
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePore volume regulated CO2 adsorption in C–C bonded porous organic frameworks†
Himan Dev
Singh
 ,
Piyush
Singh
,
Deepak
Rase
and
Ramanathan
Vaidhyanathan
*
,
Piyush
Singh
,
Deepak
Rase
and
Ramanathan
Vaidhyanathan
*
Department of Chemistry and Centre for Energy Science, Indian Institute of Science Education and Research, Dr Homi Bhabha road, Pashan, Pune, Pune 411008, India. E-mail: vaidhya@iiserpune.ac.in
First published on 20th June 2023
Abstract
Three C–C bonded porous organic polymers are assembled by reacting rigid monomers (triformylphenol, triformylphloroglucinol, triformylresorcinol) with pyrrole in a catalyst-free solvothermal reaction. The resulting black-colored amorphous polymers yield moderate Brunauer–Emmett–Teller surface areas (213 m2 g−1 to 277 m2 g−1). All the polymers exhibit the same pore size but different pore volumes. The impact of pore volume on the adsorption capacity has been investigated. The POFs show excellent CO2 adsorption properties with moderate heat of adsorption values (31.9–37.6 kJ mol−1) and good IAST selectivity for CO2/N2 and CO2/CH4 separation. The working capacity of the POFs determined using mixed gas IAST calculations shows that these POFs can preferentially adsorb CO2 from the CO2/N2 gas mixture. POFs show facile adsorption kinetics with a self-diffusion coefficient on the order of 10−9 m2 sec−1. The POFs offer excellent stability under acidic conditions and retain their working capacity after long humidity exposure. The stability of POFs under harsh acidic conditions has also been established by 273 K CO2 adsorption, which shows a negligible fall in the CO2 saturation capacity.
Introduction
The increasing atmospheric carbon dioxide concentrations due to anthropogenic emissions is a major environmental concern.1 The current level of CO2 has increased to 420 ppm and is responsible for global warming.2 Hence strategies to mitigate CO2 emissions are urgently needed. A desirable way to reduce carbon dioxide emissions into the environment is by capturing and storing carbon dioxide (CCS) from coal and gas-burning power stations.3 However, the large energy penalty in the capture process has limited the growth of CCS. The energy input necessary for the regeneration of the material is the primary cause of the high cost.3 For instance, the traditional CO2 capture method based on alkanolamine solutions produces an energy cost of about 30% of the power plant's output.4 To tackle this, creating new generation CO2 capture materials by fine-tuning the thermodynamics of the interaction between CO2 and the adsorbent is a worthy pursuit.4In this regard, porous materials have drawn interest from academia and industry across many scientific fields and have shown great promise. Porous organic frameworks (POFs) are an important class of porous materials constructed from lightweight elements linked by strong covalent bonds that show great potential in a wide range of applications, such as gas storage and separation,1,5–15 catalysis,16,17 chemical sensing,18,19 optoelectronics,20,21 and energy storage.22,23 They have shown great promise due to their low density, high porosity, exceptional thermal and chemical stability, tunable pore size, control of the number of functional groups, etc. Along with high porosity, the presence of heteroatoms plays a crucial role in high and selective CO2 capture; the heteroatoms, especially nitrogen, are known to interact with CO2via dipole–quadrupole interactions, which is why a lot of work has been carried out on amine grafting and amine impregnation to boost the CO2 capture performance.24,25
High CO2 uptake, working capacity and selectivity over other gases (particularly CO2/N2 selectivity) under humid conditions, fast adsorption kinetics, cheap production cost, and low energy consumption during adsorption and regeneration phases are crucial requirements that must be met by an effective adsorbent.26,27 While constructing a sorbent for CO2 separation, microstructural qualities such the surface area, pore volume, and pore size must be carefully taken into account in addition to surface functionalization and properties of the gas.28 Materials with pore sizes close to the kinetic diameter of the gas (3.3 Å CO2, 3.8 Å of CH4 and 3.64 Å of N2) would be more suitable due to the possibility of multiple gas–surface interactions.29 However, the correlation between pore volume and CO2 capacity is less explored and is relevant to porous polymers.30
In 2008, Thomas et al. reported the trimerization of aromatic nitriles (Covalent Triazine Frameworks). They gained much attention due to their conjugated nature, high heteroatom content and surface areas.31 CTF-0, an exceptionally stable POF with a high surface area of 2011 m2 g−1 with increased mesoporosity and depleted nitrogen content was synthesized, resulting in a CO2 uptake capacity of 4.22 mmol g−1 at 273 K, 1 bar.32 Dai and coworkers introduced methoxy groups onto a hexaazatriphenylene precursor, enabling CO2 uptake capacity of 6.3 mmol g−1 at 273 K, 1 bar with a BET surface area of 1090 m2 g−1.33 Yavuz and coworkers prepared an amide-linked polymer through the polymerization of melamine with a tri-acid chloride and noticed that the choice of solvents has a significant impact on the morphology of the polymers and, consequently, their capacity to absorb CO2.34 Wang et al. exploited aldehyde-terminated precursors and reacted with pyrrole to obtain NH-rich POFs. The polymers exhibited good CO2 uptake of 4.0 mmol g−1 and moderate CO2/N2 selectivities, thus exemplifying a simplistic approach to synthesizing highly functional polymers.35 Kanatzidis, in 2011, reported a family of polymers synthesized using Bakelite-type chemistry with uniform, microporous, spherical particles and exhibiting surface areas up to 917 m2 g−1.36 Internally, the micropores are adorned with many –OH reactive groups that can be functionalized. The POFs collect as much as 18% of their mass of CO2 at atmospheric pressure. In 2015, our group reported a triazine-resorcinol based POF with polar pore environment exhibiting very good CO2 adsorption properties along with their water repelling nature, and high stability to SOx, NOx and steam conditions.37
In this work, we have synthesized three isostructural polymers using the simplistic solvothermal reaction of pyrrole with trialdehyde-based monomers, which show the same pore size but different pore volumes. The effect of change in pore volume on the CO2 capacity is explored. And the potential of the POFs to separate CO2/N2 and CO2/CH4 has been studied. These polymers also exhibit moderate heat of adsorption and hydrophobicity, which is crucial for capturing CO2 from the humid flue gas stream without compromising the CO2 capacity.
Results and discussion
We report three isostructural microporous polymers (IISERP-POF15 (1), IISERP-POF16 (2), IISERP-POF17 (3), where IISERP stands for Indian Institute of Science Education and Research Pune, synthesized without metal catalysts. The synthesis is carried out solvothermally at 493 K in 1,4 dioxane for 48 hours (Fig. 1). Importantly, the polymer's backbone consists of C–C, which imparts exceptional thermal and chemical stability, and the catalyst-free synthesis facilitates the easy scale-up. They are characterized by solid-state magic angle spinning NMR spectroscopy; the NMR results clearly show the presence of carbon (C–OH) on the aldehyde monomer. As the number of carbons (C–OH) increases from 1 to 3, the increase in the intensity of this peak at 153 ppm is also observed (Fig. 2A). The peak corresponding to the alpha carbon of the pyrrole is also observed in the solid-state magic angle spinning NMR. However, the most interesting and characteristic peak for the polymer in the NMR appears at 65 ppm, corresponds to the aldehydic C of the monomer bound to the alpha carbon of pyrrole through the C–C bonds (Fig. 2A). IR spectroscopy confirms the presence of free OH groups in these polymers. The stretching vibration mode of the O–H bond at ∼3500 cm−1 as well as the characteristic for hydrogen bonding, broad absorbance between 3400 and 2900 cm−1, is present in all the spectra confirming the formation of the polymers (Fig. 2B). These results are in agreement with the solid-state magic angle spinning NMR. The thermo-gravimetric analysis proves the excellent thermal stability (>350 °C/623 K) for all three polymers; around 20% of the total mass is lost up to 350 °C temperature, and this can be attributed to the solvents residing in the ultra micropores of the polymers (Fig. 2C). Powder X-ray diffraction does not show any sharp peaks confirming the amorphous nature of the POFs (Fig. S1, ESI†). The FE-SEM images show a very homogeneous and similar morphology for all three polymers; they consist of spherical balls inter-grown into clusters (Fig. 2D–F and Fig. S2–S4, ESI†). TEM images also showed similar morphology with uniform micropores on the surface (Fig. S5, ESI†).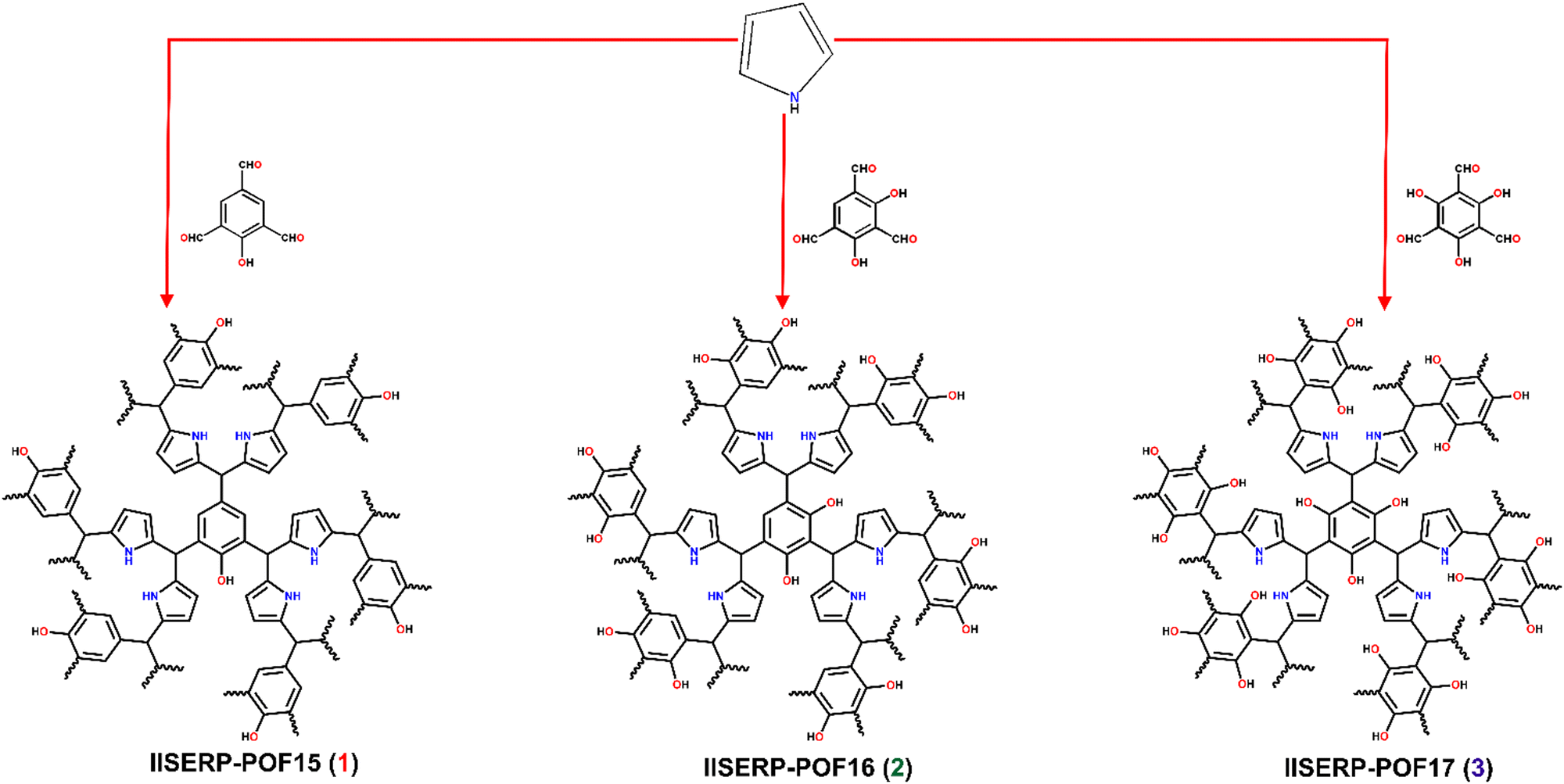 | ||
| Fig. 1 Synthesis scheme of 1, 2, and 3. | ||
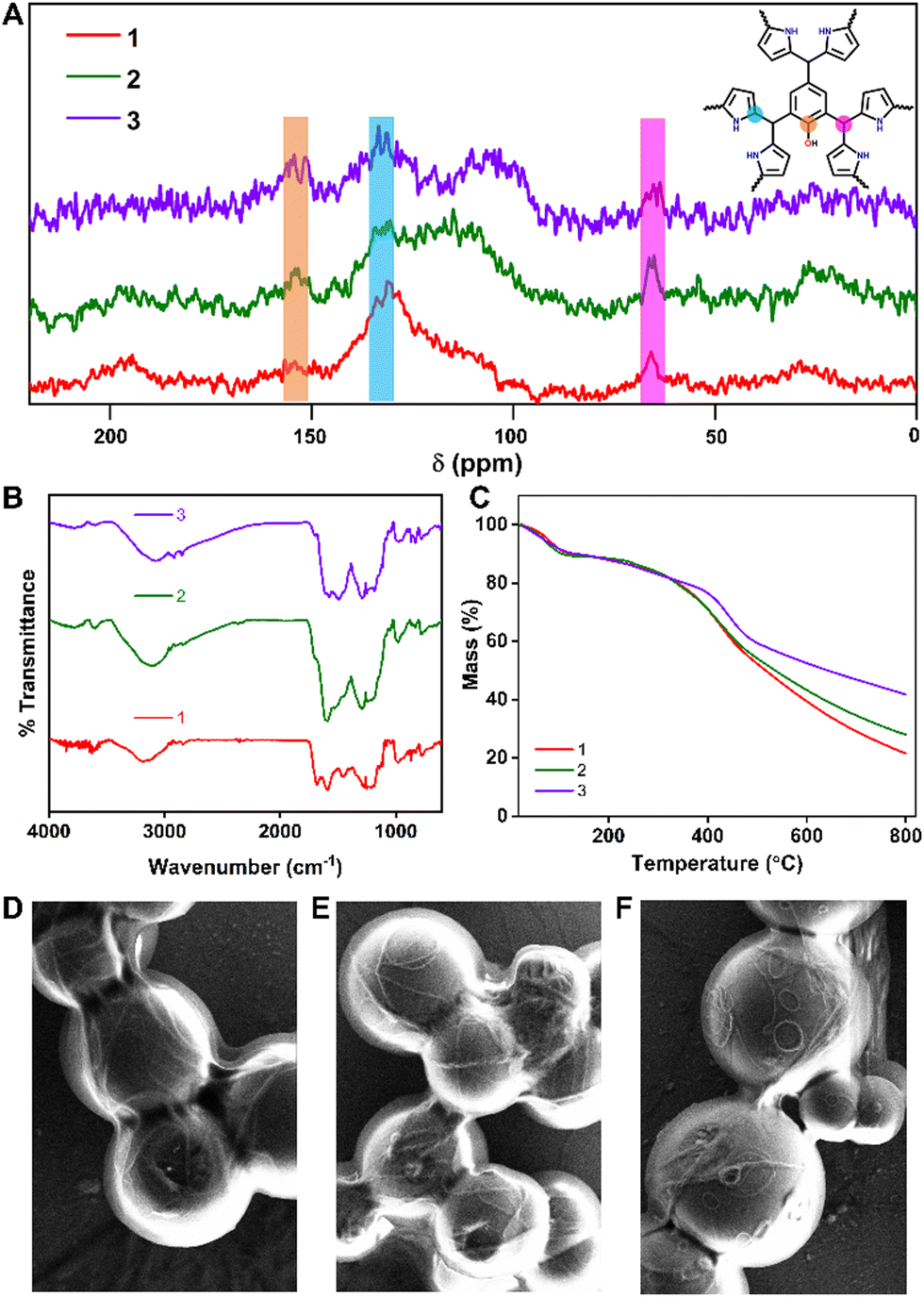 | ||
| Fig. 2 (A) Solid state NMR of POFs showing the characteristic peaks. (B) IR-spectra of the POFs showing the peaks corresponding to the different stretching frequencies. (C) Thermo-gravimetric analysis of the POFs showing the high thermal stability. (D)–(F) FE-SEM morphology of 1, 2 and 3 respectively. (SEM images with the scale bar are shown in the ESI†). | ||
The permanent porosity is established by 273 K CO2 adsorption isotherms; all three POFs show a Type I isotherm. 1, 2 and 3 show CO2 uptakes of 3, 2.6 and 2 mmol g−1 at 273 K, respectively (Fig. 3A–C). The BET fit to the isotherms yields a BET area of 277, 235 and 213 m2 g−1 and a Langmuir surface area of 345, 289, and 260 m2 g−1 for 1, 2, and 3, respectively (Fig. S6–S11, ESI†). There is a consistent decrease in the CO2 uptake capacity with the increase in the –OH groups in the polymers. The non-local density functional theory (NL-DFT) fit to the adsorption branch of the 273 K CO2 isotherm yields a pore size of 0.55 nm in all three cases. However, the pore volume reduced with the increase in the number of hydroxyl groups in the framework (Fig. 3D). Since the POFs are isostructural with the same pore size, a direct correlation between the CO2 uptake and the pore volume can be made. Hence to design better materials, it is not sufficient to have only ultra-micropores, but maintaining the pore volume on the higher side of the scale is also crucial. Based on this observation, an ideal case would be to have a material with small apertures (<0.6 nm), which allows only the selective gas molecules to enter through the molecular sieving effect, with a cage-like cavity of larger volumes that can collect large amounts of gas resulting in high working capacity and selectivity. The CO2 isotherms were also performed at 298 K and 313 K for all the POFs. The CO2 heat of adsorption was calculated by fitting the pure component isotherms to the virial model and nominal heat of adsorption values were obtained; 37.6, 35.7, and 31.9 kJ mol−1 for 1, 2 and 3, respectively, at zero coverage. They decrease linearly with an increase in the loading of gas molecules, signifying the pore-filling mode of adsorption and the absence of any specific strong binding site or the cooperativity among the guest molecules (Fig. 3E and Fig. S12–S14 and Table S1, ESI†). In order to estimate the ability of our POFs towards selective CO2 capture, we measured the pure component N2 and CH4 isotherms at ambient conditions and 0.11, 0.09 and 0.07 mmol g−1 of nitrogen and 0.55, 0.4 and 0.28 mmol g−1 of methane were adsorbed by 1, 2 and 3, respectively. The pure component adsorption isotherms were fitted using the Dual Site Langmuir (DSL) model, and IAST was employed to calculate the selectivity at different concentrations (Fig. S15–S23 and Table S2, ESI†). The CO2/N2 IAST selectivity for the flue gas composition was estimated to be 98, 222, and 171 for 1, 2, and 3, respectively. In the case of CO2/CH4, the selectivity for a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 composition is 17, 45, and 28 for 1, 2, and 3, respectively (Fig. 3F). These numbers are relatively lower compared to the CO2/N2 selectivity because of high methane uptakes as compared to nitrogen. To determine the CO2 working capacity of the polymers under the mixed gas stream (15CO2:85N2), ideal adsorbed solution theory (IAST) was employed. The mixed gas isotherms were simulated using the IAST + + software,38 which reveals that the saturation capacity of CO2 for a 15CO2:85N2 gas mixture is 0.87, 0.89 and 0.65 mmol g−1 for 1, 2 and 3, respectively (Fig. 4A).
:50 composition is 17, 45, and 28 for 1, 2, and 3, respectively (Fig. 3F). These numbers are relatively lower compared to the CO2/N2 selectivity because of high methane uptakes as compared to nitrogen. To determine the CO2 working capacity of the polymers under the mixed gas stream (15CO2:85N2), ideal adsorbed solution theory (IAST) was employed. The mixed gas isotherms were simulated using the IAST + + software,38 which reveals that the saturation capacity of CO2 for a 15CO2:85N2 gas mixture is 0.87, 0.89 and 0.65 mmol g−1 for 1, 2 and 3, respectively (Fig. 4A).
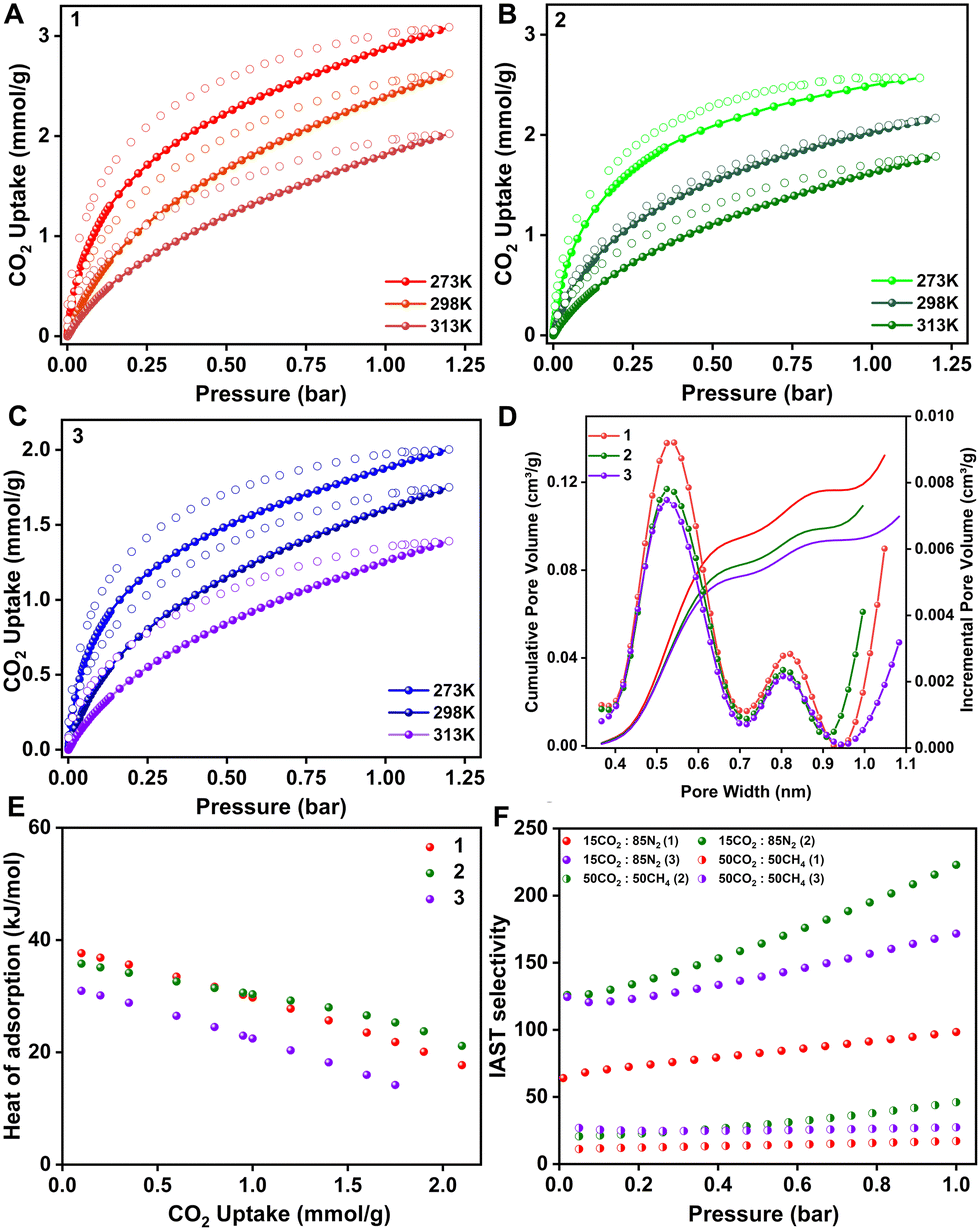 | ||
| Fig. 3 (A) Equilibrium CO2 adsorption isotherms on 1 at different temperatures. (B) Equilibrium CO2 adsorption isotherms on 2 at different temperatures. (C) Equilibrium CO2 adsorption isotherms on 3 at different temperatures. (D) Pore size and pore volume distribution of POFs obtained by NLDFT fit to the 273 K CO2 isotherm. (E) Heat of adsorption plot for POFs calculated using the Virial method. (F) IAST selectivity of POFs for CO2/N2 and CO2/CH4 gas mixtures. | ||
 | ||
| Fig. 4 (A) Binary CO2 and N2 adsorption isotherms simulated using IAST for 15CO2:85N2. (B) High resolution 273 K adsorption isotherm of 1 used in this diffusion modelling. Inset shows the CO2 self-diffusion coefficients from kinetics measurement. (C) 273 K CO2 adsorption isotherms performed on samples exposed to different conditions. (D) Gravimetric CO2–N2 cycling performed on 1. | ||
Practical CO2 separation is carried out under dynamic conditions; the adsorbent should have facile diffusion within the polymer. To determine the adsorption kinetics, the CO2 self-diffusion coefficient was calculated as a function of CO2 loading and it comes out to be 5 × 10−9 m2 s−1 (Fig. 4B), which is much superior to the traditional sorbents such as zeolites. Stability is another key aspect that determines the quality of the adsorbent. The flue gas from which we aim to capture CO2 consists of water vapors, and acidic gases such as SOx and NOx in addition to CO2 and N2.39 Since in the flue gas, CO2 and N2 are mixed with acidic gases, demonstrating the stability of adsorbents under such harsh acidic conditions would be meaningful. This is a key property of all-organic C–C bonded porous polymers which could give them an edge over MOFs or other inorganic sorbents. To verify this, we carried out 273 K CO2 adsorption on the POF samples, which were soaked in 2 M HCl for 24 hours. And no loss in the saturation CO2 capacity at 273 K was observed (Fig. 4C). Additionally, the POF samples exposed to 99% RH at 50 °C for 6 hours did not show any fall in the CO2 uptake compared to the pristine POFs (Fig. 4C), revealing their hydrolytic stability. The cycling stability of the best-performer, 1, was established by the TGA cycling experiment, which shows that the CO2 working capacity does not fall even after 12 consecutive adsorption–desorption cycles (Fig. 4D).
Water vapor in the flue gas can decompose the adsorbent if it is unstable in humid conditions.40 Since water is a polar molecule, it can interact more strongly with the binding sites present in the adsorbent, and hence the positions which are supposed to be occupied by the CO2 molecules can be taken up by the water molecules, resulting in the loss of working capacity.41,42 To avoid this, the humid flue gas stream is pre-treated to adsorb water before it is fed to the CO2-capturing column, but this adds a parasitic load on the CCS process.43 Hence if the adsorbent has water-repelling properties, it would be an additional advantage.37,44 To decipher this, water sorption isotherms were carried out at two different temperatures for all the POFs, and a linear isotherm with a low uptake for 1 was observed. However, the uptake increased going from 1 to 3; also, the shape of the isotherm changed from near-linear to Langmuir, depicting the stronger interaction of water molecules with the framework (Fig. 5A). Notably, 1, having the least number of OH functionalities, exhibits the most favorable CO2 adsorption characteristics and has the least interaction with water. In order to check the surface hydrophobicity, we performed contact angle measurements. We observed that 1 is the most hydrophobic and 3 is the least, and 2 lies in between, all with a contact angle of >90 (Fig. 5B–D). This confirms that 1 has the most affinity for water, whereas 3 has the least. The plausible reason is that the number of framework OH groups capable of hydrogen bonding with the water increases from 1 to 3. Hence an increased affinity towards the water in 3 is observed. This could also mean that the hydrophobicity and hydrophilicity can be tuned in these POFs by fixing the number of water-attracting/repelling groups.
 | ||
| Fig. 5 (A) Water sorption isotherms for 1, 2, and 3. (B)–(D) Water contact angle of 1, 2, and 3. | ||
Conclusions
In conclusion, we have synthesized three ultramicroporous POFs with similar pore sizes and different pore volumes in a catalyst-free manner. The difference in the pore volumes leads to various working capacities for CO2. They possess moderate HOA and good CO2/N2 and CH4 IAST selectivity. Water sorption isotherms and contact angle measurements on the three polymers revealed the hydrophilic/hydrophobic character of the materials, which is correlated with the number of phenolic –OH groups present in the material. The C–C bonded framework makes the polymers stable to harsh acidic conditions as well as under a high relative humidity of 99%, which is relevant to the humid flue gas capture. In the future, we aim to synthesize polymers with optimal pore sizes and the pore volume identified in this work, to access smooth adsorption–desorption kinetics. Also, making a comparable polymer with no –OH functionality would give more insights into the role of the phenolic groups in providing CO2 selectivity over water. Work is in progress.Experimental
Synthetic procedures
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge IISER Pune for the financial support. H. D. S., P. S. thank CSIR for the funding. D. R. thanks IISER Pune for the funding. We gratefully acknowledge the Air Force Office of Scientific Research under award number FA2386-21-1-4022 for the funding and support. We acknowledge the SERB (CRG/2021/008250), the MHRD-FAST (MHRD Project 150 (F. No. 5-5/2014-TS-VII & F. No. 22-2/2016-TS-II/TC)) program, the MHRD-STARS[STARS1/278] program and the “DST-Nanomission under the Thematic Unit Program” (EMR/2016/003553).References
- W. Wang, M. Zhou and D. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC.
- https://climate.nasa.gov/vital-signs/carbon-dioxide/ .
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- B. Li, Y. Zhang, R. Krishna, K. Yao, Y. Han, Z. Wu, D. Ma, Z. Shi, T. Pham, B. Space, J. Liu, P. K. Thallapally, J. Liu, M. Chrzanowski and S. Ma, J. Am. Chem. Soc., 2014, 136, 8654–8660 CrossRef CAS PubMed.
- A. Chowdhury, S. Bhattacharjee, R. Chatterjee and A. Bhaumik, J. CO2 Util., 2022, 65, 102236 CrossRef CAS.
- N. Das, R. Paul, D. Q. Dao, R. Chatterjee, K. Borah, S. Chandra Shit, A. Bhaumik and J. Mondal, ACS Appl. Nano Mater., 2022, 5, 5302–5315 CrossRef CAS.
- X. Zhu, S. M. Mahurin, S.-H. An, C.-L. Do-Thanh, C. Tian, Y. Li, L. W. Gill, E. W. Hagaman, Z. Bian, J.-H. Zhou, J. Hu, H. Liu and S. Dai, Chem. Commun., 2014, 50, 7933–7936 RSC.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- J. Lu and J. Zhang, J. Mater. Chem. A, 2014, 2, 13831–13834 RSC.
- D. Yuan, W. Lu, D. Zhao and H. C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed.
- C. F. Martín, E. Stöckel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475–5483 RSC.
- K. Huang, F. Liu and S. Dai, J. Mater. Chem. A, 2016, 4, 13063–13070 RSC.
- X. Zhang, Y.-Z. Lv, X.-L. Liu, G.-J. Du, S.-H. Yan, J. Liu and Z. Zhao, RSC Adv., 2016, 6, 76957–76963 RSC.
- A. K. Sekizkardes, P. Wang, J. Hoffman, S. Budhathoki and D. Hopkinson, Mater. Adv., 2022, 3, 6668–6686 RSC.
- A. Roy, N. Haque, R. Chatterjee, S. Biswas, A. Bhaumik, M. Sarkar and S. M. Islam, New J. Chem., 2023, 47, 6673–6684 RSC.
- S. N. Bhaduri, D. Ghosh, S. Chatterjee, R. Biswas, R. Banerjee, A. Bhaumik and P. Biswas, Inorg. Chem., 2022, 61, 18390–18399 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- B. Adhikari and S. Majumdar, Prog. Polym. Sci., 2004, 29, 699–766 CrossRef CAS.
- C. R. McNeill and N. C. Greenham, Adv. Mater., 2009, 21, 3840–3850 CrossRef CAS.
- V. Gautam, D. Rand, Y. Hanein and K. Narayan, Adv. Mater., 2014, 26, 1751–1756 CrossRef CAS PubMed.
- C. Yang, H. Wei, L. Guan, J. Guo, Y. Wang, X. Yan, X. Zhang, S. Wei and Z. Guo, J. Mater. Chem. A, 2015, 3, 14929–14941 RSC.
- D. Rase, R. Illathvalappil, H. D. Singh, P. Shekhar, L. S. Leo, D. Chakraborty, S. Haldar, A. Shelke, T. G. Ajithkumar and R. Vaidhyanathan, Nanoscale Horiz., 2023, 8, 224–234 RSC.
- M. Niu, H. Yang, X. Zhang, Y. Wang and A. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 17312–17320 CrossRef CAS PubMed.
- C. Chen, W.-J. Son, K.-S. You, J.-W. Ahn and W.-S. Ahn, Chem. Eng. J., 2010, 161, 46–52 CrossRef CAS.
- J.-B. Lin, T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Durekova, F. Akhtar, R. K. Mah, O. Ghaffari-Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. K. H. Shimizu, Science, 2021, 374, 1464–1469 CrossRef CAS PubMed.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- P. Arab, M. G. Rabbani, A. K. Sekizkardes, T. İslamoğlu and H. M. El-Kaderi, Chem. Mater., 2014, 26, 1385–1392 CrossRef CAS.
- K. S. Song, P. W. Fritz and A. Coskun, Chem. Soc. Rev., 2022, 51, 9831–9852 RSC.
- B. Lopez-Iglesias, F. Suárez-García, C. Aguilar-Lugo, A. González Ortega, C. Bartolomé, J. M. Martínez-Ilarduya, J. G. de la Campa, Á. E. Lozano and C. Álvarez, ACS Appl. Mater. Interfaces, 2018, 10, 26195–26205 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- P. Katekomol, J. R. M. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- X. Zhu, C. Tian, G. M. Veith, C. W. Abney, J. R. M. Dehaudt and S. Dai, J. Am. Chem. Soc., 2016, 138, 11497–11500 CrossRef CAS PubMed.
- S. Zulfiqar, M. I. Sarwar and C. T. Yavuz, RSC Adv., 2014, 4, 52263–52269 RSC.
- J. Yan, B. Zhang, S. Guo and Z. Wang, ACS Appl. Nano Mater., 2021, 4, 10565–10574 CrossRef CAS.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2011, 23, 1818–1824 CrossRef CAS.
- S. Nandi, U. Werner-Zwanziger and R. Vaidhyanathan, J. Mater. Chem. A, 2015, 3, 21116–21122 RSC.
- S. Lee, J. H. Lee and J. Kim, Korean J. Chem. Eng., 2018, 35, 214–221 CrossRef CAS.
- J.-Y. Lee, T. C. Keener and Y. J. Yang, J. Air Waste Manage. Assoc., 2009, 59, 725–732 CrossRef CAS PubMed.
- Y. Li and R. T. Yang, Langmuir, 2007, 23, 12937–12944 CrossRef CAS PubMed.
- L. Joos, J. A. Swisher and B. Smit, Langmuir, 2013, 29, 15936–15942 CrossRef CAS PubMed.
- J. G. Bell, M. J. Benham and K. M. Thomas, Energy Fuels, 2021, 35, 8102–8116 CrossRef CAS.
- H. A. Evans, D. Mullangi, Z. Deng, Y. Wang, S. B. Peh, F. Wei, J. Wang, C. M. Brown, D. Zhao, P. Canepa and A. K. Cheetham, Sci. Adv., 2022, 8, eade1473 CrossRef CAS PubMed.
- T. Wu, L. Shen, M. Luebbers, C. Hu, Q. Chen, Z. Ni and R. I. Masel, Chem. Commun., 2010, 46, 6120–6122 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00218g |
| This journal is © The Royal Society of Chemistry 2023 |