colleagues designed a novel microcapillary microfluidics device with a casing structure, which consisted of two cylinder microcapillaries with different inner diameters (300 and 500μm), and developed mono-disperse bullet-shaped microspheres with tunable structures.300 This deformation of microspheres was the result of the changed spatial confinement and/or the external fluid shear stress, and the bullet-shaped microspheres were found to have better embolization performance than the conventional spherical ones.
Despite the advantages of precise control of the size and distribution of microspheres, the disadvantages of microfluidics are also obvious: the production rate is slow and the required time of fabrication is positively related with the desired output volume. Therefore, great efforts must be made to improve the production efficiency of microfluidics prior to the mass production of microspheres and the further wide application in clinical practices.
2.2.3.2. Emulsion technique. The emulsion technique is an appropriate choice for batch production of HMs, and can develop large quantities of microspheres within a short period by agitating the added aqueous hydrogel precursors in the continuous oil phase. When compared with the abovementioned microfluidics, the emulsion technique provides significantly faster production rates with the use of commonly available equipment, and the size of HMs can be easily changed by adjusting the agitation parameters. In the work of Yuan et al., they prepared a series of gelatin-based HMs with porous microstructure via the combination of the emulsion technique and gradient cooling cryogelation.301 The use of the emulsion technique contributed to the highly effective harvest of numerous GelMA HMs within several hours, and then the different gradient cooling protocols resulted in different pore diameters of the microspheres (Fig. 4B). However, it is worthy of note that the size distributions of the microspheres formed through the emulsion technique (even from the same batch) are much greater, and the diameters may vary by an order of magnitude.
2.2.3.3. Extrusion fragmentation. Extrusion fragmentation is a physical fragmentation under mechanical forces that partition the hydrogels crosslinked in bulk into micro-scale components, and the size of the acquired microgels usually varies from tens of micrometers to tens of millimeters according to the extent of fragmentation. This is also a rapid technique with fast efficiency and few required resources. Compared with the above two techniques, extrusion fragmentation is free from the extensive washing steps due to the absence of oils/surfactants in the microgel fabrication, which not only avoids the decreased cell viability due to exposure to harsh oils/surfactants and repeated washing, but also retains the cytocompatibility and other original properties of the microgels.28 Different from the spherical shape based on the two abovementioned methods, the shapes of the microgels obtained from extrusion fragmentation are irregular with improved storage modulus (G′), increased pore numbers and decreased average pore sizes.298 Sinclair and co-workers reported an injectable, malleable, shear-thinning and self-healing zwitterionic injectable pellet (ZIP) microgel by repeated extrusion of the crosslinked hydrogels in bulk through micronic steel mesh, whose size could be easily tailored by tuning the mesh pore size.302 The synthesized ZIP microgel was found to have a mean diameter of 15 to 30 μm, and exhibited favorable injectability and malleability, which provided an appropriate platform for various biomedical practices including drug/cell delivery vehicle with mechanical protection and injectable tissue filler. In recent years, this technique has also been applied in the batch fabrication of hydrogel micro-strands with high-aspect-ratio from various types of bulk hydrogels for 3D-bioprinting.303 In this study, Kessel et al. deconstructed the crosslinked bulk hydrogels into micro-strands by sizing through a 40–100 μm grid, and the developed micro-strands exhibited a porous and entangled structure, and were suitable for extrusion bioprinting due to favorable rheological properties (Fig. 4C).
2.2.3.4. Electrospray. Recently, based on the principle of electrohydrodynamic printing in electronic manufacturing, some researchers have developed a novel strategy called electrospray to fabricate HMs with high uniformity and mono-dispersity. According to the report from Xie and colleagues, they successfully prepared uniform GelMA HMs (∼100 μm) based on an electro-assisted bioprinting process, which was conducted based on a printing device consisting of an electro-assisted module, a gas pressure provider and a droplet receiver.304 By adjusting the parameters of the abovementioned printing modules (such as nozzle size, voltage and gas pressure), the same device could form different printing states (including micro-dripping, shaking spindle, oscillating jet and Taylor jet) and HMs with different sizes. The experimental results demonstrated the high uniformity and mono-dispersity of the formed GelMA HMs, which exhibited excellent performance in the three representative biomedical applications of drug-controlled release, cell encapsulation and 3D bio-printing. This is an innovative strategy for HM fabrication with high efficiency and controlled deposition position, and low cost and cell damage. However, the disadvantages of this method are obvious: the multiple influencing factors, the dependence upon the high-voltage power supply, and the high difficulty in HM collection.
3. Stimuli responsiveness
From a molecular perspective, the responsiveness of smart stimuli-responsive hydrogels is mainly based on the connection or breaking of bonds that crosslink the hydrogel networks under the exposure of specific stimuli (such as temperature and pH). This process leads to the shrinkage or expansion of the hydrogel's network structures, causing changes in its physical properties (dissolution, polymerization, or collapse). Consequently, it enables the controlled release of the therapeutic agents (including drugs, cells, cytokines, and other therapeutic substances) encapsulated within the hydrogel, achieving the goal of “smart” therapy.305 This has significant implications for enhancing the targeting of treatments and mitigating the side effects associated with therapeutic agents, particularly for those with potent cytotoxicity (e.g., anticancer drugs). During the development of diseases, the physiochemical properties of the microenvironment in affected tissues undergo abnormal alterations. For instance, solid tumor tissues exhibit a significant reduction in pH compared with normal tissues, infarcted myocardial tissues show a substantial increase in the expression of matrix metalloproteinases (MMPs), and the level of reactive oxygen species (ROS) in osteoarthritis (OA) cartilage is notably elevated. These pathophysiological changes within the organisms create favorable conditions for the application of smart stimuli-responsive hydrogels in targeted therapy for specific biomedical purposes. In addition, there are some substances/linkages which can specifically respond to external stimuli (including light, ultrasound, magnetic fields, and electricity), and thus have also been investigated for the development of injectable smart stimuli-responsive hydrogels, which have exhibited notable therapeutic results in some biomedical practices. In conclusion, by leveraging the abnormal physiochemical properties of diseased tissues and incorporating specific stimulus-responsive elements, smart hydrogels hold great promise for targeted therapies in the field of biomedicine. Their potential to respond to external cues makes them versatile tools with the capacity to revolutionize certain medical treatments.3.1. Temperature responsiveness
Injectable temperature-responsive hydrogels can respond to temperature changes, and result in the sol–gel transition or the shrink/expansion/collapse of the hydrogel matrix, which is the result of the connection/fracture of the crosslinking bonds. Therefore, the main principle behind the therapeutic role of the injectable thermo-responsive hydrogel is to enable the hydrogel to gel in situ after perfect apposition to the tissue defect/cavity through the thermally induced conformational changes, in order to provide adequate mechanical support for the tissue repair, to achieve slow release of loaded drugs/cytokines, or to provide appropriate scaffolds for the migration and adhesion of host cells.306,307 As mentioned earlier, we have discussed the concept of UCST and LCST for these types of hydrogels. Notably, the most widely applied type is the LCST hydrogel, while the UCST ones are almost useless. This is because most of the phase transition temperature of UCST hydrogels is below 25 °C, which is obviously lower than the physiological temperature (30–37 °C), and thus it is difficult for UCST hydrogels to play a role in biomedical processes due to the gel–sol transition under physiological temperature.308,309 Consistent with the principle of temperature-initiated gelation, the use of thermal-sensitive materials and addition of thermal-sensitive segments are two common strategies to develop injectable temperature-responsive hydrogels. For example, the team of Lv et al. successfully prepared an injectable temperature-responsive hydrogel with the use of two biomaterials with inherent thermal sensitivity (chitosan and silk fibrin).147 Meanwhile, the addition of MgFe layered double hydroxide (LDH) significantly improved the thermal-responsiveness of this composed hydrogel, which enabled the fast in situ gelation in the tissue defects under lower temperature (from 37.4 to 32.7 °C), and then acted as a drug depot in vivo that completely fitted the targeted tissues and sustainedly release growth factors to promote osteogenesis and angiogenesis. On the other hand, Zakerikhoob et al. synthesized an injectable thermal-responsive hydrogel dressing which can gel within 27–42 °C (covering the physiological temperature) by grafting poly(N-isopropylacrylamide) (PNIPAm, a common thermal-sensitive polymers in biomedical fields) onto the chains of sodium alginate.310 The grafted PNIPAm enabled this hydrogel dressing to fill the wound completely first through injection, and then gel in situ through the response to the wound temperature. Thereafter, this hydrogel achieved the sustained release of curcumin within 72 h, and exhibited excellent alleviation of wound inflammation and promotion of wound healing.3.2. pH responsiveness
The same as temperature, change in the pH environment can also result in the contraction/expansion of the hydrogel networks, which then leads to the encapsulation/release of the loaded therapeutics; these hydrogels are called pH-responsive hydrogels. As mentioned earlier, the responsiveness towards pH is mainly determined by the weakly acidic/basic groups within hydrogel matrix. These groups donate or accept protons when the environmental pH is changed, which would disrupt the bonds supporting the networks or vary the equilibrium between electrostatic attraction and repulsion.311,312 As a result, the physicochemical properties of hydrogel matrix and the equilibrium of the inner interactions are changed, resulting in the shrinking/expansion of networks. For example, the team of Qu et al. developed a series of injectable pH-responsive hydrogels based on dibenzaldehyde-terminated PEG (PEGDA) and N-carboxyethyl chitosan (CEC), which were also endowed with good self-healing and shear-thinning properties.313 Due to the protonation of the amino groups of CEC under acidic conditions, the networks were broken faster and resulted in significantly more release of the encapsulated DOX inside or around tumor when compared with normal tissues. The consistent performances of the pH-dependent degradation and drug release both in vitro and in vivo made the hydrogels suitable for tumor therapy, which provided a promising strategy to maximize the inhibition of tumor cells while minimizing the undesirable side effects on the normal tissues.There are some dynamic covalent bonds (such as imine bonds and hydrazone bonds) that can remain stable under neutral conditions, yet would break rapidly in an acidic or basic environment. As a result, these chemical bonds have inherent responsiveness towards pH changes and thus are also well investigated for the fabrication of injectable pH-responsive hydrogels.314 For instance, in the study of Zhou et al., the authors prepared an injectable hydrogel with carboxymethyl chitosan (CMCS) and oxidized hydroxypropyl cellulose (Ox-HPC).315 Due to the reversible imine bonds between CMCS and Ox-HPC, this hydrogel exhibited favorable self-healing and shear-thinning properties, which all contributed to its good injectability. Besides, due to the inherent pH responsiveness of imine bonds, the network structures of this hybrid hydrogel were degraded faster at the pH which mimics that of tumor tissues, which resulted in the significantly accelerated release of the encapsulated phenylalanine. Furthermore, Jiang et al. developed a novel cellulose-based hydrogel based on the reversible ketoester-type acylhydrazone bonds, which enabled the newly designed hydrogels with favorable injectability, sensitive pH responsiveness, tunable mechanical property, and excellent self-healing and shear-thinning properties.242 The reported results also demonstrated the favorable pH responsiveness of this hydrogel, as nearly 100% of the encapsulated DOX was released from the hydrogel in pH 6.2 buffer within 30 days, while only 34% was released from the one in pH 7.4 buffer within the same period.
In addition, some injectable hydrogels crosslinked through the coordination bonds between multivalent metal ions and the oxygen and/or nitrogen atoms on the backbone of polymers also have responsiveness to pH changes. The coordination bonds within hydrogels exist stably in neutral conditions, yet would break rapidly once they are exposed to an acidic environment, and the hydrogel networks would collapse thereafter. Tang and co-workers prepared an injectable hydrogel with multiple functions based on the coordination between 4-arm PEG-b-polyhistidine (4PEG-PHis) and Ni2+.312 The reversible coordination bonds endowed the hydrogel with favorable injectability, shear-thinning and self-healing properties. Meanwhile, the multivalent coordination bonds exhibited excellent stability in neutral buffer (pH = 7.4), but the linkage was broken immediately as the pH value was decreased to be lower than 6.0, in which the PHis chains were protonated and the coordination with Ni2+ ions was deprived. Hence, this injectable hydrogel system was also promising for achieving pH-triggered drug release in biomedical applications.
3.3. Enzyme responsiveness
As previously mentioned, injectable hydrogels containing specific enzyme-responsive moieties can undergo enzyme-specific reactions upon injection into tissues, leading to either the formation or breakdown of the hydrogel network, resulting in reversible or irreversible gel–sol transitions, and thus achieving sensitive responses to specific stimuli. By adjusting the structure and quantity of the enzyme-responsive moieties, the responsiveness of the hydrogel can be tailored to exhibit desirable behavior in specific biological environments. Therefore, in addition to the preparation of injectable hydrogels for enzyme-initiated in situ gelation as discussed earlier, numerous enzyme-triggered hydrogels that undergo enzymatic degradation have also been extensively investigated for intelligent release of therapeutic agents carried by the hydrogel when exposed to specific enzymes. Such injectable enzyme-responsive hydrogels typically incorporate moieties (i.e., enzyme-responsive substrates) grafted onto the polymer chains of the hydrogel that match specific enzymes. These enzyme-responsive moieties can recognize and undergo enzyme-specific reactions with particular biological enzymes. Consequently, following injection into tissues, the hydrogel undergoes enzyme-catalyzed reactions with specific enzymes, leading to the disruption of the hydrogel network structure, thereby inducing stimulus-responsive drug release. For instance, Li and colleagues developed an injectable in situ-forming hydrogel (NDHM, NanoDOX-loaded HA-MMP hydrogel) based on acrylated-HA (HA-AC), DOX-loaded PDLLA-PEG-PDLLA (PDLLA, poly(D,L-lactide)) triblock copolymers (NanoDOX), and the peptide GCRDGPQGIWGQ for localized chemotherapy of oral squamous cell carcinoma (OSCC).316 Due to the overexpression of matrix metalloproteinase-2 (MMP-2) in OSCC tissues, and the presence of the GPQGIWGQ peptide sequence in the GCRDGPQGIWGQ peptide that is specifically cleaved by MMP-2, NDHM underwent faster degradation in the presence of MMP-2 after injection at the tumor site. This led to the rapid release of loaded DOX in the local tumor tissue and resulted in strong inhibition of OSCC tumor growth. Meanwhile, due to the low expression of MMP-2 in normal tissues, the undesirable side effects of DOX were significantly reduced. Moreover, other members of the MMP family, such as MMP-1 and MMP-9, have also been exploited to prepare MMP-responsive injectable hydrogels for controlled drug release in various diseases with abnormal MMP elevation to achieve “smart” therapeutic interventions.177,317,318Recently, some free fatty acids have also been investigated for the preparation of injectable enzyme-responsive hydrogels, because their molecular structures contain ester bonds that can undergo specific cleavage in the presence of esterase. In this context, Kumar et al. utilized glycerol monostearate (GMS) to prepare an injectable esterase-responsive hydrogel for the treatment of ulcerative colitis (UC).319 They first mixed GMS with budesonide (Bud), dimethyl sulfoxide, and distilled water upon heating and continuous stirring. Because GMS possesses a hydrophilic glycerol head and a hydrophobic polyethylene tail, the mixture self-assembled into a three-dimensional structured hydrogel after cooling, with the hydrophobic Bud effectively encapsulated within the hydrogel networks. According to the drug-release experiments, the encapsulated Bud in the hydrogel exhibited an esterase concentration-dependent release profile, where the release rate of Bud increased with higher esterase concentrations. This was attributed to the esterase-induced cleavage of ester bonds within the hydrogel network, leading to the accelerated hydrogel degradation. Additionally, the hydrogel demonstrated adhesion to the inflamed colonic mucosa and exhibited prolonged and sustained drug release, which significantly enhanced the therapeutic effectiveness and reduced the frequency of conventional enema treatments.
In addition, due to the susceptibility of DNA to endogenous nucleases, some DNA-based hydrogels exhibit inherent specific responsiveness to nucleases, and have been recently explored for the development of injectable enzyme-responsive hydrogels. These hydrogels rapidly degrade in the presence of endogenous nucleases, gradually breaking down into nanoscale particles, and subsequently release the carried therapeutics. As a result, these hydrogels not only possess excellent degradability (fully biodegradable), but also exhibit good penetration even in deep-seated diseased tissues and cells. The team of Zhang et al. grafted a multitude of camptothecin (CPT) onto the backbones of the phosphorothioate DNAs and assembled these modified skeletons into two types of Y-shaped building blocks.320 Subsequently, these Y-shaped building blocks were connected layer by layer to form a CPT-loaded hydrogel (CPT-DNA-Gel). Based on the Watson–Crick base pairing, CPT-DNA-Gel could autonomously form a hydrogel within 1 minute at physiological temperature (37 °C), and the hydrogel demonstrated good injectability. Moreover, due to the elevated levels of glutathione (GSH) and the presence of nucleases in tumor tissues, the disulfide bonds and DNA strands within the hydrogel were broken rapidly upon injection into the tumor tissue, leading to a substantial release of CPT. This achieved excellent localized chemotherapy efficacy, while effectively inhibiting the tumor recurrence caused by residual cancer cells after tumor resection.
