
Tunable charge-transport polarity in thienothiophene–bisoxoindolinylidene-benzodifurandione copolymers for high-performance field-effect transistors†
Zhihui
Chen‡
 ab,
Jianyao
Huang‡
a,
Weifeng
Zhang
*a,
Yankai
Zhou
ab,
Xuyang
Wei
ab,
Jinbei
Wei
a,
Yuanhui
Zheng
a,
Liping
Wang
c and
Gui
Yu
*ab
ab,
Jianyao
Huang‡
a,
Weifeng
Zhang
*a,
Yankai
Zhou
ab,
Xuyang
Wei
ab,
Jinbei
Wei
a,
Yuanhui
Zheng
a,
Liping
Wang
c and
Gui
Yu
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: zhangwf@iccas.ac.cn; yugui@iccas.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cSchool of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China
First published on 5th August 2021
Abstract
N-Type semiconducting polymers are important materials for modern electronics but limited in variety and performance. To design a new n-type polymer semiconductor requires a judicious trade-off between structural parameters involving both backbone and side-chain modifications. The appeal of backbone modification emerges from the tunable electronic structures and conformational control. To control these effects typically needs installation of substituents such as halogens. In a polymer system with high molecular complexity, a few common substituents such as methyl and methoxy groups are an underdeveloped area of chemical space. In this work, we study the substituent effects in methyl- and methoxy-substituted thienothiophene–bis(oxoindolinylidene)benzodifurandione copolymers on field-effect performances. The two substituents affect the conformations of the backbone and increase the frontier orbital energy levels of the polymers. Using such electronic effects, we are able to tune the charge transport behaviors from n-channel to ambipolar. This strategy allows further substitution patterns for backbone modification in other polymer semiconductors.
Introduction
Substituent effects are ubiquitous in all facets of organic chemistry.1 For example, bulky substituents are critical to exerting repulsive stereochemical control in asymmetric catalysis; and changing terminal substituents can promote cellular membrane permeability or add metabolic stability in drug discovery.2–4 In organic electronics, small-molecule and polymeric semiconductors are also benefited from the introduction of substituents which can construct targeted structures with increased mobilities and efficient charge injections. The importance of such substituent effects is magnified in tuning charge transport properties of semiconductors, because a modest modification in the molecular structure may lead to an uncontrollable change in macroscopic parameters.5 Currently, modifications for screening an applicable small-molecule or polymeric semiconductor require both target- and diversity-oriented synthesis—the former one is to find a certain core that holds promise for excellent performance, and the latter one probes into its relevant chemical space, a process that typically studies substituent effects. Although the screening seems tedious and time-consuming in many cases, ex post facto design guidelines learned from these trial-and-error processes can rule out unfavorable candidates for further exploration.High-mobility semiconductors are imperative in realizing modern applications, such as field-effect transistors (FETs), which promise to impact our daily life. In many applications such as logic circuits, both p- and n-type materials are needed with balanced mobilities. Although the mobilities of p-type polymeric semiconductors can rival those of amorphous or paracrystalline silicon, n-type semiconductors are still underexplored. To design a high-performance n-type semiconductor, the first concern is whether its frontier orbital energy levels match the Fermi level of the electrode. A stable electron transport material features a low-lying lowest unoccupied molecular orbital (LUMO) level close to −4.0 eV, or even lower.6–10 Electron-donating and -withdrawing substituents can readily tune this parameter; and in some cases, introducing such substituents is of necessity to ensure the energy levels are located in the optimal window. Moreover, substituents can induce intra- and intermolecular interactions to rigidify the backbone, improve aggregation, and thus facilitate charge transport. Many great strides in device performance optimization have been made for unveiling the substituent effects. Despite the well-established side-chain engineering concept,9,11,12 backbone modification by halogen and chalcogen atoms also plays vital roles in controlling the opto-electronics properties.13–15 The underlying origin includes electronic and steric effects, both of which are crucial to the charge transport processes from a mechanistic perspective. The electronic effects modulate the conjugation that provides precise control over carrier injection from electrodes to the semiconductor layer, whereas the steric effects influence the self-aggregation behavior that stems from the conformations and noncovalent interactions. Although many principles have been proposed to guide the diversity-oriented design of high-mobility materials, the substituent effects in complex molecular systems are still ill-defined because of the synergy and competition between such electronic contribution, steric repulsion, and attractive noncovalent bonding. Specifically, n-type polymeric systems typically consist of electron-withdrawing functionalities that allow for efficient electron injection and in turn enhance n-channel transport. In-depth studies on substituent effects in n-type polymer semiconductors have pushed the limits of classic design concepts, and are therefore an appealing topic for electronics applications.
In this work, we choose a polymer system with high electron affinity in an effort to probe into the substituent effects on electron mobilities. The polymers are composed of a benzodifurandione-bridged indolone (NBDO) acceptor and a substituted thienothiophene (TT) donor. NBDO contains four carbonyl groups and two pyridinic nitrogen atoms that greatly stabilize the frontier orbitals. Dai et al. for the first time reported this building block, and obtained an NBDO-based polymer semiconductor with the highest electron mobility of 3.22 cm2 V−1 s−1.16 This high electron affinity unit was then used to construct other ambipolar and n-type polymers with a maximum electron mobility exceeding 7 cm2 V−1 s−1.17–19 We introduced methyl and methoxy groups to modify the structures (hereafter referred to as DMTT for dimethylthienothiophene and MOTT for dimethoxythienothiophene). Methyl substituents can tune the electronic and morphological effects in optoelectronics,20–22 whereas methoxy substituents are used to typically form noncovalent S⋯O interactions for high-performance p-type semiconductors.23–26 Inspired by these precedent works, we note that both the NBDO acceptor and the substituted TT donor, have nearly linear linkage sites, which enable a high degree of backbone rigidity. The major object of this work lies in the steric and electronic control by introduction of the substituents, while other important structural variables, including backbone linearity and side-chain structure, are constrained to ensure the precise comparison of these substituents. We then fabricated field-effect transistors (FETs) to describe the charge transport behaviors. The results show the substituents can raise the frontier orbital energy levels and shift the charge transport polarity, both of which allows fine-tuning of the carrier mobilities for use in electronics.
