
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMutant polymerases capable of 2′ fluoro-modified nucleic acid synthesis and amplification with improved accuracy†
Trevor A.
Christensen
,
Kristi Y.
Lee
,
Simone Z. P.
Gottlieb
,
Mikayla B.
Carrier
and
Aaron M.
Leconte
 *
*
W. M. Keck Science Department of Claremont McKenna, Pitzer, and Scripps Colleges, Claremont, CA, USA. E-mail: aleconte@kecksci.claremont.edu
First published on 17th June 2022
Abstract
Nonnatural nucleic acids (xeno nucleic acids, XNA) can possess several useful properties such as expanded reactivity and nuclease resistance, which can enhance the utility of DNA as a biotechnological tool. Native DNA polymerases are unable to synthesize XNA, so, in recent years mutant XNA polymerases have been engineered with sufficient activity for use in processes such as PCR. While substantial improvements have been made, accuracy still needs to be increased by orders of magnitude to approach natural error rates and make XNA polymerases useful for applications that require high fidelity. Here, we systematically evaluate leading Taq DNA polymerase mutants for their fidelity during synthesis of 2′F XNA. To further improve their accuracy, we add mutations that have been shown to increase the fidelity of wild-type Taq polymerases, to some of the best current XNA polymerases (SFM4–3, SFM4–6, and SFP1). The resulting polymerases show significant improvements in synthesis accuracy. In addition to generating more accurate XNA polymerases, this study also informs future polymerase engineering efforts by demonstrating that mutations that improve the accuracy of DNA synthesis may also have utility in improving the accuracy of XNA synthesis.
Introduction
DNA is an important tool because of its biochemical properties, ubiquity in nature, and easy amplification without information loss. These basic properties enable DNA and RNA to serve as aptamers,1 to be used to write information in living cells,2 to serve as barcodes during drug discovery,3 and many more emerging applications. These methods are all made possible by the use of DNA polymerases, which can amplify DNA with remarkable efficiency and fidelity. However, DNA is limited by the fact that it is a natural biomolecule, making it susceptible to nucleases and other native DNA-modifying enzymes.4To overcome these limitations, researchers have sought to create genetic systems which could encode information, but which possess novel or improved properties. These new forms of DNA range from nucleic acid analogs with highly structurally divergent sugars5 to more subtle modifications, such as 2′-fluoro 2′-deoxyribonucleic acids (2′F XNA). In particular, nucleic acids with modified 2′ positions (2′ XNA) substantially increase resistance to nucleases while preserving or even expanding beneficial structural and chemical properties.6 However, natural DNA polymerases cannot synthesize 2′ XNA, requiring the engineering of mutant XNA polymerases capable of enzymatic synthesis.
Recent work has discovered a number of XNA polymerases capable of synthesizing a range of chemically diverse nucleic acids.7 A majority of these polymerases are derived from B-family DNA polymerases such as Tgo; mutants such as Tgo:TGK are capable of synthesizing up to 74 bases of supF tRNA in under 30 seconds, with measured error rates across various substrates ranging from 8 × 10−3 to 2 × 10−4.8 Other mutations found to enable XNA synthesis in Tgo have been found to similarly improve homologous B-family polymerases, showing the broad utility of these findings by enabling RNA and TNA synthesis in 9 N, Tgo, Deep Vent and KOD.9 However, the accuracy of these polymerases is limited, generally being several orders of magnitude less accurate than their wild-type DNA polymerase counterparts.
While a number of mutant XNA polymerases derived from B-family polymerases have been found to be capable of synthesizing XNA, A-family DNA polymerases, such as Taq DNA polymerase I (Taq), have also been engineered to synthesize XNA and have shown noteworthy promise. Taq is particularly interesting due to the rich body of structural10 and biochemical11 literature pertaining to it and its mutants, as well as its extensive biotechnological applications.12 While Taq mutants initially were only capable of short XNA synthesis,13 subsequent research identified additional mutations that improve the ability of Taq to synthesize XNA with dramatically improved efficiency.14 Furthermore, these syntheses can occur in the absence of manganese and superstoichiometric enzyme concentrations, which resembles cellular conditions and results in generally improved accuracy. Mutant SFM4–3 and SFM4–6 have been used successfully in synthesis of XNA, PCR amplification of partially substituted XNA, and linear amplification via polymerase chain transcription.14b,15 SFM4–3 in particular has been used in several studies to create aptamers.15a,16 While fewer applications have been demonstrated using SFP1, it has been shown to be more accurate than SFM4–617 and capable of synthesis of 2′F XNA in less than two hours.14a
Collectively, both A family and B family XNA polymerases remain inaccurate relative to natural DNA polymerases, precluding their use in applications involving the accurate synthesis of specific XNA sequences. SFM4–6 and SFP1 have been recently reported to possess error rates of 19.1 × 10−3 and 5.3 × 10−3 errors per base pair, respectively, when synthesizing 2′F XNA.17 By contrast, the measured fidelity of wild-type Taq polymerase is less than 1.5 × 10−5 errors per base pair, with modified variants of Taq designed for improved accuracy in PCR.18 To date, the overwhelming majority of applications of XNA has been to develop aptamers; however, relatively little progress has been made on applying XNA to other high-value technologies such as in vitro3,19 or in vivo2 information storage and recovery. In the long term, one can imagine orthogonal genetic systems akin to those developed for proteins and genetic circuits;20 however, significant improvements in the enzymatic synthesis of XNA must be realized before these technologies can incorporate XNA. The application of XNA to these emerging technologies has been slowed by the inaccuracy of current XNA polymerases.
To date, there are few developed approaches to engineering polymerases with improved error rates. There are numerous noteworthy examples of selections that require maintenance of a minimal level of fidelity,21 but these do not directly select for improvements in the accuracy of synthesis. For XNA synthesis, only one report of directed evolution has demonstrated indirect selection for an improvement in fidelity; in this clever example, the authors selected for enzymes which could synthesize threose nucleic acids in the absence of manganese, which led to one of the most accurate XNA polymerases identified to date.22 However, this approach is limited to enzymes that require manganese, and cannot be further applied to this enzyme. To date, there are no examples, to our knowledge, of direct selection for an improvement in the accuracy of XNA polymerases. Further, the determinants of accuracy in XNA synthesis are largely unstudied and not understood. Thus, there is a need for new approaches to engineer XNA polymerases with improved accuracy.
In contrast to XNA, a number of studies have evolved improved fidelity of natural DNA synthesis.23 Further, the mechanism of natural DNA synthesis using Taq DNA polymerase and homologous enzymes is well studied.24 Collectively, these studies have yielded a wealth of knowledge about Taq, including multiple mutations that are known to improve the accuracy of natural synthesis by Taq. While little is known about the determinants of accurate XNA synthesis, we wondered whether mutations that were previously shown to improve the accuracy of natural DNA synthesis could improve the accuracy of XNA synthesis. Here, we have tested this hypothesis by (i) evaluating the accuracy of known Taq 2′F XNA polymerases and then (ii) systematically adding mutations that improve the accuracy of natural synthesis to current XNA polymerases and evaluating the accuracy of 2′F XNA synthesis. These rationally designed mutants show improved accuracy, which should facilitate applications that require high accuracy; we show that even a small change in accuracy can lead to large improvements in the accuracy of XNA amplified via PCR. Furthermore, this is the first study to show that the determinants of accurate XNA synthesis may mirror the determinants of natural synthesis, enabling the use of prior biochemical knowledge of DNA synthesis accuracy to be leveraged to rapidly improve XNA polymerase accuracy.
Results and discussion
Accuracy of 2′F XNA synthesis by Taq XNA polymerases
Our goal in this work was to begin to develop more accurate 2′F XNA synthesis. Importantly, while 2′F modifications are less structurally divergent from natural nucleic acids than other sugar modifications, they still impart a number of beneficial properties, which is why 2′ fluoro modifications have been used to enhance therapeutic oligonucleotides.25 Further, there is evidence that fluorine modification may provide structural benefits to aptamers.15a However, to date, 2′F modifications are typically used in nucleic acids that are composed of a mixture of natural nucleotides and modified nucleotides, largely due to a lack of technologies that would allow for synthesis of fully substituted 2′F XNA. Thus, there is a need to develop tools for synthesis of fully modified 2′F XNA.While we had previously characterized SFM4–6 and SFP1 for their respective accuracies when performing 2′F XNA synthesis,17 we had also previously described that several other XNA polymerases (SFM4–3, SFM4–9, SFP4, SFP7; see Table S1, ESI† for genotypes) synthesize 2′F XNA under mild conditions as well.14a Thus, to understand the accuracies of all of these enzymes prior to engineering, we subjected all six of these enzymes to a previously described XNA polymerase fidelity assay.17
Briefly, in this assay, 2′F XNA is synthesized by the mutant enzyme, the natural DNA template and primer are removed via nuclease digestion, and, following column purification, the XNA is reverse-transcribed and amplified in a one-pot reaction by Q5 DNA polymerase. The DNA products are then submitted for next-generation DNA sequencing to quantitatively assess accuracy. A template control, which omits the XNA synthesis and reverse transcription, allows us to quantify the collective error rate of chemical synthesis of the DNA template, amplification of DNA, and next-generation DNA sequencing.
We characterized six XNA polymerases: SFP1, SFP4, SFP7, SFM4–3, SFM4–6, and SFM4–9 (Fig. 1A); all of these enzymes had been previously shown to synthesize 2′F XNA, as well as several other XNA.14a Their accuracies were measured in errors per thousand base pairs synthesized (eptbp). Among these enzymes, P1 and SFM4–3 have the lowest error rates with 5.6 and 6.9 eptbp, respectively. If accounting for errors in chemical synthesis, amplification, and sequencing using the template control, these enzymes possess error rates of approximately 2.4 eptbp and 3.7 eptbp. These values are similar to those in the most accurate systems described to date using B-family mutant polymerases.26 Considering that the enzymes described here use commercially available substrates, require short synthesis times, and the XNA can be converted back into DNA using a one-pot reaction using commercial reagents,17 these are attractive systems for applications where accuracy and efficient synthesis are important.
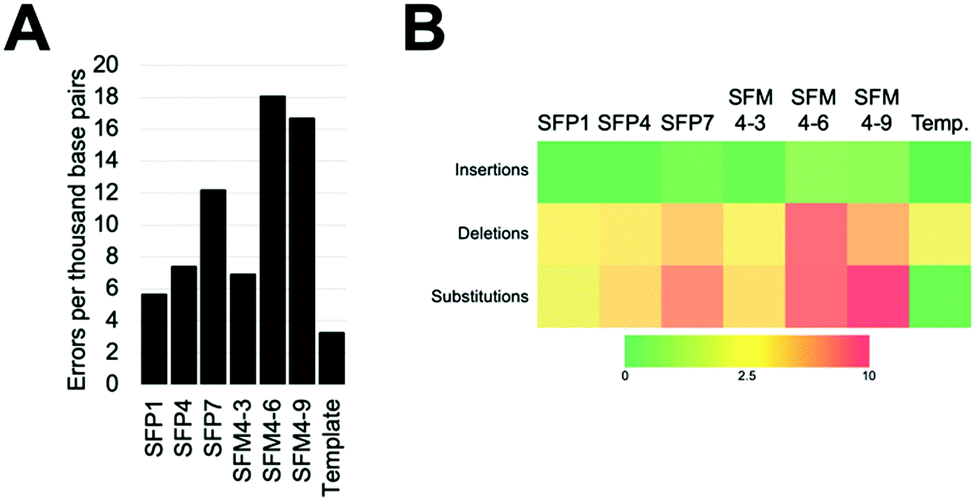 | ||
| Fig. 1 Errors created during XNA synthesis by previously discovered mutant Taq XNA polymerases. (A) Overall observed errors by polymerases. (B) Error types created by each enzyme. Template is a negative control in the absence of XNA polymerase. | ||
The most error prone mutants were SFM4–6 and SFM4–9, which create 16.7 and 18.0 eptbp, respectively. This is particularly noteworthy since SFM4–3, which is among the most accurate enzymes, and SFM4–6, which is among the least accurate enzymes, are the two mutant Taq XNA polymerases that have been used in applications most often; they have been shown to have utility in creating XNA for aptamer selections,15a,16 XNA to be used in hydrogel creation,27 and linear amplification of RNA through polymerase chain transcription (PCT).15b In spite of their usage in multiple applications, data on the accuracy of their respective XNA syntheses has been limited and has not been done comparatively to date. Our study suggests that SFM4–3 creates 2- to 3-fold fewer errors than either SFM4–6 or SFM4–9. These studies imply that, for applications where accuracy is important, or for any application involving amplification (where errors would quickly compound over multiple cycles), SFM4–3 should be chosen over SFM4–6 when possible.
To understand the types of errors these six enzymes created, we observed the number of insertions, deletions, and substitution errors each enzyme created (Fig. 1B). For all of the enzymes, substitution errors are the most commonly created errors; all enzymes possessed substitution error rates at least 2 eptbp above the template control. The prevalence of substitutions stands in contrast to the template control, which has deletions as the most abundant error type, which likely arise from chemical synthesis. The most error-prone polymerases, SFM4–6 and SFM4–9, possess higher rates of error creation relative to the other polymerases for all types of errors (insertions, deletions, substitutions). The most accurate enzymes (SFP1, SFM4–3, and SFP4) possess rates of insertions and deletions that are similar to those in the template control, showing a less than 2-fold increase in error rate for these types of errors relative to the template control. Only substitutions appear at a substantially elevated rate for these enzymes. Thus, for Taq mutant XNA polymerases, substitutions appear to be elevated for all of the enzymes, while increased insertions and deletions are observed largely in the least accurate enzymes.
To assess what types of substitution errors are most often created, we calculated the frequency of all twelve substitution errors (Table S2, ESI†). Interestingly, for all six enzymes, the most frequent substitution error for each enzyme is misincorporation of 2′F TTP against template dG. This error occurs at high rates ranging from 6.1 to 24 eptbp. Importantly, synthesis of the 2′F-T:dG mispair accounts for nearly half of substitution errors created by these enzymes (ranging from 40–59%), and constitutes a higher percentage for the more accurate enzymes. Presumably, this error occurs due to a wobble pair formed by guanosine and thymidine. Surprisingly, in contrast, the same wobble pair in the reverse context (2′F GTP misinsertion against dT) accounts for a much smaller fraction of total substitution errors; these errors compromise 7% to 18% of all substitution errors by the polymerases and occur at much lower rates ranging from 1.1 to 5.4 eptbp. What causes this asymmetry is not clear; prior data collected on reverse transcription and amplification of synthetic 2′F XNA17 suggests that this is most likely occurring during XNA synthesis.
To understand the mutational origin of these enzyme properties, we can compare the error rates, error spectra, and known amino acid mutations in each enzyme. Several mutations are shared between all six of these enzymes, which offers insights into how the error rate of XNA synthesis may be elevated above natural synthesis for these enzymes. All of the mutants share two mutations (I614E, E615G) which originate from SFM19, an enzyme which was found to synthesize short stretches of 2′ OMe XNA.13b Notably, steady-state kinetics of SFM19 did not show an elevated error rate, and so it seems unlikely that these mutations, alone, would lead to the elevated error rate of all of these enzymes. All of the enzymes also possess a mutation at E742, which sits in the vicinity of the template strand; it is possible that mutations at this position elevate the error rate overall, and further work should directly address this question. There is also the possibility that no individual mutation alone can account for the elevated error rate, and that general active site widening and/or promiscuity may cause the increase. If the latter contributes, it is likely that accuracy may be improved through systematic removal of mutations to create a minimal mutant and/or backcrossing with SFM19.
To understand the mutational origins of the variability in fidelity, we can observe the genetic differences between these enzymes. Most intriguingly, SFM4–3, which is among the more accurate enzymes, and SFM4–6 and SFM4–9, which are among the least accurate enzymes, are all identified from the same selection experiment, and they have multiple common mutations. One possibility is that SFM4–3 possesses a gain of function mutation that improves accuracy; the only unique mutation on SFM4–3 is N583S, which neighbors motif 2 in the palm domain (Fig. S1, ESI†). However, considering that SFP1 and SFP4 are similar in fidelity to SFM4–3, while not possessing mutations at N583, the more likely possibility is that SFM4–6 and SFM4–9 possess mutations that disrupt fidelity; the only mutated amino acid position that both SFM4–6 and SFM4–9 possess, but SFM4–3 does not possess, is L657M, which is adjacent to the highly conserved O-Helix (Fig. S1, ESI†), which is well known to impact fidelity.23a,d Interestingly, both 583 and 657 lie in close proximity to the n-2 position of the template strand (Fig. S1, ESI†), suggesting that these template contacts may be important for fidelity in XNA synthesis; in particular, considering the relative frequency that error-prone mutants possess insertions and deletions, it seems likely that these amino acids may play a role in this aspect of fidelity in particular.
Design and characterization of XNA polymerases with improved accuracy
Our comparative study showed that current Taq XNA polymerases possess a range of fidelities when synthesizing 2′F XNA. However, we were interested in evaluating whether we could improve the accuracy of these polymerases by leveraging mutations shown to improve natural DNA synthesis, which have an unknown impact on XNA synthesis. Thus, we identified two sets of mutations previously shown to improve the accuracy of natural synthesis. One set of three mutations (“ETL”:A661E, I665T, F667L), is located on the O-helix of the Taq polymerase and was previously shown to improve the accuracy of natural DNA synthesis threefold through increased binding of correct NTPs relative to mispairs.23a,28 We also sought to add a set of mutations in a different region of the protein; a second set of mutations (“LVL”:Q879L, H881L) is found in Motif C, which contacts the primer terminus. These mutations were originally found in a screen for increased mismatch discrimination that was used to improve the efficiency of genotyping.29To test our hypothesis, we first added the “LVL” and “ETL” mutations to SFM4–6, which is the least accurate XNA polymerase in our surveyed mutants. After cloning, expressing, and purifying the two new mutant proteins (SFM4–6:LVL and SFM4–6:ETL), we evaluated the enzyme's fidelity using the same assay used above. Gratifyingly, the addition of both the LVL and ETL mutations lowered the error rate of SFM4–6 mediated synthesis (Fig. 2A). Whereas SFM4–6 had an error rate of 18.3 eptbp, SFM4–6:LVL and SFM4–6:ETL had error rates of 11.4 eptbp and 11.3 eptbp respectively. When accounting for the template control, which contributes approximately 4 eptbp, the error rate declined approximately 2-fold upon addition of the LVL and ETL mutations.
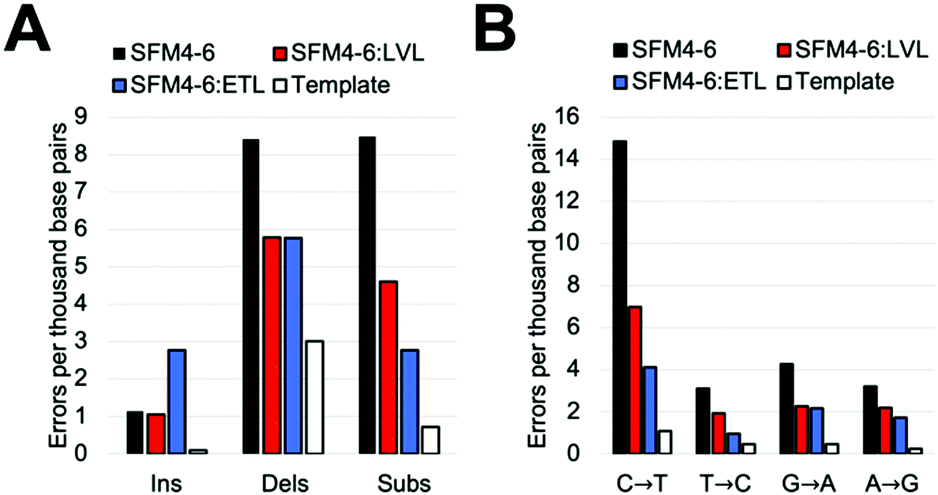 | ||
| Fig. 2 Errors created by SFM4-6, SFM4-6:LVL, and SFM4-6:ETL. (A) Error types created by each enzyme. (B) Transitions created by each enzyme. Template is a negative control in the absence of XNA polymerase. | ||
To understand what type of errors caused this decrease, we quantified the rate of insertions, deletions, and substitution errors. This decrease in total error rate was primarily caused by a reduction in substitutions (Fig. 2B); while SFM4–6 makes 8.5 substitutions per base pair, SFM4–6:LVL and SFM4–6:ETL make 4.6 and 2.8 respectively. Analysis of specific substitutions showed that every type of substitution decreased relative to SFM4–6, but they were all still higher than the template control (Table S3, ESI†). Importantly, adding LVL and ETL both reduced transition errors, the most common type of substitution error, by at least twofold on average. However, ETL had a greater impact on the C → T and T → C transition rates than LVL did, reducing them by 4.6-fold on average versus just 2.1-fold on average for LVL compared to template control.
The change in deletion error rates was remarkably consistent between SFM4–6:LVL and SFM4–6:ETL, both being a twofold reduction relative to SFM4–6 measured against the template control. Despite this improvement, the deletion error rates of all three 4–6 polymerases remain substantially higher than the template control. Surprisingly, while SFM4–6:ETL had fewer substitutions than 4–6:LVL, it had significantly more insertions than both 4–6 and 4–6:LVL (2.77 eptbp) (Fig. 2B).
To measure the fidelity of these XNA polymerases in a different sequence context, we constructed a 150 bp template with a different sequence. This new template was then used in the previously described fidelity assay to assess the fidelity of SFM4–6, SFM4–6:LVL, and SFM4–6:ETL. The mutants had very similar rates to those found in the initial assay; SFM4–6:LVL and SFM4–6:ETL possess a significantly lower error rate (13.8 and 12.5 eptbp, respectively) than the parent enzyme SFM4–6 (19.2 eptbp) (Table S4, ESI†). Generally speaking the error spectrum was also fairly similar (Table S4, ESI†). These data demonstrate that the improved accuracy obtained through addition of either the ETL or LVL mutations improve accuracy independent of the sequence context.
In their initial discovery, the LVL mutations were selected for improved discrimination against mismatched primers, which was shown to be the mechanistic source of the improved fidelity in natural synthesis.15 To understand whether the effect of LVL on SFM4–6 was similar to its effects on wild-type Taq, we evaluated whether XNA synthesis on a mismatched primer is also slowed. To evaluate this, we chemically synthesized DNA primers containing either a mismatch of T:G or G:G at the primer terminus and evaluated SFM4–6 and SFM4–6:LVL for their ability to synthesize from the matched and mismatched primer termini (Fig. 3 and Fig. S2, ESI†). SFM4–6:LVL extended the correctly paired terminus at a similar efficiency (85% extended) as its parent SFM4–6 (90%). However, SFM4–6:LVL extended far less of both the G:T and G:G mismatched primers than SFM4–6. This suggests that adding the LVL mutations to SFM4–6 similarly diminishes the rate of mismatch extension, showing that the mechanistic cause of improved fidelity in SFM4–6 may be similar to that in the native enzyme.
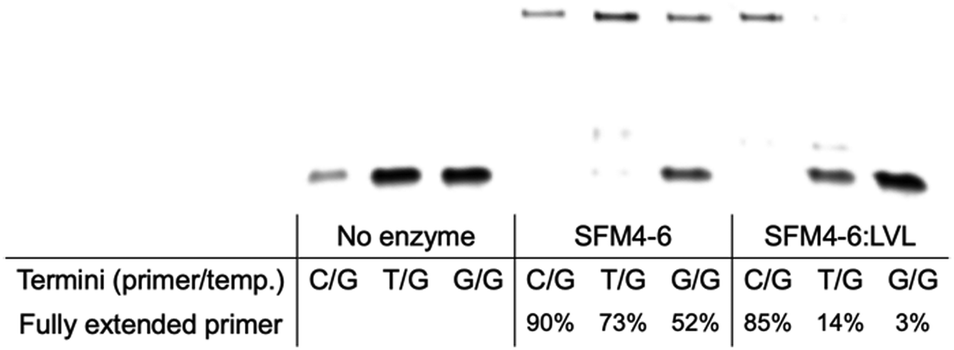 | ||
| Fig. 3 Matched and mismatched primer-termini extension by SFM4-6 and SFM4-6:LVL. Fully extended primer is measured by quantifying the band fluorescence intensity of the full-length band relative to the total fluorescence intensity in the lane. | ||
While we were encouraged to see that a diminished ability to extend incorrect primer-termini was a possible source of improved fidelity of SFM4–6:LVL, we wondered if this could potentially significantly slow enzymatic synthesis through creation of errors followed by pausing. Further, while the major product of all of the XNA syntheses was the full length product, the SFM4–6:LVL mutant did appear to display some mild increased pausing (unpublished results). To assess whether the yield of full-length product for each enzyme was quantitatively impacted by the improved accuracy, we performed a quantitative reverse transcription/amplification, as described previously.17 Briefly, in these experiments, enzymatically synthesized XNA is purified and then subjected to a qPCR. We saw no significant difference (ΔCq < 1) in the cycle threshold values between SFM4–6 and SFM4–6:ETL, but we did observe a ΔCq = 1.6 between SFM4–6 and SFM4–6:LVL (Fig. S3, ESI†). These data indicate that, under these synthesis conditions, SFM4–6:ETL is similar in efficiency to SFM4–6, but SFM4–6:LVL is slightly less efficient, which may be caused by pausing due to less extension of mismatches. However, these differences are small, and are unlikely to directly impact most applications.
We were encouraged by the improved accuracy when adding these mutations to SFM4–6, and we wondered whether a similar improvement would be attained on the two most accurate XNA polymerases that we had surveyed, SFM4–3 and SFP1. To address this, we created four mutant enzymes, each with either LVL or ETL added to either SFM4–3 or SFP1. We then assessed the accuracy of these enzymes using our fidelity assay.
Once again, adding the LVL and ETL mutations to SFM4–3 and SFP1 decreased the error rate of synthesis; in both cases, the decrease was approximately 2-fold when removing contributions from the template control (Fig. 4 and Table S5, ESI†). These magnitudes are similar to the changes from adding LVL and ETL to the SFM4–6 XNA polymerase. The resultant polymerases create errors between 5.5 and 5.6 eptbp, which is less than 2-fold higher than a parallel run template control. If accounting for the template control, the error rates are 2.3 eptbp and 2.4 eptbp, respectively. These results show that LVL and ETL can improve the error rate of synthesis for multiple XNA polymerases and, importantly, these mutations can be leveraged to create the most accurate Taq XNA polymerases identified to date.
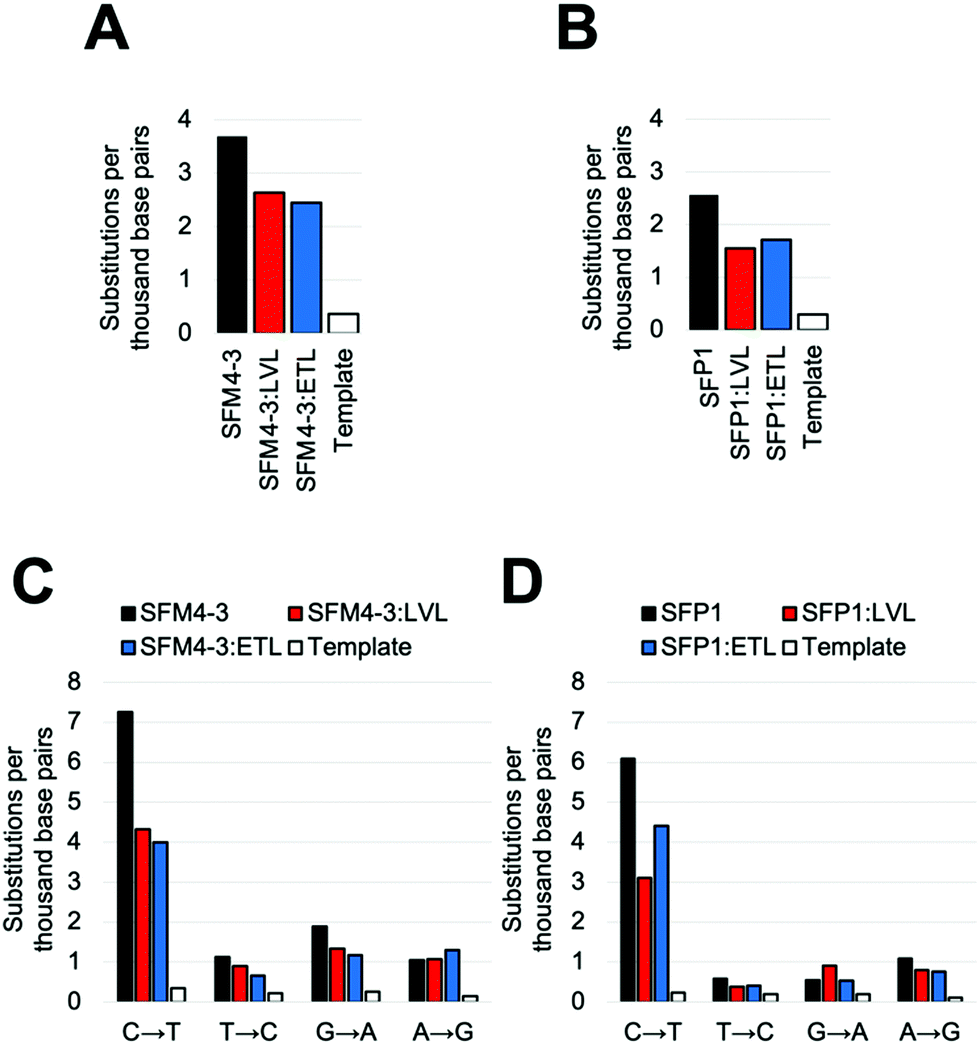 | ||
| Fig. 4 Substitution error rates during XNA synthesis by (A) SFM4-3, SFM4-3:LVL and SFM4-3:ETL and (B) SFP1, SFP1:LVL and SFP1:ETL. Transition error rate during XNA synthesis of (C) SFM4-3, SFM4-3:LVL and SFM4-3:ETL and (D) SFP1, SFP1:LVL and SFP1:ETL. | ||
Notably, for both SFP1 and SFM4–3, addition of either the LVL or the ETL mutations decreased the frequency of nearly all substitutions, insertions and deletions (Fig. 4C, D and Table S5, ESI†). Importantly, the largest decrease in error rate upon addition of ETL and LVL was for the C → T substitution error; while SFM4–3 and SFP1 make a C → T substitution error with a frequency of 7.3 and 6.1 eptbp, respectively, adding ETL or LVL reduces the frequency of this error to 4.4 eptbp or below. For the most accurate enzymes, SFP1:LVL and SFP1:ETL, the only substitution error that is observed at a frequency greater than 1 eptbp is the C → T error; all other substitutions occur at a rate below 1 eptbp. These enzymes represent a substantial improvement in accuracy for 2′F XNA synthesis.
Collectively, we have shown here that adding mutations shown to increase the accuracy of natural DNA synthesis can also improve the accuracy of 2′F XNA synthesis. This strategy was successful when adding mutations within two different conserved regions of the protein; mutations on the O-helix, which largely interacts with incoming dNTPs, and Motif C, which interacts with the primer terminus, both were able to improve the accuracy of XNA synthesis. This is noteworthy because it means that, while little is known about the determinants of XNA fidelity, scientists can leverage the vast body of knowledge on the fidelity of natural synthesis by Taq to engineer improved variants. Future studies can, and should, leverage additional antimutator mutations in other regions of the protein, such as the M-helix,23c and evaluate combinations of mutations, including LVL and ETL, to potentially reveal synergistic additions across polymerase structural elements. These may be important steps to creating XNA polymerases with high fidelity.
While this study makes it clear that addition of mutations that improve the accuracy of natural synthesis can improve the accuracy of 2′F XNA synthesis, it is unclear whether the synthesis of other types of XNA, especially those which are structurally more divergent from natural DNA than 2′F XNA, are also improved. Considering that most XNA polymerases possess an ability to use a number of altered sugar substrates,13c,26a it will be interesting to see whether these mutations that improve accuracy do so equally across different types of XNA. Furthermore, we expect these results to provide inspiration for future directed evolution experiments seeking to improve accuracy in XNA polymerases by providing valuable information on ideal amino acid positions to mutate in enzyme libraries.
PCR of partially substituted XNA with improved accuracy
Considering the improved accuracy during XNA synthesis, we wondered if the improvements would also benefit PCR performed by SFM4–3. While amplification of fully modified XNA has yet to be achieved, SFM4–3 is one of a very small number of enzymes which have been previously shown to incorporate a single modified nucleotide into DNA during a PCR.27 The resulting partially-modified nucleic acids possess beneficial properties such as nuclease resistance and expanded reactivity without the difficulties of XNA dependent XNA synthesis. To measure the effects of ETL and LVL on accuracy in this context, we performed XNA PCR containing three natural nucleoside triphosphates (dATP, dCTP, and dTTP) and one modified nucleoside triphosphate (2′F GTP) with SFM4–3, SFM4–3:LVL, and SFM4–3:ETL. To demonstrate that the new enzymes can improve the accuracy of a PCR with modified nucleotides that has previously been shown to be robust but inaccurate, we chose to reproduce a past experiment which has been performed multiple times.27When performing PCR, SFM4–3:ETL and SFM4–3:LVL are significantly more accurate than SFM4–3. Although SFM4–3 is among the most accurate XNA polymerases characterized, and has been used in PCR in multiple studies, only 35% of the DNA sequences resulting from PCR match the original DNA sequence (Fig. 5A); a control measuring the PCR amplification of the synthesized DNA template showed 96% of sequences matched the original DNA sequence. Gratifyingly, 68% of SFM4–3:LVL generated sequences and 66% of SFM4–3:ETL generated sequences matched the original sequence (Fig. 5A); this is a substantial improvement in the accuracy of amplification. Thus, especially when compounded over multiple rounds of PCR, small differences in fidelity can have large effects on the products of PCR.
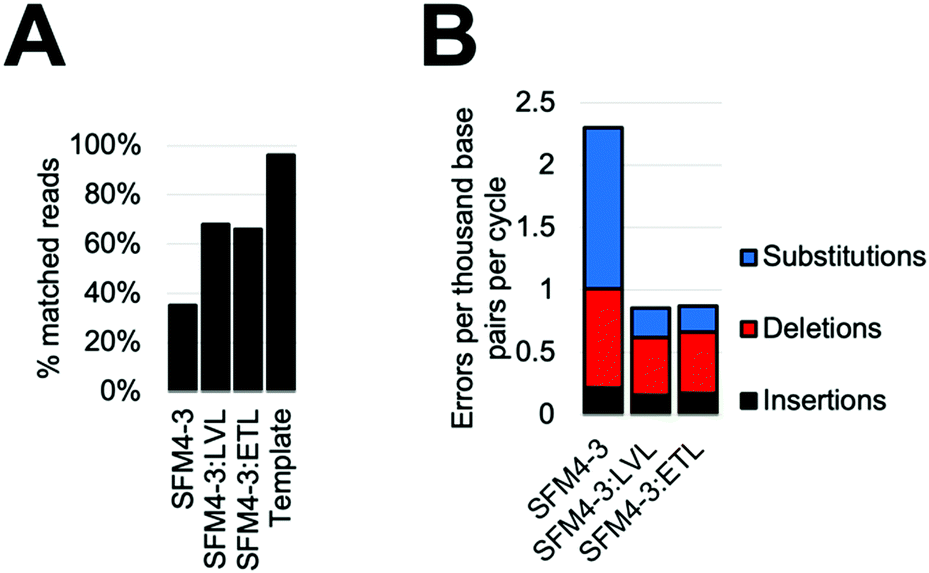 | ||
| Fig. 5 PCR with dATP, dCTP, dTTP, and 2′F dGTP by SFM4-3, SFM4-3:LVL, and SFM4-3:ETL. (A) Percentage of product sequences matching the original sequence. (B) Error types created by each enzyme; values are quantified as errors per cycle. | ||
As expected, considering that only one nucleotide is substituted, the overall error rates of the SFM4–3 mutants per cycle of the PCR were lower than those measured in the prior fidelity assay (Fig. 5B). These changes were likely caused by using a nucleotide mix containing only one 2′F-NTP; these data imply that DNA synthesis is more accurate than XNA synthesis for these enzymes. The error rates of SFM4–3:ETL and SFM4–3:LVL are 2.6-fold lower than the parent enzyme SFM4–3. The XNA PCR resulted in a relatively high proportion of insertions and deletions relative to substitution errors when compared to the prior fidelity assay (Table S6, ESI†), likely because of the altered reaction conditions. As was observed with XNA synthesis, substitutions are the most common error, and SFM4–3:ETL and SFM4–3:LVL show improved substitution rates relative to SFM4–3 (Table S6, ESI†). For all three enzymes, the most common substitution is still a C → T transition (Table S6, ESI†); however, from these data, it is not possible to differentiate whether this is from incorrect addition of dTTP against 2′F-G or incorrect addition of dATP against dC. Collectively, these data show that these newly designed XNA polymerases can be used to improve the accuracy of amplification of DNA containing modified nucleotides, and that small improvements in fidelity of XNA synthesis can lead to large differences in sequence retention during PCR. For applications using PCR of partially modified DNA, SFM4–3:LVL and SFM4–3:ETL show significantly improved accuracy due to the compounding nature of errors in PCR; these enzymes should find immediate utility in applications involving PCR.
Conclusions
Here, we have assessed the accuracy of 2′F XNA synthesis by a number of Taq XNA polymerase mutants; we have then improved these enzymes further by adding mutations previously shown to improve natural DNA synthesis fidelity to a XNA polymerase. In doing so, we have created XNA polymerases that are among the most accurate enzymes characterized to date. The effects of adding these mutations are similar across different XNA polymerases, and in many ways resemble their observed effects in wild-type Taq polymerase. These novel enzymes should facilitate applications that require highly accurate XNA polymerases, such as PCR amplification using one or more modified NTPs. While further engineering is needed to reach natural levels of accuracy, these results demonstrate that information from the substantial body of wild-type Taq polymerase literature can be used to improve XNA polymerases derived from Taq, providing a new, and efficient, approach to improving the accuracy of Taq-mediated XNA synthesis.Experimental procedures
XNA synthesis
Reactions were run using the following conditions: 40 nM 5′ IRDye700-labeled primer (AL-K017, see Table S8 (ESI†) for sequence), 80 nM template (AL-K021, see Table S8 (ESI†) for sequence), 50 mM Tris buffer (pH = 8.5, Fisher Scientific), 6.5 mM MgCl2 (Sigma Aldrich), 0.05 mg mL−1 Ac-BSA (Promega), 50 mM KCl (Sigma Aldrich), 100 μM of either 2′F dNTPs (TriLink Biotechnologies) or dNTPs (Fisher Scientific), 20 nM of enzyme. Reactions were incubated at 50 °C for 2 h; 3 μL of each reaction was removed and quenched using two volume equivalents of quenching buffer composed of 95% Formamide (Acros), 12.5 mM EDTA (Sigma Aldrich), trace amounts of Orange G powder (<1 mg, Sigma Aldrich). Assays were visualized on a 10% TGX polyacrylamide gel (Bio-Rad). Gels were imaged using Li-Cor Odyssey CLx and visualized using ImageStudio software (Li-Cor). The remaining 17 μμL of sample was digested (see next section).XNA purification, reverse transcription, and amplification
Turbo DNase was added (final concentration 0.11 U μL−1; Invitrogen) to the remaining reaction sample and incubated at 37 °C on a heat block for 40 minutes. Samples were purified using Qiaquick Nucleotide Removal Kit (Qiagen) according to the manufacturer's protocol. Purified XNA was reverse-transcribed and amplified in a PCR using barcoded primers to allow multiplexing when performing DNA sequencing using NGS (see Table S9 (ESI†) for sequences). Each 50 μL reaction contained 1 μL purified XNA or equivalent (see ESI,† for details on XNA synthesis and purification), 1× Q5 Reaction Buffer (New England Biolabs), 0.5 μM of each primer, 0.4 mM dNTPs (New England Biolabs), 6% DMSO (Fisher Scientific), milliQ purified water, and 0.02 U Q5 DNA polymerase (New England Biolabs). PCR was performed using the following cycling conditions: 98 °C for 30 s, [98 °C for 5 s, 50 °C for 15 s, 72 °C for 15 s] × 3, [98 °C for 5 s, 67 °C for 15 s, 72 °C for 15 s] × 18, 72 °C 5 min, and 4 °C hold on an Arktik Thermal Cycler (ThermoFisher). Products were visualized on 2% agarose gel containing GelRed (Biotium) and a benchtop UV transilluminator. PCR products were purified using DNA Clean and Concentrator-25 kit (Zymo Research), according to manufacturer's protocol for PCR products. DNA concentration was quantified using Qubit 3 Fluorometer and Qubit dsDNA HS Assay Kit (ThermoFisher Scientific). Samples were submitted to Genewiz for Amplicon-EZ sequencing and analyzed using a custom Python script as previously described.17 For quantitative reactions, purified products were amplified as above with addition of 1× SYBR Green (Bio-Rad) using a C-1000 thermal cycler (Bio-Rad); fluorescence was monitored after each round.Mismatch extension assay
Mismatch extension assays were carried out in a similar manner to XNA synthesis assays described above with the following modifications. Primers AL-K017, AL-K038 (T mismatch), or AL-K039 (G mismatch) were annealed with template AL-K021 as stated above (see Table S8 (ESI†) for sequences). As a control, part of the annealed solution did not contain enzyme and was added to another tube containing 2′F NTPs (final reaction concentration 200 μM). Enzyme (either SFM4–6 or LVL:SFM4–6, 5 nM reaction concentration) was added to the annealed solutions. The enzyme-annealed solutions were added to tubes with 2′F NTPs (reaction concentration 200 μM). After mixing, samples were incubated at 50 °C on a heat block for 10 minutes (SFM4–6, LVL:SFM4–6) quenched, incubated, and analyzed as stated above.PCR with modified nucleotides
PCR was performed using 200 μM of one modified nucleotide (2′F-GTP) (Trilink Biotechnologies) and 200 μM of unmodified dTTP, dATP, and dCTP (Thermo Scientific), 400 nM of primer AL-K058-T75P-for and primer AL-K059-T57P-rev (see Table S8 (ESI†) for sequences) and 200 ng template AL-K057-T75 (see Table S8 (ESI†) for sequence), using the respective XNA polymerases (2 μM) in SF buffer (50 mM Tris buffer (pH = 8.5, Fisher Scientific), 6.5 mM MgCl2 (Sigma Aldrich), 0.05 mg mL−1 Ac-BSA (Promega), 50 mM KCl (Sigma Aldrich)). The following cycling conditions were used in an Arktik Thermal Cycler (ThermoFisher Scientific): 94 °C for 2 min, [94 °C for 30 s, 49 °C for 60 s, 50 °C for 30 min] × 26 cycles, 72 °C for 99 min, and 4 °C hold. A portion of the resulting PCR products were run on 2% agarose (Research Products International) gels stained with GelRed (Biotium) to ensure their quality before being purified using a QIAquick Nucleotide Removal Kit (Qiagen).A portion of the products (1 μL) were amplified using unmodified dNTPs (200 μM) (Thermo Scientific) with barcoded NGS primers (400 nM each, see Table S9 (ESI†) for sequences). The synthesized insert (100 ng) was amplified using a different barcoded primer in parallel as a control for template errors. The following cycling conditions were used in an Arktik Thermal Cycler (ThermoFisher Scientific): 98 °C for 90 s, [98 °C for 5 s, 50 °C for 15 s, 72 °C for 20 s] × 3, [98 °C for 5 s, 66 °C for 15 s, 72 °C for 25 s] × 17, 72 °C 10 min, and 4 °C hold. The PCR products were visualized using a 2% agarose gel stained with GelRed (Biotium). Finally, these second PCR products were purified using the DNA Clean & Concentrator-25 PCR purification kit (Zymo Research) and quantified using a Qubit 3 Fluorometer (ThermoFisher Scientific) before being submitted for high-throughput sequencing (GeneWiz) using the Amplicon-EZ protocol.
For further experimental details regarding molecular cloning, enzyme expression and purification, see ESI.†
Author contributions
T. A. C., K. Y. L, S. Z. P. G., and M. B. C. executed the experiments. A. M. L. supervised the project. T. A. C. and A. M. L. wrote the manuscript. All authors contributed to the analysis of results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-1752924 CAREER award to A. M. L.).References
- J. R. Kanwar, K. Roy, N. G. Maremanda, K. Subramanian, R. N. Veedu, R. Bawa and R. K. Kanwar, Curr. Med. Chem., 2015, 22, 2539–2557 CrossRef CAS PubMed.
- T. B. Loveless, J. H. Grotts, M. W. Schechter, E. Forouzmand, C. K. Carlson, B. S. Agahi, G. Liang, M. Ficht, B. Liu, X. Xie and C. C. Liu, Nat. Chem. Biol., 2021, 17, 739–747 CrossRef CAS PubMed.
- M. A. Clark, Curr. Opin. Chem. Biol., 2010, 14, 396–403 CrossRef CAS PubMed.
- A. D. Keefe and S. T. Cload, Curr. Opin. Chem. Biol., 2008, 12, 448–456 CrossRef CAS PubMed.
- J. C. Chaput and P. Herdewijn, Angew. Chem., Int. Ed., 2019, 58, 11570–11572 CrossRef CAS PubMed.
- (a) C. Wilson and A. D. Keefe, Curr. Opin. Chem. Biol., 2006, 10, 607–614 CrossRef CAS PubMed; (b) K. Duffy, S. Arangundy-Franklin and P. Holliger, BMC Biol., 2020, 18, 112 CrossRef PubMed.
- N. Freund, M. Fürst and P. Holliger, Curr. Opin. Biotechnol., 2021, 74, 129–136 CrossRef PubMed.
- (a) C. Cozens, H. Mutschler, G. M. Nelson, G. Houlihan, A. I. Taylor and P. Holliger, Angew. Chem., Int. Ed., 2015, 54, 15570–15573 CrossRef CAS PubMed; (b) C. Cozens, V. B. Pinheiro, A. Vaisman, R. Woodgate and P. Holliger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8067–8072 CrossRef CAS PubMed.
- (a) N. Chim, C. Shi, S. P. Sau, A. Nikoomanzar and J. C. Chaput, Nat. Commun., 2017, 8, 1810 CrossRef PubMed; (b) M. R. Dunn, C. Otto, K. E. Fenton and J. C. Chaput, ACS Chem. Biol., 2016, 11, 1210–1219 CrossRef CAS PubMed.
- S. H. Eom, J. Wang and T. A. Steitz, Nature, 1996, 382, 278–281 CrossRef CAS PubMed.
- S. Ishino and Y. Ishino, Front. Microbiol., 2014, 5, 465 Search PubMed.
- R. Kranaster and A. Marx, ChemBioChem, 2010, 11, 2077–2084 CrossRef CAS PubMed.
- (a) J. L. Ong, D. Loakes, S. Jaroslawski, K. Too and P. Holliger, J. Mol. Biol., 2006, 361, 537–550 CrossRef CAS PubMed; (b) M. Fa, A. Radeghieri, A. A. Henry and F. E. Romesberg, J. Am. Chem. Soc., 2004, 126, 1748–1754 CrossRef CAS PubMed; (c) H. J. Schultz, A. M. Gochi, H. E. Chia, A. L. Ogonowsky, S. Chiang, N. Filipovic, A. G. Weiden, E. E. Hadley, S. E. Gabriel and A. M. Leconte, Biochemistry, 2015, 54, 5999–6008 CrossRef CAS PubMed.
- (a) S. L. Rosenblum, A. G. Weiden, E. L. Lewis, A. L. Ogonowsky, H. E. Chia, S. E. Barrett, M. D. Liu and A. M. Leconte, ChemBioChem, 2017, 18, 816–823 CrossRef CAS PubMed; (b) T. Chen, N. Hongdilokkul, Z. Liu, R. Adhikary, S. S. Tsuen and F. E. Romesberg, Nat. Chem., 2016, 8, 556–562 CrossRef CAS PubMed.
- (a) D. Thirunavukarasu, T. Chen, Z. Liu, N. Hongdilokkul and F. E. Romesberg, J. Am. Chem. Soc., 2017, 139, 2892–2895 CrossRef CAS PubMed; (b) T. Chen and F. E. Romesberg, J. Am. Chem. Soc., 2017, 139, 9949–9954 CrossRef CAS PubMed.
- (a) Z. Liu, T. Chen and F. E. Romesberg, Chem. Sci., 2017, 8, 8179–8182 RSC; (b) Q. Shao, T. Chen, K. Sheng, Z. Liu, Z. Zhang and F. E. Romesberg, J. Am. Chem. Soc., 2020, 142, 2125–2128 CrossRef CAS PubMed.
- A. S. Thompson, S. E. Barrett, A. G. Weiden, A. Venkatesh, M. K. C. Seto, S. Z. P. Gottlieb and A. M. Leconte, Biochemistry, 2020, 59, 2833–2841 CrossRef CAS PubMed.
- K. R. Tindall and T. A. Kunkel, Biochemistry, 1988, 27, 6008–6013 CrossRef CAS PubMed.
- (a) D. Kong, Y. Lei, W. Yeung and R. Hili, Angew. Chem., Int. Ed., 2016, 55, 13164–13168 CrossRef CAS PubMed; (b) Y. K. Sunkari, V. K. Siripuram, T. L. Nguyen and M. Flajolet, Trends Pharmacol. Sci., 2022, 43, 4–15 CrossRef CAS PubMed.
- A. Costello and A. H. Badran, Trends Biotechnol., 2021, 39, 59–71 CrossRef CAS PubMed.
- (a) D. Loakes, J. Gallego, V. B. Pinheiro, E. T. Kool and P. Holliger, J. Am. Chem. Soc., 2009, 131, 14827–14837 CrossRef CAS PubMed; (b) V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S. Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed; (c) N. Ramsay, A. S. Jemth, A. Brown, N. Crampton, P. Dear and P. Holliger, J. Am. Chem. Soc., 2010, 132, 5096–5104 CrossRef CAS PubMed.
- A. C. Larsen, M. R. Dunn, A. Hatch, S. P. Sau, C. Youngbull and J. C. Chaput, Nat. Commun., 2016, 7, 11235 CrossRef CAS PubMed.
- (a) M. Suzuki, S. Yoshida, E. T. Adman, A. Blank and L. A. Loeb, J. Biol. Chem., 2000, 275, 32728–32735 CrossRef CAS PubMed; (b) M. Strerath, C. Gloeckner, D. Liu, A. Schnur and A. Marx, ChemBioChem, 2007, 8, 395–401 CrossRef CAS PubMed; (c) E. Loh, J. Choe and L. A. Loeb, J. Biol. Chem., 2007, 282, 12201–12209 CrossRef CAS PubMed; (d) E. Loh and L. A. Loeb, DNA Repair, 2005, 4, 1390–1398 CrossRef CAS PubMed; (e) G. Raghunathan and A. Marx, Sci. Rep., 2019, 9, 590 CrossRef PubMed.
- M. F. Goodman, S. Creighton, L. B. Bloom and J. Petruska, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 83–126 CrossRef CAS PubMed.
- S. Ni, Z. Zhuo, Y. Pan, Y. Yu, F. Li, J. Liu, L. Wang, X. Wu, D. Li, Y. Wan, L. Zhang, Z. Yang, B. T. Zhang, A. Lu and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 9500–9519 CrossRef CAS PubMed.
- (a) E. Medina, E. J. Yik, P. Herdewijn and J. C. Chaput, ACS Synth. Biol., 2021, 10, 1429–1437 CrossRef CAS PubMed; (b) A. Nikoomanzar, N. Chim, E. J. Yik and J. C. Chaput, Q. Rev. Biophys., 2020, 53, e8 CrossRef CAS PubMed.
- T. Chen and F. E. Romesberg, Angew. Chem., Int. Ed., 2017, 56, 14046–14051 CrossRef CAS PubMed.
- K. Yoshida, A. Tosaka, H. Kamiya, T. Murate, H. Kasai, Y. Nimura, M. Ogawa, S. Yoshida and M. Suzuki, Nucleic Acids Res., 2001, 29, 4206–4214 CrossRef CAS PubMed.
- D. Summerer, N. Z. Rudinger, I. Detmer and A. Marx, Angew. Chem., Int. Ed., 2005, 44, 4712–4715 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00064d |
| This journal is © The Royal Society of Chemistry 2022 |