
Enhancing electrochemical performance by triggering a local structure distortion in lithium vanadium phosphate cathode for Li ion batteries†
Hyunyoung
Park‡
 a,
Wontae
Lee‡
a,
Ranjth
Thangavel
ab,
Woong
Oh
a,
Bong-Soo
Jin
c and
Won-Sub
Yoon
*ad
a,
Wontae
Lee‡
a,
Ranjth
Thangavel
ab,
Woong
Oh
a,
Bong-Soo
Jin
c and
Won-Sub
Yoon
*ad
aDepartment of Energy Science, Sungkyunkwan University, 300 Suwon-si, Gyeonggi-do 440-746, Republic of Korea. E-mail: wsyoon@skku.edu
bSchool of Energy Science and Engineering, Indian Institute of Technology Guwahati, Guwahati 781039, India
cBattery Research Center, Korea Electrotechnology Research Institute, Changwon 51543, Korea
dSKKU Institute of Energy Science and Technology (SIEST), Sungkyunkwan University, Suwon 16419, Republic of Korea
First published on 31st October 2022
Abstract
The development of high-energy Li ion batteries (LIBs) with a long cycle life is essential for meeting the energy requirements in next-generation large-scale applications. Monoclinic Li3V2(PO4)3 has emerged as a promising cathode for high-energy LIBs owing to its robust three-dimensional structure, high working voltage (>4.0 V), and high theoretical capacity (197 mA h g−1) compared with LiFePO4. Recently, attempts have been made to improve the electrochemical performance by doping various elements, among which Ti4+ is considered to be promising. However, no in-depth studies have been conducted on the cause of performance improvement in Ti-doped Li3V2(PO4)3 materials, in terms of the structure, during electrochemical reactions. Here, we demonstrate that Ti doping triggers the distortion of the VO6 octahedron and expands the space composed of Li and O ions, enhancing Li ion diffusion. Moreover, the lattice mismatch is mitigated, suppressing incomplete phase transitions and enhancing structural reversibility. Furthermore, the distortion of VO6 becomes more severe at the end of charging after Ti doping, making “the S-shaped curve” clearer during the discharge process and leading to low overpotential and high discharge capacity. These findings provide valuable opportunities for understanding electrochemically induced phase distortions and possible approaches for utilizing the distortions in high-energy-voltage cathode materials for LIBs.
1. Introduction
Li ion batteries (LIBs) have achieved considerable success in the portable electronics market, and their use is now being extended to large-scale applications such as electric vehicles and grid storage. Despite their widespread use, the improvement in their practical energy density remains limited. Developing next-generation LIBs with a cell-level energy density >350 W h kg−1 will further increase the dominance of LIBs in the battery market. Thus, extensive research has been conducted on developing new cathode chemistries with high energy density and safety. Ni-rich layered cathode materials have recently gained significant attention owing to their high energy density and low cost compared with commercial LiCoO2 cathodes.1–3 However, highly toxic nature of cobalt limits the application in the large scale application and the weak metal–oxygen bond in the layered oxide cathodes can induce oxygen loss and thermal runaway processes, triggering safety issues in state-of-the-art LIBs.4,5As an alternative, polyanionic cathodes having robust PO4 frameworks, such as LiMPO4, Li2MPO4F, and Li3M2(PO4)3 (M = transition metal), are explored as eminent candidates for high-energy batteries.6–14 The robust structure derived from the strong covalent bonds and inductive effect of the phosphate framework can help achieve a safe and long-lasting LIB even under high operating temperatures. Among them such cathodes, monoclinic Li3V2(PO4)3 (LVP) (space group: P21/n) with a high operating voltage (>3.8 V) and high capacity is a promising alternative to layered oxide cathodes.15–21
The crystal structure of monoclinic LVP constitutes a VO6 octahedron surrounded by six PO4 tetrahedrons, and each PO4 tetrahedron is surrounded by four VO6 octahedrons. The Li ions in monoclinic LVP occupy three different crystallographic sites: Li(1) is at the tetrahedral site, whereas Li(2) and Li(3) are located in highly distorted tetrahedral environments.18,19 This arrangement forms a three-dimensional framework, and Li ions located inside the cavities can enable rapid Li ion diffusion compared with that in one-dimensional phosphate cathode.15
The Li ion storage mechanism and Li ion transport pathways in LVP cathodes have been discussed extensively.22,23 For example, Morcrette et al. investigated the phase transitions and volume variations in rhombohedral and monoclinic type LVP using an operando X-ray diffraction technique. In the rhombohedral type, a Li ion can be reversibly extracted (V3+/4+ redox at 3.7 V vs. Li/Li+) from the only two Li sites. In contrast, a Li ion located in all three Li sites can be reversibly extracted in the monoclinic type through a series of complicated two-phase reactions (V3+/5+ redox) between 3.0–4.8 V to deliver a discharge capacity of 197 mA h g−1.24 Yin et al. combined neutron diffraction and 7Li nuclear magnetic resonance to observe charge ordering/disordering in lithium lattice planes during lithium extraction/insertion.18,19,25 Lee et al. employed molecular dynamics simulation and showed that the Li ion mobility in monoclinic LVP is reasonably high and strongly anisotropic in nature.26
Intrinsically, LVP exhibits poor electronic conductivity (∼2.3 × 10−8 S cm−1 at 25 °C) owing to the separated octahedral arrangement of the VO6, limiting its advantages18 hence, it requires excessive processing, such as downsizing the particles to the nanoscale to improve the electrode kinetics and constituting an ultra-thin conductive carbon network to improve the intrinsic electric conductivity.27–34 Additionally, attempts have been made to adopt transition metal dopants in the structure to enhance the electrochemical performance of LVP. Several foreign dopants have been proposed at the vanadium site, such as Al3+, Fe3+, Mg2+, Zr4+, Cr3+, and Ti4+, resulting in improvements in rate capability, cycle stability, and electrical conductivity.29,35–40 For example, Al doping stabilizes Li ion extraction/insertion at high voltage, whereas Zr doping improves electrical conductivity by narrowing the bandgap.35,38 Additionally, Ti4+ doping has been reported to decrease charge transfer resistance and boost ionic conductivity and Li ion diffusion by causing more intense disordered arrangement of Li ions, which manifest as a more sloping voltage profile curve.40,41 However, the understanding on the real-time structural evolution responsible for improved electrochemical performance is limited despite considerable improvement in electrochemical performance by using electro-inactive foreign dopants. Here, using synchrotron-based high-resolution powder diffraction (HRPD), in situ X-ray diffraction (XRD) and ex situ X-ray absorption spectroscopy (XAS) combined with a series of electrochemical analysis, we investigate the electrochemical and structural behavior of Ti-doped Li3V1.97Ti0.03(PO4)3/graphene (LVP-T) and compare with those of LVP/graphene (LVP-P). We confirm that Ti doping distorts the VO6 local environment, expanding the space around the Li ions. This local structural distortion enhances Li ion diffusion during the electrochemical reaction, suppressing lattice mismatch during Li ion removal and resulting in higher structural reversibility.
2. Experimental procedure
2.1. Synthesis of LVP-P and LVP-T
LVP-P was synthesized by applying the sol–gel method using V2O5, NH4H2PO4, and Li2CO3 as starting materials. Oxalic acid was used both as a reducing and chelating agent. Graphene oxide dispersion was used as the carbon source to improve the electronic conductivity of LVP. Initially, stoichiometric amounts of V2O5 and oxalic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mole ratio) were dissolved in water at 80 °C. After 1 h of stirring, the stoichiometric number of other precursors—NH4H2PO4, Li2CO3, and graphene oxide dispersion—were added to the solution, and the mixture was vigorously stirred to obtain the LVP gel precursor. The LVP gel precursor was subjected to two-step heat treatment at 350 °C for 4 h and 800 °C for 8 h in an Argon atmosphere. To obtain the LVP-T cathode, the same experimental procedure used for LVP-P was adopted, excluding the addition of TiO2 as the Ti precursor.
:3 mole ratio) were dissolved in water at 80 °C. After 1 h of stirring, the stoichiometric number of other precursors—NH4H2PO4, Li2CO3, and graphene oxide dispersion—were added to the solution, and the mixture was vigorously stirred to obtain the LVP gel precursor. The LVP gel precursor was subjected to two-step heat treatment at 350 °C for 4 h and 800 °C for 8 h in an Argon atmosphere. To obtain the LVP-T cathode, the same experimental procedure used for LVP-P was adopted, excluding the addition of TiO2 as the Ti precursor.
2.2 Electrode preparation and electrochemical measurements
The composition of cathodes for electrochemical characterization was as follows: active material (70 wt%), Super-P conductive carbon (20 wt%), and polyvinylidene fluoride binder (10 wt%). The electrode slurry was dispersed in N-methyl pyrrolidone (NMP) solvent, coated over Al foil, then dried at 120 °C for 12 h in a vacuum atmosphere. The electrodes were assembled in a CR2032 coin cell with lithium metal as a counter electrode and porous polypropylene Celgard film as the separator. The electrolyte used was 1.3 M LiPF6 in ethyl carbonate (EC)/diethyl carbonate (DEC) (3:7 vol/vol). Galvanostatic charge/discharge experiments were performed using a WonATech battery cycler WBCS3000S with a cutoff condition of 3.0–4.8 V. For the galvanostatic intermittent titration technique (GITT), a current rate of 0.05C based on the theoretical capacity of 197 mA h g−1 was applied for 10 min and relaxed for 3 h to attain the quasi-equilibrium state. For long-term cycling stability test, a current rate of 0.5C were applied for LVP-P and LVP-T electrodes during 100 cycles. The cyclic voltammetry (CV) was recorded at a potential range of 3.0–4.8 V at a scan rate of 0.1 mV s−1. Electrochemical impedance measurement (EIS) was also conducted after the 10th charge–discharge cycle by using a Biologic VMP3 multichannel system, and the applied frequency range was from 100 kHz to 10 mHz with an amplitude of 5 mV. The typical electrode mass and thickness were about 2.5 mg cm−2 and 30 mm, respectively. All of the electrochemical measurements were carried out at room temperature, and all capacity values were calculated based on the weight of the LVP active material.
2.3 Synchrotron-based X-ray analysis
Powder XRD patterns of the materials were obtained using a benchtop Rigaku D/MAX-2200 Ultima instrument with Cu-Kα radiation. Synchrotron-radiation-based XRD patterns of the materials were captured by using a MAR 345-image plate detector in the Pohang Accelerator Laboratory at beamline 5 A. XRD patterns were recorded using an X-ray wavelength of 0.6926 Å. The two-theta values were plotted after recalculation based on conventional Cu Kα radiation (λ = 1.5406 Å) for comparison with previous reports. XAS spectra were collected in transmission mode at beamline 10C (wide X-ray absorption fine structure) in the Pohang Accelerator Laboratory. The details of the spectroscopic electrochemical cell in the in situ XRD measurements had been previously explained by Balasubramanian et al.42The synchrotron-based XAS experiment on materials was performed in transmission mode in ionization chambers at room temperature. The storage ring was operated at 2.5 GeV. Before the measurement was performed, energy calibration for acquiring the V K-edge spectra was conducted by using the first inflection point of the spectrum of the V reference metal foil. All samples were prepared by disassembling the electrodes from the coin cells, and the electrodes were washed with diethyl carbonate (DEC) solution in an Ar filled glovebox to eliminate atmospheric contamination. The prepared electrodes were analyzed under a pressure of 3 × 10−10 torr at a resolution of 0.01 eV. The spectra were processed using the Athena software. X-ray absorption near edge structure (XANES) was obtained after normalization. The background-subtracted extended X-ray absorption fine structure (EXAFS) was k3-weighted to magnify the high-energy oscillations. It was then Fourier-transformed in the k-range from 3.0 to 11.8 Å−1.
2.4 Physical measurements
The morphology and the chemical compositions of materials were investigated by a field emission scanning electron microscopy (FE-SEM, JSM7000F, JEOL) working at 15.0 kV and energy-dispersive X-ray (EDX) mapping, respectively. The wrapping of graphene and the elements of materials were additionally characterized by transmission electron microscopy (TEM) images using a TF30ST at a 300 kV acceleration voltage and EDX mapping, respectively.3. Results and discussion
Fig. 1a shows the morphological features of LVP-P and LVP-T powders. Between the particles of LVP-P and LVP-T, notable difference of particle size and morphology is not observed, therefore, unit cell lattice change and structural behavior would be focused upon Ti doping. EDX elemental mapping studies were carried out to further investigate the distribution of each element (V, P, O, and Ti) in Fig. 1b. The Ti element is homogeneously distributed in LVP-T electrode, which clearly shows the existence of Ti atoms. The observation indicates the presence of Ti atoms in the LVP surface or within the grain boundaries. Fig. 1c shows the TEM images of LVP-P and LVP-T particles. The two images reveal the successful wrapping of graphene on the LVP surface. | ||
| Fig. 1 (a) SEM images of LVP-P and LVP-T particles and (b) energy-dispersive X-ray (EDX) mapping analysis of LVP-T particles (c) HRTEM images of LVP-P and LVP-T particles. | ||
Fig. 2a shows the HRPD patterns of LVP-P and LVP-T powders, which are indexed as a monoclinic structure (space group P21/n). The XRD pattern of LVP-P is very similar to that of LVP-P, and there is no characteristic peak of orthorhombic Li2Ti2(PO4)3 generally formed by Ti.43 Fig. S2, Tables S1 and S2† show the Rietveld refinement analysis of LVP-P and LVP-T.44 The unit cell lattice parameters of LVP-P are a = 8.609(5) Å, b = 8.594(5) Å, c = 12.039(5) Å, and V = 890.72(1) Å3, which are similar to those obtained in previous studies.18,20,23 The obtained parameters of LVP-T are as follows: a = 8.609(1) Å, b = 8.600(1) Å, c = 12.041(1) Å, and V = 891.42(1) Å3, showing no significant deviation between the two materials. There is no significant change in the size of the unit cell, but in the case of vanadium in the lattice structure, a difference occurs in the length and angle of the V–O bond, which changes the environment around the Li ion (Fig. 2b). As shown in Fig. S3, S4, Tables S3 and Table S4,† there is a difference in the local structures of Li3, Li2, and Li1 between LVP-P and LVP-T. The most notable point is that the volume of the space composed of lithium and oxygen in LVP-T is larger than that in LVP-P, which could affect the diffusion of Li ions.
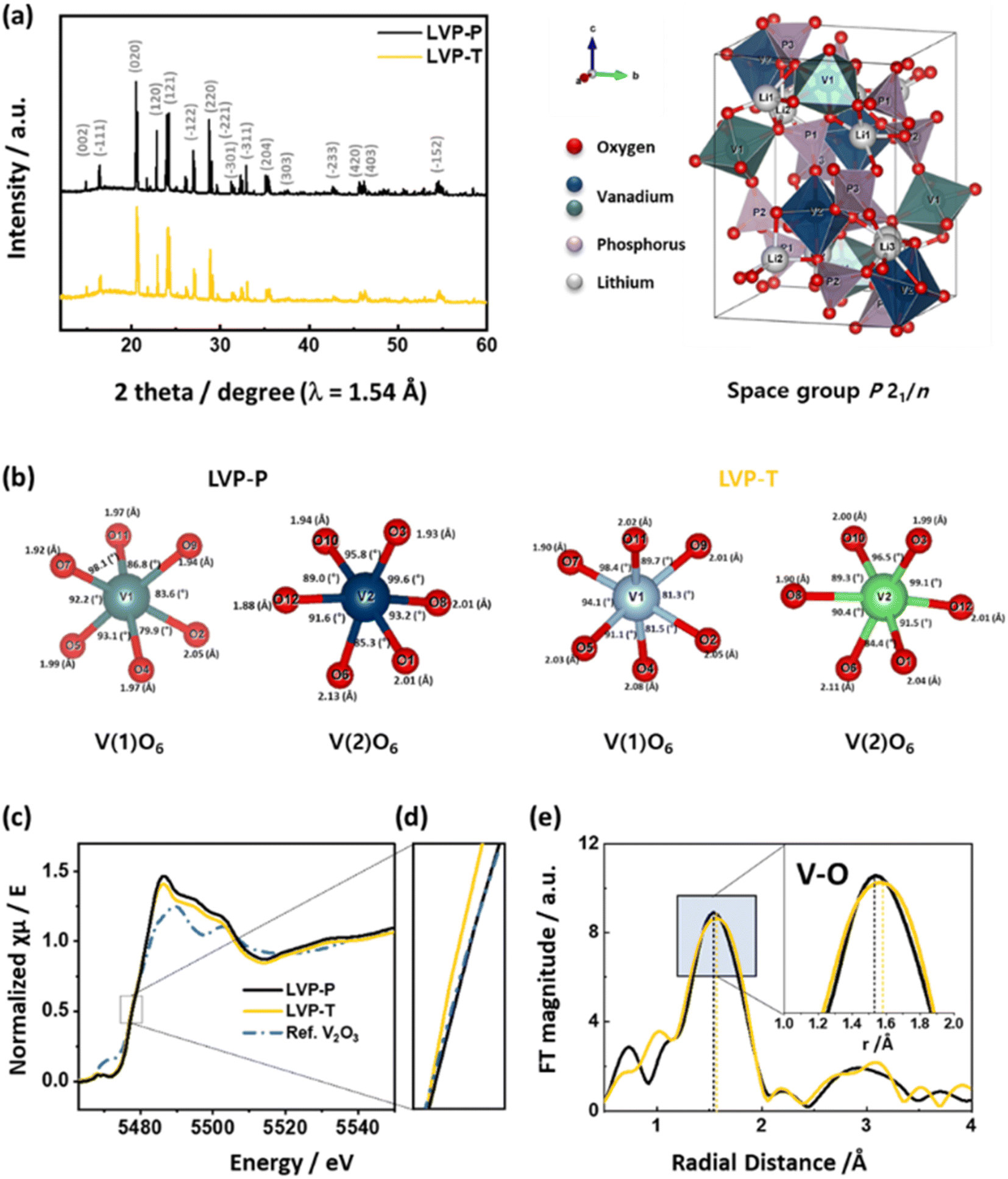 | ||
| Fig. 2 (a) HRPD patterns of LVP-P (reproduced from ref. 44 with permission from The Royal Society of Chemistry) and LVP-T. The representative unit cell structure of LVP is shown on the right-hand side of the XRD pattern. (b) Each bond length and bond angle of VO6 octahedra around the V1 and V2 sites in LVP-P and LVP-T in the crystal structures from Rietveld refinement of high-resolution powder diffraction. (c) Normalized XANES spectra and (d) magnified figure of XANES spectra at V K-edge at the pristine state of LVP-P and LVP-T with a reference of V2O3 powder. (e) k3-weighted Fourier transformed EXAFS spectra at V K-edge at the pristine state of LVP-P and LVP-T. | ||
Fig. 2c and d show normalized XANES spectra of LVP-P and LVP-T with V2O3 reference powder at the V K-edge. The V K-edge spectra of LVP-P show a nearly identical edge position to that of V2O3, indicating the oxidation state of V ions in LVP-P is 3+. However, the edge position in LVP-T is located at lower energy than in LVP-P, illustrating that the oxidation state of V ions is reduced by doped Ti4+ ions owing to its higher oxidation state than that of V3+ ions. The local structural change is further evidenced by Fourier-transformed EXAFS spectra at the V K-edge (Fig. 2e). There are changes in the peak intensity and radial distance of the 1st coordination V–O shell after Ti doping. EXAFS fitting was performed for quantitative analysis (Fig. S5†), and the radial distance and Debye–Waller factor had a deviation upon Ti doping (Table S5†). Specifically, the increase in Debye–Waller factor was observed the most in the V–O path, indicating that local distortion of VO6 octahedra was caused by Ti doping. The result of distortion around local structure around vanadium is consistent with the HRPD results of Fig. 2b and shows that Ti atoms locate in the VO6 octahedra, supported by XAS spectra at Ti K-edge in Fig. S6† and EDX mapping result of Fig. 1b. In the reports showing the effect of Ti atoms doped in the LVP materials, the oxidation state of Ti atoms is typically 4+ and the location is also at the sites of vanadium, as shown in our results.37,41–43,45
In Fig. 3a, electrochemical voltage profiles during the 1st cycle of LVP-P and LVP-T are plotted with a cutoff condition of 3.0–4.8 V at a current rate of 0.1C. The 1st charge and discharge capacities of LVP-P are 182.8 mA h g−1 and 165.1 mA h g−1, respectively, whereas those of LVP-T are 187.9 mA h g−1 and 173.6 mA h g−1, respectively. The four plateaus during the charging process indicate four phase transitions from the Li3V2(PO4)3 phase (Li3 phase) to the Li2.5V2(PO4)3 phase (Li2.5 phase), Li2V2(PO4)3 phase (Li2 phase), and Li1V2(PO4)3 phase (Li1 phase) to Li0V2(PO4)3 (Li0 phase), while the Li ion is extracted from three different Li ion sites in the order of Li3, Li1, and Li2 (Table S2†).18 In Fig. 3a(i), a longer voltage plateau at the 3rd transition and a lower voltage plateau at the last transition are observed in LVP-T compared to LVP-P. It is reported that a Li ion surrounded by four oxygen atoms bonded to V1 is extracted at the 3rd plateau. Subsequently, the Li ion is extracted from the site surrounded by oxygens bonded to V2 at the last plateau, which is the most kinetically hindered process in the delithiation processes during charging.18 During discharging, both LVP-P and LVP-T show a voltage plateau caused by a phase transition of the Li0/Li2, Li2/Li2.5, and Li2.5/Li3 phases. These plateaus are more pronounced in LVP-P than in LVP-T (Fig. 3a(ii) and (iii)). The 1st plateau of the “S-shaped curve” is reported as a result of phase distortion at the end of charging and the Li ion is inserted in two Li ion sites simultaneously.18,44 Moreover, a voltage plateau is incurred by two phase transitions when the Li ion is inserted in the Li3 site. Herein, it is inferred that the phase distortion at the end of the charge is influenced by Ti doping, affecting the shape of the voltage profile during the discharging process.
 | ||
| Fig. 3 (a) Electrochemical voltage profile of LVP-P and LVP-T between 3.0 and 4.8 V at 0.1C rate during the 1st cycle and the magnified figures of (i) the 3rd and the last plateau above ∼4.0 V during the charge; (ii) the 1st plateau (“S-shaped region”) above ∼3.7 V; (iii) the last two plateaus during the discharge under 3.8 V. Li ion diffusivity coefficient of LVP-P (reproduced from ref. 44 with permission from The Royal Society of Chemistry) and LVP-T obtained by GITT during (b) the first charge and (c) first discharge. | ||
To determine the correlation between the Li ion diffusion and voltage profile behavior, a GITT experiment, which is useful for determining the chemical diffusion coefficient of Li ions (DLi+), was conducted (Fig. S8†).46 In Fig. 3b, the diffusivity coefficients have four sudden drop points at the voltage plateau during the charging process. These drops imply that Li ion diffusion is kinetically difficult at the regions, especially in the 3rd and last plateaus. Higher diffusion coefficients during delithiation are observed in the 3rd and last plateaus for LVP-T, indicating that Li ion diffusion is improved after Ti doping. Accordingly, higher Li ion diffusion can make the voltage plateau corresponding to the 3rd phase transition longer and reduce the overpotential that occurs in the last phase transition. In Fig. 3c, the higher diffusion coefficients are also observed in the initial discharging process (∼4.05 V) and at the end of the discharging process (∼3.69 V and ∼3.59 V). This means that the smoother the voltage curve without a distinct plateau, the higher the lithium diffusion rate. That is, the disappearance of the plateau boundary would imply that Li ion diffusion is more favorable by Ti doping.41 In the second cycle, LVP-T is also more reversible and expresses a higher capacity than LVP-P (Fig. S7†).
Cyclic voltammetry measurement was carried out at a scan rate of 0.1 mV s−1 to figure out the electrochemical behavior of LVP-P and LVP-T (Fig. S9†). There are four oxidation peaks and three reduction peaks, corresponding to the relative Li ion extraction and reinsertion. The difference in oxidation and reduction peaks is derived from the initial Li ion inserting into V2(PO4)3, which is also observed in the voltage profile (Fig. 3a). LVP-T shows higher and distinct current peaks in addition to smaller potential differences between anodic and cathodic peaks, indicating the higher reversibility of the delithiation/lithiation, which is also supported by higher Li ion diffusivity coefficients of GITT results (Fig. 3b and c). EIS analysis was carried out after the 1st charge/discharge cycle at the same discharge rate in Fig. S10† and the semi-circles in the high frequency can be analyzed as an indicator of charge transfer resistance in the interface between the electrolyte and electrode. The charge transfer resistance of LVP-T is lower than that of LVP-P, indicating that Ti doping improves the charge conductivity. To compare the cycling stability of LVP-P and LVP-T, both samples were charged and discharged for 100 cycles at 0.5C (Fig. S11†). The cycle retention of LVP-T (75%) is higher than that of LVP-P (72%). The higher cycle stability of LVP-T would be derived from the higher reversibility of Li ion extraction/reinsertion and higher Li ion diffusivity trend during the cycle upon Ti doping.
In situ XRD experiments were performed while cycling at a current rate of 0.2C to find the cause of the difference in electrochemical behavior between the two materials. In Fig. 4, each phase of the two materials is represented by a specific color: the Li3 phase in blue, Li2.5 phase in green, Li2 phase in yellow, Li1 phase in orange, and Li0 phase in red. The overall structural behaviors of LVP-P and LVP-T according to the phase transitions during the electrochemical reactions are similar (Fig. 4). To quantitatively compare the structural behaviors, we calculated the corresponding lattice parameters of the LVP-P and LVP-T materials based on in situ XRD patterns (Fig. 5).
 | ||
| Fig. 4 In situ XRD patterns with time–voltage curve measured at 0.2C during the (a) 1st charge and (b) discharging process in the (−112), (210) planes of LVP-P (reproduced from ref. 44 with permission from The Royal Society of Chemistry) and LVP-T. | ||
 | ||
| Fig. 5 Change in unit cell lattice parameters obtained from in situ XRD patterns of LVP-P (unfilled; reproduced from ref. 44 with permission from The Royal Society of Chemistry) and LVP-T (filled); “a,” “b,” and “c” lattice parameter in (a) the 1st charging (delithiation) and (b) discharging (lithiation) processes. | ||
The unfilled/filled color symbol represents LVP-P and LVP-T, respectively. The most notable point during the charging process is that the difference in lattice parameters of the Li1 phase and the Li0 phase for LVP-T is smaller than that for LVP-P. This relaxed lattice mismatch can relieve the stress in the LVP system during the final phase transition. Another noteworthy point is that the “b” lattice parameter of the Li0 phase in LVP-T remains larger than that in LVP-P toward the end of charging. It can be inferred that the Ti doping effect keeps the Li ion channels wide at high voltages. Moreover, the LVP-T has only the Li0 phase at the end of the charge, whereas the LVP-P exhibits the coexistence of the Li1 and Li0 phases (Fig. 5a). This indicates that delithiation from the Li1 phase is not completed for the LVP-P material. The incomplete delithiation is derived from kinetically hindered Li ion diffusion during the last phase transition and structural distortion appearing during the phase transition.18,44 Thus, it is inferred that less lattice mismatch could enhance Li ion diffusion during phase transition and boost the completion of the phase transition.
During the discharging process (Fig. 5b), “a,” “b,” and “c” parameters increase as the Li ion is reinserted in both materials. In the case of LVP-T, the lattice parameter mismatch between the beginning and end of discharge is less than that of LVP-P. This smooth phase transition without a large gap between the Li0 phase and Li2 phase in LVP-T lowers the overpotential of the system and enhances the Li ion diffusivity as in the result of GITT (Fig. 3c). The lattice mismatch is evident in all phase transitions of LVP-P, which indicates that the kinetic disturbance of Li ion diffusion during the discharging process is more severe than that of LVP-T, resulting in a relatively delayed phase transition. Therefore, it can be confirmed that Ti doping takes an effect on suppressing incomplete phase transition during the electrochemical reaction.
To determine the effect of Ti doping on the vanadium local structure, V K-edge XAS spectra were obtained at the end of the charge and discharge states, and the corresponding spectra are given in Fig. 6. In Fig. 6a, the edge position moves toward higher energy during the charging process, indicating the oxidation of vanadium ions upon extraction of Li ions. The edge position of LVP-T is at a higher energy than that of LVP-P after the end of the charge (Fig. 6a(i)), indicating that the higher charge capacity in Fig. 3a is derived from the higher oxidation state of vanadium ions in LVP-T. In Fig. 6a(ii), the pre-edge intensity increases at the end of the charge owing to the highly distorted VO6 octahedra.44,47 In general, the dramatic increase in the intensity of the pre-edge refers to the distortion of the local structure, and there is a possibility that the local symmetry changes from octahedron to tetrahedron. However, materials having a vanadium tetrahedral local structure, such as NH4VO3, vanadinite, and CrVO4, have pre-edge intensity values of ∼1.0 in the normalized V K-edge XANES spectra.48 In contrast, LVP-P and LVP-T have relatively low pre-edge intensity values of ∼0.3. In addition, the V–O bond length at the end of the charge is ∼2.00 Å for LVP-P and ∼1.98 Å for LVP-T obtained by EXAFS fitting (Table S6†), which are larger values than the V–O bond lengths of the vanadium tetrahedral structure, from 1.66 to 1.75 Å.18,48–50 Therefore, the possibility of tetrahedral symmetry is very small; rather, the pre-edge intensity value is similar to V2O4, whose intensity is less than 0.4.51 The site symmetry of the V atom in V2O4 is C1 point group, which is the lowest symmetry among the point symmetry, forming distorted octahedra. Thus, in this study, the highly distorted VO6 octahedra are formed at the end of the charge in both LVP-P and LVP-T. In terms of the relationship between intensity and distortion, the higher pre-edge intensity of LVP-T means that the local structure of VO6 in LVP-T is more distorted than that of LVP-P at the end of the charge. During the discharging process (Fig. 6b(i) and (ii)), the edge position of both materials shifts toward lower energy and pre-edge intensity turns back to the pristine state, indicating the reduction of vanadium ions and relaxed distortion of the vanadium local structure. In Fig. 6b(i), the edge position at the end of discharge in LVP-T is nearly the same as that of the pristine state of LVP-T, whereas that of LVP-P is higher than that of the pristine state of LVP-P. This indicates that Ti doping improves the reversibility of the electrochemical reaction. Fig. 6c shows the EXAFS spectra of the pristine and fully charged states for LVP-P and LVP-T. The first peak, corresponding to the bond of V–O, shows the change in the VO6 octahedra of the host structure. The V–O bond distance of LVP-T is shorter because of the higher oxidation state of the vanadium ion compared to that of LVP-P at fully charged states, which is consistent with the XANES results. In addition, the intensity of the first coordination shell for LVP-T is lower than that for LVP-P, indicating that the local environment of V–O is more distorted in LVP-T. The spectra of the first peak is fitted in both materials to further investigate the change in V–O coordination information. In Table S6,† most V–O paths have reduced radial distance upon Ti doping, consistent with the higher oxidation state of XANES. Notable is the increase of Debye–Waller factors in most V–O paths upon Ti doping, indicating more distorted VO6 octahedra in LVP-T at the end of charge. This distortion of VO6 can further promote the phase distortion at the end of the charging process, which seems to be the cause of the clearer “S-shaped curve” during the subsequent discharging process. In Fig. 6d, EXAFS spectra of the pristine state and the end of the discharge state are plotted in both materials. The LVP-T exhibits higher reversibility in the V–O peak intensity and bond distance compared with that of LVP-P. This high reversibility is thought to be achieved by introducing Ti into the LVP system to trigger structural distortion and facilitate Li ion diffusion. In the LVP structure, the structural distortion at the last delithiation is known as the cause of the monotonous voltage curves during the discharging process.18,44 In this study, it was observed that the more severe the structural distortion, the smaller the lattice mismatch between the two phases, and the higher the Li ion mobility. Here, Ti doping is presumed to widen the Li ion pathway by distorting the VO6 local structure. Accordingly, the LVP-T shows relatively smooth voltage profile curves without distinct plateaus at the start and end of discharging process, compared with those of LVP-P.
 | ||
| Fig. 6 Normalized V K-edge XANES spectra for (a) the 1st charge (FC) and (b) the 1st discharge (FD) states for LVP-P and LVP-T. Magnified plots of XANES spectra for the region at around (i) edge half-height and (ii) pre-edge. k3-weighted Fourier-transformed V K-edge EXAFS spectra at (c) the 1st charge and (d) the 1st discharge states for LVP-P and LVP-T (inset: magnified plots of the 1st coordination peak of V–O bonds). | ||
In summary, we observed that Ti doping results in enhanced Li ion diffusion during the phase transition, and this is mainly contributed by highly distorted VO6 octahedra, widening the Li ion path. Because of the distortion, we observed an alleviation of the lattice mismatch, enhancement of the Li ion diffusion rate, and reduction in the coexistence of two phases. Consequently, local distortion of VO6 by Ti doping brought about the enhanced Li ion diffusion and more facile phase transition.
4. Conclusion
In this study, we demonstrated the structural effect of Ti doping on LVP to clarify the origin of the enhanced performance of Ti-doped LVP. Comparative analyses were performed via several electrochemical tests combined with in situ and ex situ X-ray analyses. In the Rietveld refining and XAS analysis, the VO6 octahedron is distorted after Ti doping and expands the local space composed of Li and O ions, creating an environment where Li ions are easy to diffuse. Improved Li ion diffusivity was observed in the GITT analysis, and in the structural behavior observed by in situ XRD during the electrochemical reaction, the lattice mismatch was alleviated in the case of Ti doping. Thus, Ti doping facilitates the completion of phase transition, shortening the two-phase coexistence period, which seems to have influenced a longer plateau and lower overpotential in the 1st charging process. In the V K-edge XANES and EXAFS spectra of the charged state, the VO6 distortion of the Ti-doped LVP was more severe, which further reinforced the phase distortion of the last phase transition; the distortion makes a more prominent “S-shaped curve” during the 1st discharging process. Consequently, the higher distortion causes facile phase transition derived from higher Li ion diffusion kinetics, contributing to higher structural reversibility during the electrochemical reaction. These findings provide a deeper understanding of adopting structural distortion by Ti doping in the LVP structure and suggest practical strategies for developing high-performance cathodes in Li ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work used data reproduced from ref. 44 with permission from the Royal Society of Chemistry (https://doi.org/10.1039/c9ta12435g). This work was supported by the Industrial Strategic Technology Development Program (20015809, Development of particle shape controlled nickel-based cathode material with high capacity and long cycle life) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea). Also, this work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2022R1A2B5B02002624).References
- J. S. Weaving, F. Coowar, D. A. Teagle, J. Cullen, V. Dass, P. Bindin, R. Green and W. J. Macklin, J. Power Sources, 2001, 97–98, 733–735 CrossRef CAS.
- W. Lee, S. Muhammad, C. Sergey, H. Lee, J. Yoon, Y. M. Kang and W. S. Yoon, Angew. Chem., Int. Ed., 2020, 59, 2578–2605 CrossRef CAS PubMed.
- W. Lee, S. Muhammad, T. Kim, H. Kim, E. Lee, M. Jeong, S. Son, J. H. Ryou and W. S. Yoon, Adv. Energy Mater., 2018, 8, 1–12 Search PubMed.
- K. W. Nam, S. M. Bak, E. Hu, X. Yu, Y. Zhou, X. Wang, L. Wu, Y. Zhu, K. Y. Chung and X. Q. Yang, Adv. Funct. Mater., 2013, 23, 1047–1063 CrossRef CAS.
- S. Bak, E. Hu, Y. Zhou, X. Yu, S. D. Senanayake, S. Cho, K. Kim, K. Y. Chung, X. Yang and K. Nam, Appl. Mater. Interfaces, 2014, 6, 22594–22601 CrossRef CAS PubMed.
- J. Chen, S. Wang and M. S. Whittingham, J. Power Sources, 2007, 174, 442–448 CrossRef CAS.
- D. Wang, H. Buqa, M. Crouzet, G. Deghenghi, T. Drezen, I. Exnar, N. H. Kwon, J. H. Miners, L. Poletto and M. Grätzel, J. Power Sources, 2009, 189, 624–628 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy, C. Masquelier, S. Okada and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1609–1613 CrossRef CAS.
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6, 1144–1148 CrossRef CAS.
- S. Yuvaraj, W. Oh and W. S. Yoon, J. Electrochem. Sci. Technol., 2019, 10, 1–13 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS PubMed.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed.
- C. Masquelier, C. Wurm, J. Rodríguez-Carvajal, J. Gaubicher and L. Nazar, Chem. Mater., 2000, 12, 525–532 CrossRef CAS.
- W. Lee, J. Kim, S. Yun, W. Choi, H. Kim and W. S. Yoon, Energy Environ. Sci., 2020, 13, 4406–4449 RSC.
- H. Huang, S. C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525–1528 CrossRef CAS.
- H. Huang, T. Faulkner, J. Barker and M. Y. Saidi, J. Power Sources, 2009, 189, 748–751 CrossRef CAS.
- J. Gaubicher, C. Wurm, G. Goward, C. Masquelier and L. Nazar, Chem. Mater., 2000, 12, 3240–3242 CrossRef CAS.
- S. C. Yin, H. Grondey, P. Strobel, M. Anne and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 10402–10411 CrossRef CAS PubMed.
- S. C. Yin, H. Grondey, P. Strobel, H. Huang and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 326–327 CrossRef CAS PubMed.
- M. Y. Saïdi, J. Barker, H. Huang, J. L. Swoyer and G. Adamson, Electrochem. Solid-State Lett., 2002, 5, 149–151 CrossRef.
- H. Ohkawa, K. Yoshida, M. Saito, K. Uematsu, K. Toda and M. Sato, Chem. Lett., 1999, 1017–1018 CrossRef CAS.
- M. Sato, H. Ohkawa, K. Yoshida, M. Saito, K. Uematsu and K. Toda, Solid State Ionics, 2000, 135, 137–142 CrossRef CAS.
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, 30–33 CrossRef.
- M. Morcrette, J. B. Leriche, S. Patoux, C. Wurm and C. Masquelier, Electrochem. Solid-State Lett., 2003, 6, 80–84 CrossRef.
- S.-C. Yin, P. S. Strobel, H. Grondey and L. F. Nazar, Chem. Mater., 2004, 2, 1456–1465 CrossRef.
- S. Lee and S. S. Park, J. Phys. Chem. C, 2012, 116, 25190–25197 CrossRef CAS.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294–1305 RSC.
- M. Ren, Z. Zhou, Y. Li, X. P. Gao and J. Yan, J. Power Sources, 2006, 162, 1357–1362 CrossRef CAS.
- S. Xun, J. Chong, X. Song, G. Liu and V. S. Battaglia, J. Mater. Chem., 2012, 22, 15775–15781 RSC.
- F. Wu, F. Wang, C. Wu and Y. Bai, J. Alloys Compd., 2012, 513, 236–241 CrossRef CAS.
- Q. Chen, T. Zhang, X. Qiao, D. Li and J. Yang, J. Power Sources, 2013, 234, 197–200 CrossRef CAS.
- L. L. Zhang, S. Duan, G. Peng, G. Liang, F. Zou and Y. H. Huang, J. Alloys Compd., 2013, 570, 61–64 CrossRef CAS.
- M. Choi, H. S. Kim, Y. Moo Lee, W. K. Choi and B. S. Jin, Mater. Lett., 2015, 160, 194–199 CrossRef CAS.
- J. Barker, R. K. B. Gover, P. Burns and A. Bryan, J. Electrochem. Soc., 2007, 154, A307 CrossRef CAS.
- A. R. Cho, J. N. Son, V. Aravindan, H. Kim, K. S. Kang, W. S. Yoon, W. S. Kim and Y. S. Lee, J. Mater. Chem., 2012, 22, 6556–6560 RSC.
- C. Dai, Z. Chen, H. Jin and X. Hu, J. Power Sources, 2010, 195, 5775–5779 CrossRef CAS.
- J. Xu, G. Chen, H. Zhang, W. Zheng and Y. Li, J. Appl. Electrochem., 2015, 45, 123–130 CrossRef CAS.
- K. Kalaga, F. N. Sayed, M. T. F. Rodrigues, G. Babu, H. Gullapalli and P. M. Ajayan, J. Power Sources, 2018, 390, 100–107 CrossRef CAS.
- M. Choi, K. Kang, H. S. Kim, Y. Moo Lee and B. S. Jin, RSC Adv., 2015, 5, 4872–4879 RSC.
- S. Liu, S. Li, K. Huang and Z. Chen, Acta Phys.-Chim. Sin., 2007, 23, 537–542 CrossRef CAS.
- M. Balasubramanian, X. Sun, X. Q. Yang and J. Mcbreen, J. Power Sources, 2001, 92, 1–8 CrossRef CAS.
- D. Okada, F. Nakano, K. Uematsu, H. Okawa, A. Itadani, K. Toda and M. Sato, Commun. Electrochem., 2015, 83, 828–830 CrossRef CAS.
- W. Oh, H. Park, B. S. Jin, R. Thangavel and W. S. Yoon, J. Mater. Chem. A, 2020, 8, 10331–10336 RSC.
- C. Deng, S. Zhang, S. Y. Yang, Y. Gao, B. Wu, L. Ma, B. L. Fu, Q. Wu and F. L. Liu, J. Phys. Chem. C, 2011, 115, 15048–15056 CrossRef CAS.
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569–1578 CrossRef CAS.
- S. Kim, Z. Zhang, S. Wang, L. Yang, E. J. Cairns, J. E. Penner-Hahn and A. Deb, J. Phys. Chem. C, 2016, 120, 7005–7012 CrossRef CAS.
- J. Wong, F. W. Lytle, R. P. Messmer and D. H. Maylotte, Phys. Rev. B, 1984, 30, 5596–5610 CrossRef CAS.
- H. T. Evans, Z. Kristallogr. - Cryst. Mater., 1960, 114, 257–277 CrossRef CAS.
- W. E. Baker, Am. Mineral., 1966, 51, 1712–1721 CAS.
- J. P. Gustafsson, Appl. Geochem., 2019, 102, 1–25 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06837k |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2022 |