Results and discussion
Design and synthesis
The NBDO unit has proven efficient in lowering the LUMO energy level of the polymer for electron injection; the structural modification is therefore placed on the donor moiety. To ensure the overall molecular symmetry, we introduced the substituents into 3,6-positions of thienothiophene while leaving 2,5-positions for late-stage coupling sites. We envisioned that the methyl could engage in conformational control of the backbone, whereas the electron-donating methoxy could both generate intramolecular noncovalent bonds with adjacent units and regulate the electronic effects. The attached side chains we installed on the NBDO moiety are 4-octadecyldocosyl groups, which ensure the solubility and interdigitation for the corresponding polymers.16,18Scheme 1 depicts the synthetic routes to the target NBDO-based polymers. DMTT was accessed by the ring-closure reaction of 2,5-dihydroxy-2,5-dimethyl-3-hexyne with sulfur in high pressure reactor. MOTT was obtained through a copper(II) oxide-catalyzed aromatic nucleophilic reaction of 3,6-dibromothienothiophene with sodium methoxide. Both intermediates were treated with n-butyl lithium and quenched with trimethylstannyl chloride to afford their respective crystalline, high purity tin-containing monomers.25,27 The target polymers were readily obtained through the Pd2(dba)3/P(o-tol)3-catalyzed Stille copolymerization in excellent yields. The number-average molecular weights (Mn) are 54.3, 43.6, and 32.4 kDa, and polydispersity indices (Đ) are 2.36, 1.72, and 1.93 for PNBDO-TT, PNBDO-DMTT, and PNBDO-MOTT, respectively (Fig. S1 and Table S1, ESI†). The 5% weight loss temperatures (Fig. S2, ESI†) for PNBDO-TT, PNBDO-DMTT, and PNBDO-MOTT are 407, 409, and 395 °C, respectively, indicating that these polymers are stable candidates for late-stage device optimization under annealing treatment (Table 1).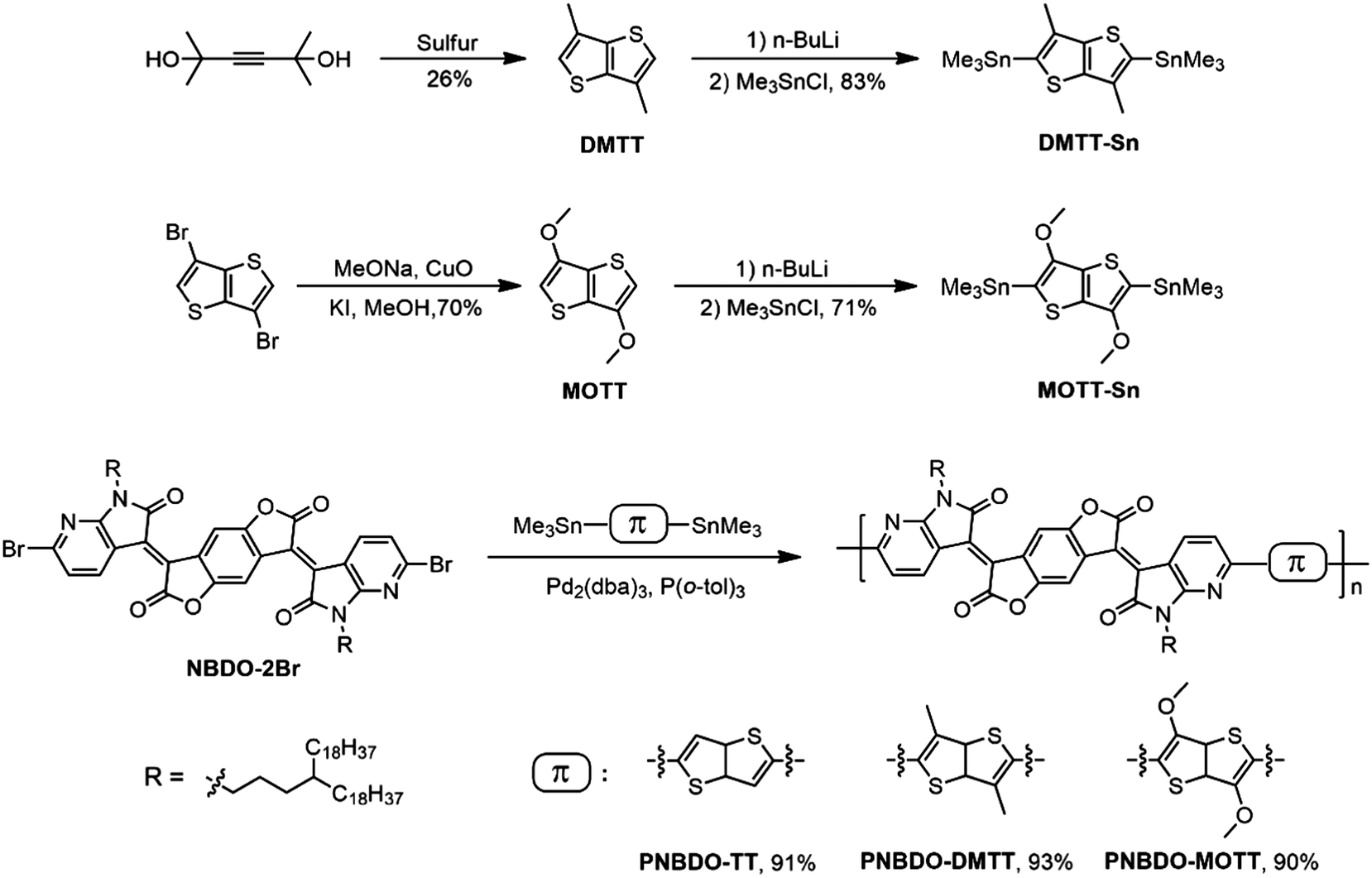 | ||
| Scheme 1 Synthetic routes and chemical structures of PNBDO-TT, PNBDO-DMTT, and PNBDO-MOTT. | ||
| Polymer | M n/Đa | λ max (nm) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (eV) c (eV) |
E HOMO (eV) | E LUMO (eV) | T dec (°C) | |
|---|---|---|---|---|---|---|---|
| Solnb | Film | ||||||
| a Determined by GPC at 150 °C. b In DCB solutions. c Calculated from the absorption edges of thin films (Eoptg = 1240λabs,onset). d Estimated by CV. EHOMO = −(4.4 + Eoxonset) eV, ELUMO = −(4.4 + Eredonset) eV. e Onset decomposition temperature (5% weight loss) measured by TGA. | |||||||
| PNBDO-TT | 54.3/2.36 | 839 | 839 | 1.35 | −5.99 | −4.07 | 407 |
| PNBDO-DMTT | 43.7/1.72 | 850 | 870 | 1.31 | −5.94 | −4.00 | 409 |
| PNBDO-MOTT | 34.5/1.93 | 897 | 908 | 1.21 | −5.75 | −4.00 | 395 |
Optical properties
To evaluate the substituent effect on optical properties, we performed UV-visible-near infrared (UV-vis-NIR) absorption spectroscopy in the 1,2-dichlorobenzene (DCB) solutions and thin films (Fig. 1 and Table 1). All spectra show broad absorption in the range of 600–1000 nm. The absorption maximum (λmax) of the unsubstituted PNBDO-TT in solution is located at 839 nm, and that in thin film does not shift. Nevertheless, its relative absorption intensity versus shoulder peaks (ca. 800 nm) is higher than that in solution spectrum, clearly indicating that the stronger π–π interactions or more orderly aggregation still occurs in PNBDO-TT thin film.28–30 For the DMTT-based polymer, the absorption maxima are 850 and 870 nm in solution and thin film, respectively. The redshift is consistent with the electronic effects that the electron-donating substituents on the donor moieties enhance the intramolecular bond polarization, leading to an increased highest occupied molecular orbital (HOMO) energy level and a narrowed energy gap. For the MOTT-based polymer, as a stronger electron donor, the installed methoxy groups further increase the absorption maxima to 897 and 908 nm in solution and film, respectively. Therefore, the degree of this destabilization effect is relevant to the electron-donating strength such that the methoxy-substituted PNBDO-MOTT exhibits a broader absorption than that of the methyl-substituted PNBDO-DMTT. From the absorption onsets, we estimated the optical gaps of TT-, DMTT-, and MOTT-based polymers to be 1.35, 1.31, and 1.21 eV, respectively. These values also agree with the electronic effects.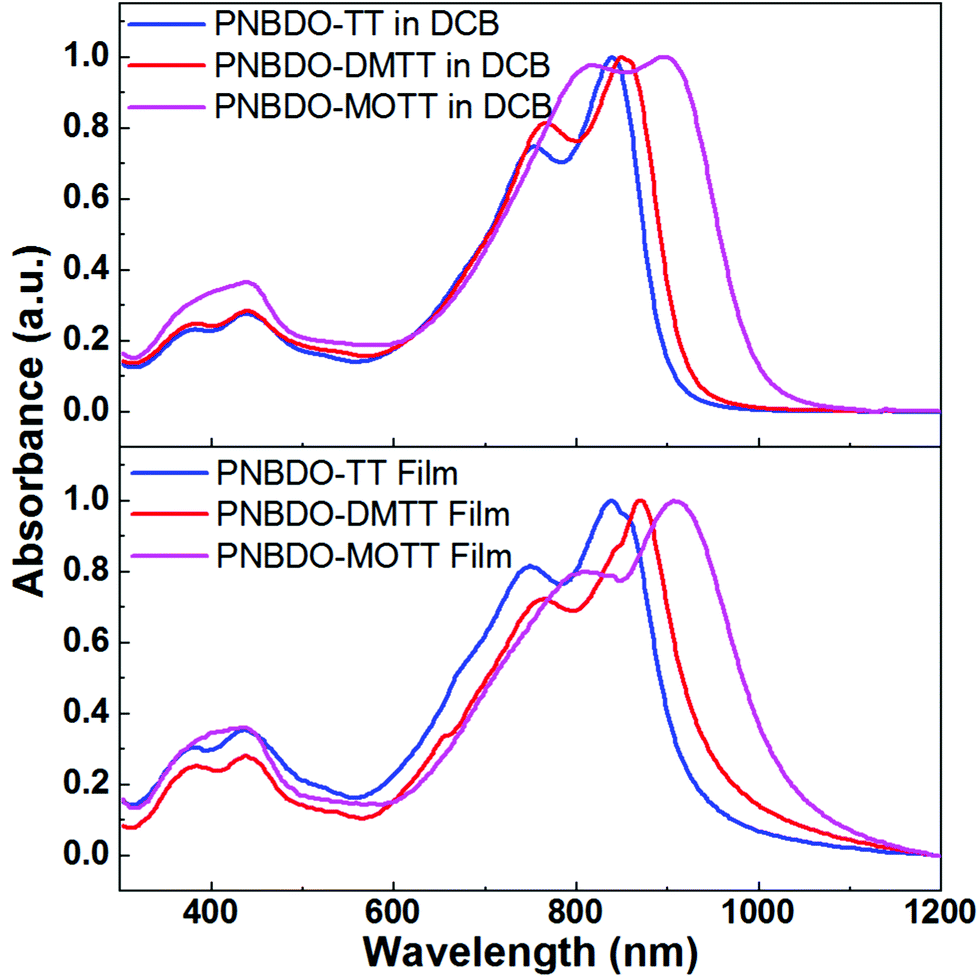 | ||
| Fig. 1 UV-vis-NIR absorption spectra of the three thienothiophene–bisoxoindolinylidene-benzodifurandione copolymers in DCB solutions and thin films. | ||
Frontier orbital energy levels
To further study the electronic effects on energy levels, we measured the conducted ultraviolet photoelectron spectroscopy (UPS) and cyclic voltammetry (CV) tests (Fig. S3, S4, Table S2, ESI† and Table 1). The UPS data indicate that the ionization energies decrease from 5.92 eV for PNBDO-TT to 5.72 eV for PNBDO-DMTT to 5.41 eV for PNBDO-MOTT. The corresponding HOMO energy levels (EUPSHOMO) are −5.92 eV, −5.72 eV, and −5.41 eV for these polymers, respectively. Consistent with the UPS data, HOMO levels estimated from the oxidation waves in CV show a similar tendency with electron-donating groups increasing the energy levels. Note that the estimated LUMO energy levels slightly increases from −4.07 eV for PNBDO-TT to −4.00 eV for PNBDO-DMTT. This energetic difference is in line with the orbital hybridization of donor and acceptor moieties, that substituents on the donor affect HOMO more significantly than LUMO. The experimental energy levels confirm the electronic tuning by substitution of methyl or methoxy groups. Compared with bithiophene-containing NBDO-based polymers, the unsubstituted TT-based polymer shows lower-lying frontier orbital energy levels, in accordance with the weaker electron-donating ability of thienothiophene.17 We infer that the LUMO energy levels of all polymers are in the optimal window of n-type materials, while the HOMO levels of the two substituted polymers may also allow hole injection.Theoretical modeling
To correlate the experimental data with theoretical electronic effects and to gain insight into the steric effects, we then studied the substituent effects using theoretical modeling. Because there is a pyridinic nitrogen atom proximal to the linking thiophene derivative, we envisage that noncovalent S⋯N interactions occur to reinforce the planarity of the backbone (Fig. 2). The single bond between the donor and acceptor may rotate to form two conformers. For the two possible conformers of PNBDO-TT, both S⋯N and CH⋯N interactions can construct planar substructures,31 whereas for the S-trans conformers of PNBDO-DMTT and PNBDO-MOTT, the lone pair of nitrogen atoms have steric repulsion with either methyl or methoxy groups. To quantify these electronic and steric effects, we calculated the energies of different conformers using potential energy scan. Simulation models were simplified using the fragments that consist of the different donor moiety and the azaindolone subunit. The molecular geometries were optimized using density functional theory at the B3LYP/6-31G(d) level.32 The dihedral angle between two subunits was set to 0–180° (syn to anti) at 10° intervals, and the 0° conformer was set as the reference in the plot. Each single-point energy was further recalculated at the MP2/cc-pVDZ level to allow a more precise prediction of noncovalent interactions.33 As shown in Fig. 3, the TT-based subunit exhibits a conventional energy diagram for highly planar molecules, with two local conformational energy minima at 0 and 180°. The S⋯N interaction leads to the energetically more stable 0° conformer. The rotational barrier of 27.5 kJ mol−1 indicates the significant planarization effect by the noncovalent interactions. The DMTT-based subunit, which contains two methyl groups on the thienothiophene, shows a smaller rotational barrier of 18.1 kJ mol−1 and a conformational ambiguity around the planar conformers. This simulated result is due to the repulsion between the attached methyl groups and the fused pyridine rings. The energetically disfavored repulsion competes against the S⋯N interaction, giving a detrimental effect on the overall planarity and conjugation of the backbone. The MOTT-based subunit also has a different conformational preference in comparison with the other two polymers. The methoxy group has an intense repulsion against the unshared electron pair of pyridinic nitrogen atoms in the 180° conformer, but strengthens the preference of the 0° conformer by formation of a weak CH⋯O interaction. These interactions give rise to an enhanced rotational barrier of 29.0 kJ mol−1 and a more significant energy difference between the 0 and 180° conformers.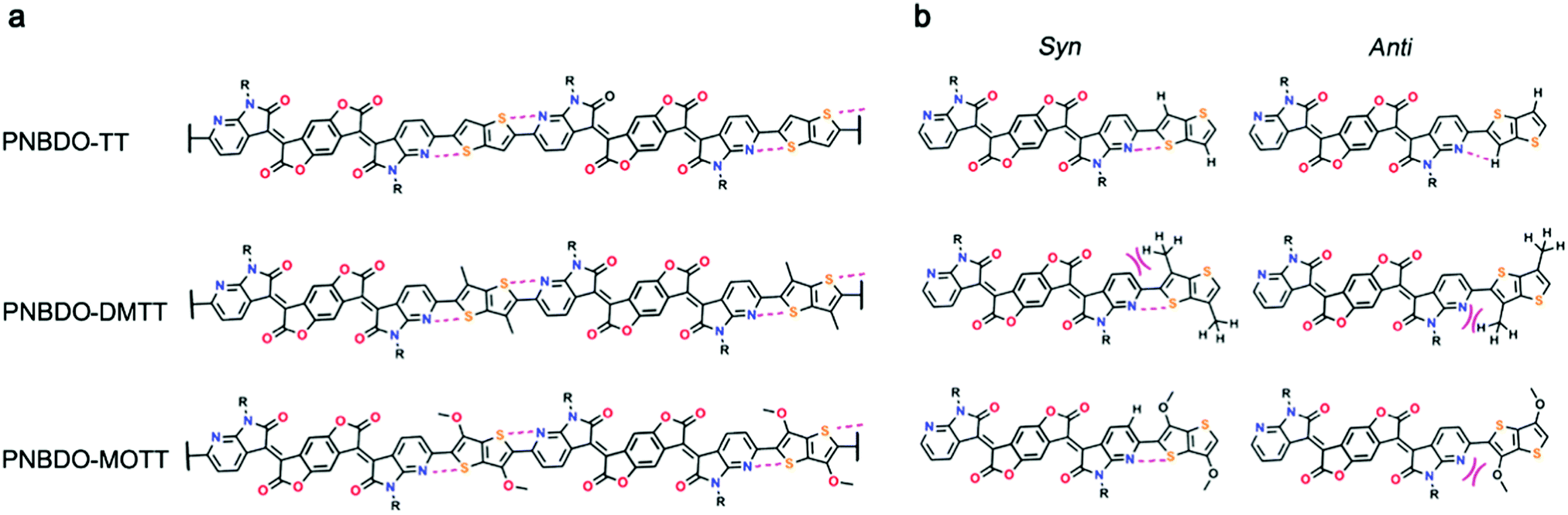 | ||
| Fig. 2 Schematic illustration of noncovalent interactions and steric repulsions in the polymer system. (a) Dimeric co-repeating unit with S⋯N interactions. (b) Different conformations of a co-repeating unit with noncovalent attractions or repulsions or both. | ||
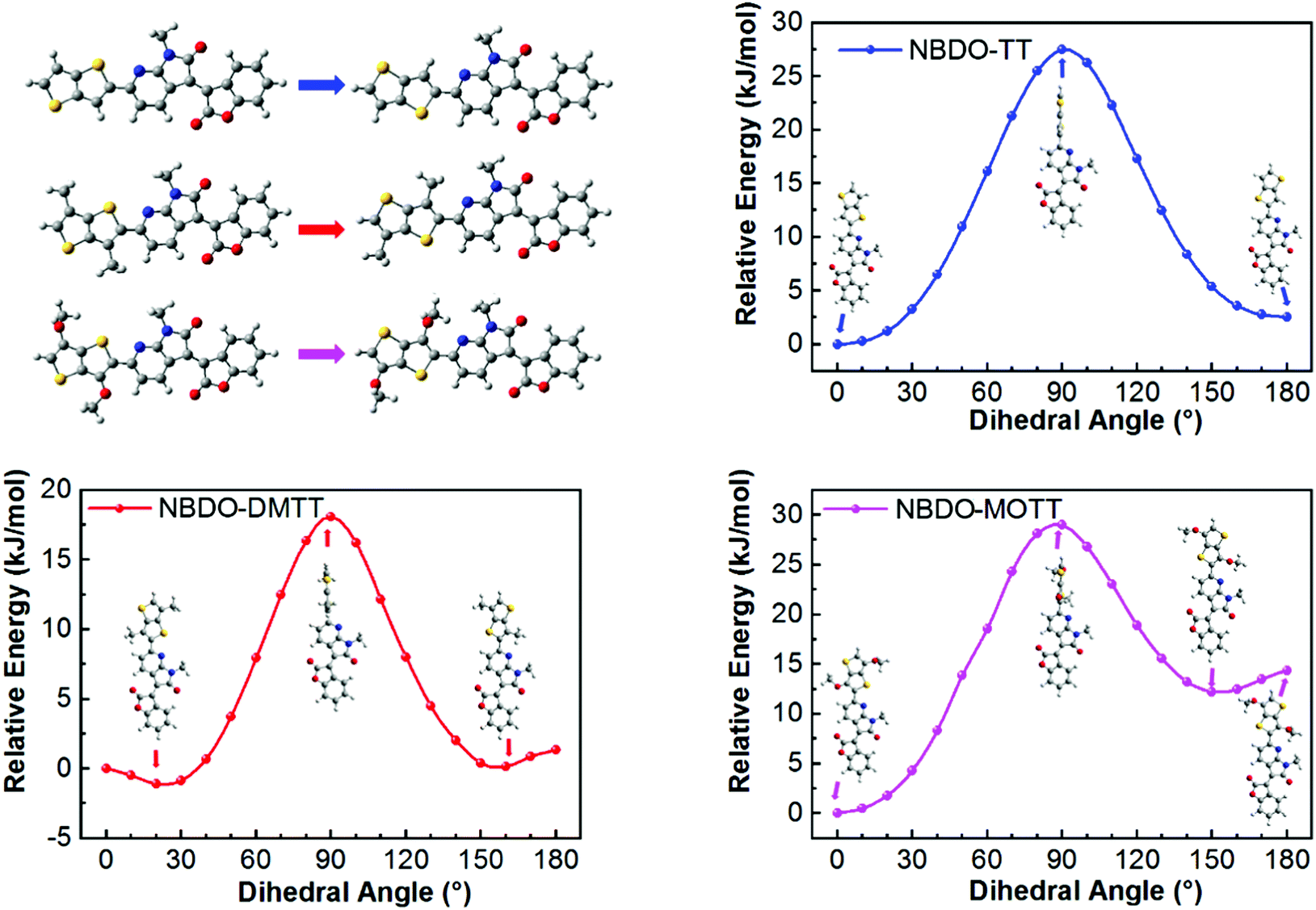 | ||
| Fig. 3 Potential energy distribution of single-bond rotation in substructures of NBDO-based polymers with different substituents. Insets are corresponding conformers at local energy minima or maxima. | ||
Notably, the methoxy groups are not oriented in a uniform manner in the optimized conformation. The methoxy groups are intrinsically flexible; their conformational preference differs depending on the neighboring molecular system. To further unveil such effect, we scanned the energy distribution regarding rotation of the methoxy group (Fig. S5, ESI†). This detailed evaluation of the methoxy group reveals that both repulsion and attraction participate in the conformational control, where steric interaction dominates the overall preference. We observed flexible conformations with dihedral angles in the range of 0–110° in the energy diagram, whatever in a NBDO-MOTT subunit or in an individual MOTT. Energetic stabilization by attractive CH⋯O interaction is tenuous because of the inferior directionality. Although the resonance effect exerts a stabilizing interaction due to electron delocalization, the repulsion between methoxy and sulfur groups in the 0° conformer slightly offsets such stabilization effect. Note that the 180° conformer shows a pronounced energy barrier due to the steric congestion twisting the co-repeating unit. These results are reasonable to clarify the overall substituent effect of methoxy groups: (1) the intrinsic flexibility of methoxy groups can affect the intermolecular packing, and (2) herein the conformational control of methoxy groups is a synergistic contribution of steric and electronic effects.
With an optimized set of torsional preferences in hand, we calculated the geometries of dimers. As expected, only methyl groups slightly twist the backbone, whereas the unsubstituted or methoxy-substituted dimers show highly planar geometries (Fig. S6, ESI†). The S⋯N interactions in these dimers are also confirmed by the electrostatic potential surface plot (Fig. S7, ESI†) and detailed reduced density gradient analyses (Fig. S8–S10, ESI†).34,35 The calculated HOMO and LUMO energy levels, as well as the tendency by substitution of methyl or methoxy groups, accord with the groups and the fused pyridine rings. The energetically disfavored repulsion competes against the S⋯N interaction, giving a detrimental effect on the overall planarity and conjugation of the backbone. The MOTT-based subunit also has a different conformational preference in comparison with the other two polymers. The methoxy group has an intense repulsion against the unshared electron pair of pyridinic nitrogen atoms in the 180° conformer, but strengthens the preference of the 0° conformer by formation of a weak CH⋯O interaction. These interactions give rise to an enhanced rotational barrier of 29.0 kJ mol−1 and a more significant energy difference between the 0 and 180° conformers.
Field-effect transistors
We then fabricated thin-film transistors based on the three polymers using the conventional solution-processing technique. We used Corning glass as the substrate and poly(methyl methacrylate) (PMMA) as the dielectric layer. The transfer and output characteristics are illustrated in Fig. 4. We observed a typical enhancement in charge transport using thermal treatment to optimize the device performance (Fig. S11, ESI†). The transistors based on PNBDO-TT only exhibit an n-type transport behavior, whereas the devices based on the other two polymers provide n-type dominated ambipolar transport behaviors. Extracting the mobility from the saturation regimes, we calculated the average electron mobilities of 1.94, 0.648, and 0.0976 cm2 V−1 s−1 for the devices based on PNBDO-TT, PNBDO-DMTT, and PNBDO-MOTT, respectively (Table 2). The highest mobilities are 2.08, 0.739, and 0.103 cm2 V−1 s−1 for PNBDO-TT, PNBDO-DMTT, and PNBDO-MOTT, respectively. The electron mobility is in line with the electron affinity of each polymer, suggesting that a slightly destabilized frontier orbital suppresses electron injection. Also observed are an increased hole injection through substitution of electron-donating functionalities, with hole mobilities increased to 0.1 cm2 V−1 s−1 for the methoxy-substituted material. It is noteworthy that the ratios of hole to electron mobility are in agreement with control over frontier orbital levels, from pure n-channel (PNBDO-TT) to near-balanced ambipolar transport (PNBDO-MOTT). These results bode well for further applications in electronics.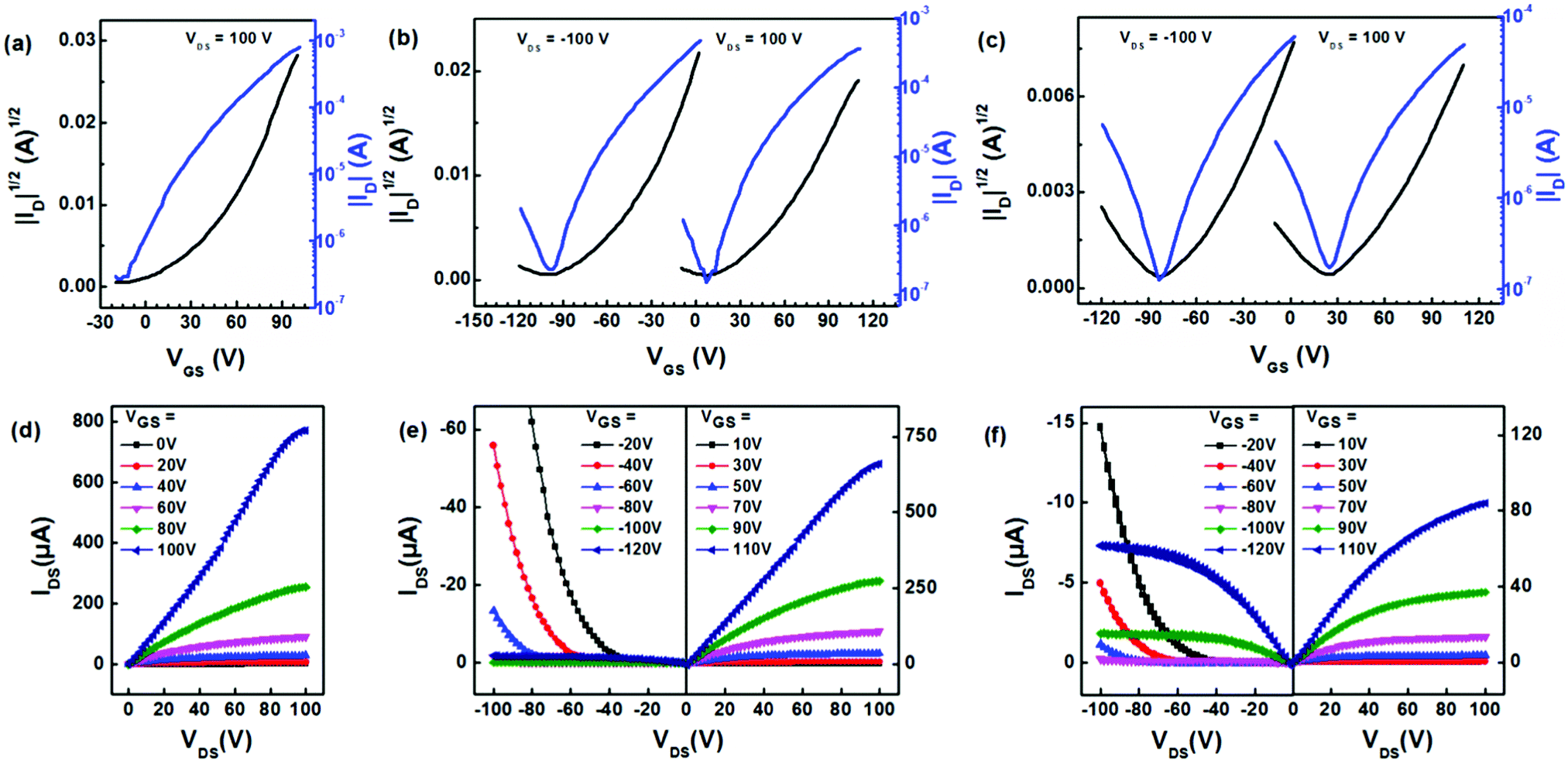 | ||
| Fig. 4 The transfer (a–c) and output (d and f) characteristics of the field-effect transistors based on three polymers measured under optimized conditions. (a and d) PNBDO-TT. (b and e) PNBDO-DMTT. (c and f) PNBDO-MOTT. | ||
| Polymer | μ avg (cm2 V−1 s−1) | μ max (cm2 V−1 s−1) | I on/Ioff | V TH (V) | |
|---|---|---|---|---|---|
| a μ avg, average mobilities calculated from 10 devices. b μ max, the maximum mobilities. | |||||
| PNBDO-TT | n-Channel | 1.94 | 2.08 | 103–104 | 43 ± 5 |
| PNBDO-DMTT | n-Channel | 0.648 | 0.739 | 103–104 | 45 ± 4 |
| p-Channel | 0.0250 | 0.0255 | 10–102 | −92 ± 5 | |
| PNBDO-MOTT | n-Channel | 0.0976 | 0.103 | 102–103 | 45 ± 4 |
| p-Channel | 0.0616 | 0.0711 | 103–104 | −87 ± 3 | |
Morphology and aggregation
The aggregation behaviors are important for intermolecular charge transport because a tightly packed chain allows faster carrier hopping in a semicrystalline, polymeric system. To detect the device optimization conditions, we first tested the surface morphology using tapping-mode atomic force microscopy (AFM) (Fig. S12, ESI†). The images all showed fiber-like grains in as-spun and annealed thin films, and the grains became larger after annealing treatment. To identify the difference in packing of the polymers with varying substituents, we performed grazing incidence wide-angle X-ray scattering (GIWAXS) measurements. Because we obtained the optimal charge transport behaviors under thermal treatment, each sample was also tested after annealing treatment. The acquired two-dimensional (2D) and 1D diffraction patterns are provided in Fig. 5 and Fig. S13, S14 (ESI†). As shown in Fig. 5, the as-spun and annealed films of the three polymers all adopt the typical edge-on dominated packing with (h00) diffraction peaks up to the fourth order in the out-of-plane direction and a characteristic (010) diffraction peak in the in-plane direction. The d–d spacing, which indicate the distance between polymer backbone through alkyl-chain interactions, are estimated to be 28.2 Å for PNBDO-TT, 27.5 Å for PNBDO-DMTT, and 28.7 Å for PNBDO-MOTT, and increase to 28.9, 28.2, 28.9 Å, respectively (Table 3). These distances with sub-Angstrom differences infer that alkyl chains are packed in a tight, interdigitated manner. The methyl and methoxy groups on the backbone do not substantially affect the overall packing motif. The π–π spacing, indicative of the distance between polymer backbones, are estimated to be 3.43–3.44 Å for the three polymers. There are significant enhancements in (010) diffraction intensities between their respective as-spun and annealed thin films of PNBDO-TT and PNBDO-DMTT, though their π–π spacing are almost the same (Fig. S14, ESI†). Therefore, the increased mobilities through thermal treatment are due mainly to the increased crystallinity. We then analyzed the coherence length using the Scherrer equation Lc = 2πK/Δq, where Lc is the coherence length, K is the shape factor (0.89), and Δq is the full width at the half-maximum of the (100) peak.36,37 In as-spun thin films, PNBDO-TT has a coherence length of 120 Å; this value increases to 228 Å after annealing. Likewise, the other two polymers also have enhanced crystallinities after annealing. We also calculate the paracrystalline disorder parameter (g) to confirm the tendency,38,39 because a reduced g factor shows a more ordered molecular packing that facilitates hopping. The g factors are estimated from the first three orders of diffraction peaks in the out-of-plane direction, and reduce from 7.38% to 4.91% for PNBDO-TT, from 8.05% to 6.42% for PNBDO-DMTT, and from 6.67% to 5.39% for PNBDO-MOTT, respectively, through annealing treatment. This parameter agrees with the enhanced charge transport properties.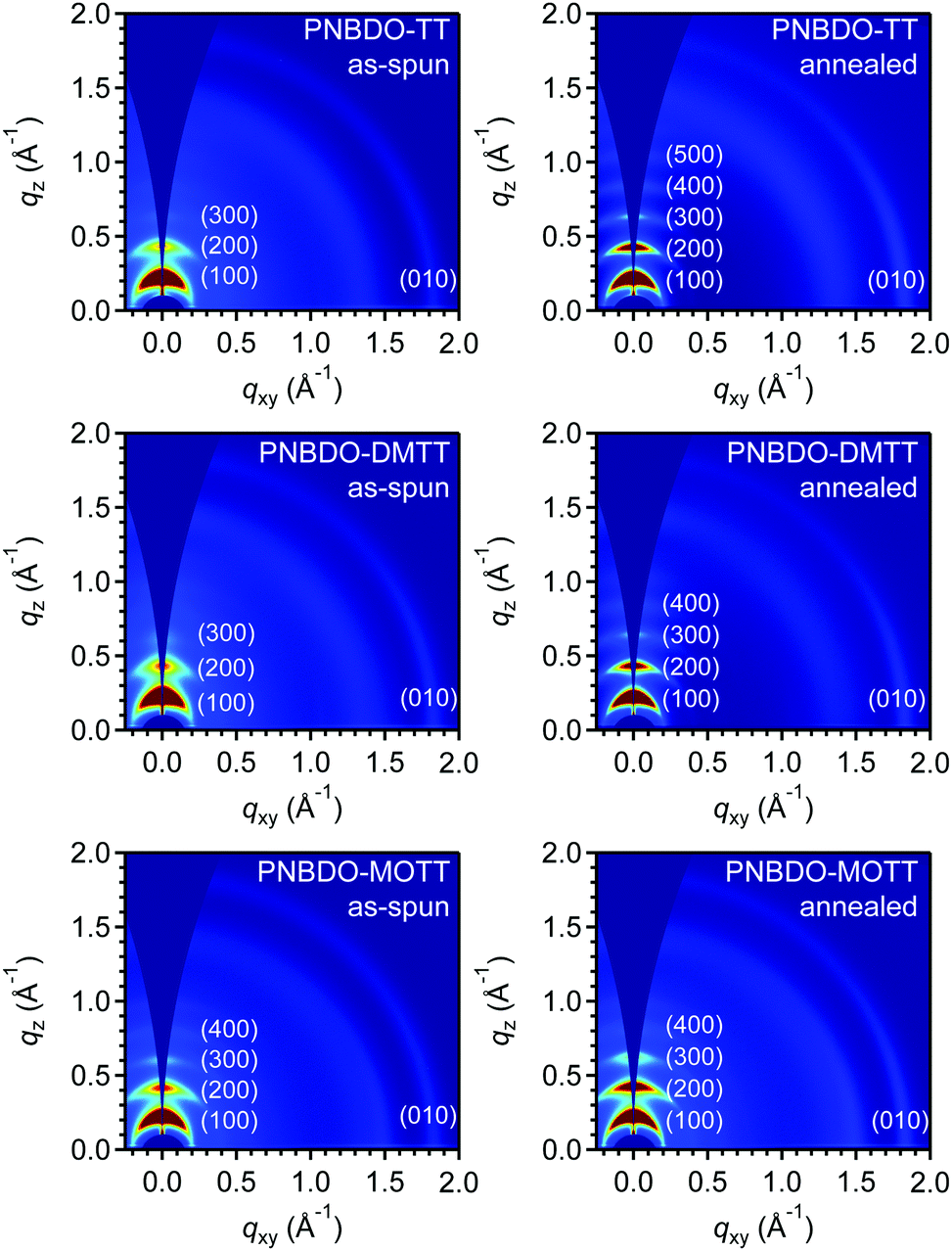 | ||
| Fig. 5 GIWAXS images of the PNBDO-TT, PNBDO-DMTT, and PNBDO-MOTT thin films. | ||
| Polymer | d–d distance (Å) | π–π distance (Å) | Coherence length (Å) | Paracrystalline disorder parameter (g) | ||||
|---|---|---|---|---|---|---|---|---|
| RT | Annealed | RT | Annealed | RT | Annealed | RT | Annealed | |
| PNBDO-TT | 28.2 | 28.9 | 3.44 | 3.44 | 120 | 228 | 7.38% | 4.91% |
| PNBDO-DMTT | 27.5 | 28.2 | 3.44 | 3.43 | 114 | 216 | 8.05% | 6.42% |
| PNBDO-MOTT | 28.7 | 28.9 | 3.43 | 3.43 | 173 | 227 | 6.67% | 5.39% |
Conclusions
In this study, we introduced two weak-to-moderate electron-donating functionalities, methyl and methoxy groups into the NBDO-TT based polymers in order to explore the substituent effects on charge transport behaviors. The unsubstituted PNBDO-TT polymer showed a unipolar n-channel behavior with the highest electron mobility up to 2.08 cm2 V−1 s−1, whereas the methyl- or methoxy-substituted polymers provided ambipolar behaviors. The results indicate that the electronic effects of electron-donating groups on the donor moiety can destabilize the frontier orbitals and allow hole injection in these molecular systems. Meanwhile, although the introduced groups exert steric control over the backbone conformations, the overall packing motif does not change according to the diffraction measurements. These less bulky functional groups are potential candidates for backbone modification of complex semiconducting polymer systems to narrow the understanding gap, leverage the theoretical insight, and guide semiconductor design.Experimental section
Material and measurements
Feedstock chemicals were purchased from Alfa, J&K, Innochem chemical or other commercial sources, and used without further purification. Anhydrous solvent (THF) was purified by distilling over sodium wire/benzophenone under inert atmosphere. (3E,7E)-3,7-bis(2-oxo-1H-pyrrolo[2,3-b]pyridin-3(2H)-ylidene)benzo[1,2-b:4,5-b′]difuran-2,6(3H,7H)-dione, 2,5-bis(trimethylstannyl)thieno[3,2-b]thiophene, 3,6-dimethylthieno[3,2-b]thiophene, and 3,6-dibromothieno[3,2-b]thiophene were prepared following the described procedures in literatures.16,25,40,41 All air- and water-sensitive reactions were performed under argon atmosphere.The 1H NMR and 13C NMR spectra of intermediates and monomers were recorded on a Bruker Fourier 300 NMR spectrometer. High temperature 1H NMR spectra of the polymer materials were recorded on a Bruker Avance III 400 NMR spectrometer at 100 °C. Mass spectrometry (MS)/high rank mass spectrometry (HRMS) were collected on a Bruker APEXII or a 9.4 T Solarix FT-ICR mass spectrometers. High temperature gel permeation chromatography (GPC) analyses were carried out on a Polymer Labs PL 220 system (concentration: 0.10 mg mL−1) using a refractive index detector at 150 °C with polystyrenes as the standard and 1,2,4-trichlorobenzene (TCB) as the eluent (flow rate: 1.00 mL min−1), respectively. Elemental analyses of polymer materials were conducted on a Carlo Erba 1106 Elemental Analyzer. Thermogravimetric analyses (TGA) were conducted on a PerkinElmer series 7 thermal analysis system under N2 at a heating rate of 10 °C min−1. Solution and thin film Absorption spectra were recorded on a Hitachi U-3010 spectrophotometer. The corresponding polymer solutions were obtained by dissolving the polymer materials in DCB and diluted to a concentration of ca. 0.03 mg mL−1, while the polymer thin films were prepared on a quartz through spin-coating method. Cyclic voltammetry (CV) curves were collected on an electrochemistry workstation at room temperature with a conventional three-electrode configuration in dry acetonitrile containing 0.1 M (n-Bu)4NPF6 using Ag/AgCl electrode and Platinum wire as reference electrode, and counter electrode, respectively. A platinum stick electrode deposited with a polymer thin layer was adopted as the working electrode. Before testing, nitrogen was bubbled into the acetonitrile solution. The energy levels were calibrated using Fc+/Fc (−4.80 eV). All UPS spectra were acquired from an Axis-Nova CJ109, Kratos, working on a polymer thin film on silicon wafer (ca. 1 × 1 cm2), with a photoelectron energy of 21.22 eV. Theoretical calculations were performed using the Gaussian09 package. The optimization of polymer dimers is calculated at the B3LYP/6-31G(d) level. Potential energy scan was conducted at 10° intervals and the single-point energy was recalculated at the MP2/cc-pVDZ level. Reduced density gradient was analyzed by the MultiWfn package.2
Film morphology was characterized using a Digital Instruments Nanoscope V atomic force microscope operated in tapping mode. Film aggregation were analysed at BL14B1 Station of Shanghai Synchrotron Radiation Facility (SSRF). To calculate the paracrystalline disorder parameter, the following equation was used: Δb = l/d[l/N + (πgh)2], where Δb is the integral breadth (b = 2 sinθ/λ = q/2π), N is the average number of Bragg planes, d is the d-spacing of the corresponding peak, h is the order of the diffraction peak, and g is the paracrystalline disorder parameter. The g value can be readily obtained from the slope of the Δb–h2 plot.
PNBDO-TT (233 mg, 91%). 1H NMR (400 MHz, d2-C2D2Cl4, δ): 9.14–8.59 (m), 8.23 (br), 7.30–7.16 (m), 6.86 (br), 6.50–5.33 (m), 4.00–3.31 (br), 2.62–2.49 (br), 1.62–0.93 (m). GPC: Mn = 53.4 kDa, Mw = 128.8 kDa, PDI = 2.36. Elemental anal. calcd for C110H170N4O6S2: C 77.32, H 10.03, N 3.28. Found: C 77.79, H 10.25, N 3.41.
PNBDO-DMTT (242 mg, 93%). 1H NMR (400 MHz, d2-C2D2Cl4, δ): 9.16–8.56 (m), 8.31 (br), 7.32–7.20 (m), 6.86 (br), 6.53–6.34 (m), 4.01–3.33 (br), 2.61–2.16 (br), 1.60–0.92 (m). GPC: Mn = 43.7 kDa, Mw = 75.3 kDa, PDI = 1.72. Elemental anal. calcd for C112H174N4O6S2: C 76.46, H 10.10, N 3.23. Found: C 76.90, H 10.33, N 3.37.
PNBDO-MOTT (238 mg, 90%). 1H NMR (400 MHz, d2-C2D2Cl4, δ): 9.16–8.29 (m), 7.43–7.20 (m), 6.89 (br), 6.54–6.34 (m), 4.32–3.47 (m), 2.56–2.30 (br), 1.59–0.90 (m). GPC: Mn = 32.5 kDa, Mw = 62.9 kDa, PDI = 1.93. Elemental anal. calcd for C112H174N4O8S2: C 76.05, H 9.92, N 3.17. Found: C 76.49, H 10.04, N 3.13.
Transistor fabrication and characterization
Organic field-effect transistors with a top-gate/bottom-contact configuration were fabricated on the 1.5 × 1.5 cm2 glass slide. These substrates were sequentially cleaned by deionized water, ethanol, and acetone, and then dried by a nitrogen gun. The 30 nm-thick Au drain/source electrodes were deposited onto the glass substrates through vacuum evaporation, and the channel length/width (L/W) of 30/1400 μm was employed. The semiconducting and dielectric layers were fabricated by sip-coating a polymer solution (3.0 mg mL−1 in DCB) and a PMMA solution (60 mg mL−1 in n-butyl acetate) and future heating at 180 and 80 °C under nitrogen atmosphere, respectively. Eventually, an Al (80 nm) gate electrode were vacuum evaporated. A Keithley 4200 was used to explore their electrical performance under air condition. The saturation mobility was extracted from the equation: IDS = (W/2L)Ciμ(VGS − Vth)2, where Ci is the capacity per unit, μ the saturation mobility, VGS the gate voltage, VDS the drain/source voltage, Vth the threshold voltage, and IDS the drain/source current.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (Grants 22075294, 21774134, 201905285, 22021002 and 51773016), Beijing Municipal Natural Science Foundation (Grant 2212054), and Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-202101). The GIWAXS tests were performed at BL14B1 Station of Shanghai Synchrotron Radiation Facility (SSRF). The authors thank the faculty for the assistance during the tests.Notes and references
- S. E. Wheeler, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS PubMed.
- D. A. Nagib and D. W. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- A. Milo, A. J. Neel, F. D. Toste and M. S. Sigman, Science, 2015, 347, 737–743 CrossRef CAS PubMed.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef.
- R. P. Fornari, P. W. Blom and A. Troisi, Phys. Rev. Lett., 2017, 118, 086601 CrossRef PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- Y. Sui, Y. Deng, T. Du, Y. Shi and Y. Geng, Mater. Chem. Front., 2019, 3, 1932–1951 RSC.
- Z. Ni, H. Wang, Q. Zhao, J. Zhang, Z. Wei, H. Dong and W. Hu, Adv. Mater., 2019, 31, 1806010 CrossRef.
- B. Kang, R. Kim, S. B. Lee, S.-K. Kwon, Y.-H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef.
- T. Lei, J.-H. Dou, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2013, 135, 12168–12171 CrossRef.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef.
- J. Tian, L. Fu, Z. Liu, H. Geng, Y. Sun, G. Lin, X. Zhang, G. Zhang and D. Zhang, Adv. Funct. Mater., 2019, 29, 1807176 CrossRef.
- N. Leclerc, P. Chávez, O. Ibraikulov, T. Heiser and P. Lévêque, Polymers, 2016, 8, 11 CrossRef PubMed.
- Z. Fei, P. Boufflet, S. Wood, J. Wade, J. Moriarty, E. Gann, E. L. Ratcliff, C. R. McNeill, H. Sirringhaus, J. S. Kim and M. Heeney, J. Am. Chem. Soc., 2015, 137, 6866–6879 CrossRef PubMed.
- Z. Lin, X. Liu, W. Zhang, J. Huang, Q. Wang, K. Shi, Z. Chen, Y. Zhou, L. Wang and G. Yu, Macromolecules, 2018, 51, 966–976 CrossRef.
- Y.-Z. Dai, N. Ai, Y. Lu, Y.-Q. Zheng, J.-H. Dou, K. Shi, T. Lei, J.-Y. Wang and J. Pei, Chem. Sci., 2016, 7, 5753–5757 RSC.
- G. Zhang, Y. Dai, L. Qiu, K. Cho, F. Ge, H. Lee and K. C. Song, Polym. Chem., 2017, 8, 2381–2389 RSC.
- K. Shi, W. Zhang, D. Gao, S. Zhang, Z. Lin, Y. Zou, L. Wang and G. Yu, Adv. Mater., 2018, 30, 1705286 CrossRef.
- K. Huang, X. Zhao, Y. Du, S. Kim, X. Wang, H. Lu, K. Cho, G. Zhang and L. Qiu, J. Mater. Chem. C, 2019, 7, 7618–7626 RSC.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. Janssen and W. Li, Adv. Mater., 2016, 28, 943–950 CrossRef PubMed.
- H. S. Kim, D. Long, Y.-Y. Noh and D.-H. Hwang, J. Mater. Chem. C, 2018, 6, 10464–10471 RSC.
- W. Li, A. Furlan, K. H. Hendriks, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2013, 135, 5529–5532 CrossRef PubMed.
- W. Zhang, Z. Mao, J. Huang, D. Gao and G. Yu, Macromolecules, 2016, 49, 6401–6410 CrossRef.
- H. Huang, Z. Chen, R. Ponce Ortiz, C. Newman, H. Usta, S. Lou, J. Youn, Y. Y. Noh, K. J. Baeg, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 10966–10973 CrossRef.
- Z. Mao, W. Zhang, J. Huang, K. Shi, D. Gao, Z. Chen and G. Yu, Polym. Chem., 2015, 6, 6457–6464 RSC.
- X. Guo, J. Quinn, Z. Chen, H. Usta, Y. Zheng, Y. Xia, J. W. Hennek, R. P. Ortiz, T. J. Marks and A. Facchetti, J. Am. Chem. Soc., 2013, 135, 1986–1996 CrossRef.
- M. Sato, A. Asami, G. Maruyama, M. Kosuge, J. Nakayama, S. Kumakura, T. Fujihara and K. Unoura, J. Organomet. Chem., 2002, 654, 56–65 CrossRef.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef PubMed.
- Y. Wang, T. Hasegawa, H. Matsumoto, T. Mori and T. Michinobu, Adv. Funct. Mater., 2017, 27, 1604608 CrossRef.
- E. Wang, Z. Ma, K. Vandewal, P. Henriksson, O. Inganäs, F. Zhang and R. A. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef PubMed.
- J. Huang, Z. Mao, Z. Chen, D. Gao, C. Wei, W. Zhang and G. Yu, Chem. Mater., 2016, 28, 2209–2218 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. Olvera de la Cruz, G. C. Schatz, L. Chen and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 10475–10483 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef PubMed.
- D. M. Smilgies, J. Appl. Crystallogr., 2009, 42, 1030–1034 CrossRef PubMed.
- J. Rivnay, S. C. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef.
- A. M. Hindeleh and R. Hosemann, J. Phys. C: Solid State Phys., 1988, 21, 4155–4170 CrossRef.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef.
- J. Chantson, H. Görls and S. Lotz, J. Organomet. Chem., 2003, 687, 39–45 CrossRef.
- J. Nakayama, H. Dong, K. Sawada, A. Lshii and S. Kumakura, Tetrahedron, 1996, 52, 471–488 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc02833b |
| ‡ Z. C. and J. H. contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |