
Cp*Rh(III)/boron hybrid catalysis for directed C–H addition to β-substituted α,β-unsaturated carboxylic acids†
Ryo
Tanaka
a,
Yuki
Hirata
a,
Masahiro
Kojima
 a,
Tatsuhiko
Yoshino
*ab and
Shigeki
Matsunaga
*ab
a,
Tatsuhiko
Yoshino
*ab and
Shigeki
Matsunaga
*ab
aFaculty of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan. E-mail: tyoshino@pharm.hokudai.ac.jp; smatsuna@pharm.hokudai.ac.jp
bGlobal Station for Biosurfaces and Drug Discovery, Hokkaido University, Sapporo 060-0812, Japan
First published on 29th November 2021
Abstract
The C–H bond addition reaction of 2-phenylpyridine derivatives with α,β-unsaturated carboxylic acids catalyzed by Cp*Rh(III)/BH3·SMe2 is reported. Activation of C–H bonds with the rhodium catalyst and activation of α,β-unsaturated carboxylic acids with the boron catalyst cooperatively work, and a BINOL-urea hybrid ligand significantly improved the reactivity. With the optimized hybrid catalytic system, various β-disubstituted carboxylic acids were obtained under mild reaction conditions.
Directed C–H functionalization is a powerful tool that enables streamlined syntheses of valuable organic molecules from readily available feedstocks.1 Trivalent group 9 transition metals bearing pentamethylcyclopentadienyl ligands (Cp*) are versatile and highly active catalysts for directed C–H functionalization.2 Among a wide variety of reported reactions that employ these catalysts, the formal conjugate addition of aromatic C–H bonds to electron-deficient olefins is an atom- and step-economical synthetic method for alkylated arenes.3,4 Although these C–H addition reactions have been well-studied over the last decade, the use of α,β-unsaturated carboxylic acids as alkylating agents has received limited attention5–7 despite the carboxy group being an important and fundamental functional group in organic synthesis. In 2019, Han, Hu, and co-workers reported the C–H alkylation of 2-phenylpyridines with coumarin-3-carboxylic acids, in which a decarboxylation takes place under the reaction conditions (Fig. 1a).5 Satoh and co-workers reported a Cp*Rh(III)-catalyzed addition of benzamide C–H bonds to the highly electron-deficient α-trifluoromethylacrylic acid in 2020 (Fig. 1a).6 In both these cases, the α,β-unsaturated carboxylic acids require an additional electron-withdrawing group. Gooßen and co-workers reported a Ru(II)-catalyzed C–H alkylation of benzoic acids, in which a sterically accessible non-substituted acrylic acid was required (Fig. 1b).7 The limitations of these reactions prompted us to seek a new catalytic system to enable redox-neutral directed C–H alkylations using more general β-substituted α,β-unsaturated carboxylic acids.
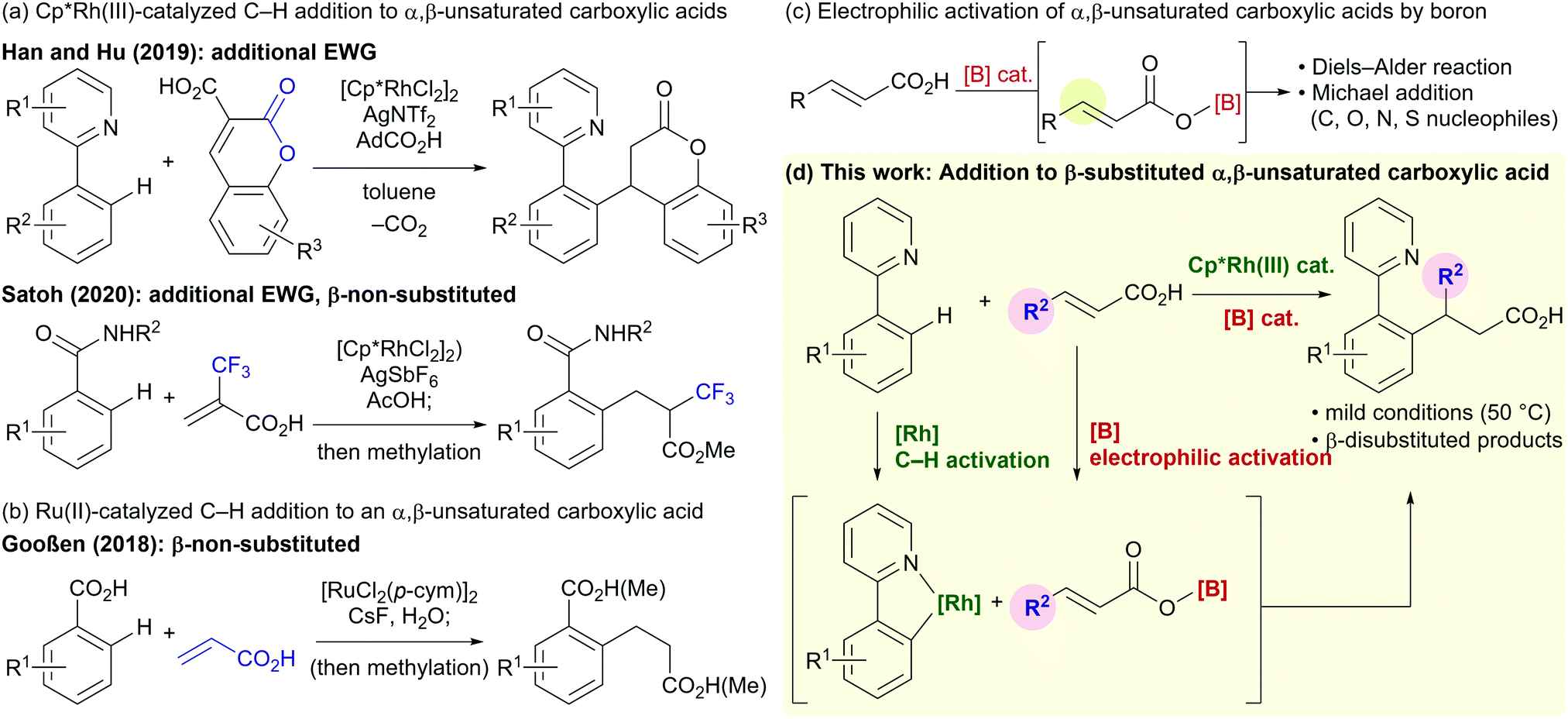 | ||
| Fig. 1 (a and b) Previous examples of directed C–H addition to α,β-unsaturated carboxylic acids; (c) boron catalysis for activation of α,β-unsaturated carboxylic acids; (d) the reaction presented in this work. | ||
Catalytic activation of a carboxy group using a boron reagent/catalyst via the formation of acyloxyboranes has emerged as an attractive method for the functionalization and derivatization of carboxylic acids.8–14 The electrophilicity of α,β-unsaturated carboxylic acids is also enhanced by this approach (Fig. 1c).9–12 In 1988, Yamamoto and co-workers reported BH3·THF-catalyzed Diels–Alder reactions between α,β-unsaturated carboxylic acids and dienes via an acyloxy–borane intermediate.9 Hall and co-workers demonstrated that aryl boronic acids are efficient catalysts for similar Diels–Alder and cycloaddition reactions.10 The Takemoto group has developed chiral catalytic systems composed of a boronic acid, thiourea, and amine for asymmetric hetero-Michael addition reactions between α,β-unsaturated carboxylic acids and a variety of nucleophiles.11 The Ishihara group has reported asymmetric Michael additions between ketones and α,β-unsaturated carboxylic acids catalyzed by a boronic acid and a chiral amine.12 Inspired by these previous studies, we envisaged that the combination of a Cp*Rh(III) catalyst and a boron catalyst would enable the directed addition of a C–H bond to various α,β-unsaturated carboxylic acids (Fig. 1d).
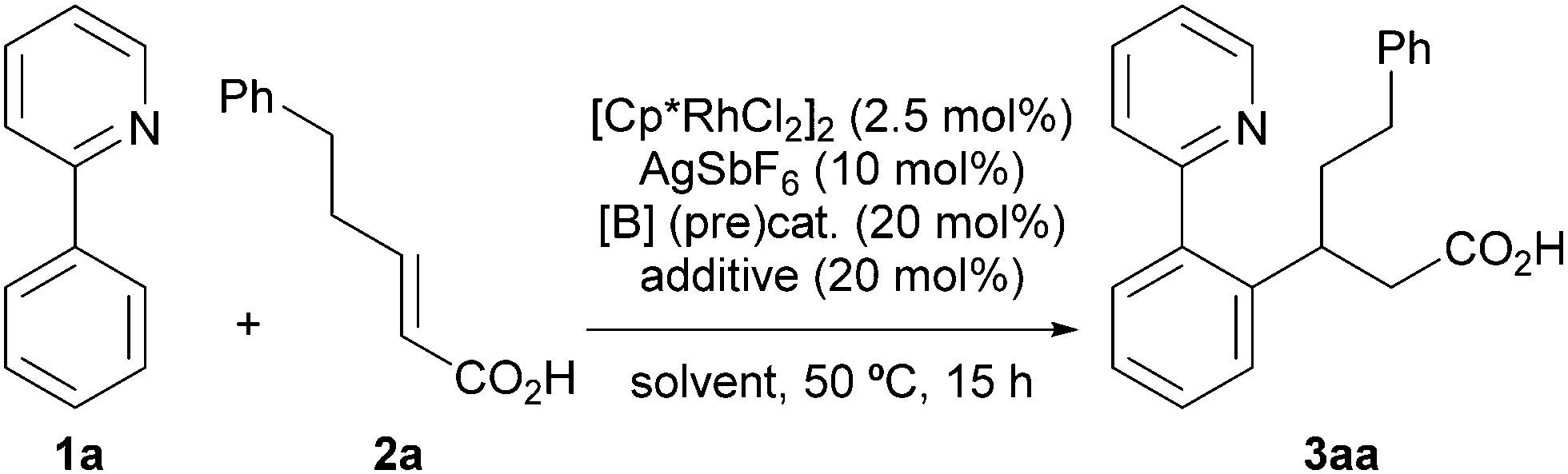
Our investigation started with the reaction of model substrates 2-phenylpyridine 1a and α,β-unsaturated carboxylic acid 2a in the presence of the metal catalyst [Cp*RhCl2]2/AgSbF6, and the boron precatalyst BH3·SMe2 (Table 1). Solvent screening (entries 1–5) revealed that the desired product 3aa was obtained when using DMF at 50 °C, albeit in a low yield (entry 5). Other solvents were not suitable for this reaction. We next examined other boron catalysts, such as B(OH)3, PhB(OH)2, and (AcO)4B2O, but the desired reaction did not proceed (entries 6–8). In the absence of BH3·SMe2, 3aa was not obtained, indicating that activation of 2a with the boron catalyst is essential for a successful reaction (entry 9). To improve the reactivity, we added a ligand for the boron catalyst. When we used Ts-L-Val (entry 10) or (S)-BINOL (entry 11), the reactivity was greatly enhanced, as reported by Shimizu, Kanai, and co-workers,14b,c and 3aa was obtained in 52% and 32% yield, respectively. We also investigated the effect of adding urea 4 (Fig. 2), which can potentially activate an acyloxyborane intermediate via hydrogen bonding.11 As expected, urea 4 improved the product yield, with respect to entry 5, to 20% (entry 12). The combination of 4 and (S)-BINOL led to a further improvement of the reactivity (entry 13). Following this promising result, we synthesized a BINOL-urea hybrid ligand 5 (Fig. 2 and see the ESI† for details) and examined its catalytic activity. To our delight, the yield increased to 86% when this new ligand was used (entry 14). We confirmed that, even when using 5, no reaction proceeded in the absence of BH3·SMe2 (entry 15), while a racemic BINOL-urea ligand (rac-5) exhibited a similar reactivity (entry 16). Under these conditions, the second C–H alkylation from 3aa was not observed possibly due to the relatively low reaction temperature (50 °C).
|
|
||||
|---|---|---|---|---|
| Entry | [B] (pre)cat. | Additive | Solvent | Yieldb (%) |
| a The reactions were run using 1a (0.1 mmol), 2a (0.1 mmol), [Cp*RhCl2]2 (2.5 mol%), AgSbF6 (10 mol%), boron catalyst (20 mol%), and an additive in the indicated solvent (0.5 M). b Determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. | ||||
| 1 | BH3·SMe2 | — | DCE | 0 |
| 2 | BH3·SMe2 | — | Toluene | 0 |
| 3 | BH3·SMe2 | — | 1,4-Dioxane | 0 |
| 4 | BH3·SMe2 | — | HFIP | 0 |
| 5 | BH3·SMe2 | — | DMF | 7 |
| 6 | B(OH)3 | — | DMF | 0 |
| 7 | PhB(OH)2 | — | DMF | 0 |
| 8 | (AcO)4B2O | — | DMF | 0 |
| 9 | — | — | DMF | 0 |
| 10 | BH3·SMe2 | Ts-L-Val | DMF | 52 |
| 11 | BH3·SMe2 | (S)-BINOL | DMF | 32 |
| 12 | BH3·SMe2 | 4 | DMF | 20 |
| 13 | BH3·SMe2 | 4 + (S)-BINOL | DMF | 48 |
| 14 | BH3·SMe2 | 5 | DMF | 86 |
| 15 | — | 5 | DMF | 0 |
| 16 | BH3·SMe2 | rac-5 | DMF | 73 |
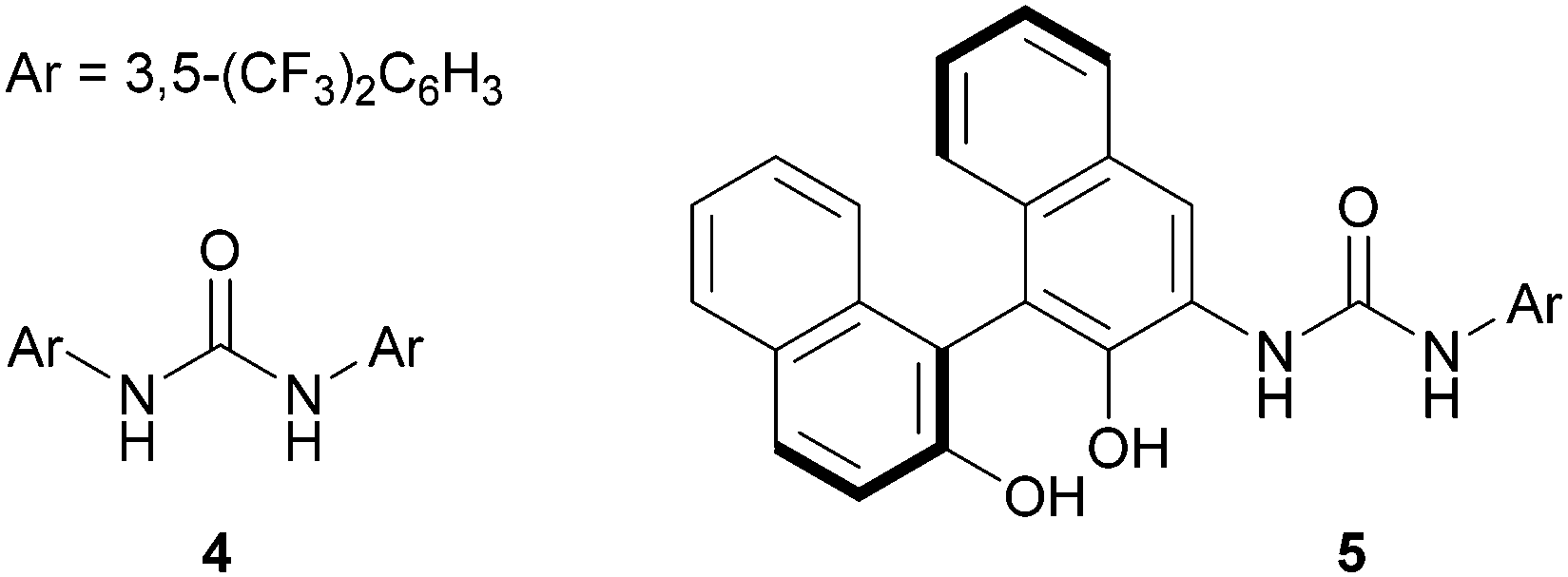 | ||
| Fig. 2 Structure of the additive/ligand used in Table 1. | ||
Under the optimized reaction conditions, we examined the scope of the 2-aryl pyridines (Scheme 1). The desired products were successfully isolated without esterification of the carboxy group. 2-Aryl pyridines bearing various substituents provided the corresponding products 3 in moderate to good yields. The reaction tolerated t-Bu (3ba, 3ja), methoxy (3ca, 3ka), halogen (3ea, 3fa, 3la), and carbonyl (3ga, 3ha, 3ma) substituents at both the para and meta positions. In the case where the substrate had a meta-substituent, the sterically less hindered C–H bond was selectively functionalized (3ia–3ma). When we performed a preparative-scale (1 mmol) reaction using 1aa as the substrate, 3aa was obtained without significant loss of yield.
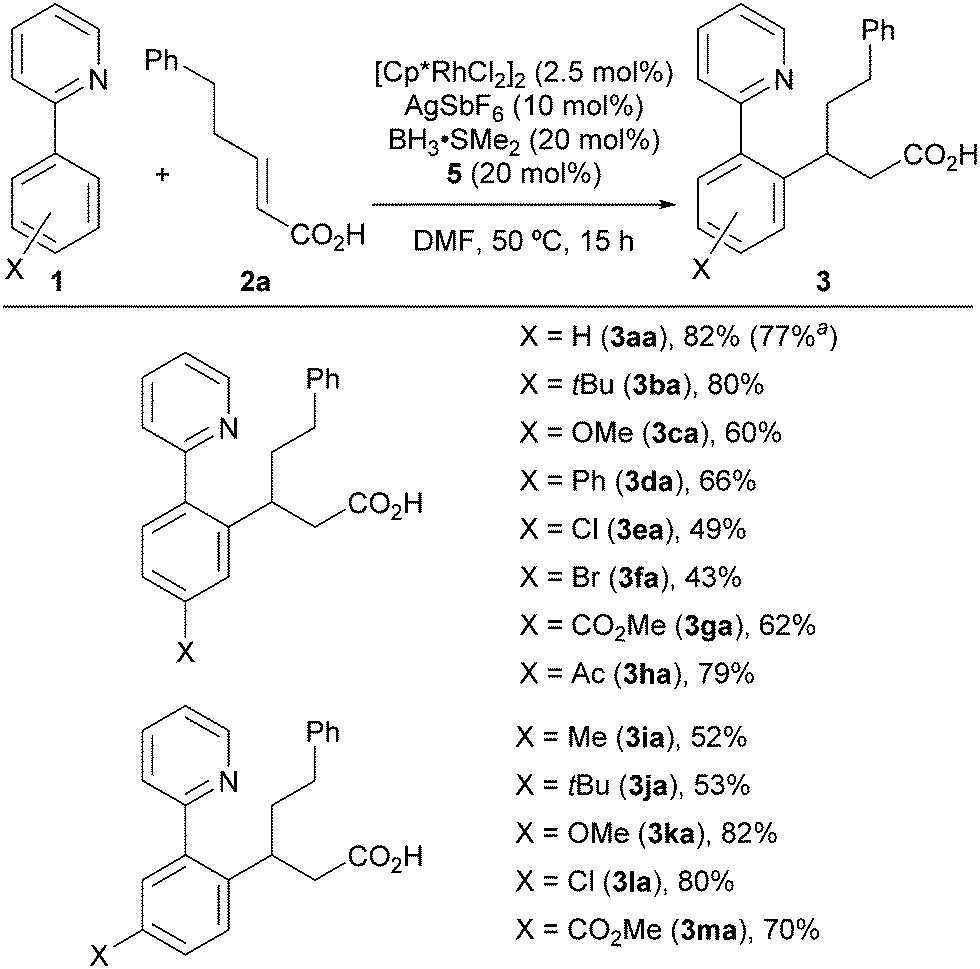 | ||
| Scheme 1 Scope of 2-aryl pyridines. The reactions were run using 1 (0.3 mmol) and 2a (0.3 mmol). a1 mmol scale. | ||
Subsequently, the scope of the α,β-unsaturated carboxylic acids was examined (Scheme 2). An α,β-unsaturated carboxylic acid bearing a linear alkyl group (2b) afforded 3ab in 59% yield. The reaction also proceeded smoothly when using a carboxylic acid bearing a sterically hindered branched alkyl group (3ac). α,β-Unsaturated carboxylic acids bearing a naphthyl group or a heterocycle, such as a thiophene or indole, are also compatible under these reaction conditions, providing the desired products in moderate to good yields (3ad–3af). A Bn-protected alcohol and a Ts-protected amine were also tolerated and afforded the desired products in moderate yields (3ag, 3ah). On the other hand, 3,3-dimethylacrylic acid, which bears two β-substituents, did not afford the desired product under the same conditions.
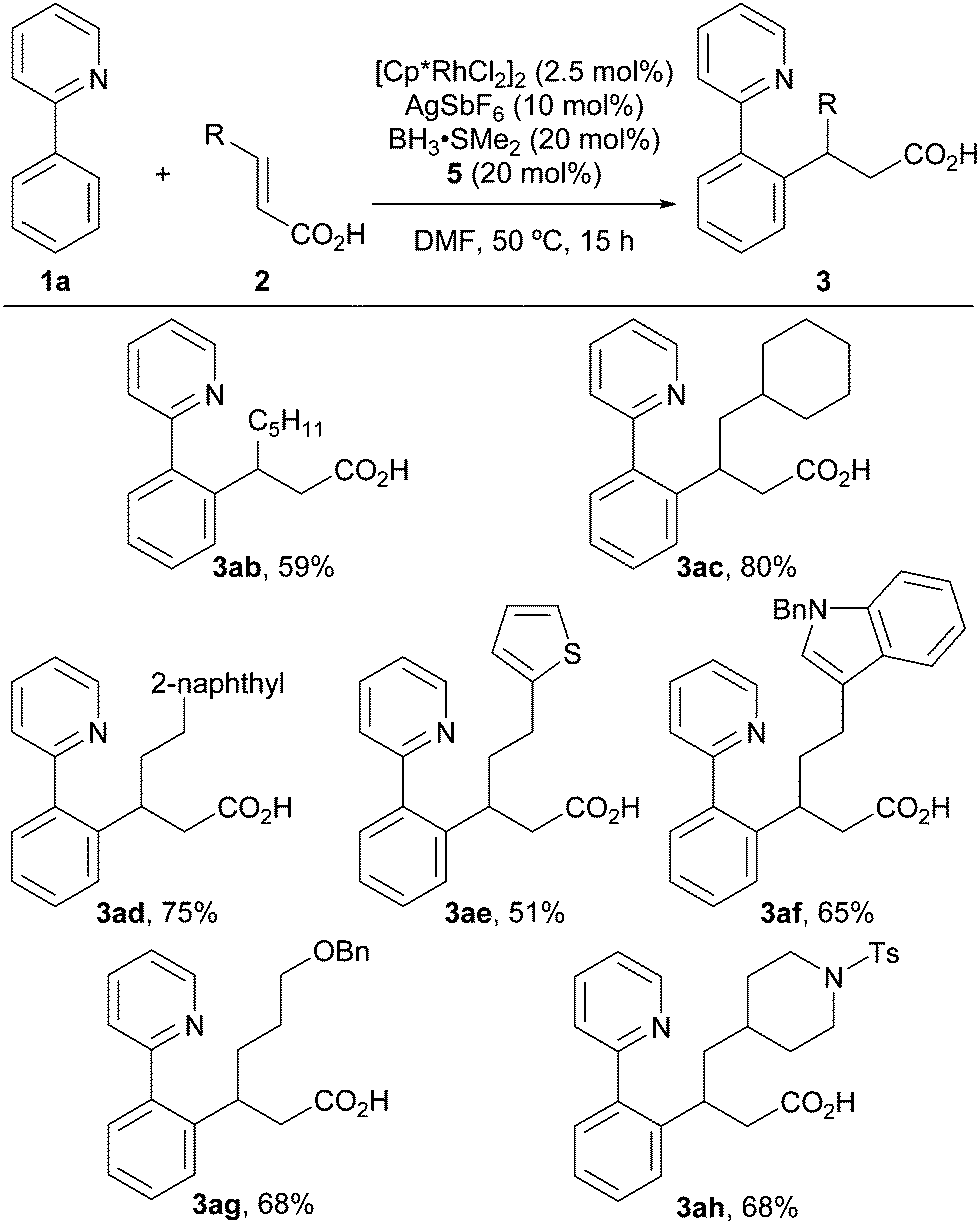 | ||
| Scheme 2 Scope of α,β-unsaturated carboxylic acids. The reactions were run using 1a (0.3 mmol) and 2 (0.3 mmol). | ||
A hypothetical catalytic cycle is shown in Fig. 3. An active rhodium species (I) is generated from [Cp*RhCl2]2 and AgSbF6 and reacts with 1 to form metallacycle IIvia deprotonative C–H activation. Meanwhile, BH3·SMe2 would react with 2 and 5 to form an active electrophile (IV), which coordinates to II and inserts into the C–Rh bond to produce III. Protonation of III and subsequent dissociation regenerates I with concomitant release of V. Finally, a B–O bond exchange reaction between V and 2 affords the product 3 and regenerates IV.9–12 Although the role of the urea functional group of 5 has not yet been experimentally proved, we expect that intramolecular hydrogen bonding with the carbonyl group in intermediate IV might enhance the reactivity of the electron-deficient C–C double bond. A computational conformation search of IV (R = CH2CH2Ph; 2a) at a the semi-empirical level suggested that most of the stable conformers involve similar intramolecular hydrogen bonding. The most stable conformation was further optimized by DFT calculations (see the ESI† for details) and the structure is shown in Fig. 4. In addition to hydrogen bonding activation of the carbonyl group by the urea, the NH moiety can interact with the phenolic oxygen, which can also enhance the Lewis acidity of the borane catalyst.15 Meanwhile, the current catalytic system, unfortunately, only produced a racemic product, which can be rationalized by the rather planar structure of IV and large distance between the binaphthyl chirality and the reactive β-carbon.
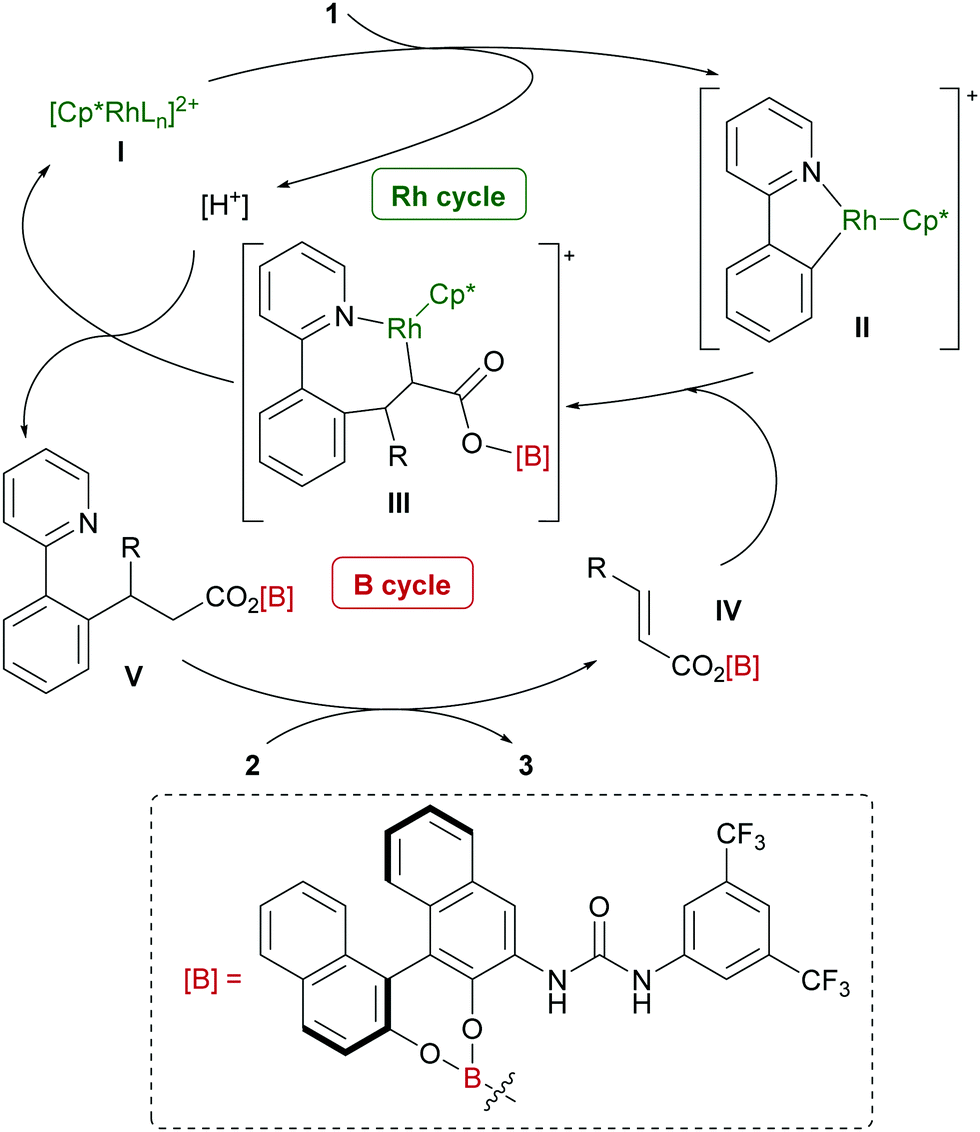 | ||
| Fig. 3 A hypothetical catalytic cycle. | ||
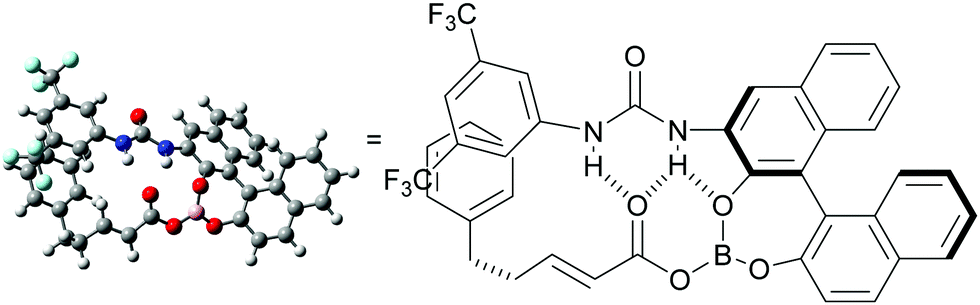 | ||
| Fig. 4 Calculated structure of acyloxyborane intermediate IV. | ||
In summary, we have demonstrated that a combination of Cp*Rh(III)/boron/BINOL-urea catalytic system enables the directed addition of an aryl C–H bond to β-substituted α,β-unsaturated carboxylic acids, which provides β-disubstituted carboxylic acids. The hybrid catalysis was essential to realize this transformation under mild reaction conditions. Although a 2-pyridyl directing group is required and additional tuning of the catalytic system and reaction conditions would be necessary for expanding the substrate scope to more synthetically valuable molecules, the current results may lead to the further development of novel hybrid catalytic systems that facilitate the use of β-substituted α,β-unsaturated carboxylic acids for metal-catalyzed C–H functionalization reactions.
This work was supported in part by JSPS KAKENHI Grant Number JP20H02730 (S. M.), JP20H04794 in Hybrid Catalysis (T. Y).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS; (b) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (c) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (d) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234 CrossRef CAS; (e) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522 CrossRef CAS; (f) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods, 2021, 1, 43 Search PubMed.
- (a) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS; (b) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS; (c) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS; (d) T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245 CrossRef CAS; (e) J. Park and S. Chang, Chem. – Asian J., 2018, 13, 1089 CrossRef CAS PubMed; (f) T. Piou and T. Rovis, Acc. Chem. Res., 2018, 51, 170 CrossRef CAS PubMed; (g) Y. Nishii and M. Miura, ACS Catal., 2020, 10, 9747 CrossRef CAS; (h) S. Shaaban, C. Davies and H. Waldmann, Eur. J. Org. Chem., 2020, 6512 CrossRef CAS; (i) T. Yoshino, S. Satake and S. Matsunaga, Chem. – Eur. J., 2020, 26, 7346 CrossRef CAS; (j) J. Mas-Roselló, A. G. Herraiz, B. Audic, A. Laverny and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 13198 CrossRef PubMed.
- (a) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS; (b) X.-Y. Shi, W.-J. Han and C.-J. Li, Chem. Rec., 2016, 16, 1178 CrossRef CAS PubMed; (c) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163 CrossRef CAS; (d) Q. Gu, Z.-J. Wu and S.-L. You, Bull. Chem. Soc. Jpn., 2021, 94, 641 CrossRef CAS.
- For early works, see: (a) L. Yang, C. A. Correia and C.-J. Li, Org. Biomol. Chem., 2011, 9, 7176 RSC; (b) L. Yang, B. Qian and H. Huang, Chem. – Eur. J., 2012, 18, 9511 CrossRef CAS; (c) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS . For other examples, see reviews in ref. 3.
- F. Han, S. Xun, L. Jia, Y. Zhang, L. Zou and X. Hu, Org. Lett., 2019, 21, 5907 CrossRef CAS PubMed.
- (a) R. Yoshimoto, Y. Usuki and T. Satoh, Chem. Lett., 2019, 48, 461 CrossRef CAS; (b) R. Yoshimoto, Y. Usuki and T. Satoh, Chem. – Asian J., 2020, 15, 802 CrossRef CAS PubMed.
- G. Zhang, F. Jia and L. J. Gooßen, Chem. – Eur. J., 2018, 24, 4537 CrossRef CAS.
- For recent reviews on boron-based catalysis, see: (a) E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945 CrossRef; (b) M. S. Taylor, Acc. Chem. Res., 2015, 48, 295–305 CrossRef CAS PubMed; (c) B. Rao and R. Kinjo, Chem. – Asian J., 2018, 13, 1279 CrossRef CAS; (d) D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475 RSC.
- K. Furuta, Y. Miwa, K. Iwanaga and H. Yamamoto, J. Am. Chem. Soc., 1988, 110, 6254s CrossRef PubMed.
- (a) R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876 CrossRef CAS PubMed; (b) H. Zheng and D. G. Hall, Tetrahedron Lett., 2010, 51, 3561 CrossRef CAS; (c) H. Zheng, R. McDonald and D. G. Hall, Chem. – Eur. J., 2010, 16, 5454 CrossRef CAS PubMed.
- (a) T. Azuma, A. Murata, Y. Kobayashi, T. Inokuma and Y. Takemoto, Org. Lett., 2014, 16, 4256 CrossRef CAS; (b) N. Hayama, T. Azuma, Y. Kobayashi and Y. Takemoto, Chem. Pharm. Bull., 2016, 64, 704 CrossRef CAS; (c) N. Hayama, R. Kuramoto, T. Földes, K. Nishibayashi, Y. Kobayashi, I. Pápai and Y. Takemoto, J. Am. Chem. Soc., 2018, 140, 12216 CrossRef CAS; (d) K. Michigami, H. Murakami, T. Nakamura, N. Hayama and Y. Takemoto, Org. Biomol. Chem., 2019, 17, 2331 RSC; (e) N. Hayama, Y. Kobayashi, E. Sekimoto, A. Miyazaki, K. Inamoto, T. Kimachi and Y. Takemoto, Chem. Sci., 2020, 11, 5572 RSC; (f) H. Murakami, A. Yamada, K. Michigami and Y. Takemoto, Asian J. Org. Chem., 2021, 10, 1097 CrossRef CAS; (g) N. Hayama, Y. Kobayashi and Y. Takemoto, Tetrahedron, 2021, 89, 132089 CrossRef CAS.
- T. Horibe, T. Hazeyama, Y. Nakata, K. Takeda and K. Ishihara, Angew. Chem., Int. Ed., 2020, 59, 17256 CrossRef CAS.
- Boron-based catalysts can activate carboxylic acids for amide bond formation. For selected examples, see: (a) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196 CrossRef CAS PubMed; (b) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124 RSC; (c) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386 CrossRef CAS PubMed; (d) H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571 CrossRef CAS.
- For activation of carboxylic acids as nucleophiles using boron catalysts, see: (a) D. Lee, S. G. Newman and M. S. Taylor, Org. Lett., 2009, 11, 5486 CrossRef CAS; (b) Y. Morita, T. Yamamoto, H. Nagai, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 7075 CrossRef CAS; (c) H. Nagai, Y. Morita, Y. Shimizu and M. Kanai, Org. Lett., 2016, 18, 2276 CrossRef CAS; (d) K. Ishizawa, H. Nagai, Y. Shimizu and M. Kanai, Chem. Pharm. Bull., 2018, 66, 231 CrossRef CAS PubMed; (e) T. Morisawa, M. Sawamura and Y. Shimizu, Org. Lett., 2019, 21, 7466 CrossRef CAS PubMed; (f) K. Sun, M. Ueno, K. Imaeda, K. Ueno, M. Sawamura and Y. Shimizu, ACS Catal., 2021, 11, 9722 CrossRef CAS; (g) T. Fujita, M. Yamane, W. M. C. Sameera, H. Mitsunuma and M. Kanai, Angew. Chem., Int. Ed., 2021, 60, 24598 CrossRef CAS.
- (a) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS; (b) K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 1561 CrossRef CAS; (c) A. K. Gupta, X. Yin, M. Mukherjee, A. A. Desai, A. Mohammadlou, K. Jurewicz and W. D. Wulff, Angew. Chem., Int. Ed., 2019, 131, 3399 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc05956d |
| This journal is © The Royal Society of Chemistry 2022 |