
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Confinement of an alkaline environment for electrocatalytic CO2 reduction in acidic electrolytes†
Xiaozhi
Li‡
ab,
Peng
Zhang‡
 abde,
Lili
Zhang
ab,
Gong
Zhang
ab,
Hui
Gao
ab,
Zifan
Pang
ab,
Jia
Yu
ab,
Chunlei
Pei
ab,
Tuo
Wang
abcd and
Jinlong
Gong
*abcd
abde,
Lili
Zhang
ab,
Gong
Zhang
ab,
Hui
Gao
ab,
Zifan
Pang
ab,
Jia
Yu
ab,
Chunlei
Pei
ab,
Tuo
Wang
abcd and
Jinlong
Gong
*abcd
aSchool of Chemical Engineering & Technology, Key Laboratory for Green Chemical Technology of Ministry of Education, Tianjin University, Tianjin 300072, China. E-mail: jlgong@tju.edu.cn
bCollaborative Innovation Center of Chemical Science & Engineering (Tianjin), Tianjin 300072, China
cHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, China
dNational Industry-Education Platform of Energy Storage, Tianjin University, 135 Yaguan Road, Tianjin 300350, China
eJoint School of National University of Singapore, Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
First published on 2nd May 2023
Abstract
Acidic electrochemical CO2 reduction reaction (CO2RR) can minimize carbonate formation and eliminate CO2 crossover, thereby improving long-term stability and enhancing single-pass carbon efficiency (SPCE). However, the kinetically favored hydrogen evolution reaction (HER) is generally predominant under acidic conditions. This paper describes the confinement of a local alkaline environment for efficient CO2RR in a strongly acidic electrolyte through the manipulation of mass transfer processes in well-designed hollow-structured Ag@C electrocatalysts. A high faradaic efficiency of over 95% at a current density of 300 mA cm−2 and an SPCE of 46.2% at a CO2 flow rate of 2 standard cubic centimeters per minute are achieved in the acidic electrolyte, with enhanced stability compared to that under alkaline conditions. Computational modeling results reveal that the unique structure of Ag@C could regulate the diffusion process of OH− and H+, confining a high-pH local reaction environment for the promoted activity. This work presents a promising route to engineer the microenvironment through the regulation of mass transport that permits the CO2RR in acidic electrolytes with high performance.
Introduction
Converting CO2 into value-added chemical feedstocks and fuels through the electrochemical CO2 reduction reaction (CO2RR) driven by renewable electricity is an appealing pathway to develop a sustainable carbon cycle.1–3 In order to boost the catalytic activity, alkaline or neutral electrolysis systems are widely employed to suppress the competing hydrogen evolution reaction (HER), which could also promote CO2 activation and C–C coupling.4–9 However, the inevitable reaction between CO2 and local/bulk OH− in alkaline or neutral electrolytes would lead to undesired CO2 consumption and crossover.10–12 This phenomenon results in low carbon utilization efficiencies (≤50% for C1 products and ≤ 25% for C2+ products) and limited energy efficiencies.13–16 Furthermore, the stability of alkaline CO2RR systems could be significantly lowered due to the accumulation of the generated carbonate.13,17 Therefore, it is essential to develop efficient CO2RR systems with high CO2 utilization efficiency and good stability.18,19Among the emerging strategies to achieve this goal, acidic CO2RR has received considerable attention.20,21 Due to the high proton concentration and the use of a proton exchange membrane (PEM), the CO2RR in acidic media offers a viable approach to reduce the formation of carbonate and eliminate CO2 crossover.22 Therefore, acidic CO2RR can break the theoretical limitation of single-pass carbon efficiency (SPCE) for alkaline CO2RR with less energy cost for product separation.23,24 Without the accumulation of carbonate, long-term stability can also be improved in acidic systems. Furthermore, higher conductivity and the economic feasibility of PEMs also make it promising for industrialization.16,25 However, the kinetically favored HER is normally predominant in acidic electrolytes, which leads to a low faradaic efficiency (FE) of CO2RR products.15,26 Therefore, it is imperative to suppress the severe HER to improve the practical viability of acidic CO2RR.
The suppression of the HER in acidic media can be realized by the design of appropriate electrocatalysts. Bimetallic catalysts have been proven to modulate CO* coverage and weaken H* adsorption through adsorbate–adsorbate interactions, thus inhibiting the HER.27 Meanwhile, the CO2RR can be further enhanced by modifying the catalyst surface with basic sites to promote CO2 adsorption.20,28 Besides the design of novel catalysts, the optimization of the microenvironment is equally critical to render acidic CO2RR feasible.29–34 For instance, a hydrophobic chemical environment has been proven to benefit the CO2RR due to the high local CO2/H2O ratio in the gas diffusion electrode (GDE).23,35 It can be realized by adding polytetrafluoroethylene nanoparticles into the catalyst layer. According to recent studies, the H+ coverage close to the cathode surface was sensitive to the K+ concentration due to the competitive adsorption of cations at the outer Helmholtz plane (OHP), and thus concentrating potassium cations in the vicinity of active sites could promote the performance of the CO2RR.16,24,36,37 Besides, controlling the formation rate of CO/OH− to compensate for the diffusion of protons from the bulk electrolyte and coating the catalyst layer with a nanoporous ion-regulatory layer to suppress the diffusion of K+ and OH− are effective ways to lower the concentration of protons near the catalyst surface.38,39 In these cases, the competitive HER from proton reduction can be effectively suppressed. Meanwhile, the crossover of CO2 is minimized because any locally generated carbonate can be converted back to CO2 by the adequate protons in the bulk acidic electrolyte.40 Overall, engineering the local reaction environment through suitable catalyst design is essential for promoting acidic CO2RR.
In this study, Ag@C electrocatalyst with Ag active sites loaded on the interior surface of hollow carbon spheres was designed, which exhibits a CO FE (FECO) of over 95% even in a strongly acidic electrolyte (pH 1.1) with improved stability. An SPCE of 46.2% was achieved at a CO2 flow rate of 2 standard cubic centimeters per minute (sccm) in virtue of the inhibited carbonate formation in acidic CO2RR. Computational modeling demonstrated that the diffusion of OH− through the hollow spheres was limited due to the presence of the porous carbon layer. Thus, the enrichment of OH− in the nanochamber brought a local alkaline environment that can effectively suppress the HER. This work highlights the importance of mass transport manipulation in engineering the reaction microenvironment for high-performance CO2RR.
Results and discussion
Synthesis and characterization of electrocatalysts
The synthetic procedure of Ag@C catalysts is illustrated in Fig. 1a. First, SiO2 spheres with an average size of ∼400 nm were synthesized as templates (Fig. S1a†). Subsequently, Ag nanoparticles were loaded on the surface of SiO2 to obtain SiO2@Ag (Fig. S1b†).41 A modified Stöber coating method was adopted to fabricate SiO2@Ag@resorcinol-formaldehyde (SiO2@Ag@RF) spheres with a core–shell structure (Fig. 1b).42 Then, SiO2@Ag@C was obtained after a calcination process under an inert atmosphere, during which RF was converted to carbon at an elevated temperature. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of SiO2@Ag@C show the full coverage of the carbon shell without Ag particles left on the outer surface (Fig. 1c and d). The as-prepared SiO2@Ag@C was finally converted into Ag@C via a chemical etching treatment. TEM observation indicates the retention of a hollow spherical morphology with a shell thickness of ∼40 nm after the successful removal of the SiO2 cores. The Ag particles (average diameter ≈ 16.1 nm) are confined inside the carbon spheres (Fig. 1e). As shown in the high-resolution TEM (HRTEM) images, the lattice spacing of the Ag@C catalyst corresponds to the (111) facet of Ag (Fig. 1f and g). The elemental mapping images of Ag@C further reveal that Ag nanoparticles are located on the inner surface of the carbon shell (Fig. 1h). The structural feature of the carbon shell was studied using N2 adsorption–desorption measurements (Fig. S2†). The Ag@C catalyst shows a porous structure with an average pore size of about 2.6 nm, which ensures sufficient reactants near the surface of the active sites (Fig. S3†). Conventional Ag/C catalysts without a core–shell structure were synthesized as the control sample (Fig. S5†). X-ray diffraction (XRD) patterns indicate the presence of metallic Ag in Ag@C and Ag/C catalysts (Fig. S6†). X-ray photoelectron spectroscopy (XPS) measurements were performed to further confirm the chemical state of the Ag active sites (Fig. S7†). The Ag 3d5/2 peak at 368.2 eV was observed, implying that the metallic Ag is the active phase in both catalysts. Both Ag@C and Ag/C catalysts possess approximately the same mass loadings (∼40%) and similar Ag nanoparticle sizes (insets of Fig. 1e, insets of Fig. S5a and Table S1†). Meanwhile, the Raman intensity ratios between D and G bands in both catalysts are similar, which implies the consistent properties of the carbon supports (Fig. S8†).42 In addition, a C@Ag catalyst with active sites loaded on the outer surface of a porous carbon layer was prepared to further demonstrate the importance of the unique structure of the Ag@C catalyst in enhancing the acidic CO2RR performance (Fig. S9†). The preparation of C@Ag followed a similar synthesis procedure to the Ag@C catalyst. | ||
| Fig. 1 Schematic illustration and morphological characterization. (a) Scheme of the synthesis process of the Ag@C catalyst. (b) SEM image of SiO2@Ag@RF. (c) SEM and (d) TEM images of SiO2@Ag@C. (e) TEM and (f and g) HRTEM images of Ag@C. The inset in (e) is the particle size distribution of Ag. (h) Energy-dispersive spectroscopy (EDS) elemental mapping images of Ag@C. | ||
CO2 electroreduction in alkaline and acidic media
The CO2RR performance was evaluated in a typical three-electrode flow cell reactor. The flow rate of electrolyte was kept constant for all the tests. As shown in Fig. 2a, both Ag@C and Ag/C catalysts exhibited a similar FECO of over 90% in the current range of 50 to 300 mA cm−2 in the alkaline electrolyte (1.0 M KOH, pH 13.7), which is consistent with previous reports.5,43,44 This result shows that an alkaline environment is beneficial for the suppression of the HER. Besides, the catalytic activity of Ag@C was tested at different CO2 flow rates from 50 to 2 sccm in the alkaline electrolyte at 200 mA cm−2 (Fig. 2b). As the flow rate decreased, the FECO showed an apparent drop due to the rapid depletion of CO2 to CO32−/HCO3− (Fig. S10†). A relatively low maximum SPCE of 20.7% was achieved at 4 sccm. | ||
| Fig. 2 CO2RR performance in alkaline and acidic media. (a) FECO on Ag@C and Ag/C at different current densities in 1.0 M KOH (pH 13.7). (b) FECO and corresponding SPCE on Ag@C at 200 mA cm−2 with different CO2 flow rates in 1.0 M KOH. (c) FECO on Ag@C and Ag/C at different current densities in 0.5 M K2SO4 (pH 1.1). (d) FECO and corresponding SPCE on Ag@C at 200 mA cm−2 with different CO2 flow rates in 0.5 M K2SO4 (pH 1.1). Error bars represent the standard deviation from at least three independent measurements. | ||
As mentioned above, acidic CO2RR may offer a viable solution to improve the carbon utilization efficiency by minimizing the formation and crossover of carbonate. After the preliminary tests in mildly acidic media (Fig. S11†), the CO2RR was further performed under strongly acidic conditions to investigate the feasibility of the Ag@C catalyst (Fig. 2c and d). An acidified 0.5 M K2SO4 solution with a pH of around 1.1 was employed as the catholyte to ensure consistent cation concentrations in both acidic and alkaline electrolysis. Besides, a relatively high electrolyte concentration contributes to the reduction of cathodic ohmic loss (Fig. S12†). As shown in Fig. 2c, the Ag@C catalyst displayed a FECO of over 95% at a current density of 300 mA cm−2, which is comparable to the performance under alkaline conditions. Meanwhile, the activity of Ag@C significantly exceeded that of Ag/C and C@Ag catalysts over the whole tested current density range (Fig. S13†). More importantly, the Ag@C catalyst exhibited relatively high FECO even at low CO2 flow rates (Fig. 2d). A high SPCE of 46.2% was achieved at 2 sccm in the acidic media, implying the advantage in minimizing carbonate formation compared with the alkaline system.
Stable operation is essential to ensure the CO2RR to be economically competitive for practical application. The stability of Ag@C in acidic and alkaline systems was examined under a constant current density of 100 mA cm−2 (Fig. 3a). An apparent deactivation of the alkaline CO2RR system was observed due to the precipitation of carbonate salt, as evidenced by the observation of the obvious salt on the backside of the gas diffusion electrode (GDE, Fig. 3b–d). In contrast, the FECO was maintained over 90% during ∼9 h of electrolysis in the acidic electrolyte. Post characterization studies demonstrate that the hollow structure of the Ag@C catalyst was well retained with negligible changes in composition (Fig. S14–S16†). The XPS data indicate the retention of metallic Ag after the long-term test (Fig. S17†), which excludes the effect of chemical state change of Ag active sites on the catalytic stability. Meanwhile, the outlet electrolyte was measured by inductively coupled plasma optical emission spectrometry (ICP-OES) after the durability test under acidic conditions. No dissolved Ag was detected (Table S2†). Therefore, the slight decline in FECO may result from the wetting of the gas diffusion layer (GDL) as suggested by Fig. 3e–g. In addition, only a minor shift in catholyte pH was observed after the long-term test in the acidic electrolyte (inset of Fig. 3a), which indicates that the stability of the electrolyte environment may also facilitate the stable operation of the whole electrolysis system. Furthermore, the stability of Ag/C in 0.5 M K2SO4 (pH 1.1) was also measured. The gradual increase of FEH2 due to the severe flooding was also observed in the Ag/C catalyst (Fig. S18a†). And the TEM image after the long-term test shows no significant change in the catalyst morphology and the particle size distribution of Ag (Fig. S18b†), which reinforces the evidence that the wetting of GDL is responsible for the degradation of acidic electrolysis.
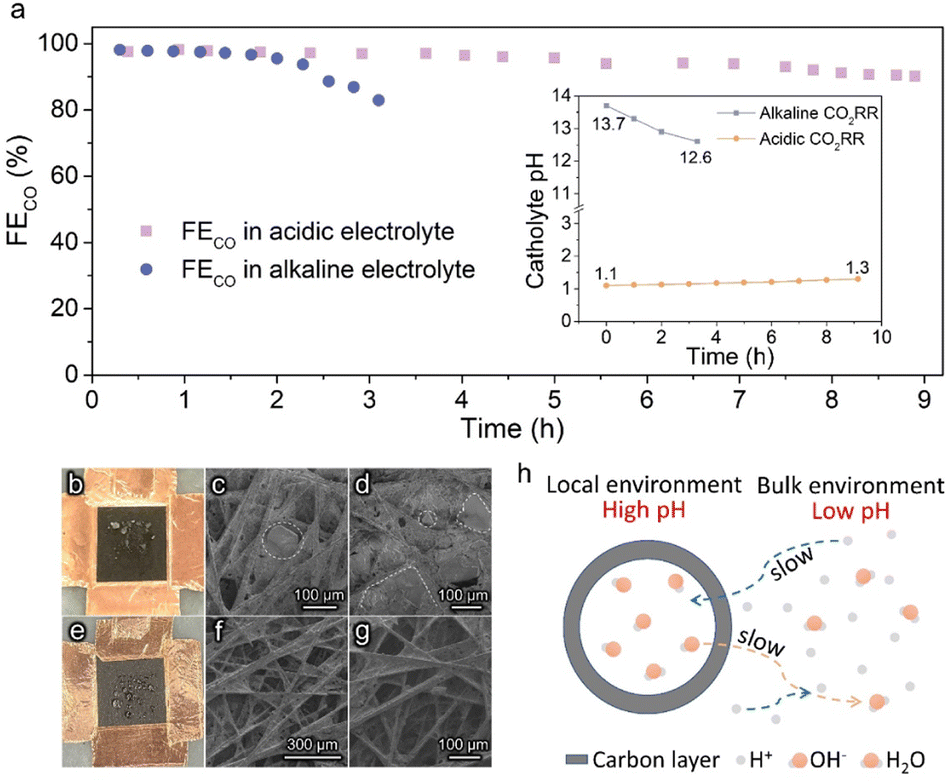 | ||
| Fig. 3 Stability performance of Ag@C in acidic and alkaline media. (a) Long-term performance of the CO2RR at 100 mA cm−2 in alkaline (1.0 M KOH, pH 13.7) and acidic (0.5 M K2SO4, pH 1.1) electrolytes. Inset shows the pH variation of the catholyte with time. (b) Photograph and (c and d) SEM images of the backside of the GDE after the stability test in 1.0 M KOH. (e) Photograph and (f and g) SEM images of the backside of the GDE after the stability test in 0.5 M K2SO4 (pH 1.1). (h) Schematic of the local reaction environment and ion transport on the Ag@C catalyst. | ||
The relatively good performance of the Ag@C catalyst in acidic electrolysis may be attributed to the rational design of its structure. As shown in Fig. 3h, it is presumed that the presence of the carbon layer confines the CO2RR within the nanochambers and provides an obstacle to the diffusion process. In the course of the CO2RR, the diffusion of H+ from the bulk electrolyte is restricted by the carbon shell. Meanwhile, OH− generated along with the reaction cannot diffuse out promptly as well. Thereby, the acidic electrolyte can be neutralized rapidly owing to the depletion of H+ and accumulation of OH−. As the reaction proceeds, the concentration gradient of OH− inside and outside the carbon layer gradually increases, which means a higher diffusion flux. When the diffusion and formation rates of OH− reach a dynamic equilibrium, a high internal concentration of OH− remains approximately constant. Ultimately, an alkaline local environment is maintained near the surface of active sites to inhibit the HER effectively.
Mass transport simulation
To further verify the proposed mechanism, mass transport simulation was adopted to explore the diffusion process in the acidic electrolyte. A sector domain was selected as the model for the calculation (Fig. 4a and S19†). The dimensions of the sector were defined according to the characterization results of SEM and TEM. In the simulations, CO2 molecules diffused to the cavity and converted into CO on the inner surface of the carbon shell.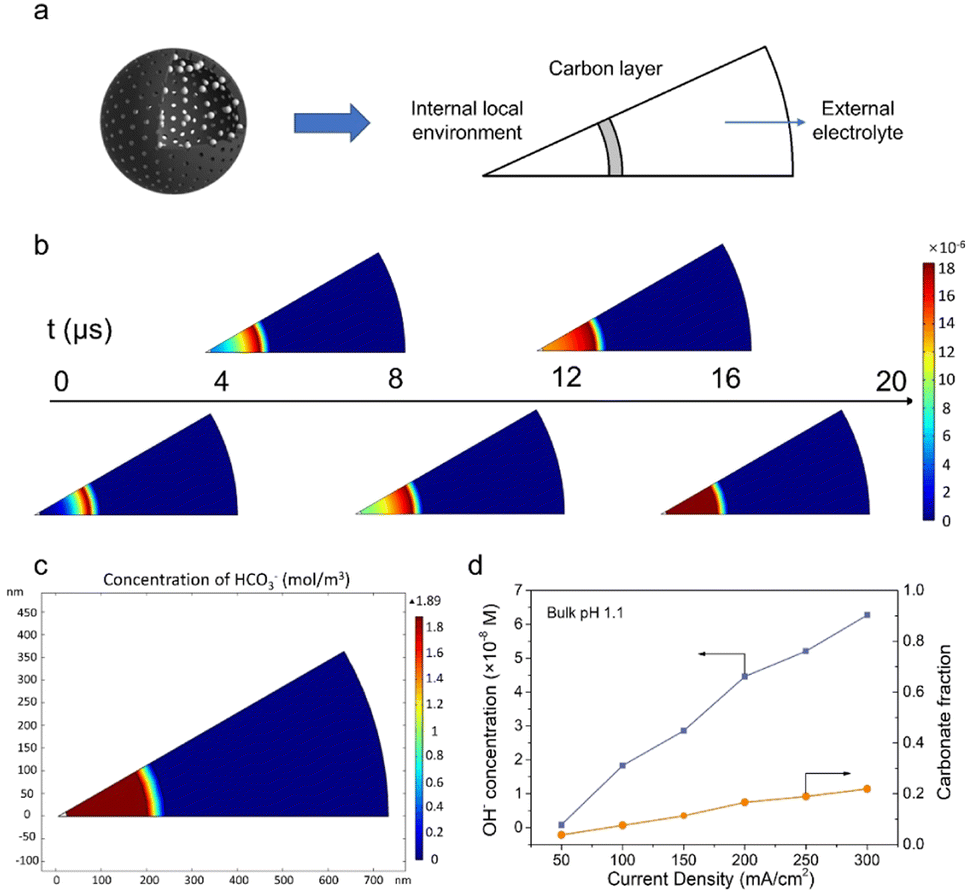 | ||
| Fig. 4 Mass transport simulations. (a) Graphical illustration of the modeling domains. (b) Variation of OH− concentration with time. (c) HCO3− concentration profile at the steady state with a current density of 100 mA cm−2. (d) OH− concentration profile and carbonate fraction at different current densities with bulk pH 1.1. The carbonate fraction is calculated using the ratio between carbonate (HCO3−, CO32−) and the sum of carbon species (HCO3−, CO32− and CO2,aq). | ||
As presented in Fig. 4b, the transient model shows the variation of OH− concentration distribution over time (Movie S1†). It is clear that the carbon layer significantly restricts the outflow of locally generated OH−, which leads to a relatively high OH− concentration inside the hollow sphere. Therefore, the high local pH could lead to an improved CO2RR performance, which is consistent with our hypothesis and experimental results. The distribution of carbonate was also assessed due to its potential impact on the system stability. An enriched carbonate concentration at the steady state was observed inside the cavity (Fig. 4c). With the increase of current density, a higher OH− concentration was obtained and consequently led to an elevated carbonate fraction (Fig. 4d and S20–S25†). However, the carbonate ratio merely stayed at a relatively low value of 0.2 even at 300 mA cm−2, in agreement with the good stability of the CO2RR. Altogether, the mass transport simulation validates the impact of the catalyst structure on tuning the local environment and promoting the CO2RR performance in the acidic electrolyte.
Conclusions
To summarize, acidic CO2RR offers a promising route to address the issues of relatively low carbon utilization efficiency and poor stability due to the inevitable formation and accumulation of carbonate in conventional alkaline systems. In this work, a stable CO2RR in the acidic electrolyte (pH 1.1) was realized without the compromise on FECO by the design of Ag@C catalysts. A high SPCE of 46.2% was achieved, which is approximately twice that of alkaline electrolysis. Mass transport simulation demonstrates that the structure of Ag@C plays a key role in regulating the diffusion pathways of H+ and OH−, consequently influencing the local pH at the surface of active sites. As a result, the confinement of an alkaline environment effectively inhibits the HER in the bulk acidic electrolyte for enhanced CO2RR performance. Overall, these findings suggest a general principle in the design of catalysts with controllable mass transport properties and emphasize the importance of microenvironment engineering for the CO2RR in acidic media.Data availability
The data that supports the findings of this study is available from the corresponding author upon reasonable request.Author contributions
J. L. G. supervised the project. P. Z. and X. Z. L. conceived the idea. X. Z. L. synthesized catalysts and conducted the CO2RR performance tests. L. L. Z. and Z. F. P. conducted mass transport simulations. G. Z., H. G., and J. Y. conducted related characterization. All the authors participated in the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Key R&D Program of China (2021YFA1501503), the National Natural Science Foundation of China (22121004 and 22108197), the Haihe Laboratory of Sustainable Chemical Transformations (CYZC202107), the TianHe Qingsuo Open Research Fund of TSYS in 2022 & NSCC-TJ, the Program of Introducing Talents of Discipline to Universities (No. BP0618007) and the Xplorer Prize for financial support.References
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, e1807166 CrossRef PubMed.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 CrossRef.
- D. Gao, W. Li, H. Wang, G. Wang and R. Cai, Trans. Tianjin Univ., 2022, 28, 245–264 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Q. Zou, R. Quintero-Bermudez, Y. J. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Norskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- N. Gupta, M. Gattrell and B. MacDougall, J. Appl. Electrochem., 2005, 36, 161–172 CrossRef.
- M. Dunwell, X. Yang, B. P. Setzler, J. Anibal, Y. Yan and B. Xu, ACS Catal., 2018, 8, 3999–4008 CrossRef CAS.
- A. J. Welch, A. Q. Fenwick, A. Böhme, H.-Y. Chen, I. Sullivan, X. Li, J. S. DuChene, C. Xiang and H. A. Atwater, J. Phys. Chem. C, 2021, 125, 20896–20904 CrossRef CAS.
- C. Chen, Y. Li and P. Yang, Joule, 2021, 5, 737–742 CrossRef.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed.
- J. E. Huang, F. W. Li, A. Ozden, A. S. Rasouli, F. P. G. de Arquer, S. J. Liu, S. Z. Zhang, M. C. Luo, X. Wang, Y. W. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- K. Xie, R. K. Miao, A. Ozden, S. Liu, Z. Chen, C. T. Dinh, J. E. Huang, Q. Xu, C. M. Gabardo, G. Lee, J. P. Edwards, C. P. O'Brien, S. W. Boettcher, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed.
- C. P. O'Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- Z. Wang, P. Hou, Y. Wang, X. Xiang and P. Kang, ACS Sustainable Chem. Eng., 2019, 7, 6106–6112 CrossRef CAS.
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- A. Ozden, F. P. García de Arquer, J. E. Huang, J. Wicks, J. Sisler, R. K. Miao, C. P. O'Brien, G. Lee, X. Wang, A. H. Ip, E. H. Sargent and D. Sinton, Nat. Sustain., 2022, 5, 563–573 CrossRef.
- X. Sheng, W. Ge, H. Jiang and C. Li, Adv. Mater., 2022, 34, e2201295 CrossRef PubMed.
- Y. Qiao, W. Lai, K. Huang, T. Yu, Q. Wang, L. Gao, Z. Yang, Z. Ma, T. Sun, M. Liu, C. Lian and H. Huang, ACS Catal., 2022, 12, 2357–2364 CrossRef CAS.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 33, 9307–9313 CrossRef CAS PubMed.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- Q. Lu, C. Chen, Q. Di, W. Liu, X. Sun, Y. Tuo, Y. Zhou, Y. Pan, X. Feng, L. Li, D. Chen and J. Zhang, ACS Catal., 2022, 12, 1364–1374 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. Jin, X. Wang and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207252 CrossRef CAS PubMed.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- J. Hong, K. T. Park, Y. E. Kim, D. Tan, Y. E. Jeon, J. E. Park, M. H. Youn, S. K. Jeong, J. Park, Y. N. Ko and W. Lee, Chem. Eng. J., 2022, 431, 133384 CrossRef CAS.
- Z. Liu, T. Yan, H. Shi, H. Pan, Y. Cheng and P. Kang, ACS Appl. Mater. Interfaces, 2022, 14, 7900–7908 CrossRef CAS PubMed.
- F. Wang, W. Zhang, H. Wan, C. Li, W. An, X. Sheng, X. Liang, X. Wang, Y. Ren, X. Zheng, D. Lv and Y. Qin, Chin. Chem. Lett., 2022, 33, 2259–2269 CrossRef CAS.
- Q. Fan, G. Bao, X. Chen, Y. Meng, S. Zhang and X. Ma, ACS Catal., 2022, 12, 7517–7523 CrossRef CAS.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- Z. Ma, Z. Yang, W. Lai, Q. Wang, Y. Qiao, H. Tao, C. Lian, M. Liu, C. Ma, A. Pan and H. Huang, Nat. Commun., 2022, 13, 7596 CrossRef CAS PubMed.
- C. J. Bondue, M. Graf, A. Goyal and M. T. M. Koper, J. Am. Chem. Soc., 2021, 143, 279–285 CrossRef CAS PubMed.
- L. Li, Z. Liu, X. Yu and M. Zhong, Angew. Chem., Int. Ed., 2023, e202300226 CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- Z. W. Deng, M. Chen and L. M. Wu, J. Phys. Chem. C, 2007, 111, 11692–11698 CrossRef CAS.
- S. Feng, W. Li, Q. Shi, Y. Li, J. Chen, Y. Ling, A. M. Asiri and D. Zhao, Chem. Commun., 2014, 50, 329–331 RSC.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- H. Hashiba, L.-C. Weng, Y. Chen, H. K. Sato, S. Yotsuhashi, C. Xiang and A. Z. Weber, J. Phys. Chem. C, 2018, 122, 3719–3726 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01040f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |