
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acceptorless dehydrogenation of 4-methylpiperidine by supported pincer-ligated iridium catalysts in continuous flow†
Kaushik
Chakrabarti
a,
Alice
Spangenberg
a,
Vasudevan
Subramaniyan
 a,
Andreas
Hederstedt
a,
Omar Y.
Abdelaziz
b,
Alexey V.
Polukeev
a,
Reine
Wallenberg
a,
Christian P.
Hulteberg
b and
Ola F.
Wendt
*a
a,
Andreas
Hederstedt
a,
Omar Y.
Abdelaziz
b,
Alexey V.
Polukeev
a,
Reine
Wallenberg
a,
Christian P.
Hulteberg
b and
Ola F.
Wendt
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: ola.wendt@chem.lu.se
bDepartment of Chemical Engineering, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden
First published on 27th July 2023
Abstract
Finding alternative and sustainable ways to produce, store and convert energy is key for reducing fossil fuel-based CO2 emissions. In this transformation, hydrogen for energy storage and hydrogen-powered fuel cells for energy conversion can play important roles. However, storage of hydrogen itself is difficult and the concept of reversible liquid organic hydrogen carriers (LOHCs) has been proposed given the advantages of using liquid storage materials. A key part in the adaption of LOHCs is the catalyst design for efficient dehydrogenation of these hydrogen-carrying species. In this study, the use of silica- and alumina-supported POCOP–Ir systems for gas phase acceptorless dehydrogenation of 4-methylpiperidine (an LOHC with 6.1 wt% hydrogen) is investigated in a continuous-flow system with a high TON. To increase stability and reactivity, a new POCOP–Ir complex with two anchors was designed and found to be highly active in the dehydrogenation of 4-methylpiperidine with ∼91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 turnovers in 45 h. In addition, this catalyst showed a maintained activity with a TOF of 1684 h−1 after 45 h.
000 turnovers in 45 h. In addition, this catalyst showed a maintained activity with a TOF of 1684 h−1 after 45 h.
Introduction
The problems associated with fossil fuels, particularly CO2 emissions, drive the world energy sector towards alternative ways to produce, store and convert energy. In this process, hydrogen-powered fuel cells are very promising, as they are non-polluting (only water is formed upon hydrogen combustion in a fuel cell to make electricity) and hydrogen possesses a high energy density by weight. However, hydrogen containment is difficult as it is a gas under atmospheric conditions and therefore has a very low volumetric energy density. A suggested solution to store and transport hydrogen is the use of liquid organic hydrogen carriers (LOHCs),1 liquids with high H2 wt% that replace the handling of hydrogen gas with that of liquids that are compatible with the current distribution system.2,3 LOHCs are covalently bonded liquid organic compounds with a high wt% of hydrogen storage capacity.4 These systems function by cycles of catalysed hydrogenation and dehydrogenation, loading the LOHC at some hydrogen source and unloading it when and where hydrogen gas is needed (Fig. 1).5–7 Therefore, a good LOHC needs a high H2 wt% as well as the ability to undergo both dehydrogenation and regeneration selectively and easily.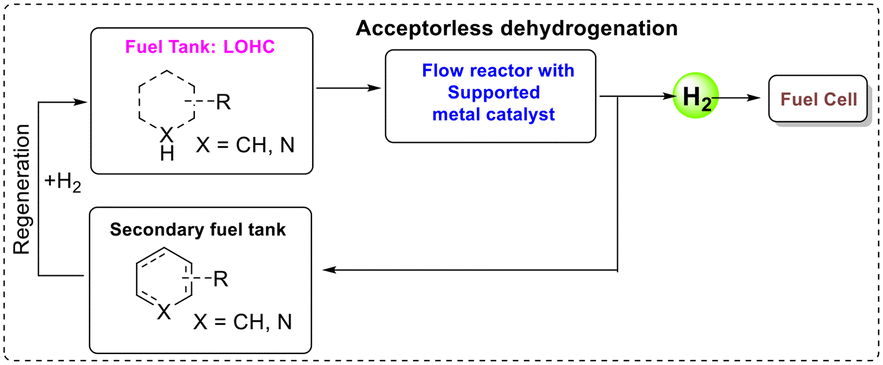 | ||
| Fig. 1 Concept of LOHCs under continuous-flow conditions. | ||
Alkane/arene LOHC systems such as cyclohexane/benzene8 and methylcyclohexane/toluene9 have been explored for these purposes, as they generally have high hydrogen carrying capacities and boiling points. However, their large enthalpies of dehydrogenation require excessive temperatures for the reaction to proceed. Recently, different short-chain alcohols (such as methanol, ethanol, isopropanol, etc.) and formic acid, glycerol, etc. were also employed for this purpose,10 but they have disadvantages, such as low boiling point and low hydrogen content. Nitrogen-containing heterocyclic compounds have shown some promise as LOHCs, as they tend to require less harsh conditions for their dehydrogenation due to their lower reaction enthalpies.11,12 The inclusion of the nitrogen atom weakens the adjacent C–H bonds and replaces one C–H bond with a weaker N–H analogue.13 Carbazoles,14 pyridines,15 pyrazines,16 indoles,17 phenazines18 and more, along with their hydrogen-rich analogues, have been investigated for LOHC purposes with some success. They have good gravimetric hydrogen-carrying capacities similar to the corresponding hydrocarbons.
A key part in the adaption of LOHCs is the catalyst design for efficient dehydrogenation of these hydrogen-carrying species. Transition metal-mediated dehydrogenation reactions have been extensively explored using various homogeneous catalytic systems.19–22 Pincer iridium complexes exhibit high activity for acceptorless dehydrogenation reactions.23–26 However, high temperature decomposition and problems of recyclability of pincer complexes in homogeneous systems are drawbacks preventing any industrial implementation. In order for LOHCs to be a practical part of existing and future infrastructure, it is probably necessary for hydrogen loading and unloading to occur in a continuous-flow-system, similar to the use of traditional fuels. This requires developing heterogeneous catalyst systems for maintaining the reactant and catalyst in separate phases, such as a solid catalyst with a liquid or gaseous reactant.27–29 A solid or solid-bound catalyst allows for the flow of substrate through the catalyst, continuously removing the formed products. In a continuous-flow reactor system, LOHCs need to pass through the catalytic reactor where hydrogen is released and directed to a fuel cell; later at the refilling station, the oxidized LOHCs can be converted back to a hydrogen-rich form. In such a process, the energy requirement for the dehydrogenation reactions will be fulfilled by a heat exchanger between the engine and the dehydrogenation reactor.
Huang et al. reported transfer dehydrogenation of alkanes using alumina-supported iridium pincer systems which showed good reactivity compared to a batch process in homogeneous medium.30 In 2018, Celik, Goldman and co-workers developed a silica-supported iridium catalytic system for continuous-flow acceptorless dehydrogenation of butane (Scheme 1).27 Later, the same group reported gas-phase butane transfer dehydrogenation by a silica-supported PCP–Ir catalyst.31 Recently, our group explored a silica-supported POCOP–Ir system for acceptorless isopropanol dehydrogenation with a high TON (Scheme 1),32 and also investigated mechanistic aspects of this reaction.33 Inspired by these excellent result in alcohol dehydrogenation, we targeted the use of N-heterocycles as LOHCs in a continuous-flow system, choosing 4-methylpiperidine/4-methylpyridine as the carrier with its substantially higher hydrogen loading of 6.1 wt%. To the best of our knowledge, continuous-flow acceptorless dehydrogenation of N-heterocycles as LOHCs using supported pincer-ligated catalytic systems has not been reported previously. Chakraborty et al. reported a homogeneous PNP–iron complex (5 mol%) which catalysed dehydrogenation of N-heterocycles using toluene or xylene as solvent,33 and Fujita et al. described 2,5-dimethylpiperazine dehydrogenation using a bidentate Ir catalytic system.16 However, most of these homogeneous systems used solvent in the dehydrogenation reaction lowering the overall loading. The methylpiperidine/methylpyridine LOHC system is known in the literature34 and was shown to be the most effective N-heterocyclic system in one study by Biniwale and co-worker.15 They used 5 wt% Pt/ACC catalyst for the dehydrogenation of 4-methylpiperidine with 23.7% conversion after 200 min at 350 °C. Recently, Oh et al. synthesised 2-(N-methylbenzyl)pyridine (MBP) and found that Pt/C or Pd/C catalyst are very effective for its dehydrogenation at 230–270 °C with 60–90% conversion.13 However, a three step synthesis of MBP and its hydrogenation to get H12-MBP is a challenging. Here we present a new supported pincer iridium catalyst that can dehydrogenate 4-methylpiperidine with excellent activity and stability.
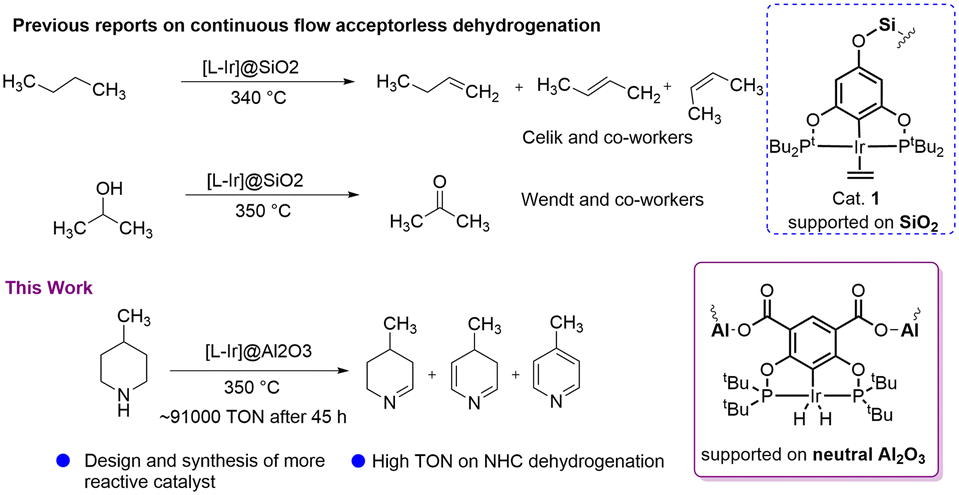 | ||
| Scheme 1 State of the art of the current work. | ||
Results and discussion
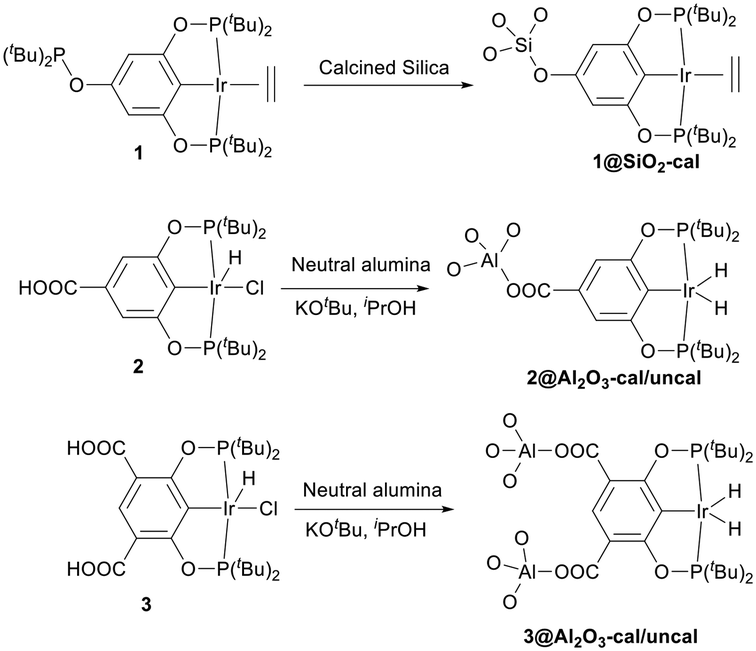
In pincer-chemistry, it is known that introducing backbone substitution may be used as a tool to tune catalytic reactivity.27,30 In addition, this backbone substitution may also act as an anchor for the metal complexes to bind with a heterogeneous support.32 Thus, longer and potentially multiple anchors can reduce any hydrophobic interactions with the surface and increase bond strength to improve the activity and stability of the catalytic system (Fig. 2). It was previously reported by our research group that silica-supported complex 1 and alumina supported complex 2 show excellent reactivity for the dehydrogenation of isopropanol in a continuous-flow system, but the alumina-based system was less active.32 However, there were problems with stability and this motivated us to develop a new iridium pincer complex with multiple anchors (complex 3).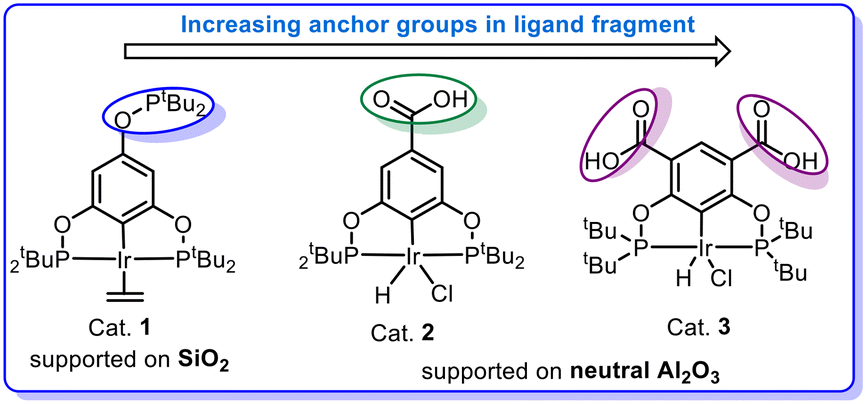 | ||
| Fig. 2 Design of new iridium pincer complex. | ||
Complex 3 was synthesized by modifying the existing protocol for complex 2. Ligand L3 was synthesized from the benzyl ester of 4,6-dihydroxyisophthalic acid via phosphorylation of the hydroxy group with di-tert-butylchlorophosphine (Scheme 2).
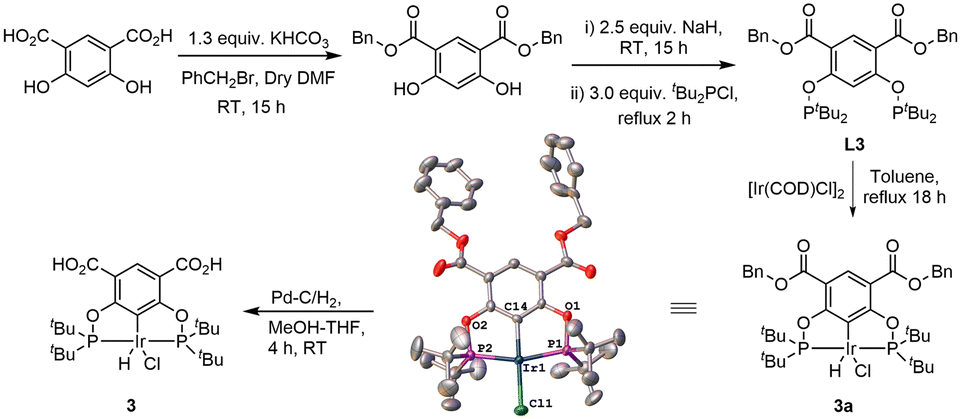 | ||
| Scheme 2 Synthetic route to 3 including the molecular structure of 3a. | ||
Precatalyst 3a was then obtained by refluxing L3 with 0.5 equiv. [Ir(COD)Cl]2 in dry toluene for 18 h. The structure was confirmed through an SXRD experiment, see ESI for details. Finally, 3 was synthesized from 3a using Pd–C/H2 in MeOH–THF (1:1) mixture of solvents. 3a and 3 were fully characterized by 1H, 13C and 31P NMR spectroscopy, IR spectroscopy and microanalysis.
Catalyst 1 was supported on calcined silica using the incipient wetness impregnation technique as described in the literature,27,32 whereas catalysts 2 and 3 were supported on alumina (calcined and uncalcined) by heating them for 2.5 h at 80 °C in isopropanol with KOtBu and the support, generating the active catalyst. After two hours, the solution had turned from a deep orange colour to almost colourless while the alumina changed colour from white to orange or pale orange, indicating immobilization of the complex and formation of catalysts 2@Al2O3-cal/uncal and 3@Al2O3-cal/uncal (Scheme 3). The BET analysis of 1@SiO2-cal and 2@Al2O3-cal has been reported previously by us.32 The BET surface area of the calcined alumina was 154 m2 g−1, and after supporting complex 3, the surface area of 3@Al2O3-cal decreased to 137 m2 g−1. The pore volume also decreased from 0.47 cm3 g−1 to 0.40 cm3 g−1, which indicates the presence of the catalyst also in the mesopores of the support. The same trend was observed for 3@Al2O3-uncal, where the BET surface area of the uncalcined alumina was 158 m2 g−1, and after supporting complex 3, the surface area of 3@Al2O3-uncal decreased slightly to 156 m2 g−1.
 | ||
| Scheme 3 Synthetic route for the supported catalysts. | ||
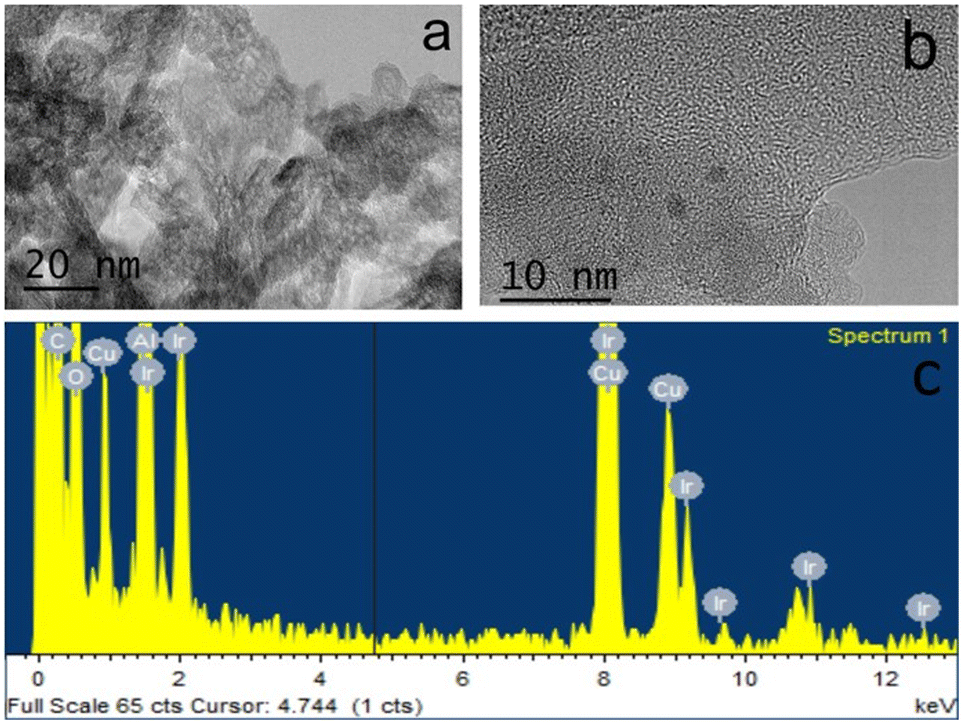
In a typical experiment, 5 mg of catalyst was supported on 150 mg of silica or 400 mg of alumina support, giving approximately 3.3 wt% of catalyst present in the silica and 1.25 wt% in the alumina support. The theoretical Ir metal content for 3@Al2O3-uncal is 0.34 wt% which matched the actual iridium content of 0.33 wt% as measured with ICP-MS analysis. 1@SiO2-cal and 2@Al2O3-cal were previously characterised.27,32 The excellent agreement between theoretical and measured iridium content suggests an effective impregnation of complex 3 on the supports. After formation of 3@Al2O3-cal/uncal, the carbonyl band at 1679 cm−1 in the IR spectrum of complex 3 undergoes a bathochromic shift, suggesting an interaction with alumina. The TEM and XEDS spectra of the prepared catalyst 3@Al2O3-cal/uncal showed an even distribution of Ir over the support and the high-resolution TEM images suggest that there is no Ir cluster formation (Fig. 3a and b). The XEDS spectra in Fig. 3c and d shows the presence of well dispersed Ir on the catalyst support.
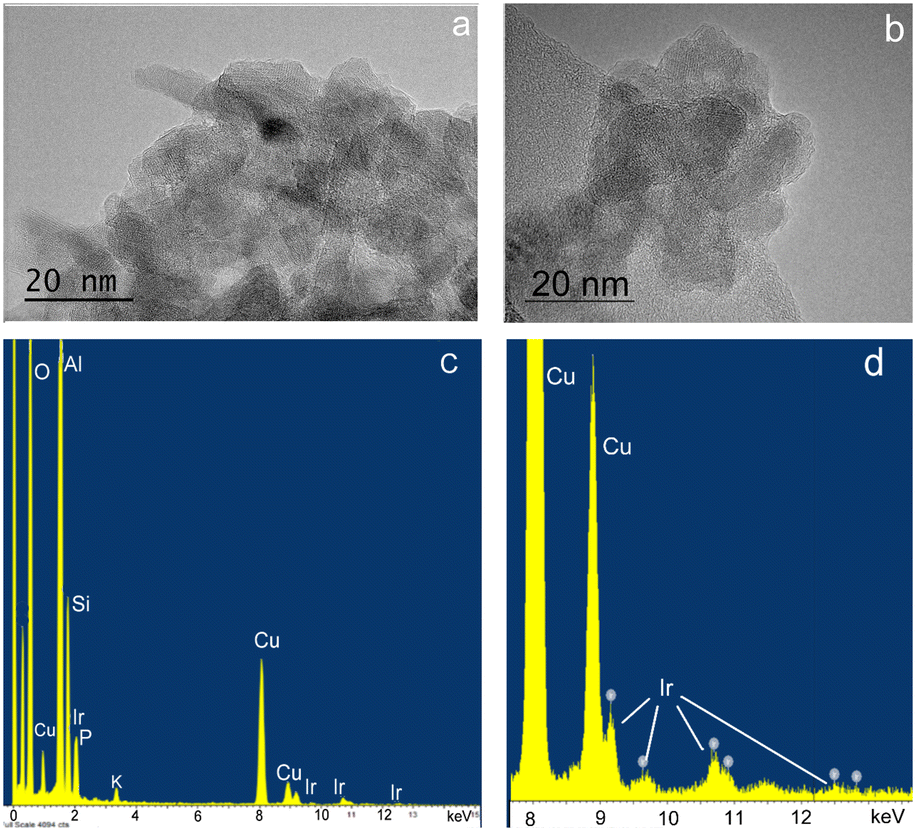 | ||
| Fig. 3 HRTEM images and XEDS spectra of 3@Al2O3-uncal (a and c) and of 3@Al2O3-cal (b and d). The full spectrum is shown in c) and expanded around the Ir-family L-peaks in d. The Cu peaks are from the sample grid. | ||
4-Methylpiperidine, with its high hydrogen loading and fairly low endothermicity, was chosen as the substrate to compare the activity and stability of the catalysts, as it had shown promising results in preliminary experiments. Both substrate and product species have high boiling points at room temperature and normal pressure, making collection easy as evaporation is low. Initially, acceptorless dehydrogenation of 4-methylpiperidine was assessed in continuous flow using the five different catalysts at 325 °C and 0.09 mL min−1 flow rate (Scheme 4 and Fig. 4).
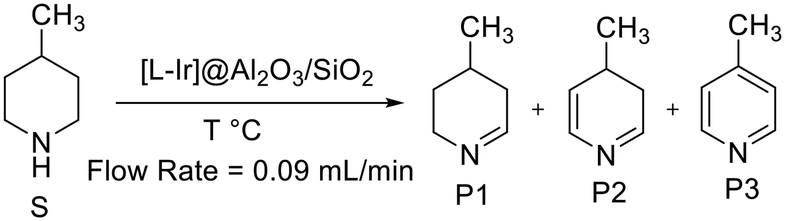 | ||
| Scheme 4 Dehydrogenation of 4-methylpiperidine with different catalysts. | ||
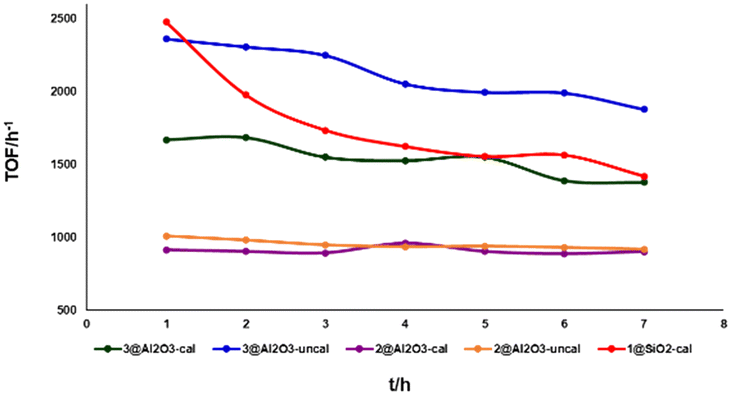 | ||
| Fig. 4 Reactivity of different catalysts at 325 °C at 0.09 mL min−1 flow rate. | ||
Initially, 1@SiO2 showed a good activity with a TOF of ∼2500 h−1. However, it loses around 50% of its reactivity within the 7 h duration of the experiment. 2@Al2O3-cal and 2@Al2O3-uncal showed moderate but steady reactivity during the initial 7 h reaction time. 3@Al2O3-cal and 3@Al2O3-uncal displayed both higher TOFs and higher total TONs after 7 h under these reaction conditions compared to their counterparts based on 2. The reactivity and stability of 3@Al2O3-uncal overall outperformed the other catalysts in the initial experiments. This supports the notion that the introduction of multiple anchor group to the ligand backbone is beneficial for improving catalytic performance. The higher activity of 3@Al2O3-uncal compared to the calcined alumina analogue may be due to the presence of more hydroxy groups at the surface of the uncalcined alumina material, binding the catalyst to the support in the desired fashion.
Having shown the conversion of 4-methylpiperidine (S) to mono-, di- and tri- dehydrogenated compounds (P1, P2, and P3), the flow reactor was modified to capture the produced hydrogen gas (Fig. S2†). The boiling points of the substrate and the three products are very high, and no additional cooling was necessary to condense the organic contents. The collected gas from the flow reaction was analysed by Raman spectroscopy (Fig. S3 and S4†), showing that the gas is pure hydrogen.35 In addition, the volume of gas produced was in good agreement with the amount expected based on the quantification of dehydrogenated products (see ESI† for further details). Feeding the hydrogen produced in the flow reaction directly to a small fuel cell showed no decrease in power over 6 h (Fig. S5†). These experiments confirm the quantitative formation of hydrogen gas from the acceptorless dehydrogenation reactions, and that the produced hydrogen is sufficiently pure to power a small fuel cell over an extended period of time.
It is expected that temperature has a substantial effect on the performance, both in terms of reactivity and stability. Using the same 0.09 mL min−1 flow rate it was found that for both 3@Al2O3-cal and 3@Al2O3-uncal the TOFs (h−1) and TONs (7 h) were improved by increasing the temperature from 300 °C to 350 °C (Fig. 5). It is noteworthy that, at lower temperature the reactivity of both catalysts is constant, whereas at higher temperature the reactivity slowly declines. This suggests that at higher temperature deactivation of the catalyst is significant, which is also supported by ICP-MS data for 3@Al2O3-uncal at different temperature after 8 h of flow experiment. The ICP-MS analyses of the spent catalyst after 8 h revealed the Ir content to be 0.23 wt% at 300 °C, 0.14 wt% at 325 °C and 0.10 wt% at 350 °C for the reaction with 3@Al2O3-uncal. This indicates that at low temperature the iridium leaching is minimized and thus the reactivity is more or less constant. We know from previous work that there is some initial leaching of physisorbed species probably explaining the decrease in metal content also at the lowest temperature.
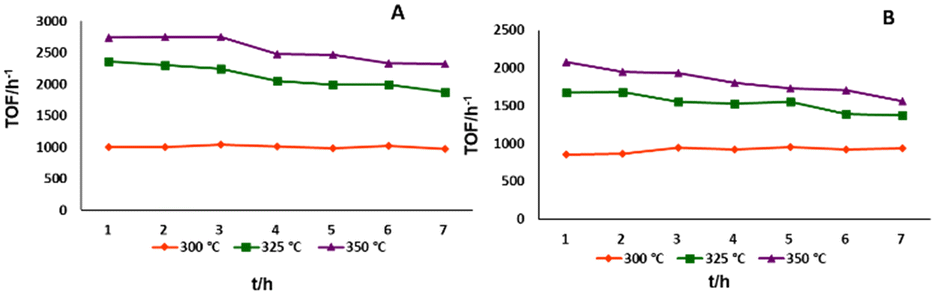 | ||
| Fig. 5 Reactivity of 3@Al2O3-uncal (A) and 3@Al2O3-cal (B) at 300 °C, 325 °C and 350 °C. | ||
Selectivity is one of the major issues in the dehydrogenation of 4-methylpiperidine (S) as it was found to occur in three consecutive steps giving a product mixture of three distinct species (P1, P2 and P3), two of which are partially dehydrogenated intermediates (P1 and P2) with P3 being the fully aromatized 4-methylpyridine. P2 consistently made up 1–2% of the reaction mixtures, with the higher concentrations (around 2%) being observed only at lower temperatures while at higher temperatures (350 °C and 325 °C) the concentration of P2 drops to <1%. The other two species, P1 and P3, were observed to constitute comparatively large fractions of the product mixtures, both increasing with temperature and with P1 consistently being the major product. This clearly indicates that the 1st dehydrogenation of 4-methylpiperidine (S), occurring over an N–C bond to produce P1, is relatively fast and followed by a comparatively slow step. The product of this slow step (P2) is then quickly dehydrogenated to form P3.
The selectivity of 2@Al2O3-cal, 3@Al2O3-cal and 3@Al2O3-uncal towards the fully aromatized product P3 over P1 and P2 were evaluated at three different temperatures and the results are summarized in Fig. 6 and Scheme 4. As can be seen, high temperatures drive the reaction towards P3 as dehydrogenation rates increase. The temperature effect on dehydrogenation of LOHCs is well explored in literature. The dehydrogenation of H12-MBP with Pt/C or Pd/C catalyst increased with higher temperature which was observed by Suh and co-workers.13
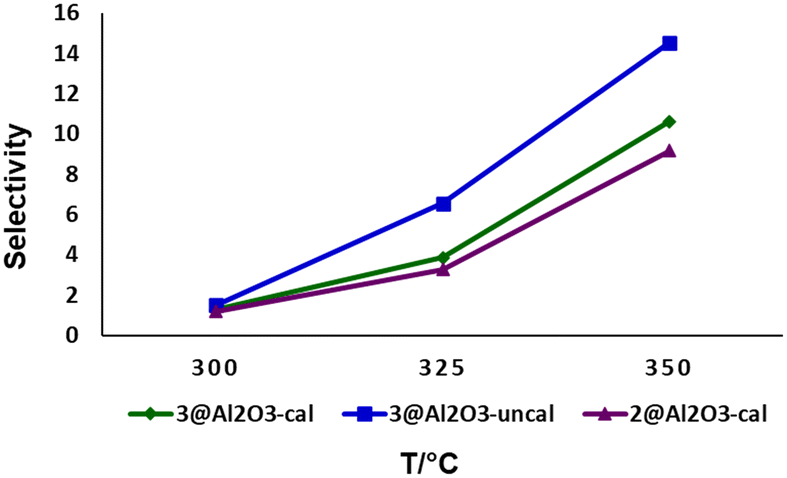 | ||
| Fig. 6 Selectivity for P3 at different temperatures using various catalysts (selectivity = [P3/(P1 + P2 + P3)] × 100), flow rate = 0.09 mL min−1. | ||
Selectivity for P3 over P1 and P2 also depended on the flow rate of 4-methylpiperidine through the reactor. Lower flow rates increase the contact time of 4-methylpiperidine with the catalyst, leading to a higher degree of dehydrogenation (Fig. 7). This phenomenon was observed for 2@Al2O3-cal, 3@Al2O3-cal and 3@Al2O3-uncal at 325 °C using flow rates between 0.02–0.2 mL min−1. It can be seen from Fig. 7A that at lower flow rates the selectivity for P3 is higher for all three catalysts. This flow rate dependence was also observed for 3@Al2O3-uncal at 350 °C (Fig. 7B).
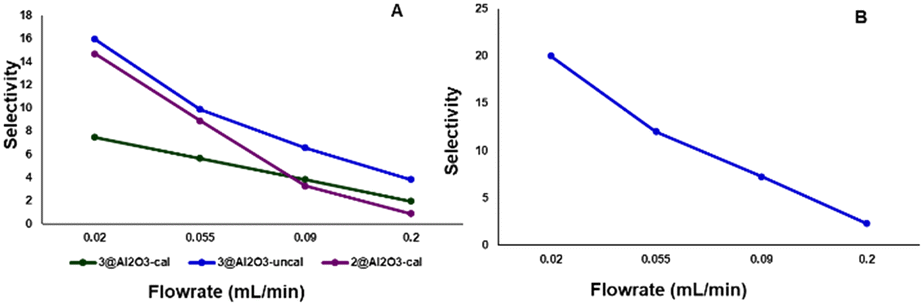 | ||
| Fig. 7 A) Selectivity for P3 at different flow rates, obtained using 2@Al2O3-cal, 3@Al2O3-cal and 3@Al2O3-uncal at 325 °C (left). B) Selectivity for P3 at different flow rates obtained using 3@Al2O3-uncal at 350 °C (right). | ||
Furthermore, it was observed that the initial TOF is strongly dependent on the flow rates of 4-methylpiperidine. As previously observed,32 the TOF is higher at higher flow rates as more 4-methylpiperidine is passed through the reactor, but the reverse is observed in case of conversion, where this decreases with higher flow rates (Fig. 8). Thus, at lower flow rates the conversion as well as the selectivity is higher, and the opposite is true for the TOF.
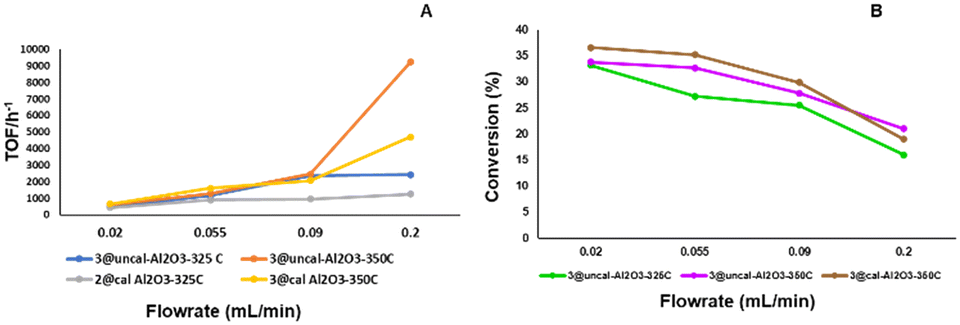 | ||
| Fig. 8 A) Initial TOF (h−1) at different flow rates, obtained using 2@Al2O3-cal-325 °C, 3@Al2O3-cal-350 °C, 3@Al2O3-uncal-325 °C and 3@Al2O3-uncal-350 °C (left). B) Initial conversion (%) at different flow rates, obtained using 3@Al2O3-cal-350 °C, 3@Al2O3-uncal-325 °C and 3@Al2O3-uncal-350 °C (right). | ||
In order to investigate the stability over longer periods and obtain an accumulated TON for 3@Al2O3-uncal, a flow reaction was performed for 45 h at 350 °C and 0.10 mL min−1 flow rate. The total TON for this experiment was ∼91000, while the TOF dropped from 2750 h−1 (after 1 h) to 1684 h−1 (after 45 h) and the selectivity dropped by 37%. Nevertheless, the catalyst showed substantial activity towards the end. According to ICP-MS analysis, the supported catalyst 3@Al2O3-uncal contained 0.07 wt% of iridium after 45 h, whereas, after 8 h the iridium content was around 0.10 wt% (compared to 0.33 wt% for the fresh catalyst). This indicates that leaching of the catalyst primarily takes place in the beginning, which is also suggested by the ICP-MS analysis of the initial fraction of the liquid mixture (0.17 wt% of iridium) obtained during the flow reaction (first hour fraction) and in agreement with our previous findings.32 A blank experiment using only calcined Al2O3 at 0.10 mL min−1 at 350 °C showed no conversion. We also obtained HRTEM images of the 3@Al2O3-uncal catalyst after 45 h on stream (Fig. 9). The image suggests very little visible particle formation, meaning that the catalyst is stable towards nanoparticle formation under these conditions and the gradual loss of activity is presumably due to leaching of molecular iridium from the support. This is in contrast to 1@SiO2, which showed substantial aggregation of nanoparticles after longer times on stream.32 We also tested the dehydrogenation reaction with the 3@Al2O3-uncal-nanoparticle catalyst. 3@Al2O3-uncal was calcined at 550 °C for 24 h under N2 atmosphere and it formed nanoparticles to as seen from the black colour of the solid. This nanoparticle catalyst was tested for 4 h at 350 °C (0.09 mL min−1 flow rate) and it showed TOF 437 h−1 for an initial 1 h, which gradually decreased to TOF 193 h−1 within 4 h. These experiments show that the molecular, supported Ir catalysts are much more reactive than Ir nanoparticles.
 | ||
| Fig. 9 A and B. HRTEM images of 3@Al2O3-uncal after 45 h reaction at 350 °C with 0.10 mL min−1 flow rate. A few particles, 2–3 nm in diameter are visible at higher magnification. The lattice spacing corresponds to metallic Ir. C. XEDS spectra of 3@Al2O3-uncal after 45 h reaction. | ||
Conclusions
A pincer complex with an aromatic backbone containing two meta-substituted carboxylate groups (3) was successfully synthesized and characterized. The complex was supported on both calcined and uncalcined Al2O3 giving an air stable neutral catalyst. Dehydrogenation of 4-methylpiperidine was performed in a continuous-flow reactor using five different metal-oxide supported catalysts. The substrate 4-methylpiperidine was converted mainly into 4-methyl-tetrahydropyridine and 4-methylpyridine, with the former accumulating to a greater extent, indicating a fast initial reaction followed by a rate-determining reaction step for the aromatization to 4-methylpyridine. The hydrogen produced is pure and can power a fuel cell for an extended period. Lower flow rates promoted the formation of the aromatic product as the contact time was increased. We found that 3@Al2O3-uncal performed the best in terms of conversion, selectivity and TOF at 325 °C and 350 °C, and the increased stability could be attributed to the double anchor. The maximum TOF for catalyst 3@Al2O3-uncal was 9200 h−1 (at 350 °C and 0.2 mL min−1); over time the cumulative TON was ca 91000 over 45 h and the catalyst was at that time still active with only a 25% decrease of activity compared to the first hour. Most of the existing literature report the use of Pt- or Pd-based heterogeneous catalyst for this dehydrogenation reaction, and the present system compares favourably to a supported Pt catalyst, which gives a lower TOF at a higher flow rate and in total a substantially lower TON. Thus, the findings of the present study are promising for the application of N-heterocycles as LOHCs.
Author contributions
This study was devised and designed by K. Chakrabarti and O. F. Wendt. The metal complexes and supported catalysts were synthesized, characterized, tested and analysed by K. Chakrabarti and A. Spangenberg. Experiments on hydrogen collection and purity were performed by V. Subramaniyan and A. Hederstedt. R. Wallenberg performed the electron microscopy analysis, and the flow reactor setup was provided by O. Y. Abdelaziz, A. V. Polukeev and C. P. Hulteberg. The manuscript was prepared by K. Chakrabarti, R. Wallenberg, O. Y. Abdelaziz and O. F. Wendt.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Olle Engkvist Foundation, the Wenner-Gren Foundation, Lund University, and the Royal Physiographic Society in Lund for financial support. We also thank the Swedish Research Council for funding the electron microscopy.Notes and references
- P. T. Aakko-Saksa, C. Cook, J. Kiviaho and T. Repo, J. Power Sources, 2018, 396, 803–823 CrossRef CAS.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 RSC.
- M. Niermann, S. Drünert, M. Kaltschmitt and K. Bonhoff, Energy Environ. Sci., 2019, 12, 290–307 RSC.
- T. He, Q. Pei and P. Chen, J. Energy Chem., 2015, 24, 587–594 CrossRef.
- E. Gianotti, M. Taillades-Jacquin, J. Rozière and D. J. Jones, ACS Catal., 2018, 8, 4660–4680 CrossRef CAS.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed.
- Z. Abdin, C. Tang, Y. Liu and K. Catchpole, iScience, 2021, 24, 102966 CrossRef CAS PubMed.
- G. Cacciola, N. Giordano and G. Restuccia, Int. J. Hydrogen Energy, 1984, 9, 411–419 CrossRef CAS.
- D. Klvana, A. Touzani, J. Chaouki and G. Bélanger, Int. J. Hydrogen Energy, 1991, 16, 55–60 CrossRef CAS.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- E. Clot, O. Eisenstein and R. H. Crabtree, Chem. Commun., 2007, 2231–2233, 10.1039/B705037B.
- A. Moores, M. Poyatos, Y. Luo and R. H. Crabtree, New J. Chem., 2006, 30, 1675–1678 RSC.
- J. Oh, K. Jeong, T. W. Kim, H. Kwon, J. W. Han, J. H. Park and Y.-W. Suh, ChemSusChem, 2018, 11, 661–665 CrossRef CAS PubMed.
- R. H. Crabtree, ACS Sustainable Chem. Eng., 2017, 5, 4491–4498 CrossRef CAS.
- S. P. Patil, A. B. Bindwal, Y. B. Pakade and R. B. Biniwale, Int. J. Hydrogen Energy, 2017, 42, 16214–16224 CrossRef CAS.
- K.-i. Fujita, T. Wada and T. Shiraishi, Angew. Chem., Int. Ed., 2017, 56, 10886–10889 CrossRef CAS PubMed.
- P. Bachmann, J. Steinhauer, F. Späth, F. Düll, U. Bauer, R. Eschenbacher, F. Hemauer, M. Scheuermeyer, A. Bösmann, M. Büttner, C. Neiß, A. Görling, P. Wasserscheid, H.-P. Steinrück and C. Papp, J. Chem. Phys., 2019, 151, 144711 CrossRef PubMed.
- D. Forberg, T. Schwob, M. Zaheer, M. Friedrich, N. Miyajima and R. Kempe, Nat. Commun., 2016, 7, 13201 CrossRef CAS PubMed.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- C. Hou, Y. Li and Z. Ke, Inorg. Chim. Acta, 2020, 511, 119808 CrossRef CAS.
- A. V. Polukeev and O. F. Wendt, Organometallics, 2017, 36, 639–649 CrossRef CAS.
- O. O. Kovalenko and O. F. Wendt, Dalton Trans., 2016, 45, 15963–15969 RSC.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- A. Friedrich and S. Schneider, ChemCatChem, 2009, 1, 72–73 CrossRef CAS.
- (a) A. V. Polukeev, R. Gritcenko, K. J. Jonasson and O. F. Wendt, Polyhedron, 2014, 84, 63–66 CrossRef CAS; (b) A. V. Polukeev, O. Y. Abdelaziz and O. F. Wendt, Organometallics, 2022, 41, 859–873 CrossRef CAS.
- S. Manojveer, S. Salahi, O. F. Wendt and M. T. Johnson, J. Org. Chem., 2018, 83, 10864–10870 CrossRef CAS PubMed.
- B. Sheludko, M. T. Cunningham, A. S. Goldman and F. E. Celik, ACS Catal., 2018, 8, 7828–7841 CrossRef CAS.
- M. H. Majeed, P. Shayesteh, P. Tunå, A. R. Persson, R. Gritcenko, L. R. Wallenberg, L. Ye, C. Hulteberg, J. Schnadt and O. F. Wendt, Chem. – Eur. J., 2019, 25, 13591–13597 CrossRef CAS PubMed.
- S. Yamaguchi, Y. Maegawa, K.-i. Fujita and S. Inagaki, ChemSusChem, 2021, 14, 1074–1081 CrossRef CAS PubMed.
- Z. Huang, M. Brookhart, A. S. Goldman, S. Kundu, A. Ray, S. L. Scott and B. C. Vicente, Adv. Synth. Catal., 2009, 351, 188–206 CrossRef CAS.
- B. Sheludko, C. F. Castro, C. A. Khalap, T. J. Emge, A. S. Goldman and F. E. Celik, ChemCatChem, 2021, 13, 407–415 CrossRef CAS.
- A. V. Polukeev, R. Wallenberg, J. Uhlig, C. P. Hulteberg and O. F. Wendt, ChemSusChem, 2022, 15, e202200085 CrossRef CAS PubMed.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- Y. Xie and D. Milstein, ACS Appl. Energy Mater., 2019, 2, 4302–4308 CrossRef CAS.
- J. Schwarz, A. Ilic, S. Kaufhold, J. Ahokas, P. Myllyperkiö, M. Pettersson and K. Wärnmark, Sustainable Energy Fuels, 2022, 6, 4388–4392 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2270316. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy00881a |
| This journal is © The Royal Society of Chemistry 2023 |