
Creating an electron-rich region on ultrafine Bi2O3 nanoparticles to boost the electrochemical carbon dioxide reduction to formate†
Jiayu
Zhan
a,
Lu-Hua
Zhang
 *a,
Xueli
Wang
a,
Yuqi
Hu
a,
Yi
Jiang
*b and
Fengshou
Yu
*a
*a,
Xueli
Wang
a,
Yuqi
Hu
a,
Yi
Jiang
*b and
Fengshou
Yu
*a
aNational-Local Joint Engineering Laboratory for Energy Conservation in Chemical Process Integration and Resources Utilization, School of Chemical Engineering and Technology, Hebei University of Technology, Tianjin 300130, P. R. China. E-mail: luhuazhang@hebut.edu.cn; yfsh@hebut.edu.cn
bCollege of Chemistry, Liaoning University, Shenyang 110036, Liaoning, China. E-mail: jiangyi@lnu.edu.cn
First published on 22nd November 2022
Abstract
Various metal–support composites have been reported for the electrochemical CO2 reduction reaction (ECRR), while the effect of synergy between the size effect and interface regulation on ECRR performance is still rarely explored. Herein, we report ultrafine Bi2O3 nanoparticles (∼1.9 nm) decorated on a nitrogen-vacancy-rich graphitic carbon nitride (Bi2O3/NV-C3N4) electrocatalyst for highly efficient ECRR to formate. The Bi2O3/NV-C3N4 electrocatalyst can achieve 95% formate selectivity at −0.9 V vs. RHE and 24 h long-term stability. Moreover, substantial formate selectivity (>80%) was gained over a wide potential range of more than 700 mV. The excellent performance was attributed to the electron transfer from NVs to Bi2O3, forming an electron-rich region on Bi2O3 and accelerating the electron transfer from Bi2O3 to the adsorbed intermediates. This work provides a strategy for enhancing the catalytic activity of bismuth oxides to achieve a high efficiency for CO2 reduction to generate liquid products.
1 Introduction
Electrochemical reduction of CO2 into high value-added products driven by renewable electricity is an effective way to facilitate CO2 recycling.1 Formate is an important liquid product that is easy to separate during preparation and can be used as a crucial chemical intermediate for industrial applications.2 Over the past few years, p-block metal Bi-based electrocatalysts have exhibited preference for formate production, but high Faraday efficiency (FE) can only be obtained at a certain potential or in a confined potential field. Once the applied potentials are more negative than the optimum value (for achieving the largest FE), FE for formate (FEformate) rapidly drops due to the significant enhancement of the competing hydrogen evolution reaction (HER).To maintain a high FEformate over a broad potential range, great efforts have been devoted to modify the structure of Bi-based electrocatalysts, considering that nanostructuring is an effective strategy to improve the performance of metal-based materials. A series of metallic bismuth (Bi0) materials with delicate nanostructures, such as Bi dendrites, Bi nanosheets, and Bi nanoflakes have been prepared. While these metallic Bi-based composites inevitably contain oxygen species, their effects on the electrochemical CO2 reduction reaction (ECRR) are not well understood.
Since it is hard to explain the role of oxygen species on the enhancement activity of CO2, Xia3 and co-workers strengthened the oxygen species in Bi-based materials in turn and found that the Bi–O structure in Bi2O3 is favorable to improve the efficiency of CO2 reduction to formate. Meanwhile, various bismuth oxide composites have also been reported. However, those catalysts usually have a large particle size, which may reach up to several micrometers. Theoretical and experimental investigations have revealed that ultrafine nanoparticles (NPs) containing numerous coordinately unsaturated sites favor the stabilization of substrates and adsorption of intermediates on the catalyst surface and therefore show great application potential in the ECRR.4 Unfortunately, there have been limited studies about bismuth oxide nanoparticles smaller than 5 nm employed for the ECRR due to the lack of an effective synthesis method. Apart from precisely tuning the size of catalysts, there is another problem in the design of catalysts: ultrafine NPs tend to agglomerate in a non-controlled manner resulting in a rapid decay in the cyclability under electrochemical conditions.
The structure design of supported catalysts offers a good reference for the enhancement of stability of small sized NPs. Graphitic carbon nitride (C3N4) has been attracting much attention and widely explored as a support for a couple of reactions.5 The higher electron density around N atoms can induce strong interactions with metal precursor ions to promote oxide growth on the support forming a high-quality oxide-support interface.6 In addition, the formed interface is proposed to redistribute electrons of the support and catalyst and therefore affects the performance of the ECRR. However, the effect of electronic state modulation of supports on ECRR performance is still rarely explored for C3N4-supported catalysts.
Herein, we report ultrafine Bi2O3 nanoparticles (∼1.9 nm) decorated on a nitrogen-vacancy-rich graphitic carbon nitride (Bi2O3/NV-C3N4) electrocatalyst for highly efficient electrochemical reduction of CO2 to formate. The Bi2O3/NV-C3N4 electrocatalyst can achieve 95% formate selectivity at −0.9 V vs. RHE and 24 h long-term stability. Moreover, substantial formate selectivity (>80%) was gained over a wide potential scope of more than 700 mV. The excellent catalytic performance was attributed to the following reasons: (i) nitrogen vacancies in C3N4 effectively lower the energy barriers for *CO2 formation and (ii) the formed metal-carbon heterojunction structure effectively accelerates electron transfer from NV-C3N4 to Bi2O3 active sites, and therefore facilitates intermediate *COO generation. This work may pave the way for bismuth oxide catalysts to be applied in different renewable energy-conversion devices.
2 Experimental
2.1 Synthesis of NV-C3N4
The synthesis of bulk C3N4 was performed by calcining melamine and urea with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 at 550 °C for 2 h in an air atmosphere. The C3N4 nanosheets were obtained by liquid exfoliation of bulk C3N4 in absolute ethanol for 3 h. NV-C3N4 was prepared by calcining the as-prepared C3N4 nanosheets in a tube furnace under an Ar atmosphere at 650 °C for 4 h.
:3 at 550 °C for 2 h in an air atmosphere. The C3N4 nanosheets were obtained by liquid exfoliation of bulk C3N4 in absolute ethanol for 3 h. NV-C3N4 was prepared by calcining the as-prepared C3N4 nanosheets in a tube furnace under an Ar atmosphere at 650 °C for 4 h.
2.2 Synthesis of Bi2O3/NV-C3N4
Bi2O3/NV-C3N4 was synthesized by the solvothermal method with NV-C3N4 as the substrate.7 Specifically, 680 mg Bi(NO3)3·5H2O was dissolved in a mixture of 24 mL ethanol, 4 mL deionized water and 12 mL ethylene glycol named solution A; 20 mg NV-C3N4 was dissolved in 20 mL ethanol named solution B. Solution A was slowly added to solution B, and the mixture was stirred in a water bath at 25 °C for 15 min and then sonicated for 15 min. The mixed solution was placed in a 100 mL Teflon-lined stainless-steel autoclave and kept at 160 °C for 5 h. After cooling to room temperature, the product was centrifuged and washed four times with deionized water and ethanol, respectively. Finally, the samples were dried in a vacuum drying oven at 50 °C for 10 h. The synthesis method of Bi2O3/C3N4 is the same as that of Bi2O3/NV-C3N4, except that NV-C3N4 is replaced by C3N4 nanosheets. The Bi2O3 nanosheets were synthesized without substrates.2.3 Electrochemical analysis
The as-prepared sample (4 mg) was evenly dispersed in a mixed solution (950 μL) with 475 μL ethanol and 475 μL deionized water. Subsequently, 50 μL Nafion (5 wt%) was added followed by ultrasonic treatment for 0.5 h forming a homogeneous ink. Finally, 50 μL ink was dropped onto carbon paper (1 × 0.5 cm2) and dried at room temperature. The electrochemical measurement was performed in a gas-tight H-type cell containing two chambers separated by a proton exchange membrane (Nafion 117) between the cathodic and anodic compartments, each chamber containing 35 mL of electrolyte and 35 mL space, and tested using a Chenhua workstation CHI 760 and an equipped three-electrode system.2.4 Product analysis
The liquid phase product was detected by ion chromatography (CIC-D120). The standard curve of formate is shown in Fig. S1.† The faradaic efficiency (FE) of formate is obtained using eqn (1):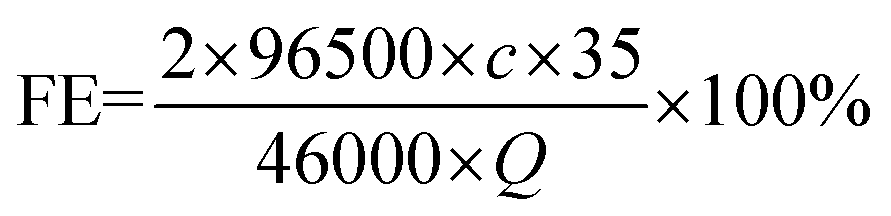 | (1) |
500 is the Faraday constant, c is the concentration of formate and Q is the number of accumulated charges.
2.5 DFT calculations
All density functional theory (DFT) calculations in this work were conducted using the Vienna ab initio simulation package (VASP).8 The ECRR calculations were performed on the catalytic materials based on the projector augmented wave (PAW) method.9 The free energy of each intermediate was calculated with the Perdew–Burke–Ernzerhof (PBE)10 functional of the generalized gradient approximation method (GGA). Convergence conditions: cutoff energy of 400 eV, convergence energy threshold of 10−5 eV, remaining forces of less than 0.01 eV Å−1. The Brillouin zone uses 3 × 1 × 1 Monkhorst–Pack k-points for sampling, and the supercell is 3 × 3 × 1. The model is separated by a 15 Å vacuum layer. All were corrected using van der Waals (vdW) forces.3 Results and discussion
The standard synthetic route of Bi2O3/NV-C3N4 is depicted in Fig. 1a. C3N4 nanosheets were first obtained by calcining a mixture of urea and melamine in air, and then by liquid exfoliation. Nitrogen-vacancy-rich C3N4 (NV-C3N4) was then obtained by annealing the as-prepared C3N4 nanosheets in Ar. Finally, Bi2O3 nanoparticles were loaded onto C3N4 and NV-C3N4 nanosheets by a simple hydrothermal method. The obtained samples were denoted as Bi2O3/C3N4 and Bi2O3/NV-C3N4, respectively. For comparison, Bi2O3 nanosheets were also synthesized without substrates. | ||
| Fig. 1 (a) Schematic diagram of the synthetic process of Bi2O3/NV-C3N4; (b) atomic structure of NV-C3N4; (c and d) TEM images of Bi2O3/NV-C3N4; Statistical analysis based on 100 Bi2O3 nanoparticles indicates that the average size of the ultrafine Bi2O3 nanoparticles is around 1.9 nm with a narrow distribution; (e) XRD patterns of Bi2O3, Bi2O3/C3N4 and Bi2O3/NV-C3N4; (f) Bi 4f spectra of Bi2O3, Bi2O3/C3N4 and Bi2O3/NV-C3N4; (g) N 1s spectra of Bi2O3/C3N4 and Bi2O3/NV-C3N4. | ||
Electron paramagnetic resonance (EPR) and X-ray photoelectron spectroscopy (XPS) measurements were first conducted to disclose the NV in C3N4. In EPR spectra (Fig. S2†), the intensity signal at g = 2.0042 of NV-C3N4 was significantly stronger than that of C3N4, indicating the successful introduction of NVs. The remarkable decrease in the N/C ratio from 1.34 (C3N4) to 1.22 (NV-C3N4), further demonstrated that significant NVs were generated on NV-C3N4 (Fig. S3a and Table S1†). The C/N atomic ratio in C3N4 is 0.743 close to 0.75 (Fig. S3b†), indicating that we have synthesized C3N4 rather than porous carbon.7,11 Deconvolution of the N 1s spectrum shows three high-resolution peaks of C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (398.4 eV), N(C)3 (400.0 eV) and C–N–H (401.5 eV). The quantitative evaluation demonstrates that the percent ratio of
C (398.4 eV), N(C)3 (400.0 eV) and C–N–H (401.5 eV). The quantitative evaluation demonstrates that the percent ratio of 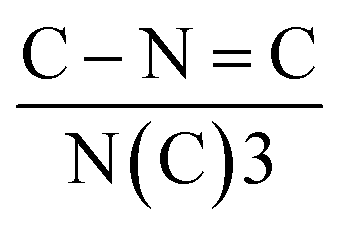 reduces from 2.14 (C3N4) to 2.00 (NV-C3N4), revealing that N–C2 (two-coordinated-N) vacancies are mainly formed on NV-C3N4 rather than N–C3 (three-coordinated-N) vacancies, as illustrated in Fig. 1b. For both C3N4 and NV-C3N4 nanosheets, a dominant (002) X-ray diffraction (XRD) peak appears at approximately 27.5°, corresponding to the interlayer reflection of the graphitic structure (Fig. S4a†). Compared with that of C3N4, the (002) peak of NV-C3N4 is relatively broadened, indicating that a disordered structure was built in the atomic arrangement of NV-C3N4.7,12 In addition, N2 desorption and adsorption isotherms (Fig. S4b†) indicate that NV-C3N4 (202.3 m2 g−1) has a larger specific surface area than C3N4 (77.3 m2 g−1) nanosheets, probably due to the introduction of NVs to generate more wrinkles. High specific surface area is generally beneficial for enhanced electrocatalytic activity, facilitating electrolyte/reactant contact by exposing more sites of geometric activity.
reduces from 2.14 (C3N4) to 2.00 (NV-C3N4), revealing that N–C2 (two-coordinated-N) vacancies are mainly formed on NV-C3N4 rather than N–C3 (three-coordinated-N) vacancies, as illustrated in Fig. 1b. For both C3N4 and NV-C3N4 nanosheets, a dominant (002) X-ray diffraction (XRD) peak appears at approximately 27.5°, corresponding to the interlayer reflection of the graphitic structure (Fig. S4a†). Compared with that of C3N4, the (002) peak of NV-C3N4 is relatively broadened, indicating that a disordered structure was built in the atomic arrangement of NV-C3N4.7,12 In addition, N2 desorption and adsorption isotherms (Fig. S4b†) indicate that NV-C3N4 (202.3 m2 g−1) has a larger specific surface area than C3N4 (77.3 m2 g−1) nanosheets, probably due to the introduction of NVs to generate more wrinkles. High specific surface area is generally beneficial for enhanced electrocatalytic activity, facilitating electrolyte/reactant contact by exposing more sites of geometric activity.
Transmission electron microscopy (TEM) images of Bi2O3/NV-C3N4 disclose that ultrafine metallic NPs with a narrow size distribution and a small average size of ∼1.9 nm are homogeneously dispersed on the porous C3N4 support (Fig. 1c and d). The elemental mapping results showed that Bi, C, N, and O elements were uniformly distributed throughout the catalyst (Fig. S5†). In addition, Bi2O3/C3N4 shares a similar morphology with Bi2O3/NV-C3N4 (Fig. S6†). XRD patterns of Bi2O3/NV-C3N4, Bi2O3/C3N4 and Bi2O3 samples reveal two main diffraction peaks at ∼28° and 43°, which can be indexed to the (201) and (002) planes of the Bi2O3 crystal phase (JCPDS No. 27-0052), respectively (Fig. 1e). These results indicate that high-density ultrafine Bi2O3 nanoparticles were successfully decorated on C3N4 and NV-C3N4 supports and both samples show similar morphology, particle size and chemical structure.
The electronic interactions between Bi2O3 and the C3N4 support were first investigated by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 1f, the gradual shifts of typical Bi 4f5/2 and 4f7/2 peaks to lower energies indicate the gradually richer electron density of Bi2O3 in Bi2O3/C3N4 and Bi2O3/NV-C3N4 samples. The larger shift implies relatively strong electron donating properties of NV-C3N4. For N 1s spectra, the energy peak positions of pyridine N in Bi2O3/C3N4 and Bi2O3/NV-C3N4 shift to higher energies compared with that in C3N4, indicating the gradually depressed electron density of N in the C3N4 support after Bi2O3 decoration (Fig. 1g). We performed UV photoelectron spectroscopy (UPS, Fig. S7†) and the results showed that the work function (Φ) value of Bi2O3/NV-C3N4 is 6.67. This value is lower than that of Bi2O3/C3N4 (6.86), which means that Bi2O3/NV-C3N4 is more efficient than Bi2O3/C3N4 for electron donation. As shown in Fig. S8,† the charge distribution at the valence band maximum (VBM) and conduction band minimum (CBM) was calculated. It is obvious that the charge density on Bi2O3/NV-C3N4 is significantly enhanced by the introduction of N vacancies. Those results mean a spontaneous electron donation from the NV-C3N4 and C3N4 supports to the Bi2O3 NPs at their coupling interface, which can potentially improve the ECRR performance.
To evaluate the ECRR performance of Bi2O3, Bi2O3/C3N4 and Bi2O3/NV-C3N4, electrochemical measurements were conducted using a three-electrode system in a tight H-type cell. The linear cyclic voltammetry (LSV) tests show that the current density of Bi2O3/NV-C3N4 is significantly higher than that of Bi2O3 and Bi2O3/C3N4, indicating that Bi2O3/NV-C3N4 has excellent CO2 reduction activity (Fig. 2a). For instance, Bi2O3/NV-C3N4 displays a current density of 18.8 mA cm−2 at −1.2 V, which was roughly 1.5 and 2.3 times larger than that of Bi2O3/C3N4 and Bi2O3, respectively. In particular, the current densities of Bi2O3/NV-C3N4 electrodes under CO2 start to accelerate at −0.8 V, which is more positive than that of the Bi2O3/C3N4 electrode (−1.0 V), indicating an improvement in the electrocatalytic activities of CO2 reduction by introducing NVs into the substrate. The formate faradaic efficiency (FEformate) analysis (Fig. 2b and S9†) indicates that all supported catalysts present an improved FEformate compared with Bi2O3 throughout the potential range. The FEformate order is Bi2O3/NV-C3N4 > Bi2O3/C3N4 > Bi2O3, which can be attributed to the interfacial effect. Bi2O3/NV-C3N4 shows the highest FEformate of 95% at −1.2 V, which is better than those of Bi2O3/C3N4 (88.2%) and Bi2O3 (81.2%). More importantly, the FEformate of Bi2O3/NV-C3N4 shows high selectivity of more than 80% over a large potential window from −1.0 to −1.7 V. The operation potential window for the high formate formation efficiency of Bi2O3/NV-C3N4 is outstanding in 0.1 M KHCO3 among the reported electrocatalysts (Table S2† and Fig. 2c).13 The FE of H2 is opposite to that of formate (Fig. S10a†), indicating that the hydrogen evolution reaction (HER) is suppressed. The FE of CO is considerably small, below 10% at all potentials (Fig. S10b†). In addition, no other liquid product was detected except for formate (Fig. S11†). The partial current densities for formate formation (jformate) of all the samples were extracted from total current densities based on FEformate (Fig. S9†). Among the three samples, Bi2O3/NV-C3N4 shows the highest current density for formate formation at the measured potential scope. For example, the jformate of Bi2O3/NV-C3N4 reaches −18 mA cm−2 at −1.2 V, which is 1.61 and 2.64 fold higher than that of Bi2O3/C3N4 and Bi2O3, respectively.
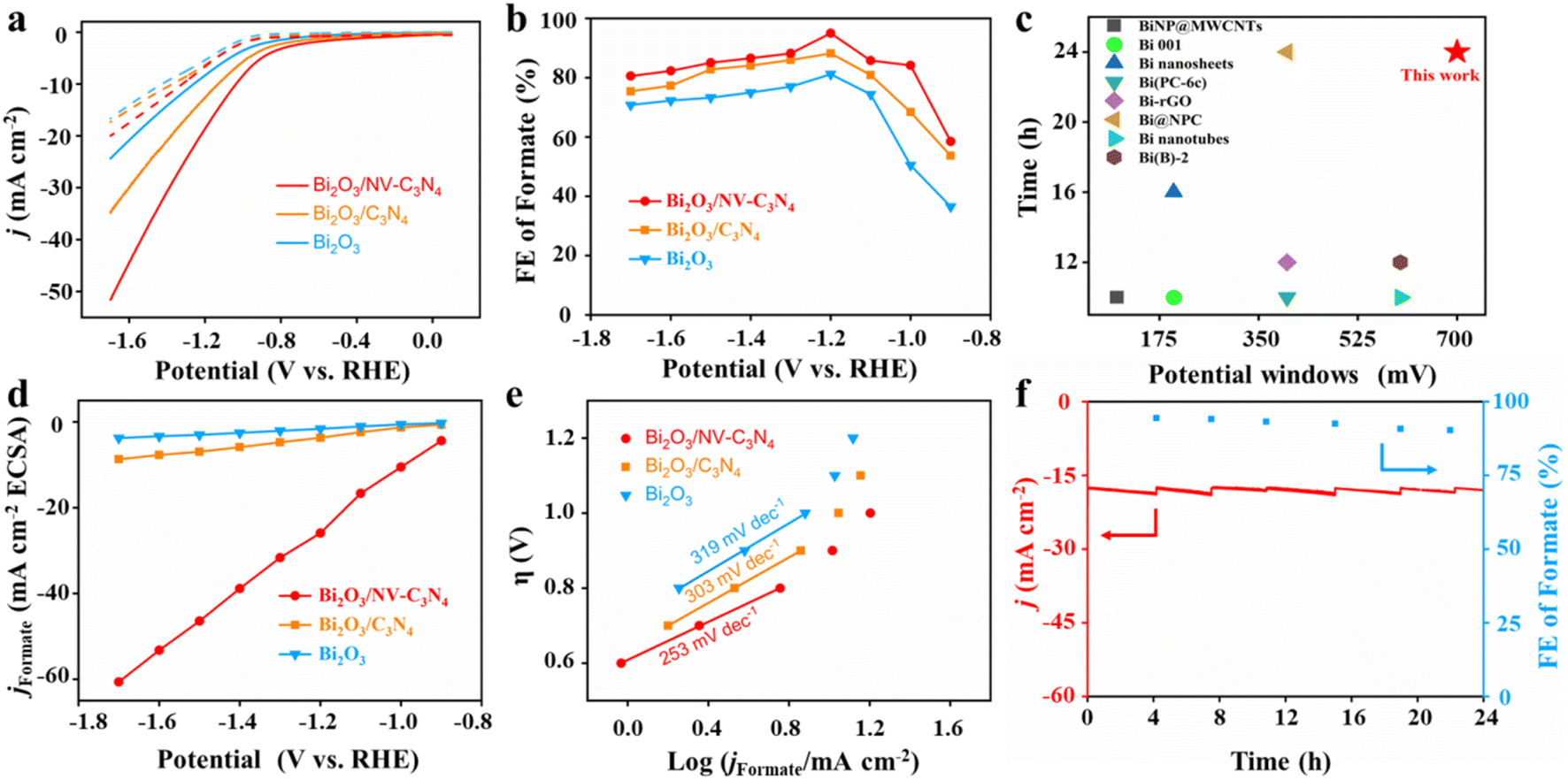 | ||
| Fig. 2 (a) LSV curves with CO2 and Ar-saturated 0.1 M KHCO3 electrolyte; (b) FEs of formate formation; (c) comparison of stability and potential windows with those of other reported electrocatalysts. (d) Partial current densities of formate normalized by ECSA; (e) Tafel slope for Bi2O3, Bi2O3/C3N4 and Bi2O3/NV-C3N4; (f) the stability of Bi2O3/NV-C3N4 at −1.2 V. | ||
To evaluate the intrinsic ECRR activities of the prepared catalysts, the electrochemical active surface areas (ECSAs) of Bi2O3, Bi2O3/C3N4 and Bi2O3/NV-C3N4 were evaluated (Fig. S12†). The capacitances of Bi2O3/NV-C3N4, Bi2O3/C3N4 and Bi2O3 are 0.69 mF cm−2, 3.01 mF cm−2 and 2.31 mF cm−2, respectively. When the current density of formate was renormalized by ECSA, the current density of Bi2O3/NV-C3N4 remained the largest at various potentials, indicating that Bi2O3/NV-C3N4 has the highest intrinsic catalytic activity (Fig. 2d). The kinetic process of the ECRR to formate transition is further explored via Tafel plots (Fig. 2e). The Tafel slope of Bi2O3/NV-C3N4 is 253 mV dec−1, much smaller than those of Bi2O3/C3N4 (303 mV dec−1) and Bi2O3 (313 mV dec−1), suggesting a more favorable reaction kinetics for the ECRR to formate on Bi2O3/NV-C3N4. The evaluation of catalytic performance suggests that the electron-rich region around Bi2O3 induced by electron transfer from supports to Bi2O3 accelerates ECRR kinetics resulting in the excellent performance of Bi2O3/NV-C3N4.
In addition to the electrocatalytic activity, stability is also a significant metric for evaluating advanced catalysts. For durable electrolysis of Bi2O3/NV-C3N4, the electrolyte was changed every four hours to test the FEformate. As shown in Fig. 2f, Bi2O3/NV-C3N4 was tested at −1.2 V for 24 h and the FEformate remains above 90%. In addition, the catalysts after electrolysis were characterized. TEM after electrolysis revealed that Bi2O3 nanoparticles exhibited slight agglomeration (Fig. S13a†), which may be the main factor for the slight performance degradation. Furthermore, XRD after electrolysis still preserves the well-preserved Bi2O3 crystal phase (Fig. S13b†), indicating the good stability of the catalyst.
To further study the ECRR mechanism and confirm the proposed reasons for the enhanced catalytic performance, we modeled three materials Bi2O3, Bi2O3/C3N4 and Bi2O3/NV-C3N4 (Fig. S14a, b† and 3a) and performed density functional theory (DFT) calculations (Fig. 3b). On the whole, the ΔGCOO of the ECRR for the three catalysts is as follows: Bi2O3 > Bi2O3/C3N4 > Bi2O3/NV-C3N4. For Bi2O3, the ΔGCOO of CO2 adsorption can reach 2.97 eV. After Bi2O3 was decorated on C3N4, the ΔGCOO was reduced by 1.70 eV, but subsequent *HCOOH formation processes cannot proceed spontaneously. The introduction of NVs on C3N4 not only lowers the ΔGCOO of *COO adsorption, but also allows the formation process of *HCOOH to proceed spontaneously. For Bi2O3/NV-C3N4, both free energies for *COO and *HCOOH adsorption were reduced, and the formation of *OCHO turns into a decisive speed step, and only 0.60 eV was needed for this step. In addition, the free energy of the HER was calculated (Fig. 3c). According to the level of the energy barriers, the degree of difficulty of spontaneity is obtained as follows: Bi2O3/NV-C3N4 > Bi2O3/C3N4 > Bi2O3, which is consistent with the whole free energy for the ECRR and the experimental results.
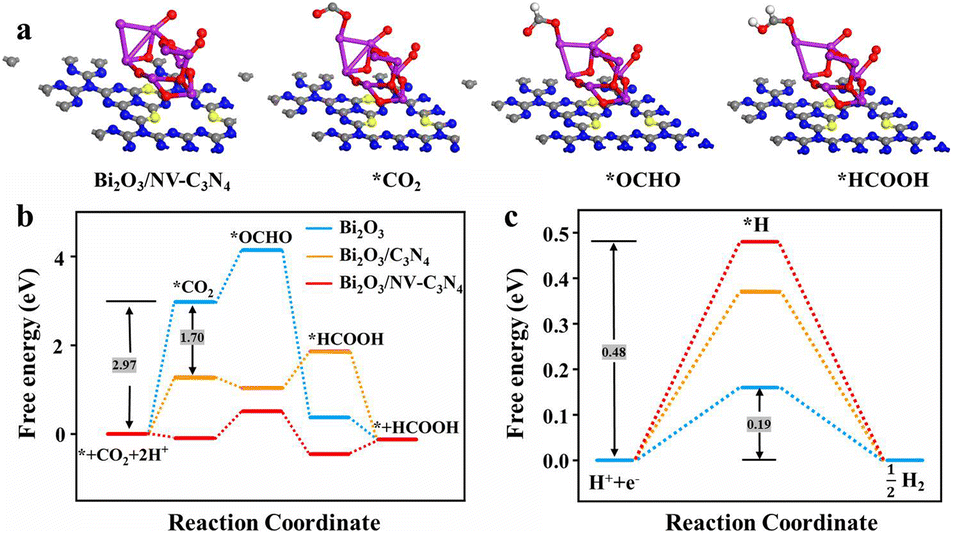 | ||
| Fig. 3 (a) Geometric structure models of three intermediates *CO2, *OCHO and *HCOOH adsorbed on Bi2O3/NV-C3N4; (b) free energy graph for CO2 conversion; (c) free energy diagrams for the HER. | ||
Conclusions
In conclusion, it has been demonstrated that the ultrafine Bi2O3 nanoparticles (∼1.9 nm) supported on graphitic carbon nitride containing nitrogen vacancies (Bi2O3/NV-C3N4) exhibit excellent selectivity and stability for the ECRR to formate. Experimental and theoretical analyses show that metal–support interfacial interactions can enrich the electron density of Bi2O3 effectively facilitating *COO generation, which is beneficial to the generation of formate. Meanwhile, the metal–support interfacial interactions can be extended to other catalyst systems to improve their catalytic performance.Author contributions
Jiayu Zhan: investigation, methodology, formal analysis, writing the original draft and DFT calculations. Lu-Hua Zhang: conceptualization, writing the original draft, writing review & editing, supervision, and funding acquisition. Xueli Wang: investigation, methodology, formal analysis, and writing the original draft. Yuqi Hu: writing review & editing. Yi Jiang: conceptualization and writing review & editing. Fengshou Yu: conceptualization, writing review & editing, supervision, and funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22008048, 21905073, 22278108, and 22008050), Hundred Talents Project of Hebei Province (No. E2019050015), Natural Science Foundation for Outstanding Youth Scholars of Hebei Province (No. B2021202061), Natural Science Foundation of Hebei Province (No. B2021202010 and B2020202066), State Key Laboratory of Fine Chemicals (KF 2108), and Open Research Fund of CNMGE Platform & NSCC-TJ.Notes and references
- (a) O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef; (b) M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2 Search PubMed; (c) P. Ding, H. Zhao, T. Li, Y. Luo, G. Fan, G. Chen, S. Gao, X. Shi, S. Lu and X. Sun, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC; (d) D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef; (e) X.-H. Zhao, Q.-S. Chen, D.-H. Zhuo, J. Lu, Z.-N. Xu, C.-M. Wang, J.-X. Tang, S.-G. Sun and G.-C. Guo, Electrochim. Acta, 2021, 367 Search PubMed; (f) H. Guo, D.-H. Si, H.-J. Zhu, Q.-X. Li, Y.-B. Huang and R. Cao, Science, 2022, 2, 295–303 Search PubMed.
- (a) N. Zouaoui, B. D. Ossonon, M. Fan, D. Mayilukila, S. Garbarino, G. de Silveira, G. A. Botton, D. Guay and A. C. Tavares, J. Mater. Chem. A, 2019, 7, 11272–11281 RSC; (b) Z. Yang, F. E. Oropeza and K. H. L. Zhang, APL Mater., 2020, 8 Search PubMed; (c) K. Van Daele, B. De Mot, M. Pupo, N. Daems, D. Pant, R. Kortlever and T. Breugelmans, ACS Energy Lett., 2021, 6, 4317–4327 CrossRef; (d) M. F. Philips, G.-J. M. Gruter, M. T. M. Koper and K. J. P. Schouten, ACS Sustainable Chem. Eng., 2020, 8, 15430–15444 CrossRef; (e) N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2019, 10, 1902338 CrossRef; (f) J. Guo, W. Zhang, L. H. Zhang, D. Chen, J. Zhan, X. Wang, N. R. Shiju and F. Yu, Adv. Sci., 2021, 8, e2102884 CrossRef.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2019, 10, 743–750 CrossRef.
- (a) R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef; (b) S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef PubMed; (c) X. Deng, D. Alfonso, T.-D. Nguyen-Phan and D. R. Kauffman, ACS Catal., 2022, 5921–5929 CrossRef; (d) X. Wang, L. H. Zhang, D. Chen, J. Zhan, J. Guo, Z. Zhang and F. Yu, ChemCatChem, 2022, 14 Search PubMed; (e) Y. Peng and S. Chen, Green Energy Environ., 2018, 3, 335–351 CrossRef; (f) W. Shang, W. Liu, X. Cai, J. Hu, J. Guo, C. Xin, Y. Li, N. Zhang, N. Wang, C. Hao and Y. Shi, Adv. Powder Mater., 2022, 100094 Search PubMed.
- (a) L. Zhang, F. Mao, L. R. Zheng, H. F. Wang, X. H. Yang and H. G. Yang, ACS Catal., 2018, 8, 11035–11041 CrossRef; (b) X. Zeng, Y. Liu, Y. Kang, Q. Li, Y. Xia, Y. Zhu, H. Hou, M. H. Uddin, T. R. Gengenbach, D. Xia, C. Sun, D. T. McCarthy, A. Deletic, J. Yu and X. Zhang, ACS Catal., 2020, 10, 3697–3706 CrossRef; (c) B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T. C. Lau, J. Bonin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef; (d) Y.-N. Gong, B.-Z. Shao, J.-H. Mei, W. Yang, D.-C. Zhong and T.-B. Lu, Nano Res., 2021, 15, 551–556 CrossRef.
- (a) Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS; (b) Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- K. Chu, Q.-q. Li, Y.-p. Liu, J. Wang and Y.-h. Cheng, Appl. Catal., B, 2020, 267 Search PubMed.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef.
- (a) B. Zhu, P. Xia, W. Ho and J. Yu, Appl. Surf. Sci., 2015, 344, 188–195 CrossRef; (b) Q. Liang, Z. Li, Y. Bai, Z.-H. Huang, F. Kang and Q.-H. Yang, Sci. China Mater., 2016, 60, 109–118 CrossRef.
- (a) Y. Kang, Y. Yang, L. C. Yin, X. Kang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed; (b) C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- (a) Y. X. Duan, K. H. Liu, Q. Zhang, J. M. Yan and Q. Jiang, Small Methods, 2020, 4 Search PubMed; (b) D. Zhang, Z. Tao, F. Feng, B. He, W. Zhou, J. Sun, J. Xu, Q. Wang and L. Zhao, Electrochim. Acta, 2020, 334 Search PubMed; (c) S. Kim, W. J. Dong, S. Gim, W. Sohn, J. Y. Park, C. J. Yoo, H. W. Jang and J.-L. Lee, Nano Energy, 2017, 39, 44–52 CrossRef CAS; (d) X. Chen, H. Chen, W. Zhou, Q. Zhang, Z. Yang, Z. Li, F. Yang, D. Wang, J. Ye and L. Liu, Small, 2021, 17, 2101128 CrossRef CAS; (e) W. Zhang, Y. Hu, L. Ma, G. Zhu, P. Zhao, X. Xue, R. Chen, S. Yang, J. Ma, J. Liu and Z. Jin, Nano Energy, 2018, 53, 808–816 CrossRef CAS; (f) K. Fan, Y. Jia, Y. Ji, P. Kuang, B. Zhu, X. Liu and J. Yu, ACS Catal., 2019, 10, 358–364 CrossRef; (g) D. Wu, J. Liu, Y. Liang, K. Xiang, X. Z. Fu and J. L. Luo, ChemSusChem, 2019, 12, 4700–4707 CrossRef CAS; (h) X. Zhang, J. Fu, Y. Liu, X.-D. Zhou and J. Qiao, ACS Sustainable Chem. Eng., 2020, 8, 4871–4876 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2se01274j |
| This journal is © The Royal Society of Chemistry 2023 |