3.4. Reactive oxygen species responsiveness
Reactive oxygen species (ROS) are a group of oxygen-containing chemical species with higher reactivity than the ground state oxygen. They play crucial roles in maintaining normal cellular physiological functions and the progression of diseases, earning them the moniker “double-edged swords” within biological systems. At physiological levels, ROS can enhance the organism's immune response and promote cell survival and proliferation. However, under pathological conditions, an excessive production of ROS beyond the cellular antioxidant capacity leads to oxidative stress, and damages lipids, proteins, and nucleic acids within the cells, causing cellular dysfunction, cell death, or even carcinogenesis.321 It is noteworthy that several typical ROS, such as hydroxyl radicals, superoxide anions, singlet oxygen, and hydrogen peroxide, play pivotal roles in the occurrence and development of numerous diseases. Consequently, they have become targeted design elements for intelligent stimulus-responsive hydrogels. Generally, a common strategy for designing injectable hydrogels with ROS responsiveness involves the incorporation of ROS-responsive linkages or moieties into the hydrogel matrix. These linkages or moieties enable the hydrogel network to remain stable in the physiological environment but undergo degradation under stimulation by high concentrations of ROS. This degradation process consumes ROS and allows for the localized ROS-responsive release of therapeutic agents at the site of the pathological tissue, achieving precise and intelligent local therapy.322Borate ester bonds are currently one of the most widely used chemical linkages with ROS-responsive capabilities. They can rapidly break in the presence of ROS, leading to the degradation of injectable hydrogels based on borate ester bonds and the subsequent release of the encapsulated therapeutic agents. Gan et al. utilized the borate ester bonds formed between PVA and a ROS-responsive compound, N1-(4-boronobenzyl)-N3-(4-boronophenyl)-N1,N1,N3,N3-tetramethylpropane-1,3-diaminium (TSPBA), to prepare an injectable hydrogel (Hgel@C5A/MΦ) with a stable 3D network structure.323 This hydrogel was designed to carry bone marrow-derived macrophages (MΦ) and a C5aR antagonist (C5A) for localized treatment of periodontitis. Given that the ROS levels in gum tissues in refractory periodontitis are significantly elevated, the injection of Hgel@C5A/MΦ into the gingival crevices triggered its degradation, leading to the on-demand release of drugs and cells in the ROS-rich periodontal microenvironment for targeted therapy (Fig. 5). Specifically, the networks of the injected Hgel@C5A/MΦ were broken due to the increased ROS, which achieved the “smart” responsive-release of the loaded C5A and MΦ and then improved the antimicrobial activity of macrophage, and finally alleviated the periodontitis. Moreover, due to the substantial generation of ROS during ischemia-reperfusion (I/R) therapy in myocardial infarction, injectable ROS-responsive hydrogels have also been applied in smart drug delivery for cardiac repair. Similarly, Li et al. utilized PVA and TSPBA to prepare an injectable hydrogel loaded with basic fibroblast growth factor (bFGF).324 The results demonstrated that the hydrogel injected into the pericardial cavity spread on the heart surface and formed an epicardial patch in situ. As there was an abundance of ROS in the myocardium at the site of I/R, the hydrogel distributed on the heart surface released its payload of bFGF predominantly in the local tissue that underwent I/R, thereby precisely reducing myocardial fibrosis, enhancing angiogenesis, and preserving cardiac function in the I/R-affected tissue. Based on the same principle, ROS-responsive hydrogels employing this strategy have also been developed for post-malignant tumor resection immunotherapy, exhibiting significant tumor-suppression effects while minimizing drug-related adverse reactions.325
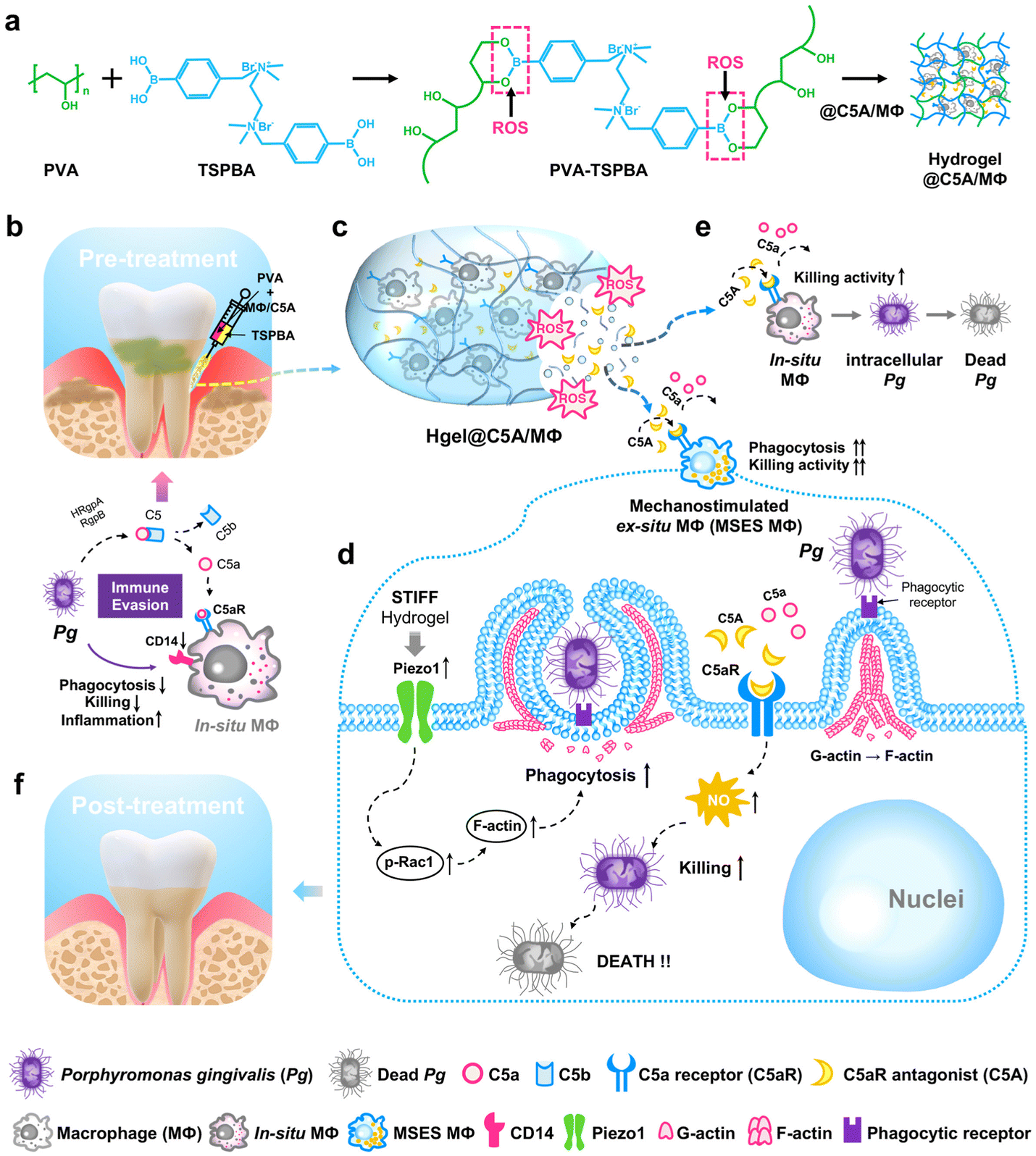 | ||
| Fig. 5 Schematic illustration of mechanism how the high-modulus ROS-sensitive hydrogel encapsulating macrophages and C5aR antagonists (Hgel@C5A/MΦ) treats periodontitis. (a) Diagram of the preparation of the PVA-TSPBA hydrogel. (b) Illustration of the administration of Hgel@C5A/MΦ and the mechanism of Pg immune evasion. (c) Release of the mechanostimulated macrophages and C5A from the Hgel@C5A/MΦ in response to the ROS-enriched periodontitis microenvironment. (d) Mechanism of improving macrophage antimicrobial activity using Hgel@C5A/MΦ. (e) Illustration of reactivating in situ macrophages to kill intracellular Pg. (f) Diagram of the final therapeutic effect of periodontitis. From ref. 323 licensed under Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0). | ||
In addition, thioketal bonds are also commonly used ROS-sensitive chemical linkages for the preparation of ROS-responsive hydrogels. Due to their susceptibility to ROS cleavage, hydrogels based on disulfide bonds enable responsive drug release in the presence of ROS. For instance, the team of Zheng et al. first encapsulated sphingosine-1-phosphate (S1P) and elamipretide (SS-31) in liposomes. Then the S1P/SS-31-loaded liposomes were incorporated into hydrogel networks, which were formed by the mixing of 4-arm-PEGsuccinimidyl glutarate ester (4-arm-PEG-SG) and poly-3-amino-4-methoxybenzoic acid with TK-NH2-modified gelatin (PAMB-G-TK). Finally, the authors successfully synthesized a novel injectable ROS-responsive hydrogel (PAMB-G-TK/4-arm-PEG-SG).326 Due to the abnormal elevation of ROS at the site of myocardial infarction (MI), the thioketal bonds within the hydrogel underwent cleavage, and led to the on-demand release of the encapsulated S1P/SS-31 liposomes at the pathological site, which then intelligently modulated the MI microenvironment and significantly improved the function of the infarcted myocardium. Similar to borate ester bonds, injectable hydrogels based on thioketal bonds have also played a positive role in “smart” treatments for localized chemotherapy and metastasis inhibition in malignant tumors.327
Recently, researchers have developed a ROS-cleavable peptide sequence which can enable the grafted hydrogels to possess ROS-responsive capabilities. Jeong et al. utilized the host–guest interactions between cyclodextrin-modified (HA-CD) and adamantane-modified HA (Ad-HA) to prepare a novel injectable hydrogel.328 Subsequently, they incorporated antimicrobial peptides (AMPs) in the form of Ad-HA-AMP conjugates into the hydrogel network of HA. Notably, the cyclic peptide linkers in the hydrogel would undergo cleavage only when exposed to ROS and MMP simultaneously. Hence, the smart hydrogel ensured that the toxic and less stable AMPs were released only at the site of bacterial infection (where both the ROS and MMP levels were significantly elevated), which greatly improved the safety of AMPs and promoted the healing process of bacteria-infected wounds.
3.5. Glucose responsiveness
Glucose is the primary source of energy for the human body, and an appropriate concentration of glucose is crucial for maintaining normal life activities. However, in some pathological conditions, such as diabetes, the excessive glucose in the body can lead to a series of complications and cause damage to tissues and organs. Currently, insulin injection has become the most used and effective treatment for patients with diabetes. Nevertheless, the pain and inconvenience caused by frequent injections and monitoring significantly impact the quality of life for these patients. Injectable glucose-responsive hydrogels can serve as a depot of insulin and release insulin on demand under high glucose stimulation, which overcomes the drawbacks of traditional insulin injections that require frequent administration. Therefore, this kind of smart hydrogel holds the potential to become the next generation of more convenient and intelligent diabetes treatment methods. Additionally, based on this glucose-responsive property, novel hydrogels have also been developed for intervening in the pathological processes of various diseases, aiming to achieve smart therapy.Currently, the glucose responsiveness of injectable hydrogels is mainly acquired through two common mechanisms. The first mechanism involves introducing phenylboronic acid (PBA) to impart glucose responsiveness to the hydrogel. This is based on the inherent ability of PBA to bind to saccharides, which can form a reversible covalent linkage with glucose-containing diol functional groups, and result in the formation of stable five-membered cyclic complexes.329 This process leads to a shifting of overall charge density of the hydrogel towards the cationically charged boron residues, and then causes significant expansion of the hydrogel network through ionic repulsion within the hydrogel. Based on this principle, glucose-responsive injectable hydrogels have been investigated for controlled drug release. The team of Dong et al. prepared an injectable hydrogel based on the reversible covalent connection between PBA and glucose under mild conditions.330 Due to the reversibility of the dynamic covalent bond, the hydrogel demonstrated excellent shear-thinning behavior and self-healing capability. Moreover, results from the drug-release experiments confirmed the glucose concentration-dependent glucose-stimulated response of the hydrogel. Specifically, in phosphate-buffered saline (PBS) solutions containing different glucose concentrations, the hydrogel released the loaded rhodamine B at a rate positively correlated with the glucose concentration (drug-release rate: 20 g L−1 > 1 g L−1 > PBS). Since PBA, as a synthetic polymer, is not easily degraded in the body and PBA-based injectable hydrogels can provide sensitive glucose responsiveness, the hydrogel holds significant potential as a long-term drug-delivery depot. Therefore, it is currently the most promising candidate for clinical translation among injectable glucose-responsive hydrogels.
Another mechanism to confer glucose responsiveness to injectable hydrogels is by embedding glucose oxidase (GOx) within the hydrogel matrix, utilizing the reaction between GOx and glucose as the transduction pathway for glucose-stimulated response. Specifically, GOx can catalyze the formation of acidic products (gluconic acid) and hydrogen peroxide (H2O2) from glucose in the presence of oxygen (O2) and water (H2O), which then leads to a decrease in pH. Subsequently, based on the pH-responsive nature of the hydrogel, the crosslinked network undergoes contraction or expansion.331,332 For example, to promote the healing of diabetic wounds, Yang et al. developed an injectable glucose-responsive hydrogel (DG@Gel).333 First, they prepared metallo-nanodrugs (DFO@MONPS) containing deferoxamine mesylate (DFO), 4,5-imidazoledicarboxylic acid (IDA), and zinc ions (Zn2+). Subsequently, DG@Gel was synthesized through the phase-transfer-mediated programmed GOx loading. According to the reported results, when injected into diabetic wounds, DG@Gel immediately responded to the hyperglycemic microenvironment by breaking down glucose into gluconic acid and H2O2 (Fig. 6). The subsequent decrease in pH led to the expansion of the crosslinked network, which facilitated the release of DFO and Zn2+ loaded within the hydrogel and exhibited synergistically antibacterial and angiogenesis-promoting effects, ultimately accelerating the healing of diabetic wounds. In addition, due to the generation of H2O2 with antimicrobial activity during the GOx-mediated glucose response process, hydrogels containing GOx have also been developed for glucose-responsive antimicrobial therapy. Zhou and colleagues utilized the Schiff base reaction between GOx-modified HA and chitosan conjugated with L-arginine (L-Arg) to prepare a novel injectable hydrogel with self-healing and antimicrobial properties (CAHG), which was applied for the treatment of infected diabetic wounds.334 In the hyperglycemic environment of infected diabetic wounds, the GOx encapsulated in the CAHG hydrogel catalyzed the production of H2O2 from glucose in the wound microenvironment, and exerted a primary antibacterial and anti-inflammatory effect. Subsequently, the generated H2O2 oxidized the coupled L-Arg in chitosan and produced nitric oxide (NO), which provided a secondary antibacterial and anti-inflammatory therapeutic effect.
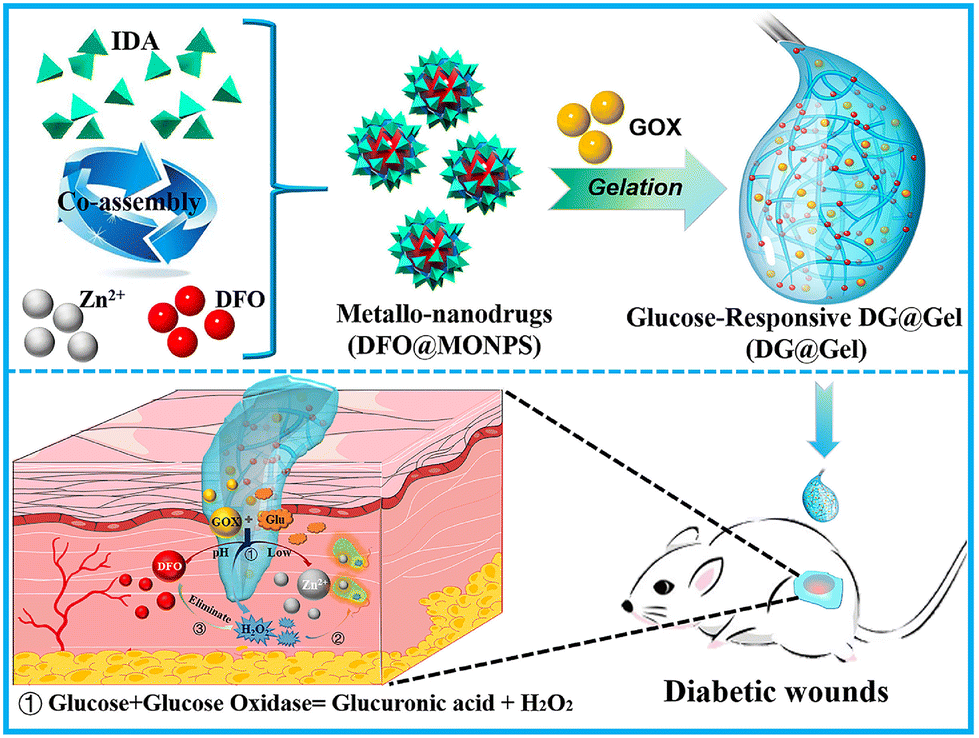 | ||
| Fig. 6 Schematics of the preparation of injectable, multifunctional, and glucose-responsive metal–organic hydrogel and its working mechanism in repairing diabetic skin wounds. Adapted with permission from ref. 333. Copyright 2021, Elsevier Ltd. | ||
3.6. Light responsiveness
Light represents an easily accessible and remotely controllable external stimulus, offering non-invasiveness, ease of management, and efficient spatiotemporal control. The properties of light-responsive hydrogels can be changed under the irradiation of light (including visible light, ultraviolet (UV), and near-infrared radiation (NIR)), which then leads to the sol–gel transition and the formation/degradation of hydrogels.335 The content related to light-initiated gelation has been discussed in the preceding text and will not be reiterated here. The following section focuses on the light responsivity of injectable hydrogels, specifically highlighting two light-triggered behaviors: the photothermal effect and photodegradation.Some substances can absorb and emit light radiation, leading to the production of heat, and such substances are commonly termed photothermal agents. Light-responsive hydrogels can be obtained by incorporating these photothermal agents into the hydrogel matrix, which is currently one of the most extensively researched and employed methods. Indocyanine green (ICG) is a representative photothermal agent and has been approved by the U.S. Food and Drug Administration (FDA). It can generate and then transfer heat to adjacent areas under NIR, which makes it suitable for drug delivery and systemic or local cancer therapy.336–338 For instance, Yang et al. incorporated ICG into the gel network formed by the directional crosslinking of sodium selenite (Se), dopamine (DA), and HA to prepare an injectable composite hydrogel (HD/Se/ICG).339 Due to the assisting crosslinking effect of Se, the retention time of ICG in the gel network was prolonged, which thereby enhanced the photothermal therapeutic effect of the hydrogel. After intra-tumoral injection, HD/Se/ICG hydrogel exhibited sufficient energy conversion efficiency under NIR and effectively inhibited the growth of breast cancer tumors in both in vitro and in vivo experiments, which made it a promising strategy for localized breast cancer treatment. In addition, some highly penetrable photothermal agents (such as black phosphorus nanoflakes, organic nanoparticles, and rare metal nanostructures) have also been developed for the preparation of light-responsive injectable hydrogels, which can provide safe photothermal activity and effectively controlled drug release.340–342
Another commonly used strategy to prepare injectable light-responsive hydrogels is grafting photosensitive functional groups onto the hydrogel chains. These functional groups possess the ability to respond to light through photochemical reactions, which can induce the phase transition of hydrogels and drug release.343,344 The o-methoxy-nitro-benzene family of monomers is the most common utilized group for photodegradable moieties, among which the most representative is o-nitrobenzyl and its derivatives, which undergo irreversible photocleavage under UV irradiation. For example, Cheng and co-workers utilized 2-nitrobenzyl-modified 4-arm PEG to synthesize an injectable hydrogel and validated its degradability under UV irradiation.345 They first modified the 4-arm PEG with 2-nitrobenzyl and phenol to obtain the hydrogel precursor solution, followed by in situ gelation in the presence of horseradish peroxidase (HRP) and H2O2. Subsequent research findings demonstrated that the introduction of 2-nitrobenzyl into the hydrogel framework resulted in the rapid degradation of the hydrogel due to the cleavage of the ester bonds of 2-nitrobenzyl under UV light exposure. Of note, the photodegradation rate of this hydrogel showed a positive correlation with the intensity of UV light (degradation rate: 100 > 50 > 10 > 0 mW cm−2), indicating the excellent light-controlled degradability of this light-responsive hydrogel. Therefore, such light-responsive injectable hydrogels have great potential in biomedical practices, especially for localized drug delivery and on-demand release applications. It is worth mentioning that the development and application of UV-induced light-responsive hydrogels are significantly limited due to the poor tissue penetration of UV light and its potential carcinogenic risk. In contrast, NIR light has excellent tissue-penetration capability, and it poses no carcinogenic risk or toxic effects to the organism. As a result, NIR-induced light-responsive hydrogels have attracted significant interest and attention in recent years.
3.7. Ultrasound responsiveness
Ultrasound is a specialized term referring to sound waves that lie beyond the range of human hearing, typically with frequencies above 20000 Hz. Due to its non-invasive nature, ease of use, controllable directionality, focused acoustic energy, strong tissue-penetration capabilities, high temporal and spatial resolution, ultrasound finds widespread applications in clinical and biomedical fields. Within the domain of injectable smart stimuli-responsive hydrogels, ultrasound is also a commonly employed exogenous stimulus. Generally, ultrasound-responsive hydrogels primarily rely on two strategies: ultrasound-induced thermal effects and mechanical forces.
Currently, the most common strategy to prepare ultrasound-responsive hydrogels involves utilizing ultrasound-induced mechanical forces to trigger the breakdown of the hydrogel network, thereby enabling the responsive release of loaded drugs. The mechanical forces generated by high-frequency vibrations of ultrasound can disrupt the physical crosslinking that maintains the 3D network structure of the hydrogel, leading to the collapse of ionically crosslinked hydrogel networks and consequently releasing the therapeutic agents encapsulated within the hydrogel. In the study conducted by Huebsch et al., they investigated the responsiveness of a series of ionically crosslinked hydrogels to ultrasound.346 They first prepared the hydrogels by ionically crosslinking alginate with Ca2+ in physiological solution. Subsequently, they subjected the hydrogels to ultrasound treatment and observed that the hydrogel structures were significantly disrupted during ultrasound exposure, but exhibited reversible and complete recovery after ultrasound cessation (similar to the untreated control group). Next, they incorporated mitoxantrone (an anthracycline anticancer drug used for treating breast cancer) into the hydrogel system to further explore the drug-release behavior of this ultrasound-responsive hydrogel. The results demonstrated that the release baseline of mitoxantrone in the control group, without ultrasound treatment, was minimal. In contrast, the release profile of mitoxantrone in the pulsed ultrasound-treated group perfectly matched the ultrasound pulse frequency, and exhibited a transient drug-release burst during ultrasound exposure. Furthermore, the authors replicated this series of ultrasound treatment experiments in different types of hydrogels and obtained consistent outcomes. Based on the same principle, Meng et al. prepared a novel injectable hydrogel by using oligo (ethylene glycol) methacrylate (OEGMA) and inorganic clay (LAPONITE®, XLS) to form the hydrogel precursor solution, into which nano-vaccines were incorporated.347 They observed that subjecting the injected hydrogel in mice to multiple rounds of ultrasound treatment resulted in repetitive gel–sol–gel transitions and pulsatile release of nano-vaccines: the ultrasound-induced crosslinking disruption caused the hydrogel to temporarily transform into a flowing sol state, which led to an explosive release of nano-vaccines; upon the cessation of the ultrasound treatment, the hydrogel spontaneously re-crosslinked and resulted in a dramatic decrease of the release rate of the nano-vaccines. This finding is particularly meaningful for preventing tumor recurrence and metastasis after tumor resection surgery, as it allows for improvement of treatment efficacy and patient compliance while reducing vaccine administration frequency. In addition to the aforementioned physically crosslinked hydrogels, recently, hydrogels based on certain dynamic covalent bonds (such as boronic ester bonds) have also been found to possess ultrasound responsiveness, and have been developed as ultrasound-responsive drug delivery systems.348
Additionally, besides inducing mechanical forces, the mechanical vibrations of ultrasound also generate heat due to high-frequency friction on the surface of objects. Therefore, ultrasound-induced thermal effects can also be utilized in the development of injectable ultrasound-responsive hydrogels. Wu and colleagues prepared an injectable block copolymer hydrogel based on methoxy-PEG (mPEG), PLGA, and 2,2′-Bis(2-oxazoline) (BOX).349 According to the reported results, the hydrogel could be easily injected through a 22-gauge needle and form a gel in situ within 1 minute in vivo, and then released both large and small-molecule drugs loaded within the networks sustainedly. When the hydrogel was exposed to ultrasound, the heat generated by ultrasound significantly enhanced the release rate of the encapsulated drugs (accelerated by ∼70-fold), and upon the cessation of ultrasound treatment, the release rate rapidly returned to the baseline level. This demonstrated that the injectable hydrogel could trigger drug release through ultrasound and represented a novel on-demand drug delivery mode based on ultrasound-induced thermal effects, which had great potential for clinical translation.
3.8. Magnetic field responsiveness
Similar to ultrasound, magnetic fields are also a type of external stimulus that possesses both non-invasiveness and excellent tissue penetrability. Magnetic field-responsive hydrogels are primarily prepared by incorporating superparamagnetic iron oxide nanoparticles (SPIONs) into the three-dimensional polymer network of hydrogels. Due to the directed movement and aggregation of SPIONs, the hydrogel network contracts under an externally applied magnetic field, which then leads to the easy extrusion and release of therapeutic agents loaded within the hydrogel. As a result, magnetic field-responsive hydrogels are commonly employed for achieving magnetically controlled drug release.350,351 For example, Kim et al. prepared a magnetic field-responsive injectable macroporous hydrogel by incorporating SPIONs and gelatin particles into the alginate-Ca2+ ionically crosslinked hydrogel network.352 They first introduced gelatin particles and SPIONs into the hydrogel network, and then increased the system temperature to 37 °C, under which the gelatin particles were dissolved due to the inherent thermal-sensitive property. Finally, a SPION-loaded hydrogel with macroporous structure, which enhanced its deformability under magnetic stimulation, was obtained (Fig. 7A). Subsequently, they applied different intensities of magnetic stimulation to the hydrogel loaded with DOX to evaluate its magnetic responsiveness. The results demonstrated that magnetic stimulation significantly accelerated the release of DOX from the hydrogel, and the release rate was positively correlated with the applied magnetic field intensity, while remaining independent of the magnetic field frequency. In vivo experiments in a tumor-bearing mouse model further confirmed the magnetically controlled drug-release behavior of DOX within the hydrogel, which then led to the substantial inhibition of tumor growth in the mice.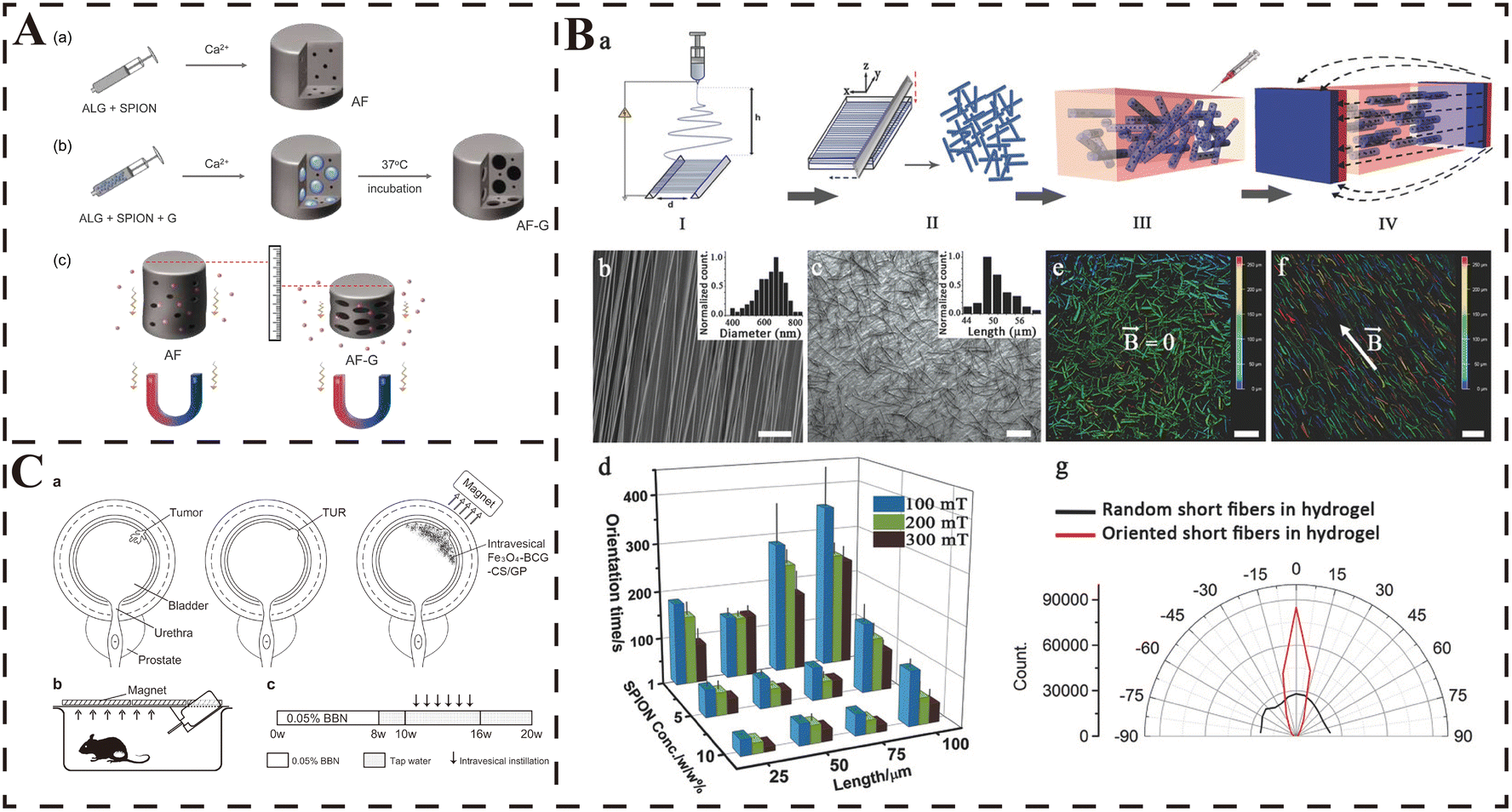 | ||
| Fig. 7 Applications of injectable magnetic field-responsive hydrogels. (A) Schematic description of the fabrication of (a) alginate ferrogel (AF) and (b) macroporous alginate ferrogel (AF-G). Alginate solution (ALG) containing superparamagnetic iron oxide nanoparticle (SPION) and gelatin particle (G) was ionically crosslinked in the presence of calcium ions, followed by incubation at 37 °C to generate the macroporous structure of AF (AF-G). (c) The improved deformation of AF-G and the resultant release of drug molecules from the gel under magnetic stimulation. Adapted with permission from ref. 352. Copyright 2019, Elsevier Ltd. (B) (a) A schematic representation of the Anisogels fabrication process. Electrospinning of aligned fibers on a parallel plate (step I), followed by embedding the fibers in an optimum cutting temperature (OCT) gel for subsequent cryosectioning. The fibers are purified and dispersed in distilled water after melting and washing off the excess of gel (step II). Randomly oriented short fibers mixed within the hydrogel precursor solution in liquid state before applying the magnetic field (step III). Fiber orientation and hydrogel solidification result in the Anisogel (step IV). (b) SEM image of aligned PLGA fibers formed on a parallel plate collector with an average diameter of 689.7 ± 88.5 nm (inset: diameter distribution histogram). (c) SEM image of 50 μm short fibers after cryosectioning (inset: length distribution histogram). Scale bars 50 μm. (d) The orientation time of magnetoresponsive short fibers with different lengths and SPION concentrations at three different magnetic fields. Depth color-coded images of magnetic fibers inside 3D fibrin hydrogels, prepared (e) in the absence of an external magnetic field and (f) in the presence of a 100 mT magnetic field. Scale bars 100 μm. (g) The angular distribution of random and oriented fibers in a 3D hydrogel corresponding to (e and f), respectively. From ref. 354 licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0). (C) (a) Schematic illustration of clinical application of the delivery system. (b) A diagram of cage for rats receiving Fe3O4-BCG-CS/GP mixture. The Fe3O4-BCG-CS/GP gel formed in bladder can be attracted and attached to the bladder wall in the magnetic field generated by the magnets on the cage. (c) Treatment protocol. TUR: transurethral resection. Adapted with permission from ref. 355. Copyright 2013, Elsevier Ltd. | ||
It is worth noting that the oscillation of SPIONs under an external magnetic field can cause a sharp increase in local temperature. Therefore, magnetic field-responsive hydrogels are also commonly utilized in magnetic hyperthermia therapy (MHT) for tumors. MHT is a novel non-invasive strategy for localized tumor treatment, which can induce deep tumor tissue hyperthermia/necrosis without penetration depth limitations under an alternating magnetic field (AMF). However, due to the inherent mobility of nanoscale particles, SPIONs are prone to leakage within the local tumor tissue, leading to undesirable heating damage to healthy tissues. Injectable hydrogels possess a stable three-dimensional network structure that can firmly anchor SPIONs within their framework, which can contribute to the accuracy and efficacy of MHT. Moreover, they significantly prolong the retention time of SPIONs at the tumor site, enabling multiple rounds of MHT with a single injection of SPIONs. Chen and colleagues covalently linked arachidonic acid (AA) and PLGA-PEG-PLGA triblock copolymers to obtain a thermosensitive hydrogel (AAGel). They then incorporated ferrimagnetic Zn0.4Fe2.6O4 nano-cubes and RSL3 (a ferroptosis inducer) into the AAGel, creating a novel injectable magnetic-responsive nanocomposite hydrogel (NPs/RSL3@AAGel).353 Due to the uniform dispersion and firm anchoring of the magnetic nano-cubes within the NPs/RSL3@AAGel, this hydrogel system enabled multiple accurate rounds of MHT under an external magnetic field after a single injection. Moreover, the release of RSL3 was promoted, which enhanced the anti-tumor efficacy of ferroptosis and ultimately led to a complete eradication of CT-26 tumors in a mouse model.
Furthermore, the highly ordered nature of the magnetic field can induce a highly organized spatial arrangement of SPIONs and materials loaded with SPIONs, which facilitates the hydrogel in achieving the desired anisotropy required for tissue engineering. It also effectively controls the behavior of cells loaded within the hydrogel network under the influence of an external magnetic field. This is particularly critical for tissue repair that requires alignment, especially in the case of neural tissue. The team of Omidinia-Anarkoli et al. developed an injectable hybrid hydrogel with magnetic-responsive short fibers and demonstrated its remarkable therapeutic efficacy in cell alignment and neural injury repair.354 They initially incorporated SPIONs into a PLGA solution, and then utilized electrospinning to produce magnetic-responsive short fibers. These fibers were subsequently mixed with the precursor solution of collagen hydrogel, and by applying a low magnetic field (≤300 mT) before gelation, the short fibers were uniformly aligned, resulting in the synthesis of the hybrid hydrogel named Anisogel (Fig. 7B). The experimental results confirmed that this magnetic field-responsive hydrogel successfully stimulated functional neural cells and fibroblasts to grow in a linear manner, and supported unidirectional propagation of neural signals along the aligned fibers. Consequently, this research offers a novel approach to exploit magnetic field-responsive hydrogels for constructing an in situ structure with controlled unidirectional alignment in vivo, promoting linear cell growth, and aiding in tissue repair with alignment (particularly for linearly oriented neural tissues like spinal cord).
In addition, the directional characteristics of magnetic field-responsive hydrogels have also been explored for targeted drug delivery in cancer therapy. For instance, in the study of Zhang et al., to enhance the efficacy of Bacillus Calmette-Guérin (BCG) in bladder cancer and reduce drug loss due to urination, they synthesized an injectable magnetic thermosensitive hydrogel loaded with BCG using Fe3O4 magnetic nanoparticles (Fe3O4-MNP), β-glycerophosphate (GP), and chitosan.355 According to the reported results, the hydrogel rapidly gelled at physiological temperature (37 °C) after injection into the bladder. Moreover, owing to the presence of SPIONs, the injected hydrogel could stay at the target location as needed under the influence of an external magnetic field, allowing continuous BCG treatment of the tumor tissue for at least 48 hours (Fig. 7C). As a result, this strategy based on magnetic-responsive hydrogel significantly prolonged the retention time of BCG in the bladder and substantially enhanced its anti-tumor efficacy.
3.9. Electricity responsiveness
Biological electrical activity plays a crucial role in maintaining normal cellular functions, intercellular communication, and numerous complex life processes, such as heartbeats, and neural signal transmission within the spinal cord and brain. Despite the high biocompatibility and resemblance to biological tissues exhibited by hydrogels, their intrinsic non-conductive properties limit their application in the repair and reconstruction of several electricity-dependent tissues. Consequently, many conductive materials, such as inorganic conductive nanomaterials, are incorporated into hydrogel matrix to confer excellent electrical conductivity, contributing to their development for various biomedical applications (including the repair of infarcted myocardium and damaged nervous systems).356,357 Furthermore, owing to the distinctive behaviors (such as contraction, expansion and bending) displayed by these modified hydrogels under electrical stimulation, some electricity-responsive hydrogels have been developed for spatiotemporally controlled drug release. Generally, introducing conductive polymers and inorganic conductive nanomaterials into the hydrogel cross-linked network represents a common strategy to confer electrical responsiveness to these hydrogels.Various conductive polymers, also known as ionic electrosensitive polymers, can undergo changes in their redox states when exposed to electrical stimulation, leading to alterations in charge distribution and electrical conductivity. Therefore, incorporating conductive polymers into hydrogel networks allows them to acquire electrical responsiveness. The most common conductive polymers are polypyrrole (PPy), polyaniline (PANI), polythiophene, and polyacrylamide (PAAm).358 Their conductivity typically depends on the conjugated system and orbital overlap. Under electrical stimulation, these electricity-responsive hydrogels achieve controlled drug release through two main mechanisms: drug expulsion from the contracting hydrogel network and drug migration in the direction of the opposite charge. For example, Ge et al. first encapsulated two hydrophobic drugs, fluorescein and roxithromycin, within polypyrrole (PPy) nanoparticles.359 Subsequently, the drug-loaded PPy nanoparticles were uniformly dispersed into a PLGA-PEG-PLGA solution to prepare the hydrogel precursor solution. Based on the thermosensitive properties of the PLGA-PEG-PLGA hydrogel, this precursor solution could be easily injected subcutaneously into FVB adult mice using a syringe at room temperature, and then gel rapidly in situ at 37 °C. In the subsequent cyclic electrical stimulation drug-release experiments (10 seconds of electrical stimulation repeated every 5 minutes), it was observed that electrical stimulation significantly enhanced the release of the encapsulated drugs from the hydrogel, and the drug-release rate showed a positive correlation with the applied voltage. In contrast, no significant release of fluorescein or roxithromycin was detected from the hydrogel without electrical stimulation. This electricity-responsive drug release was based on the electrochemical oxidation/reduction reactions of PPy nanoparticles when exposed to an electric field. The overall net charge within the polymer nanoparticles was changed during this process, and then caused the nanoparticles to undergo net contraction and led to the repulsion of non-covalently bound drug molecules. Consequently, the encapsulated drugs were released under electric field. In another study conducted by Qu et al., they prepared an injectable hydrogel (CP/OD) based on the Schiff base reaction between the PANI-grafted chitosan (CP) and oxidized dextran (OD).360 Prior to injection (before gelation), ibuprofen or amoxicillin was added to the hydrogel. Due to the continuous redox switching of CP polymers between oxidation and reduction states, the entire gel body was actually transformed into a conductor. Under the stimulation of an electric field, charged drug molecules migrated towards the electrodes carrying the opposite charge, leading to drug release from the hydrogel. Consequently, under the “ON/OFF” cyclic electrical stimulations, the CP/OD hydrogel exhibited a pulsatile drug-release behavior akin to an “ON/OFF” pattern, which achieved precise and controlled drug release from the loaded drugs within the hydrogel.
Common inorganic conductive nanomaterials used for injectable electricity-responsive hydrogels include carbon nanotubes, graphene and their derivatives. For example, Servant and co-workers prepared a hybrid hydrogel with excellent electrical responsiveness by adding methacrylic acid (MAA), N,N′-methylene bisacrylamide (MBAM) and potassium persulfate (PPS, an initiator) to an aqueous dispersion of pristine multiwalled carbon nanotubes (pMWNT).361 In the drug-release experiments using radiolabeled sucrose (14C-sucrose) as a model hydrophilic drug, a pulsatile release profile of 14C-sucrose was observed, which perfectly matched the electrical stimulation rhythm, with a consistent amount of 14C-sucrose released after each electrical stimulation. This phenomenon was attributed to the straightening and alignment of carbon nanotubes within the hydrogel matrix under electrical stimulation, which resulted in the sudden release of the drugs encapsulated in the hydrogel network. This represented the primary principle behind the electro-controlled drug release achieved by carbon nanotube-containing hydrogels. Graphene is a novel material composed of carbon atoms closely packed in a single-layer two-dimensional honeycomb lattice structure, with sp2 hybridization. It exhibits excellent electrical conductivity, thermal conductivity, optical absorption, and mechanical strength.362 Similarly, one year after the previous study, the team of Servant et al. published new research about a conductive graphene nanocomposite hydrogel (ball-milled graphene hydrogel, GBM) that also demonstrated pulsed drug release under electrical stimulation.363 Following the same principle, the GBM hydrogel was prepared through the in situ free radical polymerization of graphene, N,N′-methylene bisacrylamide (BIS), methacrylic acid (MAA), and potassium persulfate (PSS). The results showed that the incorporation of graphene significantly improved the mechanical properties of the hydrogel. Moreover, in simulated drug-release experiments using 14C-sucrose, the GBM hydrogel exhibited the same electrically controlled “ON/OFF” drug-release behavior in response to the pulsed electrical stimulation. Thus, electricity-responsive hydrogels based on inorganic conductive nanomaterials such as carbon nanotubes and graphene have immense potential for drug delivery and on-demand release applications.
4. Current biomedical applications of injectable smart stimuli-responsive hydrogels
Under healthy conditions, the structure and physicochemical properties of cells and tissues are maintained within a normal range. However, when diseases occur, many balances within the tissue microenvironment are disrupted, leading to a series of pathological processes. Throughout the development of diseases, prominent changes may occur in the tissue microenvironment, such as a decrease in the microenvironmental pH of affected tissues, imbalances in cellular redox homeostasis, disturbances in energy metabolism, and dysregulation of degradation/synthesis metabolic pathways. Based on these pathological alterations occurring in cells and tissues during disease progression, researchers have designed and applied various injectable smart stimuli-responsive hydrogels that can respond to these pathological changes. This new generation of smart stimuli-responsive hydrogels has made significant contributions to the treatment of these diseases. It is worth noting that the human body, whether in a healthy or diseased state, maintains a nearly constant temperature of ∼37 °C, which is attributed to the robust temperature control system. Therefore, injectable temperature-responsive hydrogels are currently the most widely utilized type of smart stimuli-responsive hydrogel, as they are highly versatile in various biomedical applications and enable efficient targeted drug delivery and sustained release. Herein, we mainly summarize some examples of injectable smart stimuli-responsive hydrogels that play a crucial role in biomedical applications, with the aim of assisting researchers in better understanding the potential of these smart hydrogels for disease therapy.4.1. Myocardial infarction
Cardiovascular disease is a major cause of human mortality, with myocardial infarction (MI) being a significant contributor to the high incidence of cardiac diseases in humans. Coronary artery atherosclerosis can lead to arterial stenosis and inadequate blood supply to local myocardial tissues. In the absence of established collateral circulation, coronary artery occlusion results in downstream myocardial ischemia. Prolonged ischemia for over an hour can lead to MI, and persistent MI can cause hypoxic necrosis of myocardial tissue, ultimately leading to the development of heart failure (HF).364 Notably, the levels of MMP2/9 in the myocardial tissue affected by infarction are significantly elevated, and this microenvironmental change further exacerbates the degradation of myocardial extracellular matrix (ECM). Therefore, some injectable hydrogels with MMP2/9 responsiveness have been developed for MI treatment. Chen et al. constructed a novel injectable hydrogel that responds to MMP2/9.177 The study found that this hydrogel selectively released the loaded composite gene nanocarrier (CTL4) and interleukin-4 plasmid DNA (IL4-pDNA) only at the infarcted myocardial site with MMP2/9 overexpression, which then promoted M2 polarization of macrophages at the infarct site, reduced inflammation levels, inhibited MMP expression and attenuated myocardial ECM degradation.During the occurrence of MI, the mitochondrial function within the myocardial cells is severely impaired due to the interruption of nutrients and oxygen, which is a critical factor leading to pathological ROS accumulation and the progression of MI.365 Therefore, the development of therapeutic approaches capable of reducing ROS levels and improving mitochondrial function is of significant importance in slowing down the progression of MI. Zheng and colleagues developed a ROS-responsive injectable hydrogel loaded with lipid-based nanocarriers (S1P/SS-31/Lipo) encapsulating mitochondria-targeted antioxidant SS-31 and pro-angiogenic molecule S1P.326 The thioketal links within the hydrogel network could be cleaved upon exposure to the high concentration of ROS at the infarcted myocardial site, simultaneously consuming ROS and responsively releasing S1P and SS-31, which further enhanced the mitochondrial function and promoted the vascularization in the myocardial tissue (Fig. 8).
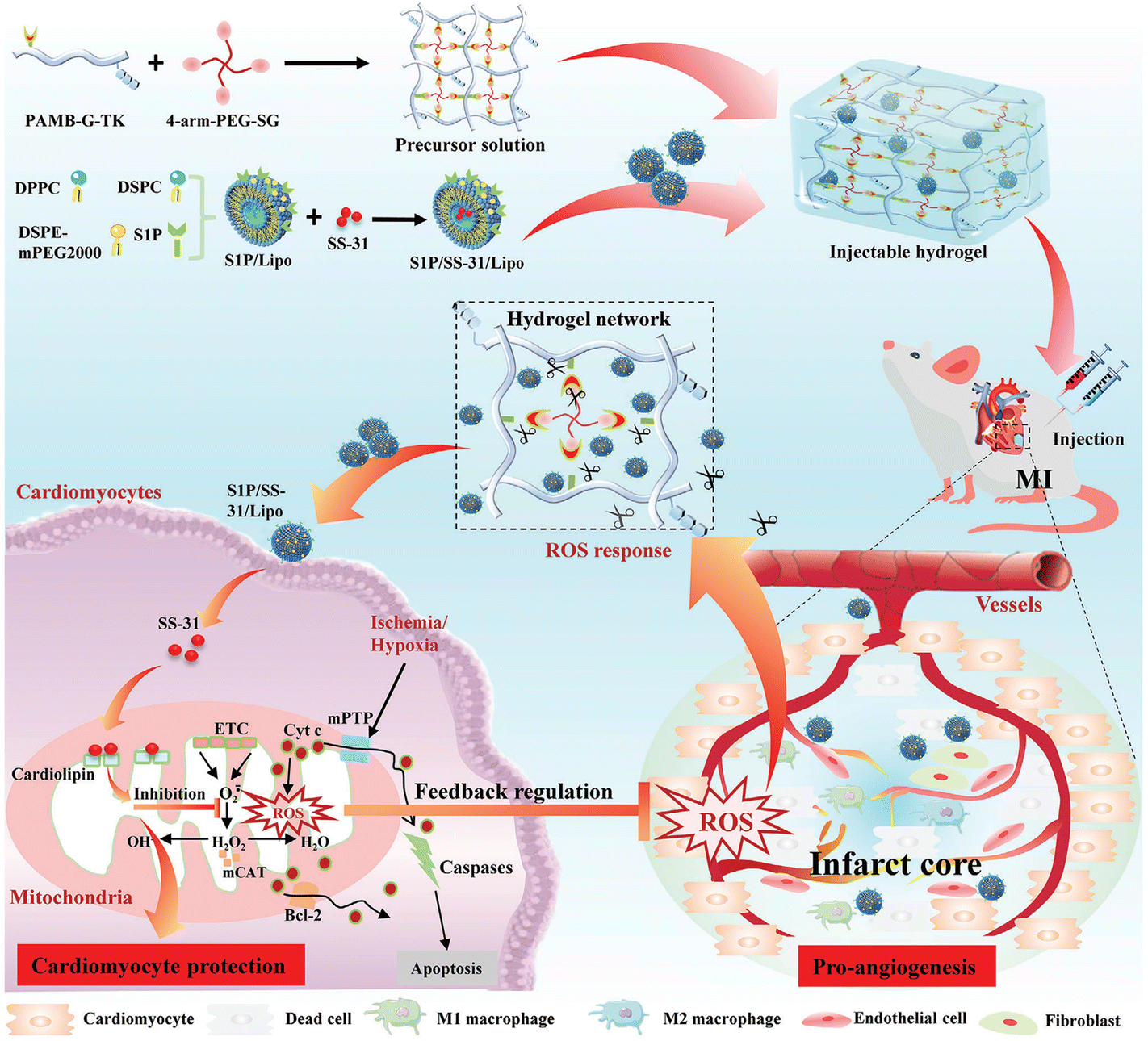 | ||
| Fig. 8 Schematic illustration of the formation and mechanism of an S1P/SS-31/Lipo-encapsulated ROS-responsive composite hydrogel for the efficient treatment of MI. Adapted with permission from ref. 326. Copyright 2022, John Wiley and Sons. | ||
In addition, due to the oxygen deprivation caused by the coronary artery occlusion, myocardial cells undergo anaerobic glycolysis, which results in the accumulation of lactate and the formation of a local acidic microenvironment at the site of MI. Therefore, in the context of MI, pH also serves as a reliable drug-controlled release signal. Taking advantage of the reduced pH in the infarcted myocardial tissue, Li et al. developed a dual-crosslinked injectable hydrogel (Gel@MSN/miR-21-5p) that could responsively release microRNA-21-5p (miR-21-5p) in an acidic pH environment for MI therapy.366 Under the acidic microenvironment of MI, the Schiff base bonds within the hydrogel were broken, and thus enabled the on-demand release of mesoporous silica nanoparticles (MSN) encapsulating miR-21-5p. This smart delivery system effectively suppressed inflammation and promoted angiogenesis in a porcine model of myocardial infarction, leading to a significant reduction in infarct size.
In conventional treatment strategies, reperfusion is considered the most effective method for treating MI. However, the process of ischemia-reperfusion (I/R) also leads to the excessive generation of ROS in myocardial tissue, which then exacerbates myocardial injury. Therefore, utilizing ROS generated during I/R to prepare intelligent and responsive injectable hydrogels is also a rational strategy for MI treatment. Li et al. developed an injectable PVA-based hydrogel loaded with bFGF to mitigate myocardial damage during I/R.324 Within the hydrogel injected into the pericardial space, the crosslinked network formed by borate ester bonds were broken in the presence of ROS, which resulted in the release of bFGF from the hydrogel. This significantly reduced myocardial fibrosis, enhanced vascularization and preserved cardiac function.
Furthermore, as the heart is an organ characterized by highly frequent bioelectric activity, the MI-induced extensive collagen fiber deposition and myocardial cell death can severely disrupt normal electrical signal conduction in the myocardial tissue, which then contributes to arrhythmias and impaired cardiac contractile function. Therefore, timely restoration of normal electrical activity in the myocardial tissue is also a potential strategy for treating MI. Zhang and colleagues developed an injectable conductive hydrogel based on the Schiff base reaction between the PPy-grafted gelatin (GP) and oxidized xanthan gum (OXG).357 After the injection into the rat myocardial scar tissue 2 days after MI, a significant decrease in myocardial fibrous tissue resistance and an acceleration of conduction velocity was observed. Additionally, the infarct area was reduced while the vascular density was increased, which then resulted in a remarkable improvement in the cardiac function of the rats.
The constant physiological temperature during MI can effectively trigger in situ gelation of temperature-responsive hydrogels. Leveraging the drug-releasing properties of hydrogel, the injected drugs can continuously exert their therapeutic effects at the local tissue, which effectively avoids drug dose-related toxicity and systemic adverse reactions caused by frequent administration. The team of Wang et al. developed a long-acting injectable temperature-responsive hydrogel loaded with Triptolide (TPL) based on the thermosensitive material Pluronic F127.367 This hydrogel gelled in situ rapidly upon injection into the body, and sustained slow release of TPL within 28 days following injection, which performed a persistent anti-inflammatory effect, protected myocardial cells, and improved cardiac function. This strategy effectively addressed the significant risk of TPL-induced liver and kidney toxicity, and reduced the dose-related toxicity resulting from the burst release of TPL locally.
4.2. Cancer
Compared with healthy cells, one of the main characteristics of tumor cells is their unrestricted proliferative ability. This feature leads to a significantly increased metabolism in tumor cells, which then results in a high demand for nutrients and oxygen in tumor tissues. Despite the remarkable angiogenic capacity, the proliferation rate of tumor cells often exceeds the rate of angiogenesis. As a result, a large proportion of tumor cells are perpetually under conditions of chronic hypoxia, which leads to anaerobic respiration and the accumulation of abundant lactate within the tumor tissue, causing localized acidification. Based on this, the decreased pH within tumor tissues often serves as an effective trigger in tumor chemical therapy. Based on the pH-responsive dynamic covalent bond (Schiff base bonds) between the dibenzaldehyde-terminated PEG (PEGDA) and N-carboxyethyl chitosan (CEC), Qu et al. constructed an injectable pH-responsive hydrogel platform for the delivery of doxorubicin (DOX) in the treatment of hepatocellular carcinoma (HCC).313 The findings demonstrated that the hydrogel exhibited pH-dependent degradation behavior and DOX release pattern: specifically, the hydrogel remained stable and exhibited negligible DOX release in the physiological pH environment, whereas, in the tumor acidic microenvironment, the hydrogel rapidly degraded and led to the significant release of DOX, which then induced tumor cell death. This approach significantly improved the targeting and effectiveness of the anti-tumor drug while reducing the off-target effects on normal tissues.The intense metabolism of tumor cells is heavily reliant on the highly efficient catalytic activity of enzymes. Therefore, some highly expressed enzymes in tumor tissues also serve as ideal “triggers” for achieving targeted tumor therapy. For instance, matrix metalloproteinase-2 (MMP-2) has been reported to be overexpressed in various types of cancer and plays a crucial role in cancer invasion, progression, metastasis, and recurrence.368 Consequently, a number of injectable smart hydrogels responsive to MMP-2 have been developed for cancer treatment. In the study of Li et al., they incorporated a peptide sequence GPQGIWGQ, which could be selectively cleaved by MMP-2, into the hydrogel crosslinking network to prepare an injectable MMP-2-responsive hydrogel for the treatment of oral squamous cell carcinoma (OSCC).316 Upon injection into the tumor, the hydrogel effectively degraded in the microenvironment with high MMP-2 expression, and then exhibited a sensitive MMP-2 responsive drug-release profile that strongly inhibited the growth of OSCC tumor. As the expression levels of MMP-2 in normal tissues were much lower than in tumor tissues, the hydrogel exhibited minimal degradation in physiological environments, which greatly reduced the side effects caused by DOX.
Furthermore, the expression of GSH (glutathione) is significantly elevated in many cancer cells, which is associated with their high intrinsic drug resistance to anticancer drugs such as platinum, doxorubicin, and alkylating agents.369,370 There is substantial evidence demonstrating that the GSH levels in cancer tissues are significantly higher than in normal tissues.371 Therefore, several injectable GSH-responsive hydrogels have also been developed for drug delivery in localized chemotherapy for cancer treatment. Liu and colleagues utilized thioctic acid and PEG to construct an injectable GSH-responsive hydrogel drug delivery system for delivering anticancer drugs in the tumor microenvironment with high GSH expression.179 The disulfide bonds in the hydrogel could undergo thiol exchange reactions with the abundant GSH in the tumor microenvironment, leading to the responsive degradation of the hydrogel matrix and release of the loaded DOX. In drug-release experiments, the hydrogel exhibited sustained DOX release for up to 80 hours under high GSH conditions. Toxicity tests further confirmed that the hydrogel only released a significant amount of DOX and effectively killed tumor cells under reductive conditions (high GSH levels). Therefore, the injectable GSH-responsive hydrogel was a promising intelligent drug delivery system for cancer treatment.
In addition to the intrinsic factors resulting from the pathological changes in tumor tissues mentioned above, some externally introduced stimuli, such as light, magnetic fields, and ultrasound, can also assist injectable intelligent responsive hydrogels in playing unique roles in cancer treatment. For instance, the injectable HA-based hydrogels loaded with photosensitizer protoporphyrin IX (PpIX) and DOX could generate a significant amount of reactive oxygen species (ROS) under near-infrared (NIR) light irradiation to kill local tumor cells. Moreover, the ROS generated by the NIR irradiation could also cleave the thioketal bonds in the hydrogel matrix, which then released the loaded DOX for further localized chemotherapy of the tumor tissue.372 Moreover, alginate hydrogels with large pores, loaded with iron oxide nanoparticles and DOX, could undergo rapid volume shrinkage upon exposure to an external magnetic field after injection into tumor tissue, which then resulted in the highly controllable local release of DOX.352 Hydrogels with ultrasound responsiveness and self-healing capabilities could achieve reversible gel–sol–gel transitions under ultrasound treatment. The gel–sol transition of the hydrogel upon exposure to ultrasound could lead to a burst release of encapsulated nanovaccines within the hydrogel, while the release was immediately terminated after the cessation of the ultrasound.347 Therefore, both ultrasound and magnetic field-responsive injectable hydrogels can offer highly spatiotemporally controlled drug delivery and release in a minimally invasive manner, with great potential for clinical translation. Meanwhile, due to the inability of cancer cells to tolerate high temperatures, some injectable intelligent responsive hydrogels based on photothermal and magnetic hyperthermia effects have made significant contributions to the field of precise and controlled tumor hyperthermia therapy.339,353 Additionally, hydrogels loaded with various therapeutics, such as anticancer drugs, cell components, multi-functional nanoparticles and immunologic agents, have been reported to play important roles in the inhibition tumor recurrence and metastasis after surgery.373
4.3. Osteoarthritis
Osteoarthritis (OA), also known as degenerative arthritis, is a progressive joint disorder characterized by the degeneration of articular cartilage. The main pathological changes in OA include cartilage wear/loss, joint space narrowing, synovial inflammation, osteophyte formation, and subchondral bone cyst formation. During the pathological process of OA, reactive oxygen species (ROS) are considered one of the main contributing factors leading to the disease. Oxidative stress caused by ROS can disrupt cartilage homeostasis, disturb chondrocyte metabolism, induce DNA damage, and ultimately lead to chondrocyte apoptosis. Consequently, advanced intra-articular drug delivery systems (DDS) based on the abnormal accumulation of ROS within the articular cartilage have been developed to trigger controlled drug release. Wu et al. synthesized a ROS-responsive copolymer (PEGDA-EDT) based on PEG diacrylate (PEGDA) and 1, 2-ethanedithiol (EDT) to prepare a nanofiber membrane.374 According to the research findings, this intelligent DDS could be “turned on” in response to excessive ROS stimulation within the joint cavity, which then enabled the sustained release of the antioxidant fucoxanthin. The prolonged release of fucoxanthin extended its residence time in the joint cavity, which was beneficial for the treatment of OA.Notably, normal joint activity demands extremely high lubrication conditions, and each movement of the joint subjects the cartilage to certain shear stress. This periodic shear force provides a novel approach for intelligent drug release in OA treatment. Therefore, Lei and colleagues developed an injectable hydrogel, loaded with celecoxib (CLX) encapsulated in nanoliposomes, based on the dynamic Schiff base bond between the aldehyde-modified HA (HA-CHO) and adipic dihydrazide-modified HA (HA-ADH).109 Since the crosslinking networks were formed by dynamic covalent bonds, the hydrogel injected into the joint cavity was disrupted and released the loaded CLX under each shear stress. When the shear stress was released, the crosslinking network re-connected and formed an updated boundary lubrication layer on the surface of the hydrogel. Consequently, this strategy not only continuously provided efficient boundary lubrication for OA cartilage, but also mitigated the inflammation by the sustained release of CLX. Moreover, based on similar principles, the same team also utilized microfluidics technology and the photopolymerization process to prepare HA-based hydrogel microspheres loaded with liposomes and rapamycin (RAPA). The hydrogel microspheres exhibited self-renewable hydration layers on their surface after shear stress to improve joint lubrication, and the sustained release of RAPA significantly increased the autophagy of chondrocytes to maintain cellular homeostasis, which then synergistically reduced joint wear and delayed the progression of OA.32
Due to their rapid clearance by the synovial lymphatic system in the joint cavity, drugs administered via intra-articular injection in traditional OA treatment have a short residence time, thus necessitating frequent injections, which then may increase patient discomfort and the risk of joint infection. Of note, injectable temperature-responsive hydrogels can serve as sustained-release platforms for these drugs to exert continuous therapeutic effects. In the study of Yi et al., they used a temperature-responsive PLGA-PEG-PLGA hydrogel to provide an injectable drug depot for interleukin-36 receptor antagonist (IL-36Ra).194 The in situ-formed hydrogel networks significantly prolonged the residence time of IL-36Ra in the joint cavity, and enabled continuous treatment of the chondrocytes within the joint cavity for up to 10 days after injection, which effectively delayed the progression of OA.
Due to the non-invasive and highly controllable nature of ultrasound, recently some researchers have explored the spatiotemporal control of drug release in the joint cavity based on an ultrasound stimulation. Jahanbekam and co-workers developed an injectable ultrasound-responsive hydrogel platform for the controlled release of hydrocortisone, which was formed based on the ultrasound-responsive Pluronic F-127 and HA and could gel in situ rapidly upon injection into the joint cavity.375 The gelled network could be transformed into a sol state temporarily and reversibly under ultrasound stimulation, leading to a significant release of hydrocortisone, while it would revert to gel state quickly after the cessation of ultrasound. As a result, it provided a novel strategy for the controlled release of drugs within the joint cavity.
4.4. Intervertebral disc degeneration
Intervertebral disc degeneration (IVDD) is the most common cause of low back pain, which is an age-related degenerative disease characterized by persistent inflammation in the local region of intervertebral discs (IVDs), gradual imbalance of synthesis/degradation metabolism, and progressive dysfunction of the nucleus pulposus (NP).376 Therefore, current practical approaches to treat IVDD involve alleviating the inflammatory response in the IVD microenvironment and regulating extracellular matrix (ECM) metabolism balance. However, as the spine is the primary weight-bearing structure of the human body, the pressure within IVDs is exceptionally high, which often leads to the extrusion of the therapeutic agents injected into the IVD and results in treatment failure. Of note, injectable temperature-responsive hydrogels can cross-link in situ in response to physiological body temperature at the injection site, which provides stable mechanical protection for the loaded therapeutic agents and thereby avoids the high-pressure-induced drug leakage. In the study of Xing et al., a decellularized extracellular matrix (dECM) derived from porcine nucleus pulposus (NP) tissue was used to load the exosomes from adipose-derived mesenchymal stem cells (ADSCs) to prepare an injectable temperature-responsive hydrogel (dECM@exo).377 This dECM@exo hydrogel formed a gel in situ and persisted stably at the injection site, which provided a growth scaffold for the host's nucleus pulposus cells (NPCs) while continuously releasing the exosomes from ADSCs (Fig. 9). This novel hydrogel significantly reduced the inflammatory levels of IVDD and improved the local ECM metabolism balance. | ||
| Fig. 9 dECM@exo synthesis and its mechanisms in the treatment of intervertebral disc degeneration. From ref. 377 licensed under Creative Commons Attribution 4.0 license (CC BY 4.0). | ||
During the course of IVDD, many matrix metalloproteinases (MMPs) are found to be overexpressed, which results in the excessive degradation of the extracellular matrix (ECM) and promotes the degeneration of IVDs.378,379 Therefore, the overexpressed MMPs in IVDD are also ideal triggers for drug release. The team of Feng et al. designed a MMP-responsive injectable hydrogel for two-stage miRNA delivery.379 After injection into the IVD, the abundant MMPs in the local fibrotic tissue first resulted in the dissociation of the hydrogel, and led to the release of polymeric micelles. Next, the released polymeric micelles responded to the MMPs again, causing the PEG shell enclosing miR-29a to detach and release miR-29a. This strategy accurately delivered miR-29a to the target NPCs within IVDD and achieved sustained inhibition of MMPs, which ultimately suppressed the fibrosis in the IVD.
Furthermore, since reactive oxygen species (ROS) also play a significant role in the progression of IVDD, several ROS-responsive injectable hydrogels have been developed for IVDD treatment.378,380 Zheng et al. prepared a novel injectable ROS-responsive hydrogel based on a ROS-responsive block polymer, methoxy PEG-b-poly (propylene sulfide) (mPEG20-b-PPS30).381 After being injected into the IVD, the hydrogel achieved localized ROS-responsive release of the loaded synthetic growth hormone-releasing hormone analog (MR409), and then significantly inhibited the secretion-related autophagy and needle puncture-induced IVDD in rats.
4.5. Tissue defects
Various factors, including inflammation, tumor, trauma and surgery, can compromise the integrity of tissues, and contribute to a variety of irregularly shaped tissue defects (e.g., bone, cartilage, and skin defects) with different depth and diameter. Due to the limited regenerative capacity of the human body, the assistance of biomaterials is often required when facing these large and complex tissue defects. Injectable hydrogels play a crucial role in the therapeutic process of tissue defect repair. First, they can fully conform to the irregular-shaped defects of the target tissues and provide essential and conforming biomechanical support during the repair process. Second, their inherent three-dimensional network structure endows them with robust capabilities for therapeutic agent loading and sustained release, making them highly promising for various therapeutic outcomes. Furthermore, their highly tunable physicochemical properties, mechanical performance, and degradation rate enable customized solutions for different types of tissue defects. Meanwhile, their advantages of minimal invasiveness, ease of operation, simple preparation process, and repeatability contribute to their wide application in the field of tissue defect repair.The most used type is injectable temperature-responsive hydrogel, as it allows the loaded therapeutics to be uniformly distributed in the pre-gel solution in vitro. Once injected into the defect site, owing to the temperature-initiated in situ gelation, the hydrogel would fill the defect completely with the evenly distributed therapeutics, and then promote the repair process and tissue reconstruction continuously with the sustained release of therapeutics. For instance, Lv et al. prepared an injectable temperature-responsive hydrogel based on chitosan and silk fibroin, and then incorporated bone morphogenetic protein-2 (BMP-2) and platelet-derived growth factor-BB (PDGF-BB) into the hydrogel.147 When this functionalized hydrogel was injected into a critical-sized calvarial defect model in New Zealand rabbits, it formed an in situ gel within approximately 150 seconds and continuously released BMP-2, PDGF-BB, Mg2+, and Fe2+ at the defect site. The hydrogel demonstrated excellent angiogenic and osteogenic properties and significantly accelerated the repair of the calvarial defect. Similarly, numerous reports have revealed the similar positive therapeutic effects of injectable temperature-responsive hydrogels in the repair process of skin, cartilage, and mandibular bone defects.310,382–384 In addition, it is worth noting that temperature-responsive hydrogels have also been explored to offer opportunities for sutureless wound closure in recent years. Liang et al. designed a temperature-responsive injectable hydrogel composed solely of sodium alginate, gelatin, protocatechualdehyde and Fe3+ for wound healing therapy.139 Based on the temperature-responsive properties of gelatin and the strong interactions within the crosslinked networks, including the interaction between the amino groups of gelatin and the carboxyl groups of sodium alginate, the interaction between the aldehyde groups of protocatechualdehyde and catechol, and the interaction between the excess aldehyde groups of protocatechualdehyde and the amino groups of gelatin, the hydrogel exhibited noticeable temperature-dependent adhesive properties: the hydrogel could be easily peeled off from the skin surface at 25 °C, while it adhered tightly to the skin at 37 °C. Notably, the hydrogel exhibited robust adhesive strength in the repeated “peel-off and re-adhesion” experiments, which provided the users with a “fault-tolerant” opportunity to remove and reposition the hydrogel adhesive in cases of misplacement. This property is of significant relevance for the future development of injectable wound adhesives for sutureless wound closure.
Among various types of skin defect repair processes, the healing of diabetic wounds is particularly unique. Chronic inflammation and slow fibrosis in the wounds of diabetic patients severely hinder the synthesis and deposition of extracellular matrix (ECM), which then contributes to the delayed wound-healing process.385 The most characteristic feature of diabetic wounds is the hyperglycemic microenvironment, which significantly inhibits the migration and proliferation of fibroblasts, suppresses immune cell function, and promotes bacterial growth. Therefore, the hyperglycemic microenvironment in diabetic wounds becomes an excellent trigger for drug release. Yang and co-workers developed an injectable glucose-responsive hydrogel composed of metal-based nanomedicines (including deferoxamine mesylate (DFO), 4,5-imidazoledicarboxylic acid (IDA), and Zn2+) and glucose oxidase (GOx) for smart treatment of diabetic wounds.333 Upon injection, the hydrogel completely covered the irregular diabetic wounds and firmly adhered to the surfaces. Subsequently, the high concentration of glucose in the microenvironment triggered the degradation of the hydrogel, and led to the generation of H2O2 and the release of Zn2+ and DFO. This hydrogel exhibited excellent synergistic antimicrobial and angiogenic effects, which finally accelerated the healing of diabetic wounds.
Additionally, the microvascular occlusion caused by diabetes leads to a reduced blood supply to the tissues surrounding the wound, resulting in severe deficiencies of oxygen, nutrients, and growth factors required for tissue healing and repair. These pathological changes can lead to increased anaerobic glycolysis and lactate accumulation in the vicinity of the wound, finally creating a local acidic microenvironment. Therefore, pH has also become a trigger for injectable smart stimuli-responsive hydrogels to promote diabetic wound healing. Jia et al. prepared a pH-responsive injectable hydrogel based on the “double H-bonds” between hyaluronic acid (HA) and collagen, and incorporated metformin (MET) for the treatment of diabetic wounds.212 Upon injection into the diabetic wounds, the hydrogel rapidly self-gelled and then responsively released the loaded MET and collagen in the local acidic microenvironment. This smart hydrogel was then demonstrated to effectively regulate macrophage polarization toward the M2 phenotype, reduced inflammation, promoted fibroblast migration and wound tissue collagen deposition, improved the remodeling process of ECM, and ultimately facilitated the closure of diabetic wounds.
4.6. Other applications
In addition to the common applications mentioned above, injectable smart stimuli-responsive hydrogels also play an important role in various specialized biomedical scenarios. For example, in some ophthalmic diseases, when the vitreous becomes turbid or diseased, it often requires replacement. However, traditional vitreous substitutes (e.g., silicone oil) have drawbacks such as emulsification and the need for secondary surgery for removal. Therefore, hydrogels that closely mimic the properties of the vitreous have emerged as ideal substitutes for the vitreous. In the study of Wang et al., they prepared a self-gelling and shear-thinning injectable hydrogel based on the dynamic Schiff base bonds between the carboxymethyl chitosan (CMCTS) and oxidized hyaluronic acid (OHA).386 The hydrogel could be injected as a liquid into the vitreous cavity and rapidly self-gelled under physiological conditions, and exhibited excellent characteristics of an ideal vitreous substitute material, including high transparency, controllable swelling properties, and exceptionally high biocompatibility. Similarly, Baker et al. utilized oxime click chemistry to prepare injectable HA-based hydrogels and developed a hydrogel substitute with properties similar to natural vitreous (including density, transparency, refractive index, and maintenance of normal intraocular pressure), which could exist stably in vivo.250 Furthermore, injectable smart stimuli-responsive hydrogels have also been developed for sustained drug delivery through intravitreal injection to treat glaucoma.47Periodontitis is one of the most common inflammatory diseases, characterized by inflammation of the gingival tissue and loss of tooth-supporting tissues. Its pathogenesis is related to the dysbiosis of the periodontal microbiome, and the persistent inflammation leads to increased levels of reactive oxygen species (ROS) in gingival tissues. To address this, Gan et al. developed a PVA-based injectable hydrogel, and the contained borate ester bonds could be cleaved in the presence of ROS.323 This contributed to the ROS-triggered release of the loaded C5a receptor antagonists (C5A) and mechanostimulated ex situ macrophages into the periodontitis-affected gingival tissues, which then effectively alleviated the periodontal inflammation and reduced the periodontitis-associated bone loss.
As the nervous system is a highly organized and strongly directional tissue, the appropriate alignment of cells and tissues after nerve injury is crucial for the functional recovery and reconstruction of neural tissues. Of note, injectable magnetic field-responsive hydrogels have exhibited excellent performances in the repair process of neural tissues. Omidinia-Anarkoli and colleagues prepared nanoscale short fibers with PLGA, which was loaded with superparamagnetic iron oxide nanoparticles (SPIONs).354 These fibers were then added to a fibrin hydrogel precursor solution, and an external magnetic field was applied prior to the gelation to orient the magnetically responsive short fibers in the solution. This smart hydrogel system facilitated the linear growth of neural cells, and provided a substantial advantage for successful alignment of cells and tissues during the neural repair process.
In addition, some specialized injectable smart stimuli-responsive hydrogels have been developed for 3D bioprinting. For example, HA-based hydrogels formed through the dynamic Schiff base bonds between hydrazide-modified HA (HA-HYD) and aldehyde-modified HA (HA-ALD) exhibited excellent shear-thinning and self-healing properties. These properties enabled smooth extrusion from the nozzle during 3D bioprinting and provided mechanical protection to the loaded cells throughout the process, which prevented the cells from dying during extrusion.387 Moreover, the light-responsive biomaterial GelMA demonstrated outstanding performance for the preparation of rapidly photopolymerizable 3D-printed niches.388
5. Conclusion and future perspectives
According to the reports summarized in this review, the materials currently used for preparing injectable smart stimuli-responsive hydrogels mainly fall into two categories: natural and synthetic materials, each with its own advantages and disadvantages. Natural materials possess excellent inherent biocompatibility and exhibit biologically active properties, biodegradability, and wide availability. However, their inherent mechanical properties are relatively poor, and they may carry potential risks of immunogenicity. Additionally, some natural materials have high production costs and lack uniformity in quality between batches. In contrast, synthetic materials offer advantages in their excellent mechanical properties and programmable synthesis, enabling highly controlled and reproducible preparation processes with lower costs. Nonetheless, their biocompatibility and biodegradability are not as favorable as those of natural materials. In order to overcome the inherent limitations of these materials, many researchers have developed hybrid hydrogels by combining two or more types of natural/synthetic materials. These hybrid hydrogels exhibited outstanding performance, such as a combination of strong mechanical properties with good biocompatibility and biodegradability. This strategy may pave the way for the next generation of advanced hydrogels: by selecting appropriate combinations of natural and synthetic materials based on specific biomedical applications, the hybrid hydrogel leverages the strengths of both materials and compensates for their respective weaknesses, thus leading to hydrogels that are better suited to meet the diverse demands of various biomedical scenarios.Due to the stable and well-defined porous network structure, hydrogels have long been extensively explored for the purpose of carrying therapeutic agents. In recent years, various physical, chemical, or combined strategies have further endowed the traditional hydrogels with the ability to carry out targeted interventions in deep tissues through simple minimally invasive injection. As a result, injectable hydrogels have made significant contributions to the advancement of minimally invasive medical treatments. Furthermore, with the rapid development of emerging chemical and manufacturing industries, the currently applied injectable hydrogels in the biomedical field are no longer merely used as simple defect fillers or tissue substitutes. They have evolved into intelligent and multifunctional therapeutic units capable of responding intelligently to various stimuli. These advanced hydrogels can “recognize” diseased tissues upon injection into the body and selectively exert their therapeutic effects. This enhances treatment targeting, reduces off-target effects on normal tissues, and thus holds great significance for the development of precision medicine.
Herein, we provide an outlook on the development of the ideal injectable smart stimuli-responsive hydrogels in the future: (1) the biocompatibility must be adequate to avoid or minimize potential inflammatory and/or immune issues after injection; (2) it must have tunable biodegradation rate, which can be adjusted to match the different regeneration rates of targeted tissues; (3) the mechanical strength of the gel should be compatible with the targeted tissue, and it has to maintain adequate mechanical stability during tissue regeneration; (4) the microstructure should be porous with interconnected pores to transport oxygen, various nutrients and biomolecules among cells through the networks, and provide enough space for the ingrowth of regenerated tissues; (5) for injectable hydrogel loaded with living cells, it should provide an ideal environment for the adhesion, growth and differentiation of both loaded and host cells; (6) the stimuli or triggers used to activate intelligent hydrogels should exhibit a high degree of specificity to prevent the “misguided” release of loaded therapeutic agents; (7) other requirements including easy preparation, non-toxicity, no toxic by-products during gelation and degradation, stimulus-responsiveness to specific pathological stimulus and spatiotemporal control.
Although the above-mentioned advanced concepts and technologies have already endowed hydrogels with remarkable smart stimuli-responsiveness, we believe that in the near future, the rapidly evolving field of emerging chemistry will provide injectable hydrogels with even more intriguing stimuli-responsive abilities, which will enable them to consistently exert positive therapeutic effects throughout unpredictable pathological processes.
Author contributions
Jiacheng Liu: investigation, methodology, project administration, resources, software, writing – original draft. Chengcheng Du: investigation, methodology, project administration, resources, writing – original draft. Wei Huang: conceptualization, funding acquisition, project administration, supervision, writing – review & editing. Yiting Lei, conceptualization, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
We acknowledge the support from the Life Science Institute of Chongqing Medical University. Jiacheng Liu deeply appreciates the enduring love, constant companionship, and unwavering encouragement of Yuanyuan Wang over the years. This study was financially supported by the National Natural Science Foundation of China, PRC (U22A20284, 82302755).References
- X. Wang, Y. Ma, F. Lu and Q. Chang, Biomater. Sci., 2023, 11, 2639–2660 RSC.
- R. Zhong, S. Talebian, B. B. Mendes, G. Wallace, R. Langer, J. Conde and J. Shi, Nat. Mater., 2023, 22, 818–831 CrossRef CAS.
- Y. Sun, L. G. Chen, X. M. Fan and J. L. Pang, Int. J. Nanomed., 2022, 17, 5001–5026 CrossRef CAS.
- P. Le Thi, D. L. Tran, T. T. Hoang Thi, Y. Lee and K. D. Park, Regener. Biomater., 2022, 9, rbac069 CrossRef PubMed.
- X. Zhao, H. Ma, H. Han, L. Zhang, J. Tian, B. Lei and Y. Zhang, Mater. Today Bio, 2022, 16, 100336 CrossRef CAS.
- S. J. Buwalda, K. W. Boere, P. J. Dijkstra, J. Feijen, T. Vermonden and W. E. Hennink, J. Controlled Release, 2014, 190, 254–273 CrossRef CAS.
- S. Li, M. Pei, T. Wan, H. Yang, S. Gu, Y. Tao, X. Liu, Y. Zhou, W. Xu and P. Xiao, Carbohydr. Polym., 2020, 250, 116922 CrossRef CAS.
- A. Hong, M. I. Aguilar, M. P. Del Borgo, C. G. Sobey, B. R. S. Broughton and J. S. Forsythe, J. Mater. Chem. B, 2019, 7, 3927–3943 RSC.
- A. Mellati, E. Hasanzadeh, M. Gholipourmalekabadi and S. E. Enderami, Mater. Sci. Eng., C, 2021, 131, 112489 CrossRef CAS PubMed.
- S. He, Z. Zhang, R. Luo, Q. Jiang, L. Yang and Y. Wang, Adv. Healthc. Mater., 2023, 12, e2300029 CrossRef.
- E. Tous, B. Purcell, J. L. Ifkovits and J. A. Burdick, J. Cardiovasc. Transl. Res., 2011, 4, 528–542 CrossRef PubMed.
- L. R. Nih, S. T. Carmichael and T. Segura, Curr. Opin. Biotechnol, 2016, 40, 155–163 CrossRef CAS.
- A. Chakraborty, S. Alexander, W. Luo, N. Al-Salam, M. Van Oirschot, S. H. Ranganath, S. Chakrabarti and A. Paul, Interdiscip. Med., 2023, e20230008 CrossRef.
- J. Van Bemmelen, Z. Anorg. Chem., 1894, 5, 466–483 CrossRef.
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- P. C. Nicolson and J. Vogt, Biomaterials, 2001, 22, 3273–3283 CrossRef CAS PubMed.
- J. H. Teichroeb, J. A. Forrest, L. W. Jones, J. Chan and K. Dalton, J. Colloid Interface Sci., 2008, 325, 157–164 CrossRef CAS.
- J. Kopeček, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5929–5946 CrossRef PubMed.
- E. Ruel-Gariépy and J. C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS.
- C. Hiemstra, Z. Zhong, P. J. Dijkstra and J. Feijen, Macromol. Symp., 2005, 224(1), 119–132 CrossRef CAS.
- S. J. Buwalda, L. Calucci, C. Forte, P. J. Dijkstra and J. Feijen, Polymer, 2012, 53, 2809–2817 CrossRef CAS.
- L. Calucci, C. Forte, S. J. Buwalda and P. J. Dijkstra, Macromolecules, 2011, 44, 7288–7295 CrossRef CAS.
- K. Nagahama, K. Fujiura, S. Enami, T. Ouchi and Y. Ohya, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6317–6332 CrossRef CAS.
- C. An, H. Li, Y. Zhao, S. Zhang, Y. Zhao, Y. Zhang, J. Yang, L. Zhang, C. Ren, Y. Zhang, J. Liu and H. Wang, Int. J. Biol. Macromol., 2023, 231, 123307 CrossRef CAS PubMed.
- A. C. Daly, L. Riley, T. Segura and J. A. Burdick, Nat. Rev. Mater., 2020, 5, 20–43 CrossRef CAS.
- C. Du and W. Huang, Nanocomposites, 2022, 8, 102–124 CrossRef CAS.
- S. Wang, S. Tavakoli, R. P. Parvathaneni, G. N. Nawale, O. P. Oommen, J. Hilborn and O. P. Varghese, Biomater. Sci., 2022, 10, 6399–6412 RSC.
- Y. Lei, Y. Wang, J. Shen, Z. Cai, Y. Zeng, P. Zhao, J. Liao, C. Lian, N. Hu and X. Luo, Adv. Funct. Mater., 2021, 31, 2105084 CrossRef CAS.
- Y. Lei, Y. Wang, J. Shen, Z. Cai, C. Zhao, H. Chen, X. Luo, N. Hu, W. Cui and W. Huang, Sci. Adv., 2022, 8, eabl6449 CrossRef CAS PubMed.
- S. Chang, S. Wang, Z. Liu and X. Wang, Gels, 2022, 8, 389 CrossRef CAS PubMed.
- B. Zhuang, T. Chen, Z. Xiao and Y. Jin, Int. J. Pharm., 2020, 577, 119048 CrossRef CAS PubMed.
- P. Mondal, I. Chakraborty and K. Chatterjee, Chem. Rec., 2022, 22, e202200155 CrossRef CAS.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- P. Mondal and K. Chatterjee, Carbohydr. Polym., 2022, 291, 119585 CrossRef CAS PubMed.
- A. P. Mathew, S. Uthaman, K. H. Cho, C. S. Cho and I. K. Park, Int. J. Biol. Macromol., 2018, 110, 17–29 CrossRef CAS PubMed.
- S. Yu, C. He and X. Chen, Macromol. Biosci., 2018, 18, e1800240 CrossRef PubMed.
- A. Mandal, J. R. Clegg, A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2020, 5, e10158 CrossRef CAS PubMed.
- Ø. Øvrebø, G. Perale, J. P. Wojciechowski, C. Echalier, J. R. T. Jeffers, M. M. Stevens, H. J. Haugen and F. Rossi, Bioeng. Transl. Med., 2022, 7, e10295 CrossRef.
- P. Bertsch, M. Diba, D. J. Mooney and S. C. G. Leeuwenburgh, Chem. Rev., 2023, 123, 834–873 CrossRef CAS PubMed.
- Z. Zheng, Y. Tan, Y. Li, Y. Liu, G. Yi, C. Y. Yu and H. Wei, J. Controlled Release, 2021, 335, 216–236 CrossRef CAS PubMed.
- O. Jeznach, D. Kołbuk and P. Sajkiewicz, J. Biomed. Mater. Res., Part A, 2018, 106, 2762–2776 CrossRef CAS PubMed.
- T. Jiang, T. Yang, Q. Bao, W. Sun, M. Yang and C. Mao, J. Mater. Chem. B, 2022, 10, 4741–4758 RSC.
- J. Wang, Y. Song, W. Xie, J. Zhao, Y. Wang and W. Yu, iScience, 2023, 26, 106577 CrossRef CAS PubMed.
- K. A. Akulo, T. Adali, M. T. G. Moyo and T. Bodamyali, Polymers, 2022, 14, 2359 CrossRef CAS PubMed.
- L. Meng, C. Shao, C. Cui, F. Xu, J. Lei and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 1628–1639 CrossRef CAS.
- X. Wang, Y. Yang, Y. Shi and F. Jia, Front. Chem., 2020, 8, 245 CrossRef PubMed.
- M. Gomez-Florit, A. Pardo, R. M. A. Domingues, A. L. Graça, P. S. Babo, R. L. Reis and M. E. Gomes, Molecules, 2020, 25, 5858 CrossRef CAS PubMed.
- G. Tang, Z. Liu, Y. Liu, J. Yu, X. Wang, Z. Tan and X. Ye, Front. Cell Dev. Biol., 2021, 9, 665813 CrossRef.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- S. Zhu, Y. Li, Z. He, L. Ji, W. Zhang, Y. Tong, J. Luo, D. Yu, Q. Zhang and Q. Bi, Front. Bioeng. Biotechnol., 2022, 10, 954501 CrossRef.
- B. Balakrishnan, N. Joshi, A. Jayakrishnan and R. Banerjee, Acta Biomater., 2014, 10, 3650–3663 CrossRef CAS PubMed.
- N. Landa, L. Miller, M. S. Feinberg, R. Holbova, M. Shachar, I. Freeman, S. Cohen and J. Leor, Circulation, 2008, 117, 1388–1396 CrossRef CAS PubMed.
- Q. Hu, Y. Nie, J. Xiang, J. Xie, H. Si, D. Li, S. Zhang, M. Li and S. Huang, Int. J. Biol. Macromol., 2023, 234, 123691 CrossRef CAS PubMed.
- M. Ghanbari, M. Salavati-Niasari, F. Mohandes, B. Dolatyar and B. Zeynali, RSC Adv., 2021, 11, 16688–16697 RSC.
- P. Wang, Y. Pu, Y. Ren, R. Yang, W. Zhang, X. Tan, W. Xue, S. Liu, S. Li and B. Chi, Carbohydr. Polym., 2022, 275, 118692 CrossRef CAS PubMed.
- E. A. Growney Kalaf, R. Flores, J. G. Bledsoe and S. A. Sell, Mater. Sci. Eng., C, 2016, 63, 198–210 CrossRef CAS PubMed.
- X. Zhang, Y. Zhu, L. Cao, X. Wang, A. Zheng, J. Chang, J. Wu, J. Wen, X. Jiang, H. Li and Z. Zhang, J. Mater. Chem. B, 2018, 6, 1951–1964 RSC.
- H. Li, Z. Hao, S. Zhang, B. Li, Y. Wang, X. Wu, Y. Hu, R. Chen, T. Chen and J. Li, Macromol. Biosci., 2023, 23, e2200481 CrossRef.
- A. F. Khan, M. Saleem, A. Afzal, A. Ali, A. Khan and A. R. Khan, Mater. Sci. Eng., C, 2014, 35, 245–252 CrossRef CAS PubMed.
- H. Zreiqat, C. R. Howlett, A. Zannettino, P. Evans, G. Schulze-Tanzil, C. Knabe and M. Shakibaei, J. Biomed. Mater. Res., 2002, 62, 175–184 CrossRef CAS PubMed.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757–2774 CrossRef CAS PubMed.
- Y. Zhu, L. Kong, F. Farhadi, W. Xia, J. Chang, Y. He and H. Li, Biomaterials, 2019, 192, 149–158 CrossRef CAS.
- T. Wu, N. Cheng, C. Xu, W. Sun, C. Yu and B. Shi, J. Biomed. Mater. Res., Part A, 2016, 104, 3004–3014 CrossRef CAS PubMed.
- J. Zhang, J. Guan, C. Zhang, H. Wang, W. Huang, S. Guo, X. Niu, Z. Xie and Y. Wang, Biomed. Mater., 2015, 10, 065011 CrossRef PubMed.
- S. Gaudreau, C. Guindi, M. Ménard, G. Besin, G. Dupuis and A. Amrani, J. Immunol., 2007, 179, 3638–3647 CrossRef CAS PubMed.
- Y. Huang, S. Onyeri, M. Siewe, A. Moshfeghian and S. V. Madihally, Biomaterials, 2005, 26, 7616–7627 CrossRef CAS PubMed.
- N. Thottappillil and P. D. Nair, Vasc. Health Risk Manage., 2015, 11, 79–91 Search PubMed.
- M. Ghorbani, J. Ai, M. R. Nourani, M. Azami, B. Hashemi Beni, S. Asadpour and S. Bordbar, Mater. Sci. Eng., C, 2017, 80, 502–508 CrossRef CAS PubMed.
- S. A. A. Ghavimi, E. S. Lungren, J. L. Stromsdorfer, B. T. Darkow, J. A. Nguyen, Y. Sun, F. M. Pfieffer, C. L. Goldstein, C. Wan and B. D. Ulery, AAPS J., 2019, 21, 41 CrossRef.
- D. Mehrotra, R. Dwivedi, D. Nandana and R. K. Singh, J. Oral Biol. Craniofac. Res., 2020, 10, 680–689 CrossRef PubMed.
- L. Deng, B. Wang, W. Li, Z. Han, S. Chen and H. Wang, Int. J. Biol. Macromol., 2022, 217, 77–87 CrossRef CAS.
- M. M. Islam, M. Shahruzzaman, S. Biswas, M. Nurus Sakib and T. U. Rashid, Bioact. Mater., 2020, 5, 164–183 Search PubMed.
- G. Guedes, S. Wang, F. Fontana, P. Figueiredo, J. Linden, A. Correia, R. J. B. Pinto, S. Hietala, F. L. Sousa and H. A. Santos, Adv. Mater., 2021, 33, e2007761 CrossRef PubMed.
- Y. Kim, Y. Hu, J. P. Jeong and S. Jung, Carbohydr. Polym., 2022, 284, 119195 CrossRef CAS PubMed.
- Y. Zhu, D. Winer, C. Goh and A. Shrestha, Biomater. Sci., 2023, 11, 2091–2102 RSC.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- J. Thomas, V. Chopra, A. Sharma, V. Panwar, S. Kaushik, S. Rajput, M. Mittal, R. Guha, N. Chattopadhyay and D. Ghosh, Int. J. Biol. Macromol., 2021, 190, 474–486 CrossRef CAS PubMed.
- K. J. Ornell, D. Lozada, N. V. Phan and J. M. Coburn, J. Mater. Chem. B, 2019, 7, 2151–2161 RSC.
- X. M. Keutgen, K. J. Ornell, A. Vogle, O. Lakiza, J. Williams, P. Miller, K. S. Mistretta, N. Setia, R. R. Weichselbaum and J. M. Coburn, Ann. Surg. Oncol., 2021, 28, 8532–8543 CrossRef PubMed.
- J. Y. Lee, S. H. Lee, H. J. Kim, J. M. Ha, S. H. Lee, J. H. Lee and B. J. Ha, Arch. Pharmacal Res., 2004, 27, 340–345 CrossRef CAS.
- Y. He, M. Sun, J. Wang, X. Yang, C. Lin, L. Ge, C. Ying, K. Xu, A. Liu and L. Wu, Acta Biomater., 2022, 151, 512–527 CrossRef CAS PubMed.
- X. Li, Q. Xu, M. Johnson, X. Wang, J. Lyu, Y. Li, S. McMahon, U. Greiser, S. A and W. Wang, Biomater. Sci., 2021, 9, 4139–4148 RSC.
- V. Castrejón-Comas, C. Alemán and M. M. Pérez-Madrigal, Biomater. Sci., 2023, 11, 2266–2276 RSC.
- Y. W. Ding, Z. Y. Wang, Z. W. Ren, X. W. Zhang and D. X. Wei, Biomater. Sci., 2022, 10, 3393–3409 RSC.
- M. K. Cowman, H. G. Lee, K. L. Schwertfeger, J. B. McCarthy and E. A. Turley, Front. Immunol., 2015, 6, 261 Search PubMed.
- Y. Wang, W. Niu, X. Qu and B. Lei, ACS Appl. Mater. Interfaces, 2022, 14, 4946–4958 CrossRef CAS PubMed.
- M. F. P. Graça, S. P. Miguel, C. S. D. Cabral and I. J. Correia, Carbohydr. Polym., 2020, 241, 116364 CrossRef PubMed.
- J. Li, N. Liu, Z. Huang, W. Wang, D. Hou and W. Wang, J. Orthop. Surg. Res., 2021, 16, 646 CrossRef PubMed.
- T. Kikani, S. Dave and S. Thakore, Int. J. Biol. Macromol., 2023, 242, 124950 CrossRef CAS PubMed.
- W. Knudson and R. S. Peterson, Chemistry and biology of hyaluronan, Elsevier, Oxford, 2004, pp. 83–124 Search PubMed.
- M. Litwiniuk, A. Krejner, M. S. Speyrer, A. R. Gauto and T. Grzela, Wounds, 2016, 28, 78–88 Search PubMed.
- G. Kogan, L. Soltés, R. Stern and P. Gemeiner, Biotechnol. Lett., 2007, 29, 17–25 CrossRef CAS PubMed.
- M. Dovedytis, Z. J. Liu and S. Bartlett, Eng. Regener., 2020, 1, 102–113 Search PubMed.
- A. Avenoso, A. D'Ascola, M. Scuruchi, G. Mandraffino, A. Calatroni, A. Saitta, S. Campo and G. M. Campo, Inflammation Res., 2018, 67, 5–20 CrossRef CAS PubMed.
- Y. S. Jung, W. Park, H. Park, D. K. Lee and K. Na, Carbohydr. Polym., 2017, 156, 403–408 CrossRef CAS PubMed.
- Y. C. Chen, W. Y. Su, S. H. Yang, A. Gefen and F. H. Lin, Acta Biomater., 2013, 9, 5181–5193 CrossRef CAS PubMed.
- Y. Li, M. Wang, M. Sun, X. Wang, D. Pei, B. Lei and A. Li, Composites, Part B, 2022, 242, 110034 CrossRef CAS.
- D. Y. Kim, H. Park, S. W. Kim, J. W. Lee and K. Y. Lee, Carbohydr. Polym., 2017, 157, 1281–1287 CrossRef CAS PubMed.
- C. B. Highley, G. D. Prestwich and J. A. Burdick, Curr. Opin. Biotechnol, 2016, 40, 35–40 CrossRef CAS PubMed.
- A. B. Csoka, G. I. Frost and R. Stern, Matrix Biol., 2001, 20, 499–508 CrossRef CAS PubMed.
- B. Tavsanli and O. Okay, Carbohydr. Polym., 2019, 208, 413–420 CrossRef CAS PubMed.
- B. Tavsanli and O. Okay, Carbohydr. Polym., 2020, 229, 115458 CrossRef CAS PubMed.
- M. Wu, J. Chen, W. Huang, B. Yan, Q. Peng, J. Liu, L. Chen and H. Zeng, Biomacromolecules, 2020, 21, 2409–2420 CrossRef CAS PubMed.
- M. R. Arkenberg, H. D. Nguyen and C.-C. Lin, J. Mater. Chem. B, 2020, 8, 7835–7855 RSC.
- M. Rizwan, A. E. G. Baker and M. S. Shoichet, Adv. Healthc. Mater., 2021, 10, e2100234 CrossRef PubMed.
- Y. Lei, X. Wang, J. Liao, J. Shen, Y. Li, Z. Cai, N. Hu, X. Luo, W. Cui and W. Huang, Bioact. Mater., 2022, 16, 472–484 CAS.
- Y. Zhu, J. Tan, H. Zhu, G. Lin, F. Yin, L. Wang, K. Song, Y. Wang, G. Zhou and W. Yi, Biomater. Sci., 2017, 5, 784–791 RSC.
- W. Y. Su, Y. C. Chen and F. H. Lin, Acta Biomater., 2010, 6, 3044–3055 CrossRef CAS PubMed.
- C. Yang, P. J. Hillas, J. A. Báez, M. Nokelainen, J. Balan, J. Tang, R. Spiro and J. W. Polarek, BioDrugs, 2004, 18, 103–119 CrossRef CAS PubMed.
- S. T. Bendtsen and M. Wei, J. Mater. Chem. B, 2015, 3, 3081–3090 RSC.
- M. H. Santos, R. M. Silva, V. C. Dumont, J. S. Neves, H. S. Mansur and L. G. Heneine, Mater. Sci. Eng., C, 2013, 33, 790–800 CrossRef CAS PubMed.
- M. A. Horn and A. W. Trafford, J. Mol. Cell. Cardiol., 2016, 93, 175–185 CrossRef CAS PubMed.
- W. Dai, L. E. Wold, J. S. Dow and R. A. Kloner, J. Am. Coll. Cardiol., 2005, 46, 714–719 CrossRef CAS PubMed.
- N. J. Blackburn, T. Sofrenovic, D. Kuraitis, A. Ahmadi, B. McNeill, C. Deng, K. J. Rayner, Z. Zhong, M. Ruel and E. J. Suuronen, Biomaterials, 2015, 39, 182–192 CrossRef CAS PubMed.
- F. S. Y. Wong, K. K. Tsang, A. M. W. Chu, B. P. Chan, K. M. Yao and A. C. Y. Lo, Biomaterials, 2019, 201, 53–67 CrossRef CAS PubMed.
- P. Sarker, D. M. Nalband, D. O. Freytes, O. J. Rojas and S. A. Khan, Biomacromolecules, 2022, 23, 4696–4708 CrossRef CAS PubMed.
- L. Wu, H. Shao, Z. Fang, Y. Zhao, C. Y. Cao and Q. Li, ACS Biomater. Sci. Eng., 2019, 5, 4272–4284 CrossRef CAS PubMed.
- T. Taguchi, L. Xu, H. Kobayashi, A. Taniguchi, K. Kataoka and J. Tanaka, Biomaterials, 2005, 26, 1247–1252 CrossRef CAS PubMed.
- E. C. Collin, S. Grad, D. I. Zeugolis, C. S. Vinatier, J. R. Clouet, J. J. Guicheux, P. Weiss, M. Alini and A. S. Pandit, Biomaterials, 2011, 32, 2862–2870 CrossRef CAS PubMed.
- L. Cen, W. Liu, L. Cui, W. Zhang and Y. Cao, Pediatr. Res., 2008, 63, 492–496 CrossRef CAS PubMed.
- Z. Chen, D. Fan and L. Shang, Biomed. Mater., 2020, 16, 012001 CrossRef PubMed.
- X. Song, C. Zhu, D. Fan, Y. Mi, X. Li, R. Z. Fu, Z. Duan, Y. Wang and R. R. Feng, Polymers, 2017, 9, 638 CrossRef PubMed.
- L. Yang, H. Wu, L. Lu, Q. He, B. Xi, H. Yu, R. Luo, Y. Wang and X. Zhang, Biomaterials, 2021, 276, 121055 CrossRef CAS PubMed.
- C. Hu, W. Liu, L. Long, Z. Wang, W. Zhang, S. He, L. Lu, H. Fan, L. Yang and Y. Wang, Biomaterials, 2022, 290, 121849 CrossRef CAS PubMed.
- Y. Guo, Z. Hu, J. Chen, Z. Zhang, Q. Liu, J. Li, J. Yang, Z. Ma, J. Zhao, J. Hu, J. Wu and Z. Chen, Int. J. Biol. Macromol., 2023, 236, 123864 CrossRef CAS PubMed.
- P. Li, M. Zhang, Z. Chen, B. Tian and X. Kang, ACS Omega, 2023, 8, 13509–13518 CrossRef CAS PubMed.
- R. Sergi, D. Bellucci and V. Cannillo, Materials, 2020, 13, 5560 CrossRef CAS PubMed.
- M. Luo, D. D. Winston, W. Niu, Y. Wang, H. Zhao, X. Qu and B. Lei, Chem. Eng. J., 2022, 431, 133596 CrossRef CAS.
- T. Chen, D. A. Small, M. K. McDermott, W. E. Bentley and G. F. Payne, Biomacromolecules, 2003, 4, 1558–1563 CrossRef CAS PubMed.
- J. Zheng, F. Zhao, W. Zhang, Y. Mo, L. Zeng, X. Li and X. Chen, Mater. Sci. Eng., C, 2018, 89, 119–127 CrossRef CAS PubMed.
- Y. Wang, M. Ma, J. Wang, W. Zhang, W. Lu, Y. Gao, B. Zhang and Y. Guo, Materials, 2018, 11, 1345 CrossRef PubMed.
- R. Haghniaz, H. Montazerian, A. Rabbani, A. Baidya, B. Usui, Y. Zhu, M. Tavafoghi, F. Wahid, H. J. Kim, A. Sheikhi and A. Khademhosseini, Adv. Healthc. Mater., 2023, e2301551, DOI:10.1002/adhm.202301551.
- J. Wang, X. Wang, Z. Liang, W. Lan, Y. Wei, Y. Hu, L. Wang, Q. Lei and D. Huang, Front. Bioeng. Biotechnol., 2023, 11, 1219460 CrossRef PubMed.
- Z. Fu, S. Xiao, P. Wang, J. Zhao, Z. Ling, Z. An, J. Shao and W. Fu, RSC Adv., 2023, 13, 10903–10913 RSC.
- D. Kim, S. Jo, D. Lee, S. M. Kim, J. M. Seok, S. J. Yeo, J. H. Lee, J. J. Lee, K. Lee, T. D. Kim and S. A. Park, Biomater. Res., 2023, 27, 60 CrossRef CAS PubMed.
- Y. Liang, H. Xu, Z. Li, A. Zhangji and B. Guo, Nano-Micro Lett., 2022, 14, 185 CrossRef CAS PubMed.
- X. Liu, J. Liu, J. Wang, T. Wang, Y. Jiang, J. Hu, Z. Liu, X. Chen and J. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 5601–5609 CrossRef CAS PubMed.
- L.-D. Koh, Y. Cheng, C.-P. Teng, Y.-W. Khin, X.-J. Loh, S.-Y. Tee, M. Low, E. Ye, H.-D. Yu and Y.-W. Zhang, Prog. Polym. Sci., 2015, 46, 86–110 CrossRef CAS.
- B. K. Bhunia and B. B. Mandal, ACS Biomater. Sci. Eng., 2019, 5, 870–886 CrossRef CAS PubMed.
- J. Hu, Y. Lu, L. Cai, K. G. Owusu-Ansah, G. Xu, F. Han, J. Bao, X. Lin and Y. Huang, Sci. Rep., 2017, 7, 2347 CrossRef PubMed.
- S. Lee, J. Choi, J. Youn, Y. Lee, W. Kim, S. Choe, J. Song, R. L. Reis and G. Khang, Biomolecules, 2021, 11, 1184 CrossRef CAS PubMed.
- R. Ziadlou, S. Rotman, A. Teuschl, E. Salzer, A. Barbero, I. Martin, M. Alini, D. Eglin and S. Grad, Mater. Sci. Eng., C, 2021, 120, 111701 CrossRef CAS PubMed.
- P. Chuysinuan, P. Nooeaid, T. Thanyacharoen, S. Techasakul, P. Pavasant and K. Kanjanamekanant, Int. J. Biol. Macromol., 2021, 193, 799–808 CrossRef CAS PubMed.
- Z. Lv, T. Hu, Y. Bian, G. Wang, Z. Wu, H. Li, X. Liu, S. Yang, C. Tan, R. Liang and X. Weng, Adv. Mater., 2023, 35, e2206545 CrossRef PubMed.
- H. Wang, Y. Jin, Y. Tan, H. Zhu, W. Huo, P. Niu, Z. Li, J. Zhang, X. J. Liang and X. Yang, Biomaterials, 2021, 275, 120992 CrossRef CAS PubMed.
- M. C. Bruno, M. C. Cristiano, C. Celia, N. d'Avanzo, A. Mancuso, D. Paolino, J. Wolfram and M. Fresta, ACS Nano, 2022, 16, 19665–19690 CrossRef CAS PubMed.
- A. D. Theocharis, S. S. Skandalis, C. Gialeli and N. K. Karamanos, Adv. Drug Delivery Rev., 2016, 97, 4–27 CrossRef CAS PubMed.
- Y. Zhang, Y. Xu and J. Gao, Biomater. Sci., 2023, 11, 3784–3799 RSC.
- M. Brown, J. Li, C. Moraes, M. Tabrizian and N. Y. K. Li-Jessen, Biomaterials, 2022, 289, 121786 CrossRef CAS PubMed.
- T. J. Keane, I. T. Swinehart and S. F. Badylak, Methods, 2015, 84, 25–34 CrossRef CAS PubMed.
- J. L. Ungerleider, T. D. Johnson, M. J. Hernandez, D. I. Elhag, R. L. Braden, M. Dzieciatkowska, K. G. Osborn, K. C. Hansen, E. Mahmud and K. L. Christman, JACC Basic Transl. Sci., 2016, 1, 32–44 CrossRef PubMed.
- J. H. Traverse, T. D. Henry, N. Dib, A. N. Patel, C. Pepine, G. L. Schaer, J. A. DeQuach, A. M. Kinsey, P. Chamberlin and K. L. Christman, JACC, 2019, 4, 659–669 Search PubMed.
- J. L. Zou, S. Liu, J. H. Sun, W. H. Yang, Y. W. Xu, Z. L. Rao, B. Jiang, Q. T. Zhu, X. L. Liu and J. L. Wu, Adv. Funct. Mater., 2018, 28, 1705739 CrossRef.
- J. S. Lee, J. Shin, H. M. Park, Y. G. Kim, B. G. Kim, J. W. Oh and S. W. Cho, Biomacromolecules, 2014, 15, 206–218 CrossRef CAS PubMed.
- A. H. Morris, H. Lee, H. Xing, D. K. Stamer, M. Tan and T. R. Kyriakides, ACS Appl. Mater. Interfaces, 2018, 10, 41892–41901 CrossRef CAS PubMed.
- T. J. Keane, J. Dziki, A. Castelton, D. M. Faulk, V. Messerschmidt, R. Londono, J. E. Reing, S. S. Velankar and S. F. Badylak, J. Biomed. Mater. Res., Part B, 2017, 105, 291–306 CrossRef CAS PubMed.
- K. Jiang, D. Chaimov, S. N. Patel, J. P. Liang, S. C. Wiggins, M. M. Samojlik, A. Rubiano, C. S. Simmons and C. L. Stabler, Biomaterials, 2019, 198, 37–48 CrossRef CAS PubMed.
- A. Basiri, M. Farokhi, M. Azami, S. Ebrahimi-Barough, A. Mohamadnia, M. Rashtbar, E. Hasanzadeh, N. Mahmoodi, M. Baghaban Eslaminejad and J. Ai, Prog. Biomater., 2019, 8, 31–42 CrossRef CAS PubMed.
- H. J. Kang, S. S. Park, G. Tripathi and B. T. Lee, Mater. Today Bio, 2022, 16, 100422 CrossRef CAS PubMed.
- H. V. Almeida, R. Eswaramoorthy, G. M. Cunniffe, C. T. Buckley, F. J. O'Brien and D. J. Kelly, Acta Biomater., 2016, 36, 55–62 CrossRef CAS PubMed.
- X. Wang, A. Ansari, V. Pierre, K. Young, C. R. Kothapalli, H. A. von Recum and S. E. Senyo, Adv. Healthc. Mater., 2022, 11, e2102265 CrossRef PubMed.
- Y. Zhu, S. Hideyoshi, H. Jiang, Y. Matsumura, J. L. Dziki, S. T. LoPresti, L. Huleihel, G. N. F. Faria, L. C. Fuhrman, R. Lodono, S. F. Badylak and W. R. Wagner, Acta Biomater., 2018, 73, 112–126 CrossRef CAS PubMed.
- J. Varshosaz, S. Masoudi, M. Mehdikhani, B. Hashemi Beni and S. Farsaei, IET Nanobiotechnol., 2019, 13, 933–941 CrossRef PubMed.
- B. Peña, M. Laughter, S. Jett, T. J. Rowland, M. R. G. Taylor, L. Mestroni and D. Park, Macromol. Biosci., 2018, 18, e1800079 CrossRef PubMed.
- G. BaoLin and P. X. Ma, Sci. China: Chem., 2014, 57, 490–500 CrossRef PubMed.
- J. P. Santerre, K. Woodhouse, G. Laroche and R. S. Labow, Biomaterials, 2005, 26, 7457–7470 CrossRef CAS PubMed.
- M. Patel, J. K. Park and B. Jeong, Biomater. Res., 2023, 27, 17 CrossRef CAS PubMed.
- L. Meng, Z. Wang, Z. Hou, H. Wang, X. Zhang, X. Zhang, X. He, X. Zhang, B. Qin, J. Li, Z. Zhang, X. Xue and Y. Wei, Front. Immunol., 2022, 13, 1064047 CrossRef CAS PubMed.
- W. Lao, L. Fan, Q. Zhang, C. Lou, H. Li, Y. Li, S. Wu, X. Li, Q. Luo, W. Zhu and X. Li, J. Mater. Chem. B, 2023, 11, 3136–3150 RSC.
- H. J. Ju, Y. B. Ji, S. Kim, H. W. Yun, J. H. Kim, B. H. Min and M. S. Kim, Macromol. Biosci., 2023, 23, e2300029 CrossRef PubMed.
- C. Li, T. Jiang, C. Zhou, A. Jiang, C. Lu, G. Yang, J. Nie, F. Wang, X. Yang and Z. Chen, Carbohydr. Polym., 2023, 299, 120198 CrossRef CAS PubMed.
- J. N. I. Balitaan, W. J. Luo, Y. W. Su, C. Y. Yu, T. Y. Wu, C. A. Chang, H. W. Jia, S. R. Lin, C. D. Hsiao and J. M. Yeh, ACS Appl. Bio Mater., 2023, 6, 552–565 CrossRef CAS PubMed.
- M. Y. Ha, D. H. Yang, S. J. You, H. J. Kim and H. J. Chun, npj Regener. Med., 2023, 8, 2 CrossRef CAS PubMed.
- W. Chen, C. Wang, W. Liu, B. Zhao, Z. Zeng, F. Long, C. Wang, S. Li, N. Lin and J. Zhou, Adv. Mater., 2023, 35, e2209041 CrossRef PubMed.
- W. Fang, L. Yang, Y. Chen and Q. Hu, Acta Biomater., 2023, 161, 50–66 CrossRef CAS PubMed.
- H. Liu, Z. Deng, T. Li, J. Bu, D. Wang, J. Wang, M. Liu, J. Li, Y. Yang and S. Zhong, Colloids Surf., B, 2022, 217, 112703 CrossRef CAS PubMed.
- H. Jia, X. Lin, D. Wang, J. Wang, Q. Shang, X. He, K. Wu, B. Zhao, P. Peng, H. Wang, D. Wang, P. Li, L. Yang, Z. Luo and L. Yang, J. Orthop. Transl., 2022, 33, 162–173 Search PubMed.
- G. Leone, M. Consumi, S. Lamponi, C. Bonechi, G. Tamasi, A. Donati, C. Rossi and A. Magnani, Mater. Sci. Eng., C, 2019, 98, 696–704 CrossRef CAS PubMed.
- M. J. Allen, J. E. Schoonmaker, T. W. Bauer, P. F. Williams, P. A. Higham and H. A. Yuan, Spine, 2004, 29, 515–523 CrossRef PubMed.
- K. Zhang, D. Wu, L. Chang, W. Duan, Y. Wang, W. Li and J. Qin, Int. J. Biol. Macromol., 2023, 230, 123294 CrossRef CAS PubMed.
- J. Xiang, Y. Bai, Y. Huang, S. Lang, J. Li, Y. Ji, B. Peng and G. Liu, J. Mater. Chem. B, 2022, 10, 7979–7994 RSC.
- W. Shi, B. Hass, M. A. Kuss, H. Zhang, S. Ryu, D. Zhang, T. Li, Y. L. Li and B. Duan, Carbohydr. Polym., 2020, 233, 115803 CrossRef CAS PubMed.
- Y. Gong, M. Chen, Y. Tan, J. Shen, Q. Jin, W. Deng, J. Sun, C. Wang, Z. Liu and Q. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 50248–50259 CrossRef CAS PubMed.
- M. Kuddushi, D. Ray, V. Aswal, C. Hoskins and N. Malek, ACS Appl. Bio Mater., 2020, 3, 4883–4894 CrossRef CAS PubMed.
- K. Yu, H. Zhou, Y. Xu, Y. Cao, Y. Zheng and B. Liang, J. Nanobiotechnol., 2023, 21, 201 CrossRef CAS PubMed.
- L. Mu, R. Dong and B. Guo, Adv. Healthc. Mater., 2023, 12, e2202699 CrossRef PubMed.
- P. Díaz-Herráez, L. Saludas, S. Pascual-Gil, T. Simón-Yarza, G. Abizanda, F. Prósper, E. Garbayo and M. J. Blanco-Prieto, J. Controlled Release, 2017, 249, 23–31 CrossRef PubMed.
- E. Garbayo, A. Ruiz-Villalba, S. C. Hernandez, L. Saludas, G. Abizanda, B. Pelacho, C. Roncal, B. Sanchez, I. Palacios, F. Prósper and M. J. Blanco-Prieto, Acta Biomater., 2021, 126, 394–407 CrossRef CAS PubMed.
- M. Endres, A. Abbushi, U. W. Thomale, M. Cabraja, S. N. Kroppenstedt, L. Morawietz, P. A. Casalis, M. L. Zenclussen, A. J. Lemke, P. Horn, C. Kaps and C. Woiciechowsky, Biomaterials, 2010, 31, 5836–5841 CrossRef CAS PubMed.
- S. Li, D. Niu, T. Shi, W. Yun, S. Yan, G. Xu and J. Yin, ACS Biomater. Sci. Eng., 2023, 9, 2625–2635 CrossRef CAS PubMed.
- Y. H. Yi, G. Chen, S. Gong, L. Z. Han, T. L. Gong, Y. X. Wang, W. H. Xu and X. Jin, ACS Biomater. Sci. Eng., 2023, 9, 1672–1681 CrossRef CAS PubMed.
- Y. Wu, X. Chang, G. Yang, L. Chen, Q. Wu, J. Gao, R. Tian, W. Mu, J. J. Gooding, X. Chen and S. Sun, Adv. Mater., 2023, 35, e2210787 CrossRef PubMed.
- P. A. Shiekh, A. Singh and A. Kumar, ACS Appl. Mater. Interfaces, 2018, 10, 18458–18469 CrossRef CAS PubMed.
- M. S. Kishta, H. H. Ahmed, M. A. M. Ali, H. A. Aglan and M. R. Mohamed, Biotech. Histochem., 2022, 97, 322–333 CrossRef CAS PubMed.
- H. Yao, J. Wang and S. Mi, Polymers, 2017, 10, 11 CrossRef PubMed.
- R. Naranjo-Alcazar, S. Bendix, T. Groth and G. Gallego Ferrer, Gels, 2023, 9, 230 CrossRef CAS PubMed.
- C. Shen, X. Zhao, Z. Ren, B. Yang, X. Wang, A. Hu and J. Hu, Int. J. Mol. Sci., 2023, 24, 4957 CrossRef CAS PubMed.
- J. Chen, J. Yang, L. Wang, X. Zhang, B. C. Heng, D. A. Wang and Z. Ge, Bioact. Mater., 2021, 6, 1689–1698 CAS.
- Z. Meng, X. Zhou, J. Xu, X. Han, Z. Dong, H. Wang, Y. Zhang, J. She, L. Xu, C. Wang and Z. Liu, Adv. Mater., 2019, 31, e1900927 CrossRef PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- H. Shih, A. K. Fraser and C. C. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 1673–1680 CrossRef CAS PubMed.
- Y. Hao, J. He, X. Ma, L. Feng, M. Zhu, Y. Zhai, Y. Liu, P. Ni and G. Cheng, Carbohydr. Polym., 2019, 225, 115257 CrossRef CAS PubMed.
- M. Patel, H. J. Lee, S. Park, Y. Kim and B. Jeong, Biomaterials, 2018, 159, 91–107 CrossRef CAS PubMed.
- Y. J. Kim and Y. T. Matsunaga, J. Mater. Chem. B, 2017, 5, 4307–4321 RSC.
- J. M. Unagolla and A. C. Jayasuriya, Appl. Mater. Today, 2020, 18, 100479 CrossRef PubMed.
- L. Yang, X. Fan, J. Zhang and J. Ju, Polymers, 2020, 12, 389 CrossRef CAS PubMed.
- J. O. Han, H. J. Lee and B. Jeong, Biomater. Res., 2022, 26, 16 CrossRef CAS PubMed.
- M.-Y. Yeh, J.-Y. Zhao, Y.-R. Hsieh, J.-H. Lin, F.-Y. Chen, R. D. Chakravarthy, P.-C. Chung, H.-C. Lin and S.-C. Hung, RSC Adv., 2017, 7, 21252–21257 RSC.
- Y. Jia, X. Zhang, W. Yang, C. Lin, B. Tao, Z. Deng, P. Gao, Y. Yang and K. Cai, J. Mater. Chem. B, 2022, 10, 2875–2888 RSC.
- A. Pourjavadi, M. Doroudian, A. Ahadpour and S. Azari, Int. J. Biol. Macromol., 2019, 126, 310–317 CrossRef CAS PubMed.
- J. Zhang, W. Lin, L. Yang, A. Zhang, Y. Zhang, J. Liu and J. Liu, Biomater. Sci., 2022, 10, 854–862 RSC.
- Y. L. Chiu, S. C. Chen, C. J. Su, C. W. Hsiao, Y. M. Chen, H. L. Chen and H. W. Sung, Biomaterials, 2009, 30, 4877–4888 CrossRef CAS PubMed.
- F. Gong, N. Yang, X. Wang, Q. Zhao, Q. Chen, Z. Liu and L. Cheng, Nano Today, 2020, 32, 100851 CrossRef CAS.
- M. Wang, B. Gao, X. Wang, W. Li and Y. Feng, Biomater. Sci., 2022, 10, 1883–1903 RSC.
- R. Chandrawati, Exp. Biol. Med., 2016, 241, 972–979 CrossRef CAS PubMed.
- L. S. Teixeira, J. Feijen, C. A. van Blitterswijk, P. J. Dijkstra and M. Karperien, Biomaterials, 2012, 33, 1281–1290 CrossRef PubMed.
- F. Lee, J. E. Chung and M. Kurisawa, Soft Matter, 2008, 4, 880–887 RSC.
- Y. Liu, R. Weng, W. Wang, X. Wei, J. Li, X. Chen, Y. Liu, F. Lu and Y. Li, Int. J. Biol. Macromol., 2020, 162, 405–413 CrossRef CAS PubMed.
- J. Zhao, L. Wang, H. Zhang, B. Liao and Y. Li, Pharmaceutics, 2022, 14, 2028 CrossRef CAS PubMed.
- N. Olov, S. Bagheri-Khoulenjani and H. Mirzadeh, Prog. Biomater., 2022, 11, 113–135 CrossRef PubMed.
- G. Li, S. Liu, Y. Chen, J. Zhao, H. Xu, J. Weng, F. Yu, A. Xiong, A. Udduttula, D. Wang, P. Liu, Y. Chen and H. Zeng, Nat. Commun., 2023, 14, 3159 CrossRef CAS PubMed.
- M. Yao, J. Zhang, F. Gao, Y. Chen, S. Ma, K. Zhang, H. Liu and F. Guan, ACS Omega, 2019, 4, 8334–8340 CrossRef CAS PubMed.
- M. Mohammadi, M. Karimi, B. Malaekeh-Nikouei, M. Torkashvand and M. Alibolandi, Int. J. Pharm., 2022, 616, 121534 CrossRef CAS PubMed.
- Y. Chao, L. Xu, C. Liang, L. Feng, J. Xu, Z. Dong, L. Tian, X. Yi, K. Yang and Z. Liu, Nat. Biomed. Eng., 2018, 2, 611–621 CrossRef CAS PubMed.
- Y. Chao, C. Liang, H. Tao, Y. Du, D. Wu, Z. Dong, Q. Jin, G. Chen, J. Xu, Z. Xiao, Q. Chen, C. Wang, J. Chen and Z. Liu, Sci. Adv., 2020, 6, eaaz4204 CrossRef CAS PubMed.
- J. Yan, Y. Miao, H. Tan, T. Zhou, Z. Ling, Y. Chen, X. Xing and X. Hu, Mater. Sci. Eng., C, 2016, 63, 274–284 CrossRef CAS PubMed.
- Z. Liu, J. Liu, X. Cui, X. Wang, L. Zhang and P. Tang, Front. Chem., 2020, 8, 124 CrossRef CAS PubMed.
- X. Yan, T. Sun, Y. Song, W. Peng, Y. Xu, G. Luo, M. Li, S. Chen, W. W. Fang, L. Dong, S. Xuan, T. He, B. Cao and Y. Lu, Nano Lett., 2022, 22, 2251–2260 CrossRef CAS PubMed.
- C. Guo and L. J. Kaufman, Biomaterials, 2007, 28, 1105–1114 CrossRef CAS PubMed.
- H. M. Yun, E. S. Lee, M. J. Kim, J. J. Kim, J. H. Lee, H. H. Lee, K. R. Park, J. K. Yi, H. W. Kim and E. C. Kim, PLoS One, 2015, 10, e0138614 CrossRef PubMed.
- M. Arjmand, A. Ardeshirylajimi, H. Maghsoudi and E. Azadian, J. Cell. Physiol., 2018, 233, 1061–1070 CrossRef CAS PubMed.
- N. Ganesh, A. Ashokan, R. Rajeshkannan, K. Chennazhi, M. Koyakutty and S. V. Nair, Tissue Eng., Part A, 2014, 20, 2783–2794 CrossRef CAS PubMed.
- E. Hasanzadeh, A. Seifalian, A. Mellati, J. Saremi, S. Asadpour, S. E. Enderami, H. Nekounam and N. Mahmoodi, Mater. Today Bio, 2023, 20, 100614 CrossRef CAS PubMed.
- S. Sathaye, A. Mbi, C. Sonmez, Y. Chen, D. L. Blair, J. P. Schneider and D. J. Pochan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 34–68 CAS.
- Z. Zhang, C. He and X. Chen, Mater. Chem. Front., 2018, 2, 1765–1778 RSC.
- Z. Fang, Y. Lv, H. Zhang, Y. He, H. Gao, C. Chen, D. Wang, P. Chen, S. Tang, J. Li, Z. Qiu, X. Shi, L. Chen, J. Yang and X. Chen, Acta Biomater., 2023, 159, 111–127 CrossRef CAS PubMed.
- J. Lou, F. Liu, C. D. Lindsay, O. Chaudhuri, S. C. Heilshorn and Y. Xia, Adv. Mater., 2018, 30, 1705215 CrossRef PubMed.
- P. K. Sharma, S. Taneja and Y. Singh, ACS Appl. Mater. Interfaces, 2018, 10, 30936–30945 CrossRef CAS PubMed.
- X. Jiang, X. Yang, B. Yang, L. Zhang and A. Lu, Carbohydr. Polym., 2021, 273, 118547 CrossRef CAS PubMed.
- G. N. Grover, R. L. Braden and K. L. Christman, Adv. Mater., 2013, 25, 2937–2942 CrossRef CAS PubMed.
- G. N. Grover and H. D. Maynard, Curr. Opin. Chem. Biol., 2010, 14, 818–827 CrossRef CAS PubMed.
- Y. Zeng, T. N. Ramya, A. Dirksen, P. E. Dawson and J. C. Paulson, Nat. Methods, 2009, 6, 207–209 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- S. Wang, G. N. Nawale, O. P. Oommen, J. Hilborn and O. P. Varghese, Polym. Chem., 2019, 10, 4322–4327 RSC.
- D. Larsen, A. M. Kietrys, S. A. Clark, H. S. Park, A. Ekebergh and E. T. Kool, Chem. Sci., 2018, 9, 5252–5259 RSC.
- M. Wendeler, L. Grinberg, X. Wang, P. E. Dawson and M. Baca, Bioconjugate Chem., 2014, 25, 93–101 CrossRef CAS PubMed.
- A. E. G. Baker, H. Cui, B. G. Ballios, S. Ing, P. Yan, J. Wolfer, T. Wright, M. Dang, N. Y. Gan, M. J. Cooke, A. Ortin-Martinez, V. A. Wallace, D. van der Kooy, R. Devenyi and M. S. Shoichet, Biomaterials, 2021, 271, 120750 CrossRef CAS PubMed.
- L. Shi, F. Wang, W. Zhu, Z. Xu, S. Fuchs, J. Hilborn, L. Zhu, Q. Ma, Y. Wang and X. Weng, Adv. Funct. Mater., 2017, 27, 1700591 CrossRef.
- M. Chen, X. Ren, L. Dong, X. Li and H. Cheng, Int. J. Biol. Macromol., 2021, 182, 1259–1267 CrossRef CAS PubMed.
- A. Tang, Y. Li, Y. Yao, X. Yang, Z. Cao, H. Nie and G. Yang, Biomater. Sci., 2021, 9, 4169–4177 RSC.
- A. Perez-San Vicente, M. Peroglio, M. Ernst, P. Casuso, I. Loinaz, H. J. Grande, M. Alini, D. Eglin and D. Dupin, Biomacromolecules, 2017, 18, 2360–2370 CrossRef CAS PubMed.
- S. M. Morozova, Gels, 2023, 9, 102 CrossRef CAS PubMed.
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137, 48668 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- X. Bai, S. Lu, Z. Cao, B. Ni, X. Wang, P. Ning, D. Ma, H. Wei and M. Liu, Carbohydr. Polym., 2017, 166, 123–130 CrossRef CAS PubMed.
- T. T. Vu, S. Yadav, O. S. Reddy, S. H. Jo, S. B. Joo, B. K. Kim, E. J. Park, S. H. Park and K. T. Lim, Pharmaceuticals, 2023, 16, 841 CrossRef CAS PubMed.
- T. T. Vu, M. Gulfam, S. H. Jo, S. H. Park and K. T. Lim, Carbohydr. Polym., 2022, 278, 118964 CrossRef CAS PubMed.
- Z. Wei, J. H. Yang, X. J. Du, F. Xu, M. Zrinyi, Y. Osada, F. Li and Y. M. Chen, Macromol. Rapid Commun., 2013, 34, 1464–1470 CrossRef CAS PubMed.
- M. Vauthier, L. Jierry, J. C. Oliveira, L. Hassouna, V. Roucoules and F. Bally-Le Gall, Adv. Funct. Mater., 2019, 29, 1806765 CrossRef.
- B. Masci, S. Pasquale and P. Thuéry, Org. Lett., 2008, 10, 4835–4838 CrossRef CAS PubMed.
- R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994–8002 CrossRef CAS PubMed.
- J. H. Lee, Biomater. Res., 2018, 22, 27 CrossRef PubMed.
- K. Goodarzi and S. S. Rao, J. Mater. Chem. B, 2021, 9, 6103–6115 RSC.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- S. Khunmanee, Y. Jeong and H. Park, J. Tissue Eng., 2017, 8, 2041731417726464 Search PubMed.
- C. F. Nising and S. Bräse, Chem. Soc. Rev., 2008, 37, 1218–1228 RSC.
- S. Summonte, G. F. Racaniello, A. Lopedota, N. Denora and A. Bernkop-Schnurch, J. Controlled Release, 2021, 330, 470–482 CrossRef CAS PubMed.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, e1800650 CrossRef PubMed.
- J. Pupkaite, J. Rosenquist, J. Hilborn and A. Samanta, Biomacromolecules, 2019, 20, 3475–3484 CrossRef CAS PubMed.
- B. Fan, D. Torres Garcia, M. Salehi, M. J. Webber, S. I. van Kasteren and R. Eelkema, ACS Chem. Biol., 2023, 18, 652–659 CrossRef CAS PubMed.
- J. Guo, H. Yao, X. Li, L. Chang, Z. Wang, W. Zhu, Y. Su, L. Qin and J. Xu, Bioact. Mater., 2023, 21, 175–193 Search PubMed.
- Y. Wang, J. Zhan, J. Huang, X. Wang, Z. Chen, Z. Yang and J. Li, Interdiscip. Med., 2023, 1, e20220005 CrossRef.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- X. Ma, R. Xing, C. Yuan, K. Ogino and X. Yan, View, 2020, 1, 20200020 CrossRef.
- Z. Z. Khaing, A. Ehsanipour, C. P. Hofstetter and S. K. Seidlits, Cells Tissues Organs, 2016, 202, 67–84 CrossRef CAS PubMed.
- C. Ligorio, J. A. Hoyland and A. Saiani, Gels, 2022, 8, 211 CrossRef CAS PubMed.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- Q. Feng, K. Wei, S. Lin, Z. Xu, Y. Sun, P. Shi, G. Li and L. Bian, Biomaterials, 2016, 101, 217–228 CrossRef CAS PubMed.
- K. L. Kiick, Soft Matter, 2008, 4, 29–37 RSC.
- T. Miyata, N. Asami and T. Uragami, Macromolecules, 1999, 32, 2082–2084 CrossRef CAS.
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797–801 CrossRef CAS PubMed.
- J.-A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973–1986 CrossRef CAS.
- A. K. Salem, F. R. A. J. Rose, R. O. C. Oreffo, X. Yang, M. C. Davies, J. R. Mitchell, C. J. Roberts, S. Stolnik-Trenkic, S. J. B. Tendler, P. M. Williams and K. M. Shakesheff, Adv. Mater., 2003, 15, 210–213 CrossRef CAS.
- C. Liu, Y. Liao, L. Liu, L. Xie, J. Liu, Y. Zhang and Y. Li, Front. Bioeng. Biotechnol., 2023, 11, 1121887 CrossRef PubMed.
- Q. Wang, Y. Qu, Z. Zhang, H. Huang, Y. Xu, F. Shen, L. Wang and L. Sun, Gels, 2022, 8, 400 CrossRef CAS PubMed.
- S. Han, Y. Park, H. Kim, H. Nam, O. Ko and J. B. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 55554–55563 CrossRef CAS PubMed.
- A. Thakur, M. K. Jaiswal, C. W. Peak, J. K. Carrow, J. Gentry, A. Dolatshahi-Pirouz and A. K. Gaharwar, Nanoscale, 2016, 8, 12362–12372 RSC.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- W. Cheng, J. Zhang, J. Liu and Z. Yu, View, 2020, 1, 20200060 CrossRef.
- X. Xu, Q. Zhang, M. Li, S. Lin, S. Liang, L. Cai, H. Zhu, R. Su and C. Yang, View, 2023, 4, 20220034 CrossRef CAS.
- Y. Xu, G. Yu, R. Nie and Z. Wu, View, 2022, 3, 20200183 CrossRef.
- Y. Wang, S. Wang, L. Li, Y. Zou, B. Liu and X. Fang, View, 2023, 4, 20220048 CrossRef.
- Z. Chen, Z. Lv, Z. Zhang, D. A. Weitz, H. Zhang, Y. Zhang and W. Cui, Exploration, 2021, 1, 20210036 CrossRef PubMed.
- D. Dendukuri, K. Tsoi, T. A. Hatton and P. S. Doyle, Langmuir, 2005, 21, 2113–2116 CrossRef CAS PubMed.
- V. G. Muir, T. H. Qazi, J. Shan, J. Groll and J. A. Burdick, ACS Biomater. Sci. Eng., 2021, 7, 4269–4281 CrossRef CAS PubMed.
- M. Samandari, F. Alipanah, S. Haghjooy Javanmard and A. Sanati-Nezhad, Sens. Actuators, B, 2019, 291, 418–425 CrossRef CAS.
- Q.-W. Cai, X.-J. Ju, C. Chen, Y. Faraj, Z.-H. Jia, J.-Q. Hu, R. Xie, W. Wang, Z. Liu and L.-Y. Chu, Chem. Eng. J., 2019, 370, 925–937 CrossRef CAS.
- Z. Yuan, X. Yuan, Y. Zhao, Q. Cai, Y. Wang, R. Luo, S. Yu, Y. Wang, J. Han, L. Ge, J. Huang and C. Xiong, Small, 2021, 17, e2006596 CrossRef PubMed.
- A. Sinclair, M. B. O'Kelly, T. Bai, H. C. Hung, P. Jain and S. Jiang, Adv. Mater., 2018, 30, e1803087 CrossRef PubMed.
- B. Kessel, M. Lee, A. Bonato, Y. Tinguely, E. Tosoratti and M. Zenobi-Wong, Adv. Sci., 2020, 7, 2001419 CrossRef CAS PubMed.
- M. Xie, Q. Gao, H. Zhao, J. Nie, Z. Fu, H. Wang, L. Chen, L. Shao, J. Fu, Z. Chen and Y. He, Small, 2019, 15, e1804216 CrossRef PubMed.
- Z. Li, Y. Zhou, T. Li, J. Zhang and H. Tian, View, 2022, 3, 20200112 CrossRef CAS.
- Z. Piao, J. K. Park and B. Jeong, ACS Appl. Mater. Interfaces, 2023, 15, 17688–17695 CrossRef CAS PubMed.
- M. H. Park, M. K. Joo, B. G. Choi and B. Jeong, Acc. Chem. Res., 2012, 45, 424–433 CrossRef CAS PubMed.
- N. Ferreira, L. Ferreira, V. Cardoso, F. Boni, A. Souza and M. Gremião, Eur. Polym. J., 2018, 99, 117–133 CrossRef CAS.
- L. Klouda, Eur. J. Pharm. Biopharm., 2015, 97, 338–349 CrossRef CAS.
- M. Zakerikhoob, S. Abbasi, G. Yousefi, M. Mokhtari and M. S. Noorbakhsh, Carbohydr. Polym., 2021, 271, 118434 CrossRef CAS PubMed.
- S. Samimi Gharaie, S. M. H. Dabiri and M. Akbari, Polymers, 2018, 10, 1317 CrossRef PubMed.
- Q. Tang, D. Zhao, Q. Zhou, H. Yang, K. Peng and X. Zhang, Macromol. Rapid Commun., 2018, 39, e1800109 CrossRef.
- J. Qu, X. Zhao, P. X. Ma and B. Guo, Acta Biomater., 2017, 58, 168–180 CrossRef CAS PubMed.
- Y. Yang, W. Zeng, P. Huang, X. Zeng and L. Mei, View, 2021, 2, 20200042 CrossRef CAS.
- Y. Zhou, Z. Zhai, Y. Yao, J. C. Stant, S. L. Landrum, M. J. Bortner, C. E. Frazier and K. J. Edgar, Carbohydr. Polym., 2023, 300, 120213 CrossRef CAS PubMed.
- W. Li, C. Tao, J. Wang, Y. Le and J. Zhang, RSC Adv., 2019, 9, 31264–31273 RSC.
- Y. Yang, W. Zheng, W. Tan, X. Wu, Z. Dai, Z. Li, Z. Yan, Y. Ji, Y. Wang, W. Su, S. Zhong, Y. Li, Y. Sun, S. Li and W. Huang, Acta Biomater., 2023, 157, 321–336 CrossRef CAS.
- H. K. Noddeland, M. Lind, K. Petersson, F. Caruso, M. Malmsten and A. Heinz, Biomacromolecules, 2023, 24, 3203–3214 CrossRef CAS PubMed.
- A. Kumar, Kanika, V. Kumar, A. Ahmad, R. K. Mishra, A. Nadeem, N. Siddiqui, M. M. Ansari, S. S. Raza, K. K. Kondepudi and R. Khan, ACS Biomater. Sci. Eng., 2023, 9, 4781–4793 CrossRef CAS.
- J. Zhang, Y. Guo, G. Pan, P. Wang, Y. Li, X. Zhu and C. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 21441–21449 CrossRef CAS.
- Z. He, Q. Xu, B. Newland, R. Foley, I. Lara-Saez, J. F. Curtin and W. Wang, J. Mater. Chem. B, 2021, 9, 6326–6346 RSC.
- H. J. Lee and B. Jeong, Small, 2020, 16, e1903045 CrossRef PubMed.
- Z. Gan, Z. Xiao, Z. Zhang, Y. Li, C. Liu, X. Chen, Y. Liu, D. Wu, C. Liu, X. Shuai and Y. Cao, Bioact. Mater., 2023, 25, 347–359 CAS.
- Z. Li, D. Zhu, Q. Hui, J. Bi, B. Yu, Z. Huang, S. Hu, Z. Wang, T. Caranasos, J. Rossi, X. Li, K. Cheng and X. Wang, Adv. Funct. Mater., 2021, 31, 2004377 CrossRef CAS.
- S. Sun, W. Gu, H. Wu, Q. Zhao, S. Qian, H. Xiao, K. Yang, J. Liu, Y. Jin, C. Hu, Y. Gao, H. Xu, H. Liu, J. Ji and Y. Chen, Adv. Funct. Mater., 2022, 32, 2205038 CrossRef CAS.
- Z. Zheng, C. Lei, H. Liu, M. Jiang, Z. Zhou, Y. Zhao, C. Y. Yu and H. Wei, Adv. Healthc. Mater., 2022, 11, e2200990 CrossRef PubMed.
- S. X. Chen, J. Zhang, F. Xue, W. Liu, Y. Kuang, B. Gu, S. Song and H. Chen, Bioact. Mater., 2023, 21, 86–96 CAS.
- S. H. Jeong, S. Cheong, T. Y. Kim, H. Choi and S. K. Hahn, ACS Appl. Mater. Interfaces, 2023, 15, 16471–16481 CrossRef CAS.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- Y. Dong, W. Wang, O. Veiseh, E. A. Appel, K. Xue, M. J. Webber, B. C. Tang, X. W. Yang, G. C. Weir, R. Langer and D. G. Anderson, Langmuir, 2016, 32, 8743–8747 CrossRef CAS PubMed.
- K. Podual, F. Doyle III and N. Peppas, Polymer, 2000, 41, 3975–3983 CrossRef CAS.
- K. Podual, F. J. Doyle III and N. A. Peppas, J. Controlled Release, 2000, 67, 9–17 CrossRef CAS PubMed.
- J. Yang, W. Zeng, P. Xu, X. Fu, X. Yu, L. Chen, F. Leng, C. Yu and Z. Yang, Acta Biomater., 2022, 140, 206–218 CrossRef CAS.
- X. Zhou, B. Zhao, L. Wang, L. Yang, H. Chen, W. Chen, H. Qiao and H. Qian, J. Controlled Release, 2023, 359, 147–160 CrossRef CAS PubMed.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, e1807333 CrossRef PubMed.
- L. Boni, G. David, A. Mangano, G. Dionigi, S. Rausei, S. Spampatti, E. Cassinotti and A. Fingerhut, Surg. Endosc., 2015, 29, 2046–2055 CrossRef PubMed.
- E. P. Porcu, A. Salis, E. Gavini, G. Rassu, M. Maestri and P. Giunchedi, Biotechnol. Adv., 2016, 34, 768–789 CrossRef CAS PubMed.
- H. Wang, X. Li, B. W. Tse, H. Yang, C. A. Thorling, Y. Liu, M. Touraud, J. B. Chouane, X. Liu, M. S. Roberts and X. Liang, Theranostics, 2018, 8, 1227–1242 CrossRef CAS PubMed.
- M. Yang, S. Y. Lee, S. Kim, J. S. Koo, J. H. Seo, D. I. Jeong, C. Hwang, J. Lee and H. J. Cho, J. Controlled Release, 2020, 324, 750–764 CrossRef CAS PubMed.
- X. Zhu, H. Zhang, H. Huang, Y. Zhang, L. Hou and Z. Zhang, Nanotechnology, 2015, 26, 365103 CrossRef.
- H. Zhang, X. Zhu, Y. Ji, X. Jiao, Q. Chen, L. Hou, H. Zhang and Z. Zhang, J. Mater. Chem. B, 2015, 3, 6310–6326 RSC.
- G. Yang, X. Wan, Z. Gu, X. Zeng and J. Tang, J. Mater. Chem. B, 2018, 6, 1622–1632 RSC.
- F. Andrade, M. M. Roca-Melendres, E. F. Durán-Lara, D. Rafael and S. Schwartz Jr., Cancers, 2021, 13, 1164 CrossRef CAS PubMed.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS.
- Y. Cheng, C. He, K. Ren, Y. Rong, C. Xiao, J. Ding, X. Zhuang and X. Chen, Macromol. Rapid Commun., 2018, 39, e1800272 CrossRef PubMed.
- N. Huebsch, C. J. Kearney, X. Zhao, J. Kim, C. A. Cezar, Z. Suo and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9762–9767 CrossRef CAS PubMed.
- Z. Meng, Y. Zhang, J. She, X. Zhou, J. Xu, X. Han, C. Wang, M. Zhu and Z. Liu, Nano Lett., 2021, 21, 1228–1237 CrossRef CAS PubMed.
- W. Sun, H. Jiang, X. Wu, Z. Xu, C. Yao, J. Wang, M. Qin, Q. Jiang, W. Wang, D. Shi and Y. Cao, Nano Res., 2018, 12, 115–119 CrossRef.
- C. H. Wu, M. K. Sun, Y. Kung, Y. C. Wang, S. L. Chen, H. H. Shen, W. S. Chen and T. H. Young, Ultrason. Sonochem., 2020, 62, 104875 CrossRef CAS PubMed.
- B. Y. Shin, B. G. Cha, J. H. Jeong and J. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 31372–31380 CrossRef CAS PubMed.
- H. Kim, H. Park, J. W. Lee and K. Y. Lee, Carbohydr. Polym., 2016, 151, 467–473 CrossRef CAS PubMed.
- C. Kim, H. Kim, H. Park and K. Y. Lee, Carbohydr. Polym., 2019, 223, 115045 CrossRef CAS PubMed.
- X. Chen, H. Wang, J. Shi, Z. Chen, Y. Wang, S. Gu, Y. Fu, J. Huang, J. Ding and L. Yu, Biomaterials, 2023, 298, 122139 CrossRef CAS PubMed.
- A. Omidinia-Anarkoli, S. Boesveld, U. Tuvshindorj, J. C. Rose, T. Haraszti and L. De Laporte, Small, 2017, 13, 1702207 CrossRef.
- D. Zhang, P. Sun, P. Li, A. Xue, X. Zhang, H. Zhang and X. Jin, Biomaterials, 2013, 34, 10258–10266 CrossRef CAS.
- Y. Liu, J. Liu, S. Chen, T. Lei, Y. Kim, S. Niu, H. Wang, X. Wang, A. M. Foudeh, J. B. Tok and Z. Bao, Nat. Biomed. Eng., 2019, 3, 58–68 CrossRef CAS PubMed.
- L. Zhang, T. Li, Y. Yu, K. Shi, Z. Bei, Y. Qian and Z. Qian, Bioact. Mater., 2023, 20, 339–354 CAS.
- H. Song, Y. Wang, Q. Fei, D. H. Nguyen, C. Zhang and T. Liu, Exploration, 2022, 2, 20220006 CrossRef PubMed.
- J. Ge, E. Neofytou, T. J. Cahill 3rd, R. E. Beygui and R. N. Zare, ACS Nano, 2012, 6, 227–233 CrossRef CAS PubMed.
- J. Qu, X. Zhao, P. X. Ma and B. Guo, Acta Biomater., 2018, 72, 55–69 CrossRef CAS PubMed.
- A. Servant, L. Methven, R. P. Williams and K. Kostarelos, Adv. Healthc. Mater., 2013, 2, 806–811 CrossRef CAS PubMed.
- N. O. Weiss, H. Zhou, L. Liao, Y. Liu, S. Jiang, Y. Huang and X. Duan, Adv. Mater., 2012, 24, 5782–5825 CrossRef CAS PubMed.
- A. Servant, V. Leon, D. Jasim, L. Methven, P. Limousin, E. V. Fernandez-Pacheco, M. Prato and K. Kostarelos, Adv. Healthc. Mater., 2014, 3, 1334–1343 CrossRef CAS PubMed.
- M. Tibaut, D. Mekis and D. Petrovic, Cardiovasc. Hematol. Agents Med. Chem., 2017, 14, 150–159 CrossRef PubMed.
- M. N. Sack, F. Y. Fyhrquist, O. J. Saijonmaa, V. Fuster and J. C. Kovacic, J. Am. Coll. Cardiol., 2017, 70, 196–211 CrossRef CAS PubMed.
- Y. Li, X. Chen, R. Jin, L. Chen, M. Dang, H. Cao, Y. Dong, B. Cai, G. Bai, J. J. Gooding, S. Liu, D. Zou, Z. Zhang and C. Yang, Sci. Adv., 2021, 7, eabd6740 CrossRef CAS PubMed.
- K. Wang, K. Zhu, Z. Zhu, F. Shao, R. Qian, C. Wang, H. Dong, Y. Li, Z. Gao and J. Zhao, J. Nanobiotechnol., 2023, 21, 227 CrossRef CAS PubMed.
- S. Aznavoorian, B. A. Moore, L. D. Alexander-Lister, S. L. Hallit, L. J. Windsor and J. A. Engler, Cancer Res., 2001, 61, 6264–6275 CAS.
- S. Tsuchida and K. Sato, Crit. Rev. Biochem. Mol. Biol., 1992, 27, 337–384 CrossRef CAS PubMed.
- K. D. Tew, Cancer Res., 1994, 54, 4313–4320 CAS.
- E. Ferruzzi, R. Franceschini, G. Cazzolato, C. Geroni, C. Fowst, U. Pastorino, N. Tradati, J. Tursi, R. Dittadi and M. Gion, Eur. J. Cancer, 2003, 39, 1019–1029 CrossRef CAS PubMed.
- X. Xu, Z. Zeng, Z. Huang, Y. Sun, Y. Huang, J. Chen, J. Ye, H. Yang, C. Yang and C. Zhao, Carbohydr. Polym., 2020, 229, 115394 CrossRef CAS PubMed.
- Y. Feng, Z. Zhang, W. Tang and Y. Dai, Exploration, 2023, 20220173 CrossRef PubMed.
- J. Wu, Z. Qin, X. Jiang, D. Fang, Z. Lu, L. Zheng and J. Zhao, npj Regener. Med., 2022, 7, 66 CrossRef CAS PubMed.
- S. Jahanbekam, N. Mozafari, A. Bagheri-Alamooti, S. Mohammadi-Samani, S. Daneshamouz, R. Heidari, N. Azarpira, H. Ashrafi and A. Azadi, Int. J. Biol. Macromol., 2023, 240, 124449 CrossRef CAS PubMed.
- Y. Wang, Y. Wu, B. Zhang, C. Zheng, C. Hu, C. Guo, Q. Kong and Y. Wang, Biomaterials, 2023, 298, 122132 CrossRef CAS PubMed.
- H. Xing, Z. Zhang, Q. Mao, C. Wang, Y. Zhou, X. Zhou, L. Ying, H. Xu, S. Hu and N. Zhang, J. Nanobiotechnol., 2021, 19, 264 CrossRef CAS PubMed.
- X. Xu, D. Wang, C. Zheng, B. Gao, J. Fan, P. Cheng, B. Liu, L. Yang and Z. Luo, Theranostics, 2019, 9, 2252–2267 CrossRef CAS PubMed.
- G. Feng, Z. Zha, Y. Huang, J. Li, Y. Wang, W. Ke, H. Chen, L. Liu, Y. Song and Z. Ge, Adv. Healthc. Mater., 2018, 7, e1800623 CrossRef PubMed.
- H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS PubMed.
- Q. Zheng, H. Shen, Z. Tong, L. Cheng, Y. Xu, Z. Feng, S. Liao, X. Hu, Z. Pan, Z. Mao and Y. Wang, Theranostics, 2021, 11, 147–163 CrossRef CAS PubMed.
- R. L. Sala, M. Y. Kwon, M. Kim, S. E. Gullbrand, E. A. Henning, R. L. Mauck, E. R. Camargo and J. A. Burdick, Tissue Eng., Part A, 2017, 23, 935–945 CrossRef CAS PubMed.
- L. Lei, Z. Liu, P. Yuan, R. Jin, X. Wang, T. Jiang and X. Chen, J. Mater. Chem. B, 2019, 7, 2722–2735 RSC.
- T. M. D. Le, H. T. T. Duong, T. Thambi, V. H. Giang Phan, J. H. Jeong and D. S. Lee, Biomacromolecules, 2018, 19, 3536–3548 CrossRef CAS PubMed.
- N. Fransén-Pettersson, N. Duarte, J. Nilsson, M. Lundholm, S. Mayans, Å. Larefalk, T. D. Hannibal, L. Hansen, A. Schmidt-Christensen, F. Ivars, S. Cardell, R. Palmqvist, B. Rozell and D. Holmberg, PLoS One, 2016, 11, e0159850 CrossRef PubMed.
- S. Wang, J. Chi, Z. Jiang, H. Hu, C. Yang, W. Liu and B. Han, Carbohydr. Polym., 2021, 256, 117519 CrossRef CAS PubMed.
- L. L. Wang, C. B. Highley, Y. C. Yeh, J. H. Galarraga, S. Uman and J. A. Burdick, J. Biomed. Mater. Res., Part A, 2018, 106, 865–875 CrossRef CAS PubMed.
- E. Jirigala, B. Yao, Z. Li, Y. J. Zhang, C. Zhang, L. T. Liang, F. L. Zhang, X. Y. Yuan, X. L. Duan, W. Song, M. D. Zhang, Y. Kong, X. B. Fu and S. Huang, Mil. Med. Res., 2023, 10, 17 CAS.
Footnote |
| † These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |